Moorestown, New Jersey, United States


PUBLISHED23 March 2023
AstraZeneca’s Calquence (acalabrutinib), a next generation, selective Bruton’s tyrosine kinase (BTK) inhibitor, has been conditionally approved in China for the treatment of adult patients with mantle cell lymphoma (MCL) who have received at least one prior therapy. This is the first approved indication for Calquence in China.
The conditional approval by the National Medical Products Administration (NMPA) was based on positive results from two clinical trials, including the ACE-LY-004 global Phase II trial in adults with relapsed or refractory MCL and a Phase I/ II trial in Chinese patients with relapsed or refractory MCL and other B-cell malignancies.1,2 Continued approval for this indication may be contingent upon verification of ongoing randomised controlled confirmatory trials.
MCL is typically an aggressive, rare form of non-Hodgkin lymphoma (NHL) that accounts for between 2-6% of all patients diagnosed with NHL in China. Patients are generally diagnosed around 60 years of age, often at later stages of the disease.3
Jun Zhu, Chief Physician, Department of Lymphatic Oncology, Peking University Cancer Hospital, Beijing, said: “Mantle cell lymphoma progresses rapidly and responds poorly to conventional treatment such as immunochemotherapy. Before the emergence of BTK inhibitors, there were few satisfactory treatment options for patients. The next-generation BTK inhibitor Calquence has higher target selectivity, fewer side effects, and a higher response rate compared to currently available treatments. This approval of Calquence in China can provide a new treatment option which can better benefit patients with this disease.”
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “This approval for Calquence offers people living with mantle cell lymphoma in China an effective and tolerable new treatment option to help control their disease. As the first approval in China for Calquence, it is also an exciting step forward for AstraZeneca in blood cancers, enabling us to help more patients across the globe gain access to innovative treatments.”
Results from the ACE-LY-004 Phase II trial showed at a median follow up of 15.2 months, investigator-assessed overall response rate (ORR) with Calquence was 80.6% (95% confidence interval [CI] 72.6-87.2), with a complete response (CR) achieved in 39.5% of patients with relapsed or refractory MCL (95% CI 30.9-48.7).1 Longer-term follow-up data showed at 38.1 months, patients treated with Calquence remained progression-free for a median of 22 months, with median overall survival (OS) of 59.2 months (95% CI 36.5-NE).4
Additionally, results from a Phase I/II trial conducted in China showed Calquence achieved a 82.4% ORR, with a CR achieved in 35.3% of patients with MCL based on a blinded independent central-review (BICR) analysis (95% CI 65.5-93.2). Calquence reduced the risk of disease progression or death by 51.5% (95% CI 33.3-67.0) at 12 months, with an estimated duration of response (DOR) of 65.5% (95% CI 66.6-93.3). The median DOR was not reached.2
The safety and tolerability of Calquence in these trials was consistent with that observed in previous clinical trials.1,2,4
Calquence is approved for the treatment of chronic lymphocytic leukaemia (CLL) and small lymphocytic lymphoma (SLL) in the US and Japan and is approved for the treatment of CLL in the EU and in several other countries worldwide in the treatment-naïve and relapsed or refractory settings. Calquence is also approved in the US and several other countries for the treatment of adult patients with MCL who have received at least one prior therapy. Calquence is not currently approved for the treatment of MCL in Japan or the EU.
Notes
Mantle cell lymphoma (MCL)
MCL is a rare B-cell malignancy subtype and is typically clinically aggressive with a poor prognosis.3 The epidemiology of MCL in Asia is not well documented, with few published datasets describing the incidence and outcomes.3 MCL accounts for between 2-6% of all patients diagnosed with NHL China.3 Patients are generally diagnosed around 60 years of age and often at later stages of disease (Stage III or IV).3
ACE-LY-004
ACE-LY-004 is an open-label, single-arm Phase II clinical trial evaluating Calquence in adult patients with relapsed or refractory MCL.5 Patients in the trial were adults with MCL and an Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 2 or lower who had relapsed or were refractory to 1-5 prior therapies, had no prior BTK/BCL-2 inhibitor exposure, and did not require warfarin/vitamin K antagonists. Patients received oral Calquence 100mg twice-daily until progressive disease or toxicity. ORR (investigator-assessed partial response or better per Lugano classification), DOR, progression-free survival (PFS), OS and safety were assessed.1
Phase I/II trial in Chinese patients
The Phase I/II trial is an open-label, multicentre clinical trial evaluating pharmacokinetics, safety and efficacy of Calquence in adult Chinese patients with relapsed or refractory MCL and other advanced B-cell malignancies.2 In Phase I, patients with relapsed or refractory B-cell malignancies received a single dose of Calquence 100mg orally followed by a two-day washout period and subsequent treatment with Calquence 100mg orally twice daily in 28-day cycles, until progressive disease (PD) or treatment discontinuation (TD) for any other reason. In Phase II, patients with relapsed or refractory MCL and ECOG status of 2 or lower received Calquence 100mg orally twice daily in 28-day cycles until PD or TD. The primary efficacy endpoint was ORR per Lugano classification for NHL assessed by BICR. Secondary endpoints were investigator-assessed ORR, BICR- and investigator-assessed time to response, DOR, PFS, OS and adverse events. 2
Calquence
Calquence (acalabrutinib) is a next-generation, selective inhibitor of BTK. Calquence binds covalently to BTK, thereby inhibiting its activity.6 In B cells, BTK signalling results in activation of pathways necessary for B-cell proliferation, trafficking, chemotaxis and adhesion.
Calquence is approved for the treatment of CLL and SLL in the US, approved for CLL in the EU and many other countries worldwide and approved in Japan for relapsed or refractory CLL and SLL.
As part of an extensive clinical development programme, AstraZeneca is currently evaluating Calquence in more than 20 company-sponsored clinical trials. Calquence is being evaluated for the treatment of multiple B-cell blood cancers, including CLL, MCL, diffuse large B-cell lymphoma, Waldenström’s macroglobulinaemia, marginal zone lymphoma and other haematologic malignancies.
AstraZeneca in haematology
AstraZeneca is pushing the boundaries of science to redefine care in haematology. We have expanded our commitment to patients with haematologic conditions, not only in oncology but also in rare diseases with the acquisition of Alexion, allowing us to reach more patients with high unmet needs. By applying our deep understanding of blood cancers, leveraging our strength in solid tumour oncology and delivering on Alexion’s pioneering legacy in complement science to provide innovative medicines for rare diseases, we are pursuing the end-to-end development of novel therapies designed to target underlying drivers of disease.
By targeting haematologic conditions with high unmet medical needs, we aim to deliver innovative medicines and approaches to improve patient outcomes. Our goal is to help transform the lives of patients living with malignant, rare and other related haematologic diseases, shaped by insights from patients, caregivers and physicians to have the most meaningful impact.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company’s focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Mar. 23, 2023 6:24 AM ET
By: Ravikash, SA News Editor
China' National Medical Products Administration (NMPA) granted conditional approval to AstraZeneca's (NASDAQ:AZN) Calquence to treat adult patients with mantle cell lymphoma (MCL) who have received at least one prior therapy.

March 23, 2023
NORTH CHICAGO, Ill., March 23, 2023 /PRNewswire/ -- AbbVie (NYSE: ABBV) today announced topline results from a Phase 2 study of upadacitinib (RINVOQ®, 30 mg) given alone or as combination therapy (ABBV-599) with a Bruton's Tyrosine Kinase inhibitor (elsubrutinib, 60 mg), once daily in patients with moderately to severely active systemic lupus erythematosus (SLE).1 The SLEek study met the primary endpoint of SLE Responder Index (SRI-4) and steroid dose less than or equal to 10 mg prednisone equivalent once per day at week 24 in the upadacitinib 30 mg group.1,2 Based on the results, AbbVie is advancing its clinical program of upadacitinib in SLE to Phase 3.
"Systemic lupus erythematosus is a very unpredictable life-long condition and the way it affects a patient can change over time. Therefore, there is a critical need for additional treatment options," said Roopal Thakkar, M.D., senior vice president, development and regulatory affairs and chief medical officer, AbbVie. "With a quarter-century of experience and commitment to the treatment of rheumatic diseases, our focus remains on areas of high unmet need like systemic lupus erythematosus, and we look forward to further evaluation of the potential benefits that upadacitinib could bring to patients."
A total of 341 participants enrolled in the Phase 2 study and were subsequently divided into five experimental groups according to treatment regimen (upadacitinib in combination with placebo; upadacitinib, at two different doses, combined with elsubrutinib; elsubrutinib in combination with placebo; placebo only).1 The primary outcome measure was achievement of the SRI-4 with a steroid dose less than or equal to 10 mg prednisone equivalent once daily at week 24.1 SRI-4 is defined as a greater or equal to 4-point reduction in SLE Disease Activity Index 2000 score without worsening of the overall condition or the development of significant disease activity in new organ systems.1
The safety results for the upadacitinib 30 mg arm of the study were generally consistent with the known safety profile of upadacitinib, with no new safety signals identified.2-7 Types of adverse events reported with upadacitinib combined with elsubrutinib were similar to those reported for patients treated with upadacitinib alone.2 Full results from the study will be presented at a future medical congress. Use of upadacitinib and elsubrutinib in SLE are not approved and their safety and efficacy have not been evaluated by regulatory authorities.
Additional information about the study can be found at www.clinicaltrials.gov under the identifier NCT03978520.
About Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is a complex, multi-organ, autoimmune disorder characterized by the production of pathogenic autoantibodies and tissue deposition of immune complexes.8,9 In SLE, the body's immune system attacks healthy tissue of the musculoskeletal system, skin, kidneys, and other critical organs, leading to symptoms such as fatigue, joint pain and impaired function.8,9 The prevalence of SLE is higher in women compared to men, and SLE occurs more frequently in people of color.10,11
About RINVOQ® (upadacitinib)
Discovered and developed by AbbVie scientists, RINVOQ is a selective and reversible JAK inhibitor that is being studied in several immune-mediated inflammatory diseases.3-7,12,13,14,15,16,17,18,19 Based on enzymatic and cellular assays, RINVOQ demonstrated greater inhibitory potency for JAK-1 vs JAK-2, JAK-3 and TYK-2.3 The relevance of inhibition of specific JAK enzymes to therapeutic effectiveness and safety is not currently known.3
Phase 3 trials of RINVOQ in rheumatoid arthritis, atopic dermatitis, psoriatic arthritis, axial spondyloarthritis, Crohn's disease, ulcerative colitis, giant cell arteritis and Takayasu arteritis are ongoing.3-7,12-19 The use of upadacitinib in systemic lupus erythematosus is not approved and its safety and efficacy have not been evaluated by regulatory authorities.
US Indications and Important Safety Information about RINVOQ® (upadacitinib)3
USES
RINVOQ is a prescription medicine used to treat:
It is not known if RINVOQ is safe and effective in children with juvenile idiopathic arthritis, psoriatic arthritis, ulcerative colitis, ankylosing spondylitis, or non-radiographic axial spondyloarthritis.
RINVOQ is safe and effective in children 12 years of age and older weighing at least 88 pounds (40 kg) with atopic dermatitis.
It is not known if RINVOQ is safe and effective in children under 12 years of age with atopic dermatitis.
About AbbVie in Rheumatology
For more than 20 years, AbbVie has been dedicated to improving care for people living with rheumatic diseases. Anchored by a longstanding commitment to discovering and delivering transformative therapies, we pursue cutting-edge science that improves our understanding of promising new pathways and targets, ultimately helping more people living with rheumatic diseases reach their treatment goals. For more information, visit AbbVie in rheumatology.
About AbbVie
AbbVie's mission is to discover and deliver innovative medicines that solve serious health issues today and address the medical challenges of tomorrow. We strive to have a remarkable impact on people's lives across several key therapeutic areas: immunology, oncology, neuroscience, eye care, virology, women's health and gastroenterology, in addition to products and services across our Allergan Aesthetics portfolio. For more information about AbbVie, please visit us at www.abbvie.com. Follow @abbvie on Twitter, Facebook, LinkedIn or Instagram.
SOURCE AbbVie
Mar. 23, 2023 9:02 AM ET
By: Dulan Lokuwithana, SA News Editor
AbbVie (NYSE:ABBV) added ~1% pre-market Thursday after announcing that its interleukin-23 inhibitor Skyrizi met the primary and all secondary endpoints in a Phase 3 trial for adults with ulcerative colitis, a disease in the large intestine.
https://seekingalpha.com/news/3950354-abbvie-gains-skyrizi-succeeds-ulcerative-colitis

March 22, 2023 at 1:14 PM EDTPDF Version
— First regulatory approval for Incyte PD-1 inhibitor based on the results of the POD1UM-201 trial
— Zynyz is also being studied in additional tumor types and in combination with other Incyte pipeline compounds
WILMINGTON, Del.--(BUSINESS WIRE)--Mar. 22, 2023-- Incyte (Nasdaq:INCY) today announced that the U.S. Food and Drug Administration (FDA) has approved Zynyz™ (retifanlimab-dlwr), a humanized monoclonal antibody targeting programmed death receptor-1 (PD-1), for the treatment of adults with metastatic or recurrent locally advanced Merkel cell carcinoma (MCC). The Biologics License Application (BLA) for Zynyz for this indication has been approved under accelerated approval by the U.S. FDA based on tumor response rate and duration of response (DOR). Continued approval of Zynyz for this indication may be contingent on verification and description of clinical benefit in confirmatory trials.
MCC is a rare and aggressive type of skin cancer that frequently appears as a single, painless, reddish-purple skin nodule on the head, neck and arms in skin exposed to sunlight1. MCC tends to grow quickly and has a high rate of metastatic disease, leading to a poor prognosis2,3. The estimated five-year overall survival (OS) rate is 14% in patients with MCC who present with distant metastatic disease3. MCC impacts less than 1 per 100,000 people in the U.S., but incidence rates are rapidly rising, especially in adults over the age of 654,5.
“More than a third of patients with MCC present with regional or distant metastases, which are associated with high rates of mortality,” said Dr. Shailender Bhatia, University of Washington and Fred Hutchinson Cancer Center. “The approval of Zynyz offers healthcare providers another first-line treatment option against MCC that can result in durable responses in patients with metastatic disease, and I look forward to having Zynyz in our treatment portfolio for these difficult-to-treat patients.”
The FDA approval was based on data from the POD1UM-201 trial, an open-label, multiregional, single-arm study that evaluated Zynyz in adults with metastatic or recurrent locally advanced MCC who had not received prior systemic therapy for their advanced disease. Among chemotherapy-naïve patients (n=65), Zynyz monotherapy resulted in an objective response rate (ORR) of 52% (95% confidence interval [CI]: 40-65) as determined by independent central review (ICR) using RECIST v1.1. Complete response was seen in 12 patients (18%), and 22 patients (34%) showed partial response. Among the responding patients, the duration of response (DOR) ranged from 1.1 to 24.9+ months, and 76% (26/34) experienced a DOR of six months or longer, and 62% (21/34) experienced a DOR of 12 months or longer by landmark analysis.
Serious adverse reactions occurred in 22% of patients receiving Zynyz. The most frequent serious adverse reactions (≥ 2% of patients) were fatigue, arrhythmia and pneumonitis. Permanent discontinuation of Zynyz due to an adverse reaction occurred in 11% of patients. The most common (≥10%) adverse reactions that occurred in patients receiving Zynyz were fatigue, musculoskeletal pain, pruritus, diarrhea, rash, pyrexia and nausea.
“Zynyz offers patients and healthcare professionals an additional first-line anti-PD-1 option for patients with metastatic or recurrent locally advanced MCC, which can be a challenging and aggressive disease to treat,” said Hervé Hoppenot, Chief Executive Officer, Incyte. “Incyte is grateful to the investigators and patients around the world who participated in the POD1UM-201 trial. We continue to study the potential of Zynyz in additional tumor types and in combination with other Incyte pipeline compounds.”
Incyte is committed to supporting patients and removing barriers to access medicines. Eligible patients in the U.S. who are prescribed Zynyz have access to IncyteCARES (Connecting to Access, Reimbursement, Education and Support), a comprehensive program offering personalized patient support, including financial assistance and ongoing education and additional resources. More information about IncyteCARES is available by visiting www.incytecares.com or calling 1-855-452-5234.
About POD1UM
The POD1UM (PD1 Clinical Program in Multiple Malignancies) clinical trial program for retifanlimab includes POD1UM-201 and several other Phase 1, 2 and 3 studies for patients with solid tumors, including registration-directed POD1UM trials evaluating retifanlimab as a monotherapy for patients with microsatellite instability-high endometrial cancer and squamous cell carcinoma of the anal canal (SCAC); and in combination with platinum-based chemotherapy for patients with non-small cell lung cancer and SCAC.
About POD1UM-201
POD1UM-201 (NCT03599713) is an open label, multiregional, single arm study evaluating retifanlimab in patients with metastatic or recurrent locally advanced Merkel cell carcinoma (MCC) who had not received prior systemic therapy for their advanced disease.
Patients received Zynyz 500 mg intravenously every four weeks until disease progression, unacceptable toxicity, for up to 24 months. Tumor response assessments were performed every eight weeks for the first year of therapy and 12 weeks thereafter.
The primary endpoint was objective response rate (ORR) as determined by independent central radiographic review using RECIST v1.1. Secondary endpoints included duration of response (DOR), disease control rate (DCR), progression-free survival (PFS) and overall survival (OS); safety and pharmacokinetics.
For more information about the study, please visit: https://clinicaltrials.gov/ct2/show/NCT03599713.
About Zynyz™ (retifanlimab-dlwr)
Zynyz (retifanlimab-dlwr), is an intravenous PD-1 inhibitor indicated in the U.S. for the treatment of adult patients with metastatic or recurrent locally advanced Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
Zynyz is marketed by Incyte in the U.S. In 2017, Incyte entered into an exclusive collaboration and license agreement with MacroGenics, Inc. for global rights to retifanlimab.
Zynyz is a trademark of Incyte.
Please see the full Prescribing Information for ZYNYZ for additional Important Safety Information.
About Incyte
Incyte is a Wilmington, Delaware-based, global biopharmaceutical company focused on finding solutions for serious unmet medical needs through the discovery, development and commercialization of proprietary therapeutics. For additional information on Incyte, please visit Incyte.com and follow @Incyte.
View source version on businesswire.com: https://www.businesswire.com/news/home/20230322005618/en/
Source: Incyte
Mar. 22, 2023 2:09 PM ET
Incyte Corporation (INCY), MGNX
By: Dulan Lokuwithana, SA News Editor

Thursday, March 16, 2023 - 07:30pmView pdf
Pfizer and Astellas announce positive topline results from Phase 3 EMBARK trial
NEW YORK and TOKYO, March 16, 2023 – Pfizer Inc. (NYSE: PFE) and Astellas Pharma Inc. (TSE: 4503, President and CEO: Kenji Yasukawa, Ph.D., “Astellas”) today announced positive topline results from the Phase 3 EMBARK trial evaluating XTANDI® (enzalutamide) in men with non-metastatic hormone-sensitive prostate cancer (nmHSPC; also known as non-metastatic castration-sensitive prostate cancer or nmCSPC) with high-risk biochemical recurrence (BCR). Patients enrolled in the trial were randomized to one of three study arms: XTANDI plus leuprolide, placebo plus leuprolide, or XTANDI monotherapy. The study met its primary endpoint with a statistically significant and clinically meaningful improvement in metastasis-free survival (MFS) for patients treated with XTANDI plus leuprolide versus placebo plus leuprolide.
At the time of the analysis, a positive trend in the key secondary endpoint of overall survival (OS) was also observed, but these data were not yet mature. Patients in the trial will be followed for a subsequent final OS analysis. The study also met a key secondary endpoint with a statistically significant and clinically meaningful improvement in MFS for patients treated with XTANDI monotherapy versus placebo plus leuprolide. Additional key secondary endpoints reached statistical significance, including time to prostate-specific antigen (PSA) progression and time to first use of new antineoplastic therapy. Other secondary endpoints are being analyzed. No new safety signals have been observed to date in the preliminary safety analysis, which is consistent with the established safety profile of XTANDI.
“As the only novel hormone therapy approved for three disease states of prostate cancer in the U.S., XTANDI has impacted hundreds of thousands of men,” said Chris Boshoff, M.D., Ph.D., Chief Development Officer, Oncology and Rare Disease, Pfizer Global Product Development. “The topline findings from EMBARK are highly encouraging and we look forward to engaging with health authorities to potentially bring XTANDI to men with non-metastatic hormone-sensitive prostate cancer with high-risk biochemical recurrence.”
“While current treatment options for localized prostate cancer are intended to be curative, some men remain at higher risk for biochemical recurrence following primary treatment, which may result in metastases,” said Ahsan Arozullah, M.D., MPH, Senior Vice President and Head of Development Therapeutic Areas, Astellas. “The EMBARK trial is the first study to demonstrate a statistically significant improvement in MFS using the combination of XTANDI plus leuprolide in men with this stage of disease.”
Detailed results from EMBARK will be presented at a future medical meeting. These data will also be discussed with regulatory authorities, including the U.S. Food and Drug Administration (FDA), to support a potential regulatory submission for XTANDI in this indication.
About EMBARK
The Phase 3, randomized, double-blind, placebo-controlled, multi-national trial enrolled 1,068 patients with non-metastatic hormone-sensitive prostate cancer (nmHSPC; also known as non-metastatic castration-sensitive prostate cancer or nmCSPC) with high-risk biochemical recurrence (BCR) at sites in the United States, Canada, Europe, South America, and the Asia-Pacific region. Patients who were considered high-risk BCR had a prostate-specific antigen (PSA) doubling time ≤ 9 months, serum testosterone ≥ 150 ng/dL (5.2 nmol/L), and screening PSA by the central laboratory ≥ 1 ng/mL if they had a radical prostatectomy (with or without radiotherapy) as primary treatment for prostate cancer or at least 2 ng/mL above the nadir if they had radiotherapy only as primary treatment for prostate cancer. Patients in the EMBARK trial were randomized to receive enzalutamide 160 mg daily plus leuprolide, enzalutamide 160 mg as a monotherapy, or placebo plus leuprolide.
The primary endpoint of the trial was metastasis-free survival (MFS) for enzalutamide plus leuprolide and placebo plus leuprolide. MFS is defined as the duration of time in months between randomization and the earliest objective evidence of radiographic progression by central imaging or death. For more information on the EMBARK (NCT02319837) trial go to www.clinicaltrials.gov.
XTANDI has not been approved for the treatment of patients with nmHSPC with high-risk BCR.
About Non-Metastatic Hormone-Sensitive Prostate Cancer with High-Risk Biochemical Recurrence
Non-metastatic hormone- (or castration-) sensitive prostate cancer (nmHSPC or nmCSPC) means there is no detectable evidence of the cancer spreading to distant parts of the body (metastases) with conventional radiological methods (CT/MRI) and the cancer still responds to medical or surgical treatment to lower testosterone levels.1,2 Of men who have undergone definitive prostate cancer treatment, including radical prostatectomy, radiotherapy, or both, an estimated 20-40% will experience a biochemical recurrence (BCR) within 10 years.3 About 9 out of 10 men with high-risk BCR will develop metastatic disease, and 1 in 3 will die as a result of the recurrence.3 The EMBARK trial focused on men with high-risk BCR. Per the EMBARK protocol, patients with nmHSPC with high-risk BCR are those initially treated by radical prostatectomy or radiotherapy, or both, with a PSA doubling time ≤ 9 months. High-risk BCR patients with a PSA doubling time of ≤ 9 months have a higher risk of metastases and death.4
About XTANDI® (enzalutamide)
XTANDI (enzalutamide) is an androgen receptor signaling inhibitor. The recommended dosage of XTANDI is 160 mg (capsules or tablets) administered orally once daily with or without food. XTANDI is a standard of care that has received regulatory approvals for use in men with mHSPC, mCRPC, and nmCRPC in the United States and for one or more of these indications in more than 100 countries, including the European Union and Japan. More than 720,000 patients have been treated with XTANDI globally.5
Please see Full Prescribing Information for additional safety information.
About Pfizer Oncology
At Pfizer Oncology, we are committed to advancing medicines wherever we believe we can
make a meaningful difference in the lives of people living with cancer. Today, we have an
industry-leading portfolio of 24 approved innovative cancer medicines and biosimilars that generated $12.1 billion in revenue in 2022, including the best-selling therapies for metastatic breast cancer and prostate cancer. Our in-line portfolio is focused on four broad, key areas where we have pioneered several breakthroughs: breast cancer, genitourinary cancer, hematology, and precision medicine, and we are advancing an extensive pipeline of 33 programs in clinical development.
About Astellas
Astellas Pharma Inc. is a pharmaceutical company conducting business in more than 70 countries around the world. We are promoting the Focus Area Approach that is designed to identify opportunities for the continuous creation of new drugs to address diseases with high unmet medical needs by focusing on Biology and Modality. Furthermore, we are also looking beyond our foundational Rx focus to create Rx+® healthcare solutions that combine our expertise and knowledge with cutting-edge technology in different fields of external partners. Through these efforts, Astellas stands on the forefront of healthcare change to turn innovative science into VALUE for patients. For more information, please visit our website at https://www.astellas.com/en.
About the Pfizer/Astellas Collaboration
In October 2009, Medivation, Inc., which is now part of Pfizer (NYSE:PFE), and Astellas (TSE: 4503) entered into a commercial agreement to jointly develop and commercialize XTANDI® (enzalutamide) in the United States, while Astellas has responsibility for manufacturing and all additional regulatory filings globally, as well as commercializing the product outside the United States. Pfizer receives alliance revenues as a share of U.S. profits and receives royalties on sales outside the U.S.
Mar. 17, 2023 6:30 AM ET
Pfizer Inc. (PFE), ALPMF, ALPMY
By: Ravikash, SA News Editor
Pfizer (NYSE:PFE) and Astellas Pharma's (OTCPK:ALPMF) (OTCPK:ALPMY) Xtandi met the main goal of a phase 3 trial to treat men with non-metastatic hormone-sensitive prostate cancer (nmHSPC).

March 22, 2023 at 7:00 AM EDT Back
Approval extends Evkeeza to children aged 5 to 11 with homozygous familial hypercholesterolemia (HoFH), an inherited condition characterized by extremely high low-density lipoprotein cholesterol (LDL-C)
48% reduction in LDL-C from baseline at week 24 when Evkeeza was added to other lipid-lowering therapies in the pivotal trial
TARRYTOWN, N.Y., March 22, 2023 (GLOBE NEWSWIRE) -- Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) today announced the U.S. Food and Drug Administration (FDA) has extended the approval of Evkeeza® (evinacumab-dgnb) as an adjunct to other lipid-lowering therapies to treat children aged 5 to 11 with homozygous familial hypercholesterolemia (HoFH). Evkeeza is the first angiopoietin-like 3 (ANGPTL3) inhibitor treatment indicated for children as young as 5 years old to control dangerously high levels of low-density lipoprotein cholesterol (LDL-C) caused by HoFH. Evkeeza was initially approved as an adjunct to other lipid-lowering therapies in those aged 12 years and older with HoFH in February 2021.
“At the Family Heart Foundation, we know that children with homozygous familial hypercholesterolemia, and those caring for them, often live in fear of what the future holds as they contend with the dangerously high levels of bad cholesterol, or LDL-C, caused by this genetic disorder,” said Mary McGowan, M.D., Chief Medical Officer of the Family Heart Foundation. “Only 5% of rare diseases actually have an FDA-approved treatment. With this FDA approval, the HoFH community now has a much-needed treatment for young children, potentially making it possible for many to achieve recommended LDL-C levels much earlier in the course of this rare disease. This is a hopeful development for those living with HoFH.”
HoFH is an ultra-rare inherited condition that affects approximately 1,300 people in the U.S. and is the most severe form of familial hypercholesterolemia (FH). HoFH occurs when two copies of the FH-causing genes are inherited, one from each parent, resulting in dangerously high levels (usually >400 mg/dL) of LDL-C. Those living with HoFH are at risk for premature atherosclerotic disease and cardiac events even in their teenage years. Many patients are not diagnosed or are only diagnosed later in life.
“Guidelines recommend screening all children at high risk for homozygous familial hypercholesterolemia starting at age 2. However, until now, a positive diagnosis was often met with the frustration of having limited treatment options to help these children,” said Carissa M. Baker-Smith, M.D., MPH, Co-Director of Nemours Cardiac Center Cardiovascular Research and Innovation Program, Director of Nemours Cardiac Center Pediatric Preventive Cardiology, pediatric cardiologist, and a trial investigator. “By adding Evkeeza to standard lipid-lowering therapies in this pivotal trial, children were able to reduce their LDL-C, with the vast majority able to achieve declines of nearly 50%. These are clinically meaningful results that physicians should consider when developing a treatment approach for these young patients.”
Despite treatment with other lipid-lowering therapies, children entered the Phase 3 trial with an average LDL-C level of 264 mg/dL, more than twice the target (<110 mg/dL) for pediatric patients with HoFH. With the addition of Evkeeza, children were able to reduce their LDL-C by 48% at week 24 on average, meeting the trial’s primary endpoint. Significant reductions were also observed in other key secondary endpoints including levels of apolipoprotein B (ApoB), non-high-density lipoprotein cholesterol (non-HDL-C) and total cholesterol.
The safety profile of Evkeeza observed in these patients (n=20) was consistent with the safety profile observed in adults and pediatric patients aged 12 years and older, with the additional adverse reaction of fatigue. Fatigue was reported in 3 (15%) patients. The most common adverse events (AEs) occurring in >15% of patients were COVID-19 (n=15), pyrexia (n=5), headache (n=4), throat pain (oropharyngeal pain, n=4) as well as upper abdominal pain, diarrhea, vomiting, fatigue, nasopharyngitis, rhinitis and cough (all n=3). Most reported AEs were mild or moderate, and none led to study discontinuation.
“Since it was first approved, Evkeeza has become the standard of care for homozygous familial hypercholesterolemia in those aged 12 years or older. We’re gratified that now children as young as 5 years old have the potential to benefit from this treatment,” said George D. Yancopoulos, M.D., Ph.D., President and Chief Scientific Officer at Regeneron. “As a first-in-class medicine for this relentless disease, Evkeeza exemplifies the promise of genetics-based research to transform treatment paradigms. Evkeeza’s journey from target discovery to treatment innovation was only made possible due to our long-term investment in genetics research and monoclonal antibody technologies, and this remains a central tenet of our science-first approach to this day.”
Regeneron is committed to helping patients who have been prescribed Evkeeza access their medication. Regeneron's myRARE™ patient support program offers financial assistance to eligible patients who need help with the out-of-pocket cost of Evkeeza. Under the program, eligible patients with commercial insurance may pay as little as $0 in out-of-pocket costs for Evkeeza. In addition, myRARE™ offers resources to help patients and healthcare providers get started with Evkeeza including product information, insurance benefit verification, community resources and appointment reminders. For more information, call 1-833-EVKEEZA (833-385-3392) or visit www.EVKEEZA.com.
The FDA evaluated the supplemental biologics license for Evkeeza in this indication under Priority Review, which is reserved for medicines that represent potentially significant improvements in efficacy or safety in treating serious conditions.
The safety and effectiveness of Evkeeza have not been established in patients with other causes of hypercholesterolemia, including those with heterozygous familial hypercholesterolemia (HeFH). The effect of Evkeeza on cardiovascular morbidity and mortality has not been determined.
About the Pivotal Pediatric Trial
The three-part, single-arm, open-label trial evaluated Evkeeza added to other lipid-lowering therapies in pediatric patients with HoFH aged 5 to 11 years. Part A (n=6) was a Phase 1b trial designed to assess the pharmacokinetics (PK), safety and tolerability of Evkeeza. Part B (n=14) evaluated the efficacy of Evkeeza during a 24-week treatment period and enrolled patients with an average age of 9 years. Among them, 86% were on statins, 93% were on ezetimibe, 50% were on LDL apheresis and 14% were on lomitapide. Patients received Evkeeza 15 mg/kg every four weeks delivered intravenously alongside their lipid-lowering treatment regimen. The primary endpoint was change in LDL-C at week 24. Secondary endpoints included the effect of Evkeeza on other lipid parameters (i.e., apolipoprotein B, non-high-density lipoprotein cholesterol, lipoprotein[a] and total cholesterol), efficacy by mutation status, safety and tolerability, immunogenicity and PK.
Patients who completed Part A or B were allowed to continue treatment in Part C (n=20), an ongoing Phase 3 extension trial. Parts A, B and C were not designed to evaluate the effect of Evkeeza on cardiovascular events.
About Evkeeza® (evinacumab)
Evkeeza was invented using Regeneron’s VelocImmune® technology and is a fully human monoclonal antibody that binds to and blocks the function of angiopoietin-like 3 (ANGPTL3), a protein that inhibits lipoprotein lipase (LPL) and endothelial lipase (EL) and regulates circulating lipids, including LDL-C.
Regeneron scientists discovered the angiopoietin gene family more than two decades ago. Human genetics research published in New England Journal of Medicine in 2017 by scientists from the Regeneron Genetics Center® found that patients whose ANGPTL3 gene did not function properly (called a “loss-of function mutation”) have significantly lower levels of key blood lipids, including LDL-C, and that this is associated with a significantly lower risk of coronary artery disease.
The generic name for Evkeeza in its approved U.S. indications is evinacumab-dgnb, with dgnb the suffix designated in accordance with Nonproprietary Naming of Biological Products Guidance for Industry issued by the U.S. FDA.
Regeneron is responsible for the development and distribution of Evkeeza in the U.S. and is collaborating with Ultragenyx to clinically develop, commercialize and distribute Evkeeza outside of the U.S.
About Regeneron’s VelocImmune Technology
Regeneron’s VelocImmune technology utilizes a proprietary genetically engineered mouse platform endowed with a genetically humanized immune system to produce optimized fully human antibodies. When Regeneron's President and Chief Scientific Officer George D. Yancopoulos was a graduate student with his mentor Frederick W. Alt in 1985, they were the first to envision making such a genetically humanized mouse, and Regeneron has spent decades inventing and developing VelocImmune and related VelociSuite® technologies. Dr. Yancopoulos and his team have used VelocImmune technology to create a substantial proportion of all original, FDA-approved fully human monoclonal antibodies currently available. This includes Evkeeza® (evinacumab-dgnb), REGEN-COV® (casirivimab and imdevimab), Dupixent® (dupilumab), Libtayo® (cemiplimab-rwlc), Praluent® (alirocumab), Kevzara® (sarilumab) and Inmazeb® (atoltivimab, maftivimab and odesivimab-ebgn).
Please see full Prescribing Information, including Patient Information.
About Regeneron
Regeneron (NASDAQ: REGN) is a leading biotechnology company that invents, develops and commercializes life-transforming medicines for people with serious diseases. Founded and led for 35 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to nine FDA-approved treatments and numerous product candidates in development, almost all of which were homegrown in our laboratories. Our medicines and pipeline are designed to help patients with eye diseases, allergic and inflammatory diseases, cancer, cardiovascular and metabolic diseases, pain, hematologic conditions, infectious diseases and rare diseases.
Regeneron is accelerating and improving the traditional drug development process through our proprietary VelociSuite® technologies, such as VelocImmune®, which uses unique genetically humanized mice to produce optimized fully human antibodies and bispecific antibodies, and through ambitious research initiatives such as the Regeneron Genetics Center, which is conducting one of the largest genetics sequencing efforts in the world.
For more information, please visit www.Regeneron.com or follow @Regeneron on Twitter.
Mar. 22, 2023 7:54 AM ET
Regeneron Pharmaceuticals, Inc. (REGN), RARE
By: Ravikash, SA News Editor1 Comment
The U.S. Food and Drug Administration (FDA) approved the expanded use of Regeneron Pharmaceuticals' (NASDAQ:REGN) Evkeeza as an adjunct to other lipid-lowering therapies to treat children aged five to 11 years with homozygous familial hypercholesterolemia (HoFH).

3/20/2023
Darolutamide approved for additional prostate cancer indication in China
ORION CORPORATION
PRESS RELEASE
20 March 2023 at 09:00 EET
Darolutamide approved for additional prostate cancer indication in China
The Chinese National Medical Products Administration (NMPA) has approved the oral androgen receptor inhibitor (ARi) darolutamide in combination with docetaxel for the treatment of patients with metastatic hormone-sensitive prostate cancer (mHSPC). Darolutamide is already approved in China for the treatment of patients with non-metastatic castration-resistant prostate cancer (nmCRPC), who are at high risk of developing metastatic disease.
The approval is based on the positive results from the Phase III ARASENS trial, which demonstrated that darolutamide plus androgen deprivation therapy (ADT) in combination with docetaxel significantly reduced the risk of death by 32.5% compared to ADT with docetaxel, in patients with metastatic hormone-sensitive prostate cancer. Additionally, the darolutamide combination showed consistent benefits across clinically relevant secondary endpoints, with the overall incidence of treatment-emergent adverse events being similar between treatment arms. These results were published in The New England Journal of Medicine.1
Darolutamide is being investigated in a broad development program with three additional ongoing or planned large clinical studies, to evaluate its potential across prostate cancer patients from the early- to the late-stage of this disease. This includes the ARANOTE Phase III trial evaluating darolutamide plus androgen deprivation therapy (ADT) versus ADT alone for mHSPC.
Darolutamide is developed jointly by Orion and Bayer.
About the ARASENS Trial
The ARASENS trial is a randomized, Phase III, multi-center, double-blind, placebo-controlled trial which was prospectively designed to investigate the efficacy and safety of oral darolutamide, an androgen receptor inhibitor (ARi), plus androgen deprivation therapy (ADT) and the chemotherapy docetaxel in patients with metastatic hormone-sensitive prostate cancer (mHSPC). A total of 1,306 newly diagnosed patients were randomized in a 1:1 ratio to receive 600 mg of darolutamide twice a day or matching placebo, plus ADT and docetaxel.
The primary endpoint of this trial was overall survival (OS). Secondary endpoints included time to castration-resistant prostate cancer (CRPC), time to pain progression, time to first symptomatic skeletal event (SSE), time to initiation of subsequent anticancer therapy, all measured at 12‐week intervals, as well as adverse events (AEs) as a measure of safety and tolerability. Results from this trial were published in the New England Journal of Medicine.1 A plain language summary publication of these data was published in Future Oncology.2 The ARASENS trial demonstrated that darolutamide plus androgen deprivation therapy (ADT) and docetaxel significantly reduced the risk of death by 32.5% compared to ADT plus docetaxel.1 Improvements in the secondary endpoints supported the benefit observed in the primary endpoint, overall survival.1
About Metastatic Hormone-Sensitive Prostate Cancer
Prostate cancer is the second most commonly diagnosed malignancy in men worldwide. In 2020, an estimated 1.4 million men were diagnosed with prostate cancer, and about 375,000 died from the disease worldwide.3
At the time of diagnosis, most men have localized prostate cancer, meaning their cancer is confined to the prostate gland and can be treated with curative surgery or radiotherapy. Upon relapse, when the disease will metastasise or spread, or if the disease is newly diagnosed, but has already spread, the disease is hormone-sensitive and androgen deprivation therapy (ADT) is the cornerstone of treatment for this hormone-sensitive disease. Current treatment options for men with metastatic hormone-sensitive prostate cancer (mHSPC) include hormone therapy, such as ADT, androgen receptor pathway inhibitors plus ADT or a combination of docetaxel chemotherapy and ADT. Despite these treatments, a large proportion of men with mHSPC will eventually experience progression to metastatic castration-resistant prostate cancer (mCRPC), a condition with limited survival.
About darolutamide
Darolutamide is an oral androgen receptor inhibitor (ARi) with a distinct chemical structure that binds to the receptor with high affinity and exhibits strong antagonistic activity, thereby inhibiting the receptor function and the growth of prostate cancer cells. The low potential for blood-brain barrier penetration for darolutamide is supported by preclinical models and neuroimaging data in healthy humans. This is supported by the overall low incidence of central nervous system (CNS)-related adverse events (AEs) compared to placebo as seen in the ARAMIS Phase III trial4 and the maintained verbal learning and memory observed in the darolutamide arm of the Phase II ODENZA trial.5
The product is approved under the brand name Nubeqa® in more than 80 countries around the world for the treatment of patients with non-metastatic castration-resistant prostate cancer (nmCRPC), who are at high risk of developing metastatic disease. It is also approved for the treatment of patients with metastatic hormone-sensitive prostate cancer (mHSPC) in a number of markets including the U.S., Japan, EU and China. Filings in other regions are underway or planned by Bayer. The compound is also being investigated in further studies across various stages of prostate cancer, including in the ARANOTE Phase III trial evaluating darolutamide plus androgen deprivation therapy (ADT) versus ADT alone for metastatic hormone-sensitive prostate cancer (mHSPC), as well as the Australian and New Zealand Urogenital and Prostate Cancer Trials Group (ANZUP) led international Phase III co-operative group DASL-HiCaP (ANZUP1801) trial evaluating darolutamide as an adjuvant treatment for localised prostate cancer with very high risk of recurrence. Information about these trials can be found at www.clinicaltrials.gov. In addition, a study to explore the potential of darolutamide in the early setting for patients who have experienced a rise in their prostate specific antigen (PSA) levels following surgery or radiation, is also planned.
Mar. 21, 2023 4:55 AM ET
Bayer Aktiengesellschaft (BAYRY), BAYZF
By: Ravikash, SA News Editor
China's National Medical Products Administration (NMPA) approved Bayer (OTCPK:BAYRY) (OTCPK:BAYZF) and Orion Corporation's oral drug Nubeqa, in combination with docetaxel, to treat patients with metastatic hormone-sensitive prostate cancer (mHSPC).

03/09/2023
– Clinical data submitted are consistent with global data and support enfortumab vedotin as a platinum-free option in patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1/L1 inhibitor and platinum-based chemotherapy –
TOKYO & BOTHELL, Wash.--(BUSINESS WIRE)-- Astellas Pharma Inc. (TSE:4503, President and CEO: Kenji Yasukawa, Ph.D., “Astellas”) and Seagen Inc. (Nasdaq: SGEN) today announced that the Center for Drug Evaluation (CDE) of the China National Medical Products Administration (NMPA) has accepted the Biologics License Application (BLA) for enfortumab vedotin for the treatment of patients with locally advanced or metastatic urothelial cancer (la/mUC) who received prior treatment with a PD-1/L1 inhibitor and platinum-based chemotherapy.
“In China, there were nearly 86,000 new cases of bladder cancer in 2020, and we are working with the NMPA to seek approval for enfortumab vedotin for patients with advanced stage disease,” said Ahsan Arozullah, M.D., M.P.H., Senior Vice President and Head of Development Therapeutic Areas, Astellas. “Enfortumab vedotin has become a second- and third-line treatment option for many patients around the world with previously treated locally advanced or metastatic urothelial cancer, and an approval in China may bring this therapy to those patients.”
The BLA submission for enfortumab vedotin is based on data from the EV-203 study (NCT04995419), a single-arm, open-label, multicenter Phase 2 study of enfortumab vedotin in Chinese patients with la/mUC who previously received a PD-1/L1 inhibitor and platinum-based chemotherapy. Results showed that EV-203 met its primary endpoint, showing statistical significance in objective response rate (ORR) by independent review committee (IRC) for patients treated with enfortumab vedotin alone compared to historical controls. Efficacy and pharmacokinetic data from the study are in line with global data, and EV-203 is a bridging study to EV-301, a Phase 3 randomized study that has supported global registrations of enfortumab vedotin, and EV-201 Cohort 1.
Please see Important Safety Information, including BOXED WARNING, at the end of this press release for further safety information regarding enfortumab vedotin including serious skin reactions.
Enfortumab vedotin alone and in combination with other therapies is the subject of a robust clinical development program aimed at addressing unmet medical needs across the continuum of urothelial cancer and in other solid tumors.
About Bladder and Urothelial Cancer
Globally, approximately 573,000 new cases of bladder cancer and 212,000 deaths are reported annually.1 Urothelial cancer accounts for 90% of all bladder cancers and can also be found in the renal pelvis, ureter and urethra.2 Approximately 12% of cases are locally advanced or metastatic urothelial cancer at diagnosis.3
In China, the incidence rate of bladder cancer in 2020 ranked 12th among all cancers, with an estimated 85,649 new cases that year. The five-year prevalence of bladder cancer in China is estimated to be 16.26/100,000 cases, or 235,393 cases.4
About the EV-203 Trial
The EV-203 trial (NCT04995419) is a Phase 2, multicenter, single-arm bridging study in China designed to evaluate the efficacy, safety and pharmacokinetic performance of enfortumab vedotin as treatment for patients in China. A total of 40 patients were enrolled in the study.
About the EV-301 Trial
The EV-301 trial (NCT03474107) is a global, multicenter, open-label, randomized Phase 3 trial designed to evaluate enfortumab vedotin versus physician's choice of chemotherapy (docetaxel, paclitaxel or vinflunine) in 608 patients with locally advanced or metastatic urothelial cancer who were previously treated with a PD-1/L1 inhibitor and platinum-based chemotherapies. The primary endpoint is overall survival, and secondary endpoints include progression-free survival, overall response rate, duration of response and disease control rate, as well as assessment of safety/tolerability and quality-of-life parameters.
About the EV-201 Trial
The EV-201 trial (NCT03219333) is a single-arm, multi-cohort, multicenter, pivotal phase 2 clinical trial of enfortumab vedotin for patients with locally advanced or metastatic urothelial cancer who have been previously treated with a PD-1 or PD-L1 inhibitor, including those who have also been treated with a platinum-containing chemotherapy (Cohort 1) and those who have not received a platinum-containing chemotherapy in this setting and who are ineligible for cisplatin (Cohort 2). The trial enrolled 125 patients in Cohort 1 and 89 patients in Cohort 2 at multiple centers internationally. The primary endpoint is confirmed objective response rate per blinded independent central review. Secondary endpoints include assessments of duration of response, disease control rate, progression-free survival, overall survival, safety and tolerability.
Results of EV-301 and EV-201 Cohort 2 clinical trials supported the full and supplemental approval of PADCEV® (enfortumab vedotin-ejfv) by the U.S. Food and Drug Administration in July 2021. Additionally, results from EV-301 and EV-201 Cohort 1 serve as core data to support the Marketing Authorization Applications for enfortumab vedotin in the global market, including the European Union, Japan and Singapore.
About PADCEV
PADCEV (enfortumab vedotin-ejfv) is a first-in-class antibody-drug conjugate (ADC) that is directed against Nectin-4, a protein located on the surface of cells and highly expressed in bladder cancer.5 Nonclinical data suggest the anticancer activity of PADCEV is due to its binding to Nectin-4-expressing cells followed by the internalization and release of the anti-tumor agent monomethyl auristatin E (MMAE) into the cell, which result in the cell not reproducing (cell cycle arrest) and in programmed cell death (apoptosis).6
PADCEV (enfortumab vedotin-ejfv) U.S. Indication & Important Safety Information
BOXED WARNING: SERIOUS SKIN REACTIONS
Indication
PADCEV® is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer (mUC) who:
For more information, please see the full Prescribing Information including BOXED WARNING for PADCEV here .
About Astellas
Astellas Pharma Inc. is a pharmaceutical company conducting business in more than 70 countries around the world. We are promoting the Focus Area Approach that is designed to identify opportunities for the continuous creation of new drugs to address diseases with high unmet medical needs by focusing on Biology and Modality. Furthermore, we are also looking beyond our foundational Rx focus to create Rx+® healthcare solutions that combine our expertise and knowledge with cutting-edge technology in different fields of external partners. Through these efforts, Astellas stands on the forefront of healthcare change to turn innovative science into VALUE for patients. For more information, please visit our website at https://www.astellas.com/en.
About Seagen
Seagen Inc. is a global biotechnology company that discovers, develops and commercializes transformative cancer medicines to make a meaningful difference in people’s lives. Seagen is headquartered in the Seattle, Washington area, and has locations in California, Canada, Switzerland and the European Union. For more information on the company’s marketed products and robust pipeline, visit www.seagen.com and follow @SeagenGlobal on Twitter.
About the Astellas and Seagen Collaboration
Astellas and Seagen are co-developing enfortumab vedotin under a 50:50 worldwide development and commercialization collaboration. In the United States, Astellas and Seagen co-promote enfortumab vedotin under the brand name PADCEV® (enfortumab vedotin-ejfv). In the Americas outside the US, Seagen holds responsibility for commercialization activities and regulatory filings. Outside of the Americas, Astellas holds responsibility for commercialization activities and regulatory filings.
Source: Seagen Inc.
Mar. 10, 2023 6:21 AM ET
Seagen Inc. (SGEN), ALPMY, ALPMF
By: Ravikash, SA News Editor

Mar 17, 2023
Basel, March 16, 2023 — Novartis today announced the U.S. Food and Drug Administration (FDA) granted approval for Tafinlar® (dabrafenib) + Mekinist® (trametinib) for the treatment of pediatric patients 1 year of age and older with low-grade glioma (LGG) with a BRAF V600E mutation who require systemic therapy. The FDA also approved liquid formulations of Tafinlar and Mekinist, marking the first time a BRAF/MEK inhibitor has been developed in a formulation suitable for patients as young as one year of age. These approvals make Tafinlar + Mekinist the first and only approved combination targeted therapy to treat pediatric patients with BRAF V600E LGG.
“Pediatric cancer research is vital to uncover new treatment methods for a population,” said Dr. Eric Bouffet, MD, FRCPC, Principal Investigator of the TADPOLE clinical trial and Associate Scientist Emeritus at The Hospital for Sick Children (SickKids). “Developing targeted therapies based on the unique genetic features of a patient’s tumor is the future of pediatric cancer care.”
This FDA approval of Tafinlar + Mekinist is based on results from the Phase II/III TADPOLE trial (NCT02684058) that showed patients randomized to receive Tafinlar + Mekinist experienced a statistically significant improvement in overall response rate (ORR) of 47% (CI: 35-59%) compared to 11% (CI: 3-25%) for those randomized to receive chemotherapy. At a median follow-up of 18.9 months, median progression-free survival (PFS) was 20.1 months with Tafinlar + Mekinist (CI: 12.8 months-not estimable) compared to 7.4 months with chemotherapy (CI: 3.6-11.8 months, hazard ratio=0.31 [CI: 0.17-0.55] [p<0.001]).
“It is more important than ever to test for genetic mutations in patients living with low-grade glioma. This FDA approval may offer new hope to pediatric patients living with BRAF V600E low-grade glioma,” said Dr. Roger Packer, senior vice president of the Center for Neurosciences and Behavioral Medicine at Children’s National Hospital. “This has the potential to change the way healthcare providers treat these pediatric patients, offering a significant advancement compared to chemotherapy.”
The safety profile of Tafinlar + Mekinist observed in this study was consistent with the known safety profile in other approved indications. The most common adverse reactions (>=15%) were pyrexia (68%), rash (51%), headache (47%), vomiting (34%), musculoskeletal pain (34%), fatigue (33%), diarrhea (29%), dry skin (26%), nausea (25%), hemorrhage (25%), abdominal pain (25%), dermatitis acneiform (22%), dizziness (15%), upper respiratory tract infection (15%) and weight increased (15%). These data were highlighted as part of an official press briefing and oral presentation at the 2022 American Society of Clinical Oncology (ASCO) Annual Meeting.
“This new indication for Tafinlar + Mekinist is a potential new standard of care treatment option for young patients with this form of brain cancer with a BRAF V600E mutation, in formulations specifically designed for them,” said Reshema Kemps-Polanco, Executive Vice President, US Oncology at Novartis. “We are thankful for the families, including children and adolescents, that participated in the clinical trial that led to this approval and whose bravery has led to a new hope for children living with this serious brain cancer.”
LGG is the most common pediatric brain cancer. BRAF V600 mutations are present in 15-20% of pediatric LGGs and are associated with poor survival outcomes and less favorable response to chemotherapy4. BRAF mutations have been identified as drivers of cancer growth across a wide range of solid tumors, and often have limited treatment options4,5.
Full prescribing information for Tafinlar + Mekinist can be found at https://www.novartis.us/sites/www.novartis.us/files/tafinlar.pdf and https://www.novartis.us/sites/www.novartis.us/files/mekinist.pdf.
About Tafinlar + Mekinist
The combination of Tafinlar + Mekinist, the worldwide targeted therapy leader in BRAF/MEK-inhibition research and patients reached, may help to slow tumor growth by blocking signals associated with the BRAF and MEK kinases that are implicated in the growth of various types of cancer1,2,4,5. Tafinlar + Mekinist has been studied in more than 6,000 BRAF-positive patients in more than 20 ongoing and completed trials, including in pediatric patients 1 year of age and older, and has been prescribed to more than 200,000 patients worldwide6.
This FDA approval is the sixth for Tafinlar + Mekinist, which is indicated across multiple BRAF V600 solid tumors, including melanoma, thyroid cancer and lung cancer1,2.
https://www.us.tafinlarmekinist.com/
Indication and Important Safety Information
TAFINLAR and MEKINIST are prescription medicines that can be used in combination to treat people with a type of skin cancer called melanoma:
TAFINLAR and MEKINIST are prescription medicines that can be used in combination to help prevent melanoma that has a certain type of abnormal “BRAF” gene from coming back after the cancer has been removed by surgery.
TAFINLAR and MEKINIST are prescription medicines that can be used in combination to treat a type of lung cancer called non-small cell lung cancer (NSCLC) that has spread to other parts of the body (metastatic NSCLC), and that has a certain type of abnormal “BRAF V600E” gene.
TAFINLAR and MEKINIST are prescription medicines that can be used in combination to treat a type of thyroid cancer called anaplastic thyroid cancer (ATC):
TAFINLAR and MEKINIST are prescription medicines that can be used in combination to treat solid tumors in adults and children 6 years of age and older:
The effectiveness of TAFINLAR and MEKINIST in these patients is based on 2 adult studies and 1 pediatric study that measured 2 types of response to treatment (response rate and duration of response). No clinical information is available to show if these patients treated with TAFINLAR and MEKINIST live longer or if their symptoms improve. Ongoing studies exist to determine how TAFINLAR and MEKINIST works over a longer period.
TAFINLAR and MEKINIST are prescription medicines that can be used in combination to treat a type of brain tumor called glioma in children 1 year of age and older
TAFINLAR, in combination with MEKINIST, is not for use in treating people with colorectal cancer or wild-type BRAF solid tumors. MEKINIST should not be used to treat people who already have received a BRAF inhibitor for treatment of their melanoma and it did not work or is no longer working.
Your health care provider will perform a test to make sure that TAFINLAR and MEKINIST, in combination, are right for you.
It is not known if TAFINLAR used in combination with MEKINIST is safe and effective in children younger than 6 years of age.
TAFINLAR and MEKINIST, in combination, may cause serious side effects such as the risk of new cancers, including both skin cancer and nonskin cancer. Patients should be advised to contact their health care provider immediately for any skin changes, including a new wart, skin sore, or bump that bleeds or does not heal, or a change in the size or color of a mole.
When TAFINLAR is used in combination with MEKINIST, it can cause serious bleeding problems, especially in the brain or stomach, that can lead to death. Patients should be advised to call their health care provider and get medical help right away if they have any signs of bleeding, including headaches, dizziness, or feeling weak, coughing up blood or blood clots, vomiting blood or their vomit looks like “coffee grounds,” or red or black stools that look like tar.
MEKINIST, alone or in combination with TAFINLAR, can cause inflammation of the intestines or tears in the stomach or intestines that can lead to death. Patients should report to their health care provider right away if they have any of the following symptoms: bleeding, diarrhea (loose stools) or more bowel movements than usual, stomach-area (abdomen) pain or tenderness, fever, or nausea.
TAFINLAR, in combination with MEKINIST, can cause blood clots in the arms or legs, which can travel to the lungs and can lead to death. Patients should be advised to get medical help right away if they have the following symptoms: chest pain, sudden shortness of breath or trouble breathing, pain in their legs with or without swelling, swelling in their arms or legs, or a cool or pale arm or leg.
The combination of TAFINLAR and MEKINIST can cause heart problems, including heart failure. A patient’s heart function should be checked before and during treatment. Patients should be advised to call their health care provider right away if they have any of the following signs and symptoms of a heart problem: feeling like their heart is pounding or racing, shortness of breath, swelling of their ankles and feet, or feeling lightheaded.
TAFINLAR, in combination with MEKINIST, can cause severe eye problems that can lead to blindness. Patients should be advised to call their health care provider right away if they get: blurred vision, loss of vision, or other vision changes, seeing color dots, halo (seeing blurred outline around objects), eye pain, swelling, or redness.
TAFINLAR, in combination with MEKINIST, can cause lung or breathing problems. Patients should be advised to tell their health care provider if they have new or worsening symptoms of lung or breathing problems, including shortness of breath or cough.
Fever is common during treatment with TAFINLAR in combination with MEKINIST but may also be serious. In some cases, chills or shaking chills, too much fluid loss (dehydration), low blood pressure, dizziness, or kidney problems may happen with the fever. Patients should be advised to call their health care provider right away if they get a fever.
Rash and other skin reactions are common side effects of TAFINLAR in combination with MEKINIST. In some cases, these rashes and other skin reactions can be severe or serious, may need to be treated in a hospital, or lead to death. Patients should be advised to call their health care provider if they get any of the following symptoms: blisters or peeling of skin, mouth sores, blisters on the lips or around the mouth or eyes, high fever or flu-like symptoms, and/or enlarged lymph nodes.
Some people may develop high blood sugar or worsening diabetes during treatment with TAFINLAR in combination with MEKINIST. For patients who are diabetic, their health care provider should check their blood sugar levels closely during treatment. Their diabetes medicine may need to be changed. Patients should be advised to tell their health care provider if they have increased thirst, urinate more often than normal, or produce an increased amount of urine.
TAFINLAR may cause healthy red blood cells to break down too early in people with glucose-6-phosphate dehydrogenase deficiency. This may lead to a type of anemia called hemolytic anemia, where the body does not have enough healthy red blood cells. Patients should be advised to tell their health care provider if they have yellow skin (jaundice), weakness or dizziness, or shortness of breath.
TAFINLAR, in combination with MEKINIST, can cause new or worsening high blood pressure (hypertension). A patient’s blood pressure should be checked during treatment. Patients should be advised to tell their health care provider if they develop high blood pressure, their blood pressure worsens, or if they have severe headache, lightheadedness, blurry vision, or dizziness.
For women of reproductive potential, TAFINLAR and MEKINIST, in combination, can harm your unborn baby. Your health care provider will do a test to see if you are pregnant before starting treatment with TAFINLAR and MEKINIST in combination. Use effective birth control (contraception) during treatment with TAFINLAR and MEKINIST in combination, and for 4 months after stopping treatment with TAFINLAR and MEKINIST.
Men (including those who have had a vasectomy) should use condoms during sexual intercourse during treatment with TAFINLAR and MEKINIST and for at least 4 months after the last dose of TAFINLAR and MEKINIST.
The most common side effects for patients with metastatic melanoma receiving the combination are pyrexia, nausea, rash, chills, diarrhea, headache, vomiting, hypertension, arthralgia, peripheral edema, and cough. The most common side effects for patients with stage III melanoma as adjuvant therapy receiving the combination are pyrexia, tiredness, nausea, headache, rash, chills, diarrhea, vomiting, arthralgia, and myalgia. The most common side effects for patients with NSCLC receiving the combination are pyrexia, tiredness, nausea, vomiting, diarrhea, dry skin, decreased appetite, edema, rash, chills, hemorrhage, cough, and dyspnea. The most common side effects for adults with solid tumors that cannot be removed by surgery or have spread to other parts of the body who are receiving the combination are fever, tiredness, nausea, rash, chills, headache, bleeding, cough, vomiting, constipation, diarrhea, muscle and or joint aches, and swelling of arms and legs. The most common side effects for children with solid tumors that cannot be removed by surgery or have spread to other parts of the body who are receiving the combination are fever, rash, vomiting, tiredness, dry skin, cough, diarrhea, acnearea, headache, stomach- (abdomen) pain, nausea, bleeding, constipation, and skin infection around fingernails or toenails. The most common side effects of in children 1 year of age and older with low-grade glioma receiving the combination include fever, rash, headache, vomiting, muscle and bone pain, tiredness, dry skin, diarrhea, nausea, bleeding, stomach area (abdomen) pain, and acne.
Please see full Prescribing Information for TAFINLAR and MEKINIST at https://www.novartis.us/sites/www.novartis.us/files/tafinlar.pdf and https://www.novartis.us/sites/www.novartis.us/files/mekinist.pdf.
About Novartis
Novartis is reimagining medicine to improve and extend people’s lives. We deliver high-value medicines that alleviate society’s greatest disease burdens through technology leadership in R&D and novel access approaches. In our quest to find new medicines, we consistently rank among the world’s top companies investing in research and development. About 106,000 people of more than 140 nationalities work together to bring Novartis products to nearly 800 million people around the world. Find out more at https://www.novartis.com
Mar. 17, 2023 5:45 AM ET
By: Ravikash, SA News Editor
The U.S. Food and Drug Administration (FDA) approved the expanded use of Novartis' (NYSE:NVS) Tafinlar plus Mekinist to treat children one year of age and older with low-grade glioma (LGG) having a BRAF V600E mutation, requiring systemic therapy.

Thursday, March 16, 2023 - 03:18pm View pdf
NEW YORK--(BUSINESS WIRE)-- Pfizer Inc. (NYSE: PFE) announced today that the U.S. Food and Drug Administration’s (FDA) Antimicrobial Drugs Advisory Committee (AMDAC) voted 16 to 1 that available data support the safety and effectiveness of PAXLOVID™ (nirmatrelvir tablets and ritonavir tablets) for the treatment of mild-to-moderate COVID-19 in adult patients who are at high risk for progression to severe illness. The AMDAC's vote, while not binding, will be considered by the FDA when making its decision regarding the potential approval of PAXLOVID.
“We believe it is critical for adults who are at high risk of progression to severe COVID-19 to have access to safe and effective treatment options, like PAXLOVID, to help prevent avoidable hospitalizations and deaths,” said James Rusnak, Senior Vice President and Chief Development Officer, Internal Medicine, Anti-infectives and Hospital, Pfizer. “We are encouraged by the AMDAC’s positive vote today. The outcome is well supported by the strong safety and efficacy data seen both in our clinical trials and in a growing base of real-world evidence, showing that PAXLOVID helps to reduce the risk of hospitalization or death for high-risk adult patients regardless of vaccination status.”
The AMDAC based its vote on the totality of scientific and real-world evidence shared by Pfizer, including safety and efficacy data from the EPIC (Evaluation of Protease Inhibition for COVID-19) clinical development program. This included results from the Phase 2/3 EPIC-HR study (Evaluation of Protease Inhibition for COVID-19 in High-Risk Patients), which enrolled unvaccinated, non-hospitalized adults, aged 18 years and older, with confirmed COVID-19 who are at increased risk of progressing to severe disease. The data showed an 86% reduction in risk of COVID-19-related hospitalization or death from any cause through Day 28 in patients treated with PAXLOVID within 5 days of symptoms onset, compared to placebo. The vote was further supported by results from a secondary endpoint of the Phase 2/3 EPIC-SR study (Evaluation of Protease Inhibition for COVID-19 in Standard-Risk Patients) which showed the effectiveness of Paxlovid in a sub-group of non-hospitalized adults, aged 18 years and older, with confirmed COVID-19 who had at least one risk factor for progression to severe disease and who were fully vaccinated.
In addition, real-world evidence presented to the AMDAC showed that PAXLOVID’s clinical profile in the post-authorization setting is consistent with the safety and efficacy conclusions from the EPIC clinical program, including observations made when the Omicron variant and its lineages were the predominant forms of SARS-CoV-2 in circulation. This real-world evidence also shows the effectiveness of PAXLOVID among vaccinated patients and patients who developed natural immunity.1,2,3
COVID-19 continues to cause significant burden in the U.S. as case rates fluctuate and new variants and sub-variants emerge, regardless of virulence. Approximately 4,000‒5,000 hospital admissions and 500‒600 deaths are caused by the virus each day in the U.S., as of January 2023.4 With more than 200 million adults in the U.S. at high risk of severe COVID-19, there is a critical need for treatment options in this population.5 According to the U.S. Centers for Disease Control and Prevention (CDC), factors which could put someone at high risk of severe COVID-19 include any of the following: being aged 50 and older, obesity, diabetes (type 1 and type 2), heart conditions, smoking (current or former), physical inactivity, chronic kidney or liver disease, and cancer, among others.6
If approved by the FDA, PAXLOVID could be the first U.S. FDA-approved oral treatment for COVID-19. The target Prescription Drug User Fee Act (PDUFA) action date for a decision by the FDA is May 2023. Under the FDA emergency use authorization (EUA), PAXLOVID is currently authorized for use in, and remains available to, adults and pediatric patients (12 years of age and older weighing at least 40 kg) at high risk of progression to severe COVID-19. In the U.S., more than 10 million treatment courses of PAXLOVID have been prescribed to date.7
Pfizer continues to gather pediatric data from the ongoing clinical trial EPIC-Peds (Evaluation of Protease Inhibition for COVID-19 in Pediatric Patients) and intends to submit a supplemental New Drug Application (NDA) to support the FDA approval of PAXLOVID in children at a future date. In February 2023, the European Commission (EC) granted standard Marketing Authorization (MA) of PAXLOVID for the treatment of COVID-19 in adults who do not require supplemental oxygen and who are at increased risk of the disease becoming severe.
About PAXLOVID™ (nirmatrelvir tablets and ritonavir tablets)
PAXLOVID is a SARS-CoV-2 main protease (Mpro) inhibitor (also known as SARS-CoV-2 3CL protease inhibitor) therapy. It was developed to be administered orally so that it can be prescribed early after infection, potentially helping patients avoid severe illness (which can lead to hospitalization and death). Nirmatrelvir, which originated in Pfizer laboratories, is designed to block the activity of the Mpro, an enzyme that the coronavirus needs to replicate. Co-administration with a low dose of ritonavir helps slow the metabolism, or breakdown, of nirmatrelvir in order for it to remain active in the body for longer periods of time at higher concentrations to help combat the virus.
Nirmatrelvir is designed to inhibit viral replication at a stage known as proteolysis, which occurs before viral RNA replication. In preclinical studies, nirmatrelvir did not demonstrate evidence of mutagenic DNA interactions.
Current variants of concern can be resistant to treatments that work by binding to the spike protein found on the surface of the SARS-CoV-2 virus. PAXLOVID, however, works intracellularly by binding to the highly conserved Mpro (3CL protease) of the SARS-CoV-2 virus to inhibit viral replication. Nirmatrelvir has shown consistent in vitro antiviral activity against the variants Alpha, Beta, Delta, Gamma, Lambda, Mu, and Omicron BA.1, BA.2, BA.2.12.1, BA.4, BA.4.6, BA.5, BF.7, BQ.1.11, BQ.1 and XBB.1.5. Work is ongoing to evaluate activity against recently identified variants as they become available for testing.
PAXLOVID is generally administered at a standard dose of 300 mg (two 150 mg tablets) of nirmatrelvir with one 100 mg tablet of ritonavir, taken together twice-daily for five days. One carton contains five blister packs of PAXLOVID, as co-packaged nirmatrelvir tablets with ritonavir tablets, providing all required doses for a full five-day treatment course. The dose for patients with moderate renal impairment (eGFR ≥30 to <60 mL/min) should be reduced to 150 mg nirmatrelvir (one 150 mg tablet) with 100 mg ritonavir (one 100 mg tablet), with both tablets taken together twice daily for five days (PAXLOVID is not recommended in patients with severe renal impairment [eGFR <30 mL/min]).
For more information, please visit www.PAXLOVID.com.
U.S. FDA Emergency Use Authorization Statement
PAXLOVID has not been approved, but has been authorized for emergency use by FDA under an EUA, for the treatment of adults and pediatric patients (12 years of age and older weighing at least 40 kg) with a current diagnosis of mild-to-moderate coronavirus disease 2019 (COVID-19) and who are at high risk for progression to severe COVID-19, including hospitalization or death.
The emergency use of PAXLOVID is only authorized for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of drugs and biological products during the COVID-19 pandemic under Section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the declaration is terminated or authorization revoked sooner.
AUTHORIZED USE
The U.S. Food and Drug Administration (FDA) has issued an Emergency Use Authorization (EUA) for the emergency use of the unapproved product PAXLOVID for the treatment of adults and pediatric patients (12 years of age and older weighing at least 40 kg) with a current diagnosis of mild-to-moderate coronavirus disease 2019 (COVID-19) and who are at high risk for progression to severe COVID-19, including hospitalization or death.
Please see Fact Sheet for Healthcare Providers and Fact Sheet for Patients, Parents, and Caregivers.
About Pfizer: Breakthroughs That Change Patients’ Lives
At Pfizer, we apply science and our global resources to bring therapies to people that extend and significantly improve their lives. We strive to set the standard for quality, safety and value in the discovery, development and manufacture of health care products, including innovative medicines and vaccines. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments and cures that challenge the most feared diseases of our time. Consistent with our responsibility as one of the world's premier innovative biopharmaceutical companies, we collaborate with health care providers, governments and local communities to support and expand access to reliable, affordable health care around the world. For more than 170 years, we have worked to make a difference for all who rely on us. We routinely post information that may be important to investors on our website at www.Pfizer.com. In addition, to learn more, please visit us on www.Pfizer.com and follow us on Twitter at @Pfizer and @Pfizer News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
View source version on businesswire.com: https://www.businesswire.com/news/home/20230316005635/en/
Source: Pfizer Inc.
Mar. 16, 2023 3:30 PM ET
By: Dulan Lokuwithana, SA News Editor5 Comments
Update 3.30 PM EST: Adds remarks from Pfizer (NYSE:PFE)

March 10, 2023 6:45 am ET
RAHWAY, N.J. & KINGSTON, Ontario--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside of the United States and Canada, and the Canadian Cancer Trials Group (CCTG) today announced that the Phase 2/3 CCTG IND.227/KEYNOTE-483 trial evaluating KEYTRUDA, Merck’s anti-PD-1 therapy, in combination with chemotherapy met its primary endpoint of overall survival (OS) for the first-line treatment of patients with unresectable advanced or metastatic malignant pleural mesothelioma. IND.227 was sponsored by CCTG, in collaboration with investigators in Italy (co-sponsored by National Cancer Institute of Naples - NCIN), and France (co-sponsored by The French Cooperative Thoracic Intergroup - IFCT); Merck provided KEYTRUDA and support for the trial. At the final analysis of the study, KEYTRUDA plus chemotherapy showed a statistically significant and clinically meaningful improvement in OS compared to chemotherapy alone in these patients. The safety profile of KEYTRUDA in combination with chemotherapy in this study was consistent with previously reported studies. Results will be presented at an upcoming medical meeting and discussed with regulatory authorities worldwide.
“Malignant pleural mesothelioma is a rapidly progressing cancer that develops in the lining of the lungs and has a poor prognosis,” said Dr. Eliav Barr, senior vice president, head of global clinical development and chief medical officer, Merck Research Laboratories. “Patients are in need of new treatments that can improve survival outcomes, and these positive results support the potential of KEYTRUDA in combination with chemotherapy as a first-line treatment for patients with the most common form of malignant mesothelioma.”
“There have been few treatment advances for patients with malignant pleural mesothelioma, which can be challenging to treat through surgery and radiation alone,” said Dr. Quincy Chu, CCTG’s study chair of the IND.227 trial/KEYNOTE-483 trial. “The results from the trial have the potential to make a difference for patients with this disease who have had limited treatment options available to them.”
Merck has an extensive clinical development program in lung cancer and is advancing multiple registration-enabling studies, with research directed at earlier stages of disease and novel combinations.
About IND.227/KEYNOTE-483
IND.227/KEYNOTE-483 is a randomized, open-label, randomized Phase 2/3 trial (ClinicalTrials.gov, NCT02784171) sponsored and conducted by the Canadian Cancer Trials Group (CCTG) in collaboration with National Cancer Institute of Naples (NCIN) and Intergroupe Francophone de Cancérologie Thoracique (IFCT). Support for the trial was provided by Merck. The trial evaluated KEYTRUDA in combination with chemotherapy for the treatment of patients with unresected advanced malignant pleural mesothelioma. The primary endpoint of the study is OS, and secondary endpoints include progression-free survival (PFS) and objective response rate (ORR) as assessed by blinded independent central review (BICR) per Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 modified for mesothelioma, safety and quality of life. The Phase 3 part of the trial enrolled 440 patients who were randomized to receive:
About malignant mesothelioma
Malignant mesothelioma is a type of cancer that starts in the linings of certain parts of the body, including the chest, abdomen, heart and testicles. Worldwide, it is estimated there were more than 30,000 new cases of malignant mesothelioma diagnosed and more than 26,000 deaths from the disease in 2020. Pleural mesothelioma, which develops in the lining of the lungs, is the most common form of malignant mesothelioma, accounting for about 75% of all cases. Malignant pleural mesothelioma often progresses rapidly, and the five-year survival rate is only 12%. Although incidence of malignant mesothelioma has gradually declined in the United States, continued use of and exposure to asbestos around the world has resulted in increasing global rates of this aggressive disease.
About the Canadian Cancer Trials Group
The Canadian Cancer Trials Group (CCTG) is a cancer clinical trials research cooperative that runs phase I-III trials to test anti-cancer and supportive therapies at over 85 hospitals and cancer centres across Canada. From their operations centre at Queen's University, CCTG has supported more than 600 trials enrolling 100,000 patients from 40 countries on 6 continents through a global network of 20,000 investigators and clinical trial staff. CCTG is a national program of the Canadian Cancer Society and their aim is to improve survival and quality of life for all people with cancer. CCTG IND.227 was supported by a grant from the Canadian Cancer Society (CCS) (707213). For further information, please visit the CCTG website: www.cctg.ca.
About KEYTRUDA® (pembrolizumab) injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.
Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [tumor proportion score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
KEYTRUDA, as a single agent, is indicated as adjuvant treatment following resection and platinum-based chemotherapy for adult patients with stage IB (T2a ≥4 cm), II, or IIIA NSCLC.
Additional Indications for KEYTRUDA in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of adult and pediatric (12 years and older) patients with stage IIB, IIC, or III melanoma following complete resection.
Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [Combined Positive Score (CPS) ≥1] as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy. KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Carcinoma
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
Non-muscle Invasive Bladder Cancer
KEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with MSI-H central nervous system cancers have not been established.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC) as determined by an FDA-approved test.
Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or gastroesophageal junction (GEJ) (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
KEYTRUDA, in combination with chemotherapy, with or without bevacizumab, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
KEYTRUDA is indicated for the adjuvant treatment of patients with RCC at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions.
Endometrial Carcinoma
KEYTRUDA, as a single agent, is indicated for the treatment of patients with advanced endometrial carcinoma that is MSI-H or dMMR, as determined by an FDA-approved test, who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation.
Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
Merck’s focus on cancer
Our goal is to translate breakthrough science into innovative oncology medicines to help people with cancer worldwide. At Merck, the potential to bring new hope to people with cancer drives our purpose and supporting accessibility to our cancer medicines is our commitment. As part of our focus on cancer, Merck is committed to exploring the potential of immuno-oncology with one of the largest development programs in the industry across more than 30 tumor types. We also continue to strengthen our portfolio through strategic acquisitions and are prioritizing the development of several promising oncology candidates with the potential to improve the treatment of advanced cancers. For more information about our oncology clinical trials, visit www.merck.com/clinicaltrials.
About Merck
At Merck, known as MSD outside of the United States and Canada, we are unified around our purpose: We use the power of leading-edge science to save and improve lives around the world. For more than 130 years, we have brought hope to humanity through the development of important medicines and vaccines. We aspire to be the premier research-intensive biopharmaceutical company in the world – and today, we are at the forefront of research to deliver innovative health solutions that advance the prevention and treatment of diseases in people and animals. We foster a diverse and inclusive global workforce and operate responsibly every day to enable a safe, sustainable and healthy future for all people and communities. For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf .
Source: Merck & Co., Inc.
Mar. 10, 2023 7:12 AM ET
By: Dulan Lokuwithana, SA News Editor1 Comment

Friday, March 10, 2023 - 06:45am
ZAVZPRET is the first and only calcitonin gene-related peptide (CGRP) receptor antagonist nasal sprayfor the acute treatment of migraine in adults
Expands Pfizer’s migraine portfolio, which includes an oral therapy for both acute and preventive treatment, to further meet the needs of people living with this debilitating disease
NEW YORK--(BUSINESS WIRE)-- Pfizer Inc. (NYSE: PFE) today announced the U.S. Food and Drug Administration (FDA) has approved ZAVZPRET™ (zavegepant), the first and only calcitonin gene-related peptide (CGRP) receptor antagonist nasal spray for the acute treatment of migraine with or without aura in adults. In its pivotal Phase 3 study, ZAVZPRET was statistically superior to placebo on the co-primary endpoints of pain freedom and freedom from most bothersome symptom at two hours post-dose. The pivotal study also demonstrated pain relief as early as 15 minutes in a prespecified secondary endpoint versus placebo.
“The FDA approval of ZAVZPRET marks a significant breakthrough for people with migraine who need freedom from pain and prefer alternative options to oral medications,” said Angela Hwang, Chief Commercial Officer, President, Global Biopharmaceuticals Business, Pfizer. “ZAVZPRET underscores Pfizer’s commitment to delivering an additional treatment option to help people with migraine gain relief and get back to their daily lives. Pfizer will continue to build its migraine franchise to further support the billions of people worldwide impacted by this debilitating disease.”
The FDA approval is based on two pivotal randomized, double-blind, placebo-controlled studies that established the efficacy, tolerability and safety profiles of ZAVZPRET for the acute treatment of migraine. In these studies, ZAVZPRET was statistically superior to placebo on the co-primary endpoints of pain freedom (defined as a reduction of moderate or severe headache pain to no headache pain) and freedom from most bothersome symptom at two hours post-dose (defined as the absence of the self-identified most bothersome symptom). The pivotal Phase 3 study published in The Lancet Neurology found ZAVZPRET showed broad efficacy by also demonstrating statistically significant superiority to placebo across 13 of 17 prespecified secondary outcome measures, including early time point endpoints (e.g., 15 and 30-minute pain relief and return to normal function at 30 minutes), return to normal function at 2 hours, and durable efficacy endpoints (e.g., 2-24 and 2-48 hour sustained pain freedom and sustained pain relief). On the 14th endpoint, return to normal function at 15 minutes post-dose, the difference between ZAVZPRET and placebo was not significant. Consequently, in keeping with the trial’s statistical analysis plan, the remaining secondary endpoints were not formally tested.
“When a migraine hits, it has a significant negative impact on a person’s daily life,” said Kathleen Mullin, M.D., Associate Medical Director at New England Institute for Neurology & Headache. “Among my migraine patients, one of the most important attributes of an acute treatment option is how quickly it works. As a nasal spray with rapid drug absorption, ZAVZPRET offers an alternative treatment option for people who need pain relief or cannot take oral medications due to nausea or vomiting, so they can get back to normal function quickly.”
ZAVZPRET was well tolerated in clinical trials. The most common adverse reactions reported in at least 2% of patients treated with ZAVZPRET and at a frequency greater than placebo were taste disorders (includes dysgeusia and ageusia), nausea, nasal discomfort and vomiting. ZAVZPRET is contraindicated in patients with a history of hypersensitivity to zavegepant or to any of its components. Hypersensitivity reactions, including facial swelling and urticaria, have occurred with ZAVZPRET in clinical studies.
ZAVZPRET is anticipated to be available in pharmacies in July 2023.
About Migraine
Nearly 40 million people in the United States suffer from migraine1 and the World Health Organization classifies migraine as the second leading cause of disability in the world.2 Migraine is characterized by debilitating attacks lasting four to 72 hours with multiple symptoms, including pulsating headaches of moderate to severe pain intensity often associated with nausea or vomiting, and/or sensitivity to sound (phonophobia) and sensitivity to light (photophobia).3
About CGRP Receptor Antagonism
Small molecule CGRP receptor antagonists represent a novel class of drugs for the treatment of migraine. For acute treatment, this unique mode of action offers an alternative to other agents, including those patients who have contraindications to the use of triptans or who have a poor response to triptans or are intolerant to them. CGRP signal-blocking therapies have not been associated with medication overuse headache (MOH) or rebound headache, which can limit the clinical utility of other acute treatments.
About ZAVZPRET
Zavegepant is a third generation, high affinity, selective and structurally unique, small molecule CGRP receptor antagonist and the only CGRP receptor antagonist in clinical development with both intranasal and oral formulations.
INDICATION
ZAVZPRET™ (zavegepant) is indicated for the acute treatment of migraine with or without aura in adults.
Limitations of Use: ZAVZPRET is not indicated for the preventive treatment of migraine.
Please click here for full Prescribing Information.
About Pfizer: Breakthroughs That Change Patients’ Lives
At Pfizer, we apply science and our global resources to bring therapies to people that extend and significantly improve their lives. We strive to set the standard for quality, safety and value in the discovery, development and manufacture of health care products, including innovative medicines and vaccines. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments and cures that challenge the most feared diseases of our time. Consistent with our responsibility as one of the world's premier innovative biopharmaceutical companies, we collaborate with health care providers, governments and local communities to support and expand access to reliable, affordable health care around the world. For more than 170 years, we have worked to make a difference for all who rely on us. We routinely post information that may be important to investors on our website at www.pfizer.com. In addition, to learn more, please visit us on and follow us on Twitter at @Pfizer and @Pfizer News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
View source version on businesswire.com: https://www.businesswire.com/news/home/20230309005795/en/
Source: Pfizer Inc.
Mar. 10, 2023 7:01 AM ET
By: Ravikash, SA News Editor13 Comments

FDA Advisory Committee votes in favour of the clinical benefit of Roche’s Polivy combination for people with previously untreated diffuse large B-cell lymphoma
March 09, 2023
Basel, 10 March 2023 - Roche (SIX: RO, ROG; OTCQX: RHHBY) announced today that the U.S. Food and Drug Administration (FDA) Oncologic Drugs Advisory Committee (ODAC) voted 11 to 2 in favour of Polivy® (polatuzumab vedotin-piiq) in combination with Rituxan® (rituximab) plus cyclophosphamide, doxorubicin and prednisone (R-CHP) for the treatment of people with previously untreated diffuse large B-cell lymphoma (DLBCL). The ODAC provides the FDA with independent opinions and recommendations from outside medical experts though the recommendations are not binding. The FDA is expected to make a final decision on its review of the supplemental Biologics License Application (sBLA) for Polivy in this indication by 2 April 2023.
“Today’s committee decision to recognise the potential of this Polivy combination as a first-line treatment option is important since four in ten people with diffuse large B-cell lymphoma relapse or do not respond to initial treatment,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “We believe the clinical benefit demonstrated in the POLARIX study may improve outcomes for many people with newly diagnosed DLBCL and look forward to continued collaboration with the FDA to make this treatment option available in the US.”
More than 60 countries have approved this Polivy combination for the treatment of adult patients with previously untreated DLBCL, including in the EU, UK, Japan, Canada and China. Polivy in combination with R-CHP was recently added to the National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology (NCCN Guidelines®) as a category 1, preferred regimen for first-line DLBCL.
DLBCL is an aggressive, hard-to-treat disease and is the most common form of non-Hodgkin lymphoma in the US. Limited progress has been made in improving patient outcomes in previously untreated DLBCL over the last two decades. Polivy in combination with R-CHP is the first treatment in 20 years to show a significant improvement in progression-free survival (PFS) over the standard of care, MabThera/Rituxan in combination with cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP), in this setting.
The sBLA submission is based on pivotal data from the phase III POLARIX study, which demonstrated a statistically significant and clinically meaningful improvement in PFS with Polivy plus R-CHP compared to standard-of-care R-CHOP in first-line DLBCL. The risk of disease progression, relapse or death was reduced by 27% with Polivy plus R-CHP compared with R-CHOP (hazard ratio [HR] 0.73; 95% confidence interval [CI]: 0.57–0.95; p<0.02). Safety outcomes were consistent with those seen in previous clinical trials, and the safety profile was comparable for Polivy plus R-CHP versus R-CHOP, including rates of Grade 3-4 adverse events (AEs; 57.7% versus 57.5%), serious AEs (34.0% versus 30.6%), Grade 5 AEs (3.0% versus 2.3%), and AEs leading to dose reduction (9.2% versus 13.0%).
Polivy in combination with bendamustine and MabThera/Rituxan is currently approved in more than 80 countries worldwide for the treatment of adults with relapsed or refractory DLBCL after one or more prior therapies, including in the US under FDA accelerated approval, as a readily available, fixed-duration treatment option .
About the POLARIX study
POLARIX [NCT03274492] is an international phase III, randomised, double-blind, placebo-controlled study evaluating the efficacy, safety and pharmacokinetics of Polivy® (polatuzumab vedotin-piiq) plus MabThera®/Rituxan® (rituximab), cyclophosphamide, doxorubicin and prednisone (R-CHP) versus MabThera/Rituxan, cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP) in people with previously untreated diffuse large B-cell lymphoma. Eight-hundred and seventy-nine patients were randomised 1:1 to receive either Polivy plus R-CHP plus a vincristine placebo for six cycles, followed by MabThera/Rituxan for two cycles; or R-CHOP plus a Polivy placebo for six cycles, followed by two cycles of MabThera/Rituxan. The primary outcome measure is progression-free survival as assessed by the investigator using the Lugano Response Criteria for malignant lymphoma. POLARIX is being conducted in collaboration with The Lymphoma Study Association and The Lymphoma Academic Research Organisation.
About diffuse large B-cell lymphoma
Diffuse large B-cell lymphoma (DLBCL) is the most common form of non-Hodgkin lymphoma (NHL), accounting for about one in three cases of NHL.1 DLBCL is an aggressive (fast-growing) type of NHL.1 While it is generally responsive to treatment in the frontline, as many as 40% of people will relapse or have refractory disease, at which time salvage therapy options are limited and survival is short.2,3 Approximately 160,000 people worldwide are estimated to be diagnosed with DLBCL each year.4
About Polivy® (polatuzumab vedotin-piiq)
Polivy is a first-in-class anti-CD79b antibody-drug conjugate (ADC). The CD79b protein is expressed specifically in the majority of B-cells, an immune cell impacted in some types of non-Hodgkin lymphoma (NHL), making it a promising target for the development of new therapies. Polivy binds to cancer cells such as CD79b and destroys these B-cells through the delivery of an anti-cancer agent, which is thought to minimise the effects on normal cells. Polivy is being developed by Roche using Seagen ADC technology and is currently being investigated for the treatment of several types of NHL.
https://www.polivy.com/
About Roche in haematology
Roche has been developing medicines for people with malignant and non-malignant blood diseases for more than 20 years; our experience and knowledge in this therapeutic area runs deep. Today, we are investing more than ever in our effort to bring innovative treatment options to patients across a wide range of haematologic diseases. Our approved medicines include MabThera®/Rituxan® (rituximab), Gazyva®/Gazyvaro® (obinutuzumab), Polivy® (polatuzumab vedotin-piiq), Venclexta®/Venclyxto® (venetoclax) in collaboration with AbbVie, Hemlibra® (emicizumab) and Lunsumio® (mosunetuzumab). Our pipeline of investigational haematology medicines includes T-cell engaging bispecific antibodies glofitamab, targeting both CD20 and CD3, and cevostamab, targeting both FcRH5 and CD3; Tecentriq® (atezolizumab), a monoclonal antibody designed to bind with PD-L1 and crovalimab, an anti-C5 antibody engineered to optimise complement inhibition. Our scientific expertise, combined with the breadth of our portfolio and pipeline, also provides a unique opportunity to develop combination regimens that aim to improve the lives of patients even further.
About Roche
Founded in 1896 in Basel, Switzerland, as one of the first industrial manufacturers of branded medicines, Roche has grown into the world’s largest biotechnology company and the global leader in in-vitro diagnostics. The company pursues scientific excellence to discover and develop medicines and diagnostics for improving and saving the lives of people around the world. We are a pioneer in personalised healthcare and want to further transform how healthcare is delivered to have an even greater impact. To provide the best care for each person we partner with many stakeholders and combine our strengths in Diagnostics and Pharma with data insights from the clinical practice.
In recognising our endeavour to pursue a long-term perspective in all we do, Roche has been named one of the most sustainable companies in the pharmaceuticals industry by the Dow Jones Sustainability Indices for the thirteenth consecutive year. This distinction also reflects our efforts to improve access to healthcare together with local partners in every country we work.
Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan.
For more information, please visit www.roche.com.
All trademarks used or mentioned in this release are protected by law.
NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibilities for their application or use in any way.
Mar. 10, 2023 5:22 AM ET
Roche Holding AG (RHHBY), RHHBF
By: Ravikash, SA News Editor
Roche (OTCQX:RHHBY) (OTCQX:RHHBF) said a panel of the U.S. Food and Drug Administration (FDA) voted in favor of approval of a combination treatment consisting of its medicine Polivy to treat a type of blood cancer.

PUBLISHED9 March 2023 9 March 2023 07:00 GMT
Positive high-level results from a planned interim analysis of the AEGEAN Phase III, placebo- controlled trial showed that treatment with AstraZeneca’s Imfinzi (durvalumab) in combination with neoadjuvant chemotherapy before surgery and as adjuvant monotherapy after surgery demonstrated a statistically significant and clinically meaningful improvement in event-free survival (EFS) versus neoadjuvant chemotherapy alone followed by surgery for patients with resectable early-stage (IIA-IIIB) non-small cell lung cancer (NSCLC).
Results from the final pathologic complete response (pCR) and major pathologic response (mPR) analyses were consistent with previously announced positive results. The trial will continue as planned to assess key secondary endpoints including disease-free survival (DFS) and overall survival (OS).
Each year there are an estimated 2.2 million people diagnosed with lung cancer globally with 80-85% of patients diagnosed with NSCLC, the most common form of lung cancer.1-3 Approximately 25-30% of all patients with NSCLC are diagnosed early enough to have surgery with curative intent.4-5 However, only around 56-65% of patients with Stage II disease will survive for five years.6 This decreases to 41% for patients with Stage IIIA and 24% for patients with Stage IIIB disease, reflecting a high unmet medical need.6
John V. Heymach, MD, PhD. Professor and Chair Thoracic/Head and Neck Medical Oncology, The University of Texas MD Anderson Cancer Center, said: “Treating patients early with durvalumab both before and after surgery delivers a significant and clinically meaningful benefit in resectable non-small cell lung cancer, where new options are urgently needed to offer patients the best chance of long-term survival. The AEGEAN results provide compelling evidence that this novel durvalumab regimen can drive improved outcomes in this curative-intent setting.”
Susan Galbraith, Executive Vice President, Oncology R&D, AstraZeneca, said: “Patients with resectable non-small cell lung cancer face unacceptably high rates of recurrence, despite treatment with chemotherapy and surgery. We have shown that adding Imfinzi both before and after surgery significantly increased the time patients live without recurrence or progression events. We will continue to follow patients for overall survival.”
Imfinzi was well tolerated and no new safety concerns were observed in the neoadjuvant and adjuvant settings. Further, adding Imfinzi to neoadjuvant chemotherapy was consistent with the known profile for this combination and did not increase complications or adverse events, or compromise patients' ability to undergo surgery versus chemotherapy alone.
These data will be presented at a forthcoming medical meeting and shared with global health authorities.
AstraZeneca has a comprehensive portfolio of approved and potential new medicines in development for patients with lung cancer. In addition to these results, the Company is also announcing today that Tagrisso (osimertinib) met a secondary endpoint of OS in the ADAURA Phase III trial in early-stage (IB, II and IIIA) epidermal growth factor receptor-mutated (EGFRm) NSCLC after complete tumour resection with curative intent.
Notes
Lung cancer
Lung cancer is the leading cause of cancer death among both men and women, accounting for about one-fifth of all cancer deaths.1 Lung cancer is broadly split into NSCLC and small cell lung cancer (SCLC).2 The majority of NSCLC patients are diagnosed with advanced disease while approximately 25-30% present with resectable disease at diagnosis.4-5 Early-stage lung cancer diagnoses are often only made when the cancer is found on imaging for an unrelated condition.7-8
For patients with resectable tumours, the majority eventually develop recurrence despite complete tumour resection and adjuvant chemotherapy.9
AEGEAN
AEGEAN is a randomised, double-blind, multi-centre, placebo-controlled global Phase III trial evaluating Imfinzi as perioperative treatment for patients with resectable Stage IIA-IIIB (Eighth Edition AJCC Cancer Staging Manual) NSCLC, irrespective of PD-L1 expression. Perioperative therapy includes treatment before and after surgery, also known as neoadjuvant/adjuvant therapy. In the trial, 802 patients were randomised to receive a 1500mg fixed dose of Imfinzi plus chemotherapy or placebo plus chemotherapy every three weeks for four cycles prior to surgery, followed by Imfinzi or placebo every four weeks (for up to 12 cycles) after surgery. Patients with known EGFR or ALK genomic tumour aberrations were excluded from the primary efficacy analyses.
In the AEGEAN trial, the primary endpoints were pCR, defined as no viable tumour in the resection specimen (including lymph nodes) following neoadjuvant therapy, and EFS, defined as the time from randomisation to an event like tumour recurrence, progression precluding definitive surgery, or death. Key secondary endpoints were mPR, defined as residual viable tumour of less than or equal to 10% in the resected primary tumour following neoadjuvant therapy, DFS, OS, safety and quality of life. The final pathologic response analyses were performed after all patients had the opportunity for surgery and pathology assessment per the trial protocol. The trial enrolled participants in 264 centres in more than 25 countries including in the US, Canada, Europe, South America and Asia.
Imfinzi
Imfinzi (durvalumab) is a human monoclonal antibody that binds to the PD-L1 protein and blocks the interaction of PD-L1 with the PD-1 and CD80 proteins, countering the tumour's immune-evading tactics and releasing the inhibition of immune responses.
Imfinzi is the only approved immunotherapy and the global standard of care in the curative-intent setting of unresectable, Stage III NSCLC in patients whose disease has not progressed after chemoradiation therapy based on the PACIFIC Phase III trial.
Imfinzi is also approved in the US, EU, Japan, China and many other countries around the world for the treatment of extensive-stage SCLC based on the CASPIAN Phase III trial. In an exploratory analysis in 2021, updated results from the CASPIAN trial showed Imfinzi plus chemotherapy tripled patient survival at three years versus chemotherapy alone. Additionally, Imfinzi is approved in combination with a short course of Imjudo (tremelimumab) and chemotherapy for the treatment of metastatic NSCLC in the US, EU and Japan based on the POSEIDON Phase III trial.
In addition to its indications in lung cancer, Imfinzi is also approved in combination with chemotherapy in locally advanced or metastatic biliary tract cancer in the US, EU, Japan and several other countries; in combination with Imjudo in unresectable hepatocellular carcinoma in the US, EU and Japan; and in previously treated patients with advanced bladder cancer in several countries.
Since the first approval in May 2017, more than 150,000 patients have been treated with Imfinzi.
AstraZeneca has several ongoing registrational trials focused on testing Imfinzi in earlier stages of lung cancer, including in resectable NSCLC (ADJUVANT BR.31) and unresectable NSCLC (PACIFIC-2, 4, 5, 8 and 9), and in limited-stage SCLC (ADRIATIC).
As part of a broad development programme, Imfinzi is being tested as a single treatment and in combinations with other anti-cancer treatments for patients with SCLC, NSCLC, bladder cancer, several gastrointestinal (GI) cancers, ovarian cancer, endometrial cancer and other solid tumours.
AstraZeneca in lung cancer
AstraZeneca is working to bring patients with lung cancer closer to cure through the detection and treatment of early-stage disease, while also pushing the boundaries of science to improve outcomes in the resistant and advanced settings. By defining new therapeutic targets and investigating innovative approaches, the Company aims to match medicines to the patients who can benefit most.
The Company's comprehensive portfolio includes leading lung cancer medicines and the next wave of innovations, including Tagrisso and Iressa (gefitinib); Imfinzi and Imjudo; Enhertu (trastuzumab deruxtecan) and datopotamab deruxtecan in collaboration with Daiichi Sankyo; Orpathys (savolitinib) in collaboration with HUTCHMED; as well as a pipeline of potential new medicines and combinations across diverse mechanisms of action.
AstraZeneca is a founding member of the Lung Ambition Alliance, a global coalition working to accelerate innovation and deliver meaningful improvements for people with lung cancer, including and beyond treatment.
AstraZeneca in immuno-oncology (IO)
AstraZeneca is a pioneer in introducing the concept of immunotherapy into dedicated clinical areas of high unmet medical need. The Company has a comprehensive and diverse IO portfolio and pipeline anchored in immunotherapies designed to overcome evasion of the anti-tumour immune response and stimulate the body’s immune system to attack tumours.
AstraZeneca aims to reimagine cancer care and help transform outcomes for patients with Imfinzi as a single treatment and in combination with Imjudo as well as other novel immunotherapies and modalities. The Company is also exploring next-generation immunotherapies like bispecific antibodies and therapeutics that harness different aspects of immunity to target cancer.
AstraZeneca is boldly pursuing an innovative clinical strategy to bring IO-based therapies that deliver long-term survival to new settings across a wide range of cancer types. With an extensive clinical programme, the Company also champions the use of IO treatment in earlier disease stages, where there is the greatest potential for cure.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Mar. 09, 2023 4:46 AM ET
By: Ravikash, SA News Editor
AstraZeneca (NASDAQ:AZN) said data from a phase 3 trial showed that Imfinzi-based therapy before and after surgery significantly increased the time patients with lung cancer lived without the disease recurring or progressing.
The late stage study, dubbed AEGEAN, is evaluating Imfinzi (durvalumab) as a perioperative therapy for patients with resectable stage 2A-3B non-small cell lung cancer (NSCLC), irrespective of PD-L1 expression.

PUBLISHED9 March 2023
Positive high-level results from the ADAURA Phase III trial showed AstraZeneca’s Tagrisso (osimertinib) demonstrated a statistically significant and clinically meaningful improvement in overall survival (OS), a key secondary endpoint, compared to placebo in the adjuvant treatment of patients with early-stage (IB, II and IIIA) epidermal growth factor receptor-mutated (EGFRm) non-small cell lung cancer (NSCLC) after complete tumour resection with curative intent.
In May 2020, AstraZeneca announced Tagrisso demonstrated a statistically significant and clinically meaningful improvement in disease-free survival (DFS) in this setting. In September 2022, updated results demonstrated a median DFS of nearly five and a half years.
Per the ADAURA trial protocol, patients on placebo that recurred with metastatic disease had the opportunity to receive open-label Tagrisso.
Roy S. Herbst, MD, PhD, Deputy Director and Chief of Medical Oncology at Yale Cancer Center and Smilow Cancer Hospital, New Haven, Connecticut, and principal investigator in the ADAURA Phase III trial, said: “These new survival data for osimertinib reinforce the unprecedented ADAURA disease-free survival results and confirm its potential to extend patients’ lives in early-stage disease. The ADAURA results provide powerful evidence that osimertinib offers the best possible care for patients with early-stage EGFR-mutated non-small cell lung cancer who historically faced high rates of recurrence and previously had no targeted options after surgery.”
Susan Galbraith, Executive Vice President, Oncology R&D, AstraZeneca, said: “The ADAURA trial brought the first targeted medicine to patients with early-stage EGFR-mutated non-small cell lung cancer. Today, these exciting overall survival results validate adjuvant Tagrisso as the standard of care in this setting and reinforce the importance of early diagnosis and testing for EGFR mutation in lung cancer.”
The safety and tolerability of Tagrisso in the ADAURA trial were consistent with its established profile and no new safety concerns were reported.
These new ADAURA OS results in the early-stage resectable setting add to the extensive body of evidence for Tagrisso in EGFRm NSCLC which has now shown a statistically significant and clinically meaningful OS benefit in both the early adjuvant and late-stage metastatic settings. The data will be presented at a forthcoming medical meeting.
Each year there are an estimated 2.2 million people diagnosed with lung cancer globally with 80-85% of patients diagnosed with NSCLC, the most common form of lung cancer.1-3 Approximately 25-30% of all patients with NSCLC are diagnosed early enough to have surgery with curative intent.4‑5 Further, 73% of patients with Stage IB and 56-65% of patients with Stage II disease will survive for five years.6 This decreases to 41% for patients with Stage IIIA and 24% for patients with Stage IIIB disease, reflecting a high unmet medical need.6
AstraZeneca has several ongoing registrational trials focused on testing Tagrisso in earlier stages of lung cancer, including in the neoadjuvant resectable setting (NeoADAURA), in the Stage IA2-IA3 adjuvant resectable setting (ADAURA2), and in the Stage III locally advanced unresectable setting (LAURA).
Tagrisso is approved to treat early-stage lung cancer in more than 90 countries, including in the US, EU, China and Japan, and additional global regulatory reviews are ongoing. Tagrisso is also approved for the 1st-line treatment of patients with locally advanced or metastatic EGFRm NSCLC and for the treatment of locally advanced or metastatic EGFR T790M mutation-positive NSCLC in the US, EU, China, Japan and many other countries.
AstraZeneca has a comprehensive portfolio of approved and potential new medicines in development for patients with lung cancer. In addition to these results, the Company has also announced today positive results from the AEGEAN Phase III trial of Imfinzi (durvalumab) in combination with neoadjuvant chemotherapy before surgery and as adjuvant monotherapy after surgery in Stage IIA-IIIB resectable NSCLC.
Notes
Lung cancer
Lung cancer is the leading cause of cancer death among both men and women, accounting for about one-fifth of all cancer deaths.1 Lung cancer is broadly split into NSCLC and small cell lung cancer.2 The majority of all NSCLC patients are diagnosed with advanced disease while approximately 25-30% present with resectable disease at diagnosis.4‑5 Early-stage lung cancer diagnoses are often only made when the cancer is found on imaging for an unrelated condition.7‑8
For patients with resectable tumours, the majority eventually develop recurrence despite complete tumour resection and adjuvant chemotherapy.9
Approximately 10-15% of NSCLC patients in the US and Europe, and 30-40% of patients in Asia have EGFRm NSCLC.10-12 These patients are particularly sensitive to treatment with an EGFR-tyrosine kinase inhibitor (EGFR-TKI) which block the cell-signalling pathways that drive the growth of tumour cells.13
ADAURA
ADAURA was a randomised, double-blind, placebo-controlled, global Phase III trial in the adjuvant treatment of 682 patients with Stage IB, II, IIIA EGFRm NSCLC following complete tumour resection and, at physicians’ and patients’ discretion, adjuvant chemotherapy. Patients were treated with Tagrisso 80mg once-daily oral tablets or placebo for three years or until disease recurrence.
The trial was enrolled in more than 200 centres across more than 20 countries, including the US, Europe, South America, Asia and the Middle East. The primary endpoint was DFS in Stage II and IIIA patients and key secondary endpoints included DFS in Stage IB, II and IIIA patients, and OS in both the primary and overall populations.
Though the primary data readout was originally anticipated in 2022, data from the trial were reported early following a recommendation from an Independent Data Monitoring Committee (IDMC) based on its determination of overwhelming efficacy.
Tagrisso
Tagrisso (osimertinib) is a third-generation, irreversible EGFR-TKI with proven clinical activity in NSCLC, including against central nervous system metastases. Tagrisso (40mg and 80mg once-daily oral tablets) has been used to treat nearly 700,000 patients across its indications worldwide and AstraZeneca continues to explore Tagrisso as a treatment for patients across multiple stages of EGFRm NSCLC.
In addition to investigating Tagrisso in early-stage disease, AstraZeneca is also studying the medicine in combination with chemotherapy in locally advanced and metastatic EGFRm NSCLC (FLAURA2). The Company is also researching ways to address tumour mechanisms of resistance through the SAVANNAH and ORCHARD Phase II trials, and the SAFFRON Phase III trial, which test Tagrisso given concomitantly with savolitinib, an oral, potent and highly selective MET TKI, as well as other potential new medicines.
AstraZeneca in lung cancer
AstraZeneca is working to bring patients with lung cancer closer to cure through the detection and treatment of early-stage disease, while also pushing the boundaries of science to improve outcomes in the resistant and advanced settings. By defining new therapeutic targets and investigating innovative approaches, the Company aims to match medicines to the patients who can benefit most.
The Company’s comprehensive portfolio includes leading lung cancer medicines and the next wave of innovations, including Tagrisso (osimertinib) and Iressa (gefitinib); Imfinzi (durvalumab) and Imjudo (tremelimumab); Enhertu (trastuzumab deruxtecan) and datopotamab deruxtecan in collaboration with Daiichi Sankyo; Orpathys (savolitinib) in collaboration with HUTCHMED; as well as a pipeline of potential new medicines and combinations across diverse mechanisms of action.
AstraZeneca is a founding member of the Lung Ambition Alliance, a global coalition working to accelerate innovation and deliver meaningful improvements for people with lung cancer, including and beyond treatment.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company’s focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Mar. 09, 2023 5:25 AM ET
By: Ravikash, SA News Editor1 Comment
AstraZeneca (NASDAQ:AZN) said Tagrisso showed survival benefit in patients with a type of lung cancer who had undergone surgery, in a phase 3 trial.
March 8, 2023Download PDF
RIDGEFIELD, Conn. and INDIANAPOLIS, March 8, 2023 /PRNewswire/ -- The U.S. Food and Drug Administration (FDA) accepted a supplemental New Drug Application (sNDA) for Jardiance® (empagliflozin) investigating a potential new indication to lower blood sugar along with diet and exercise in children 10 years and older with type 2 diabetes, Boehringer Ingelheim and Eli Lilly and Company (NYSE: LLY) announced.
"There are clear unmet needs for young people living with type 2 diabetes, which has nearly doubled in prevalence in people aged 10-19 over the past two decades," said Mohamed Eid, M.D., M.P.H., M.H.A., vice president, Clinical Development & Medical Affairs, Cardio-Renal-Metabolism & Respiratory Medicine, Boehringer Ingelheim Pharmaceuticals, Inc. "We look forward to working closely with the FDA during the review process and while we await a decision on our efforts to bring another potential treatment option to children 10 years and older with type 2 diabetes."
The sNDA is based on the results from the DINAMO phase III trial, in which Jardiance was associated with a statistically significant reduction in the primary endpoint of change from baseline in A1c at 26 weeks compared with placebo in participants aged 10-17 years with type 2 diabetes. When added to other baseline treatments (diet, exercise, metformin and/or insulin), Jardiance 10 mg and 25 mg pooled doses reduced A1c by 0.84% compared with placebo at week 26 (95% CI −1.50 to −0.19; P=0.012). Reduction in A1c in participants treated with Tradjenta® (linagliptin) was not statistically significant when compared with placebo. A numerical reduction of 0.34% (P=0.2935) was observed. Results were presented during the International Diabetes Federation World Diabetes Congress 2022.
Overall, the safety data in DINAMO was consistent with the previously known safety profile of Jardiance.
Initially approved in 2014, Jardiance is a once-daily tablet used along with diet and exercise to lower blood sugar in adults with type 2 diabetes; and to reduce the risk of cardiovascular death in adults with type 2 diabetes and known cardiovascular disease. Jardiance is also indicated to reduce the risk of cardiovascular death and hospitalization for heart failure in adults with heart failure. Jardiance is not for patients with type 1 diabetes, or to improve glycemic control in adults with type 2 diabetes with an eGFR <30 mL/min/1.73 m2. Jardiance is contraindicated in people with hypersensitivity to empagliflozin or any of the excipients in Jardiance, and in patients on dialysis. Please see additional Important Safety Information below.
"Impacting an estimated 39,000 people under the age of 20, type 2 diabetes is a growing health issue for young people in the U.S.," said Jeff Emmick, M.D., Ph.D., vice president, Product Development, Lilly. "If approved, Jardiance would be a new oral treatment option for type 2 diabetes in children 10 years and older in the U.S., making this application acceptance an important step forward as we add to the body of knowledge for this vulnerable patient population, for whom oral treatment options have been limited."
About DINAMO: DIabetes study of liNAgliptin and eMpagliflozin in children and adOlescents
DINAMO (NCT03429543) is a multicenter, randomized, double-blind, parallel group phase III trial that enrolled participants aged 10-17 years with type 2 diabetes (A1c 6.5% to 10.5%) previously treated with metformin or insulin. The primary endpoint was change from baseline in A1c at 26 weeks. Of the 262 participants screened, 158 were randomly assigned to treatment with Jardiance (10 mg) (n=52), Tradjenta (5 mg) (n=53) or placebo (n=53) once daily. Participants in the Jardiance group who did not have A1c below 7.0% by week 12 were re-randomized to either remain on 10 mg or increase to 25 mg. Participants in the placebo group were reassigned at week 26 to Tradjenta 5 mg or Jardiance 10 mg or 25 mg. All participants were treated with diet and exercise plus metformin and/or insulin (or no background if metformin intolerant). Safety was assessed until week 52.
What is JARDIANCE?
JARDIANCE is a prescription medicine used to:
JARDIANCE is not for people with type 1 diabetes. It may increase their risk of diabetic ketoacidosis (increased ketones in the blood or urine).
JARDIANCE is not for use to lower blood sugar in adults with type 2 diabetes who have severe kidney problems, because it may not work.
IMPORTANT SAFETY INFORMATION
Do not take JARDIANCE if you are allergic to empagliflozin or any of the ingredients in JARDIANCE.
Do not take JARDIANCE if you are on dialysis.
For more information, please see Prescribing Information and Medication Guide.
Boehringer Ingelheim and Eli Lilly and Company
In January 2011, Boehringer Ingelheim and Eli Lilly and Company announced an Alliance that centers on compounds representing several of the largest diabetes treatment classes. Depending on geographies, the companies either co-promote or separately promote the respective molecules each contributing to the Alliance. The Alliance leverages the strengths of two of the world's leading pharmaceutical companies to focus on patient needs. By joining forces, the companies demonstrate their commitment, not only to the care of people with diabetes, but also to investigating the potential to address areas of unmet medical need.
About Boehringer Ingelheim
Boehringer Ingelheim is working on breakthrough therapies that improve the lives of humans and animals. As a leading research-driven biopharmaceutical company, the company creates value through innovation in areas of high unmet medical need. Founded in 1885 and family-owned ever since, Boehringer Ingelheim takes a long-term perspective. Around 52,000 employees serve more than 130 markets in the three business areas, Human Pharma, Animal Health, and Biopharmaceutical Contract Manufacturing. Learn more at www.boehringer-ingelheim.com/us.
About Lilly
Lilly unites caring with discovery to create medicines that make life better for people around the world. We've been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world's most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer's disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we're motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram and LinkedIn.
![]()
View original content to download multimedia:https://www.prnewswire.com/news-releases/us-fda-accepts-supplemental-new-drug-application-for-jardiance-for-children-10-years-and-older-with-type-2-diabetes-301765746.html
SOURCE Eli Lilly and Company
Mar. 08, 2023 9:38 AM ET
By: Ravikash, SA News Editor
PUBLISHED6 March 2023 06 March 2023 07:00 GMT
Positive high-level results from an analysis of the ongoing DESTINY-PanTumor02 Phase II trial showed AstraZeneca and Daiichi Sankyo’s Enhertu (trastuzumab deruxtecan) met the prespecified target for objective response rate (ORR) and demonstrated durable response across multiple HER2-expressing advanced solid tumours in heavily pretreated patients.
Enhertu is a specifically engineered HER2-directed antibody drug conjugate (ADC) being jointly developed and commercialised by AstraZeneca and Daiichi Sankyo.
The DESTINY-PanTumor02 Phase II trial is evaluating the efficacy and safety of Enhertu in patients with locally advanced, unresectable, or metastatic previously treated, HER2-expressing solid tumours not eligible for curative therapy, including biliary tract, bladder, cervical, endometrial, ovarian, pancreatic, and rare cancers. The primary endpoint of the trial is investigator-assessed confirmed ORR and investigator-assessed duration of response (DoR) is a key secondary endpoint.
The data will be presented at an upcoming medical meeting and shared with global regulatory authorities.
HER2 is a tyrosine kinase receptor protein expressed on the surface of various tissue cells throughout the body and is involved in normal cell growth.1,2 In some cancer cells, HER2 expression is amplified or the cells have activating mutations.1,3 While HER2-directed therapies have been used to treat breast, gastric and lung cancers, more research is needed evaluating their potential role in treating other HER2-expressing tumour types.2,4-6
Cristian Massacesi, Chief Medical Officer and Oncology Chief Development Officer, AstraZeneca, said, “Enhertu has already demonstrated its potential to improve outcomes for patients with HER2-targetable breast, gastric and lung cancers, and these positive initial results in other tumour settings with significant unmet need are very encouraging. The DESTINY-PanTumor02 results mark an important step forward in our understanding of the potential role of Enhertu across multiple HER2-expressing tumour types.”
Ken Takeshita, Global Head, R&D, Daiichi Sankyo, said, “The clinically meaningful responses seen in the DESTINY-PanTumor02 trial reaffirm our belief in the potential of Enhertu across multiple HER2-expressing cancers. The results seen so far across multiple cohorts of the trial will inform next steps of our broad development programme as we look to bring this important medicine to as many patients as quickly as possible.”
The safety profile observed in patients treated with Enhertu in the DESTINY-PanTumor02 trial was consistent with that seen in other trials of Enhertu with no new safety signals identified.
Notes
DESTINY-PanTumor02
DESTINY-PanTumor02 is a global, multicentre, multi-cohort, open-label Phase II trial evaluating the efficacy and safety of Enhertu (5.4mg/kg) for the treatment of HER2-expressing tumours, including biliary tract cancer, bladder cancer, cervical cancer, endometrial cancer, ovarian cancer, pancreatic cancer and rare tumours.
The primary efficacy endpoint of DESTINY-PanTumor02 is confirmed ORR as assessed by investigator. Secondary endpoints include DoR, disease control rate, progression-free survival, overall survival, safety, tolerability and pharmacokinetics.
DESTINY-PanTumor02 has enrolled 268 patients at multiple sites in Asia, Europe and North America. For more information about the trial, visit ClinicalTrials.gov.
Enhertu
Enhertu is a HER2-directed ADC. Designed using Daiichi Sankyo’s proprietary DXd ADC technology, Enhertu is the lead ADC in the oncology portfolio of Daiichi Sankyo and the most advanced programme in AstraZeneca’s ADC scientific platform. Enhertu consists of a HER2 monoclonal antibody attached to a topoisomerase I inhibitor payload, an exatecan derivative, via a stable tetrapeptide-based cleavable linker.
Enhertu (5.4mg/kg) is approved in more than 40 countries for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received a (or one or more) prior anti-HER2-based regimen, either in the metastatic setting or in the neoadjuvant or adjuvant setting, and have developed disease recurrence during or within six months of completing therapy based on the results from the DESTINY-Breast03 trial.
Enhertu (5.4mg/kg) is approved in more than 30 countries for the treatment of adult patients with unresectable or metastatic HER2-low (immunohistochemistry [IHC] 1+ or IHC 2+/in-situ hybridisation [ISH]-) breast cancer who have received a prior systemic therapy in the metastatic setting or developed disease recurrence during or within six months of completing adjuvant chemotherapy based on the results from the DESTINY-Breast04 trial.
Enhertu (5.4mg/kg) is approved under accelerated approval in the US for the treatment of adult patients with unresectable or metastatic non-small cell lung cancer whose tumours have activating HER2 (ERBB2) mutations, as detected by an FDA-approved test, and who have received a prior systemic therapy based on the results from the DESTINY-Lung02 trial. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
Enhertu (6.4mg/kg) is approved in more than 30 countries for the treatment of adult patients with locally advanced or metastatic HER2-positive gastric or gastroesophageal junction adenocarcinoma who have received a prior trastuzumab-based regimen based on the results from the DESTINY-Gastric01 trial and/or DESTINY-Gastric02 trial.
Enhertu development programme
A comprehensive global development programme is underway evaluating the efficacy and safety of Enhertu monotherapy across multiple HER2-targetable cancers. Trials in combination with other anticancer treatments, such as immunotherapy, are also underway.
Daiichi Sankyo collaboration
Daiichi Sankyo Company, Limited (TSE: 4568) [referred to as Daiichi Sankyo] and AstraZeneca entered into a global collaboration to jointly develop and commercialise Enhertu (a HER2-directed ADC) in March 2019, and datopotamab deruxtecan (DS-1062; a TROP2-directed ADC) in July 2020, except in Japan where Daiichi Sankyo maintains exclusive rights. Daiichi Sankyo is responsible for the manufacturing and supply of Enhertu and datopotamab deruxtecan.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Mar. 06, 2023 4:55 AM ET
AstraZeneca PLC (AZN), DSKYF, DSNKY
By: Ravikash, SA News Editor
AstraZeneca (NASDAQ:AZN) and Daiichi Sankyo (OTCPK:DSKYF) (OTCPK:DSNKY) said their drug Enhertu met the target for objective response rate (ORR) and showed durable response in multiple HER2-expressing advanced solid tumors in heavily pretreated patients in an ongoing phase 2 trial.
FDA Accepts BioMarin's Supplemental New Drug Application to Expand Use of VOXZOGO® (vosoritide) for Injection to Treat Children with Achondroplasia Under the Age of 5
Mar 7, 2023
The Supplemental New Drug Application is Based on Positive Results from Global Randomized Phase 2 Study in Infants and Young Children
FDA set PDUFA Target Action Date of October 21, 2023
SAN RAFAEL, Calif., March 7, 2023 /PRNewswire/ -- BioMarin Pharmaceutical Inc. (Nasdaq: BMRN), a global biotechnology company dedicated to transforming lives through genetic discovery, announced today that the U.S. Food and Drug Administration (FDA) has accepted the company's supplemental New Drug Application (sNDA) for VOXZOGO® (vosoritide) for injection to expand treatment in the United States to include children with achondroplasia under the age of 5. Achondroplasia is the most common form of disproportionate short stature.
The FDA has set a PDUFA target action date of October 21, 2023 for the sNDA.
"We are pleased that the FDA has accepted our sNDA and are working closely with the agency to facilitate completion of the review in a timely manner," said Hank Fuchs, M.D., president, Worldwide Research and Development at BioMarin. "There are currently no approved pharmacological treatments in the United States for children under 5 with achondroplasia and this approval could potentially extend access to all children with achondroplasia, whose growth plates are not closed."
The sNDA is supported by results from a Phase 2 randomized, double-blind, placebo-controlled clinical trial, which demonstrated similar safety and efficacy profiles in children under 5 years of age as compared to those ages 5 years and older.
In January, the European Medicines Agency (EMA) validated BioMarin's application for extension of indication for VOXZOGO to treat children with achondroplasia under the age of 2. Approval of the submissions would mean VOXZOGO could potentially be prescribed as early as birth for more than 1,000 additional children eligible for treatment for achondroplasia.
VOXZOGO is the first FDA and EMA approved treatment for children with achondroplasia with open epiphyses (bone growth plates).
About VOXZOGO® (vosoritide) for Injection
In patients with achondroplasia, endochondral bone growth, an essential process by which bone tissue is created, is negatively regulated due to a gain of function mutation in fibroblast growth factor receptor 3 gene (FGFR3). VOXZOGO, a C-type natriuretic peptide (CNP) analog, represents a new class of therapy, which acts as a positive regulator of the signaling pathway downstream of FGFR3 to promote endochondral bone growth.
Through BioMarin's broad clinical development program, the company has enrolled 250 children with achondroplasia from eight countries in seven clinical studies evaluating the safety and efficacy of VOXZOGO.
VOXZOGO is approved in the U.S. and indicated to increase linear growth in pediatric patients with achondroplasia who are 5 years of age and older with open epiphyses. This indication is approved under accelerated approval based on an improvement in annualized growth velocity. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s). To fulfill this post-marketing requirement, BioMarin intends to use the ongoing open-label extension studies compared to available natural history.
VOXZOGO is also approved in the EU, Brazil, and Australia in children with achondroplasia who are 2 years of age and older with open growth plates. It is also approved in Japan in children from birth who have achondroplasia with open growth plates.
About Achondroplasia
Achondroplasia, the most common form of skeletal dysplasia leading to disproportionate short stature, is characterized by slowing of endochondral ossification, which results in disproportionate short stature and disordered architecture in the long bones, spine, face, and base of the skull. This condition is caused by a change in the fibroblast growth factor receptor 3 gene (FGFR3), a negative regulator of bone growth.
More than 80% of children with achondroplasia have parents of average stature and have the condition as the result of a spontaneous change in the gene. The worldwide incidence rate of achondroplasia is about one in 25,000 live births. VOXZOGO is being studied in children whose growth plates are still "open," typically those under 18 years of age. Approximately 25% of people with achondroplasia fall into this category.
VOXZOGO U.S. Important Safety Information
What is VOXZOGO used for?
What is the most important safety information about VOXZOGO?
What are the most common side effects of VOXZOGO?
How is VOXZOGO taken?
About BioMarin
Founded in 1997, BioMarin is a global biotechnology company dedicated to transforming lives through genetic discovery. The company develops and commercializes targeted therapies that address the root cause of the genetic conditions. BioMarin's unparalleled research and development capabilities have resulted in eight transformational commercial therapies for patients with rare genetic disorders. The company's distinctive approach to drug discovery has produced a diverse pipeline of commercial, clinical, and pre-clinical candidates that address a significant unmet medical need, have well-understood biology, and provide an opportunity to be first-to-market or offer a substantial benefit over existing treatment options. For additional information, please visit www.biomarin.com.
BioMarin® and VOXZOGO® are registered trademarks of BioMarin Pharmaceutical Inc.
SOURCE BioMarin Pharmaceutical Inc.
Mar. 07, 2023 5:58 PM ET
BioMarin Pharmaceutical Inc. (BMRN)
By: Anuron Mitra, SA News Editor

Final cumulative pooled IBD safety data support the longstanding safety profile of STELARA across all IBD approved indications
Additional long term extension data demonstrate more than half of STELARA-treated patients with ulcerative colitis achieved clinical remission, clinical response, and/or demonstrated endoscopic improvement at four years
SPRING HOUSE, PENNSYLVANIA, March 4, 2023 – The Janssen Pharmaceutical Companies of Johnson & Johnson today announced final pooled long-term safety results for STELARA® (ustekinumab) through five years in adults with moderately to severely active Crohn’s disease (CD) and four years in adults with moderately to severely active ulcerative colitis (UC), as well as final four-year clinical and endoscopic outcomes from the UNIFI long-term extension (LTE) study evaluating the efficacy of STELARA for the treatment of adults with moderately to severely active UC.1,2 These data are a part of Janssen’s 22 oral and poster presentations at the 18th Congress of the European Crohn’s and Colitis Organization (ECCO), taking place in Copenhagen, Denmark, March 1-4.
“These data reinforce the known efficacy and safety profile of STELARA, and demonstrate it can be an effective long-term treatment option for patients living with moderately to severely active ulcerative colitis,” said UNIFI study author Waqqas Afif, M.D., Associate Professor, Department of Medicine, Division of Experimental Medicine and Division of Gastroenterology at McGill University Health Centre in Montreal, Canada.a “Importantly, clinical and endoscopic outcomes reinforce the durable efficacy of STELARA, as we remain committed to developing therapies that provide patients with lasting remission.”
Final STELARA long-term pooled safety analysis (Oral presentation OP39):1
A final pooled safety analysis of six Phase 2/3 IBD studies included 2,575 patients treated with STELARA and a total of 4,826 patient-years (PY) of follow-up.
Final UNIFI LTE clinical and endoscopy outcomes through four years from STELARA treatment (Oral presentation OP15):2 Results from the UNIFI LTE study, among 205 adult patientsb with a history of moderate to severe UC who had achieved clinical response to treatment with intravenous (IV) STELARA, were randomized to STELARA 90 mg every eight weeks (q8w) or every 12 weeks (q12w)c at baseline of the maintenance study, and continued treatment in the LTE, showed that at week 200:d
“These long-term studies underscore Janssen’s commitment to developing novel therapies addressing unmet medical need,” said Jan Wehkamp, M.D., Ph.D., Vice President, Gastroenterology Disease Area Leader, Janssen Research & Development, LLC. “Our findings reinforce our confidence in STELARA as a therapy of choice for patients seeking lasting relief from inflammatory bowel disease."
Editor’s Notes:
a. Dr. Afif received grant support from Janssen. He has not been compensated for any media work.
b. Patients were randomized to STELARA at maintenance baseline and continued treatment in the LTE, who either had Mayo score data (including endoscopy) at week 200 or had experienced treatment failure2
c. q12w dosing is not currently approved for STELARA in the U.S.; current approved dosing is a subcutaneous 90 mg dose eight weeks after initial intravenous dose, then every eight weeks thereafter3
d. Patients who had treatment failure (i.e., had ostomy or colectomy or discontinued STELARA due to lack of therapeutic effect or worsening UC) before week 200 were also included, and were imputed as nonresponders.2
e. Clinical remission is defined as a Mayo score ≤2 points and no individual subscore >12
f. Clinical response is defined as a decrease in Mayo score of ≥30% and ≥3 points from induction baseline with either a decrease in rectal bleeding subscore of ≥1 from induction baseline or a rectal bleeding subscore of 0 or 12
g. Modified Mayo score (without Physician’s Global Assessment subscore) response is defined as a decrease in modified Mayo score of ≥30% and ≥2 points from induction baseline with either a decrease in rectal bleeding subscore of ≥1 from induction baseline or a rectal bleeding subscore of 0 or 12
h. Endoscopic improvement, endoscopic healing, or mucosal healing is defined as an endoscopy subscore of 0 or 12
About UNIFI (NCT02407236)2,4
UNIFI was a Phase 3 protocol designed to evaluate the safety and efficacy of STELARA induction and maintenance dosing for the treatment of moderately to severely active ulcerative colitis in adults who demonstrated an inadequate response to or were unable to tolerate conventional (i.e., corticosteroids, immunomodulators) or biologic (i.e., one or more TNF blockers or vedolizumab) therapies. Both the induction and maintenance studies were randomized, double-blind, placebo-controlled, parallel group, multi-center studies.
The induction study was of at least 8 weeks duration for each participant. Participants achieving clinical response in the induction study were eligible for the maintenance study. The maintenance study was 44 weeks in duration. The primary endpoint of the induction study was clinical remission at week 8, and the primary endpoint for the maintenance study was clinical remission at week 44 among responders to a single intravenous (IV) STELARA infusion (130 mg or ~6 mg/kg). Overall, 523 IV STELARA induction responders were randomized to subcutaneous (SC) maintenance therapy (175 SC placebo; 172 STELARA 90 mg q12w; 176 STELARA 90 mg q8w). 284 STELARA patients who completed week 44 entered the LTE. Placebo patients were discontinued after week 44 unblinding. The long-term extension of UNIFI followed eligible participants for an additional three years upon completion of the maintenance study.
Starting at week 56, randomized patients with UC worsening could adjust to q8w dosing. Outcomes based on the Mayo score (including endoscopy assessed by a local reader) were evaluated in the final efficacy visit at week 200. Patients who had treatment failure (i.e., had ostomy or colectomy, or discontinued STELARA due to lack of therapeutic effect or worsening UC) before week 200 were also included, and were imputed as non-responders.
About Ulcerative Colitis
Ulcerative Colitis (UC) is a chronic disease of the large intestine, also known as the colon, in which the lining of the colon becomes inflamed and develops tiny open sores, or ulcers, that produce pus and mucus.5 It is the result of the immune system’s overactive response.5 Symptoms vary, but may include loose and more urgent bowel movements, persistent diarrhea, abdominal pain, bloody stool, loss of appetite, weight loss and fatigue.6
About Crohn’s Disease
CD is one of the two main forms of IBD, which affects an estimated three million Americans.7 CD is a chronic inflammatory condition of the gastrointestinal tract with no known cause, but the disease is associated with abnormalities of the immune system that could be triggered by a genetic predisposition, diet or other environmental factors.8
Symptoms of CD can vary, but often include abdominal pain and tenderness, frequent diarrhea, rectal bleeding, weight loss and fever.9
About STELARA® (ustekinumab)3
STELARA® (ustekinumab), a human interleukin (IL)-12 and IL-23 antagonist, is approved in the United States for the treatment of: 1) adults and children six years and older with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy; 2) adults and children six years and older with active psoriatic arthritis; 3) adult patients (18 years and older) with moderately to severely active Crohn’s disease; 4) adult patients (18 years and older) with moderately to severely active ulcerative colitis.
The Janssen Pharmaceutical Companies of Johnson & Johnson maintain exclusive worldwide marketing rights to STELARA.
https://www.stelarainfo.com/ulcerative-colitis
Please click to read the full Prescribing Information and Medication Guide for STELARA® and discuss any questions you have with your doctor.
About the Janssen Pharmaceutical Companies of Johnson & Johnson
At Janssen, we’re creating a future where disease is a thing of the past. We’re the Pharmaceutical Companies of Johnson & Johnson, working tirelessly to make that future a reality for patients everywhere by fighting sickness with science, improving access with ingenuity, and healing hopelessness with heart. We focus on areas of medicine where we can make the biggest difference: Cardiovascular, Metabolism & Retina; Immunology; Infectious Diseases & Vaccines; Neuroscience; Oncology; and Pulmonary Hypertension.
Learn more at www.janssen.com.
Follow us at www.twitter.com/JanssenGlobal.
Janssen Research & Development, LLC is a part of the Janssen Pharmaceutical Companies of Johnson & Johnson.
Mar. 04, 2023 12:21 PM ET
Johnson & Johnson (JNJ)By: Jonathan Block, SA News Editor3 Comments
March 7, 2023 at 1:00 AM EST
More than 300,000 people in the U.S. suffer from CSU that is inadequately controlled by antihistamines
TARRYTOWN, N.Y. and PARIS, March 07, 2023 (GLOBE NEWSWIRE) -- Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) and Sanofi today announced that the U.S. Food and Drug Administration (FDA) has accepted for review the supplemental Biologics License Application (sBLA) for Dupixent® (dupilumab) to treat adults and adolescents aged 12 years and older with chronic spontaneous urticaria (CSU) that is not adequately controlled with the current standard of care, H1 antihistamine treatment. The target action date for the FDA decision is October 22, 2023.
CSU is an inflammatory skin condition driven in part by type 2 inflammation, which causes sudden and debilitating hives and swelling on the skin. Swelling, called angioedema, may occur most commonly on the face, hands and feet, but can also affect the throat and upper airways. CSU is typically treated with H1 antihistamines, medicines that target histamine-1 receptors on cells to control symptoms of urticaria. However, the disease remains uncontrolled in up to 50% of patients, who are left with limited alternative treatment options. These individuals continue to experience symptoms, including persistent itch or burning sensations that can be debilitating and significantly impact quality of life.
The sBLA is supported by data from two Phase 3 trials (LIBERTY-CUPID Studies A and B) evaluating Dupixent in two different patient populations with uncontrolled CSU. Study A was conducted in CSU patients who were uncontrolled on standard-of-care antihistamines with efficacy and safety data supporting the submission, while Study B was conducted in CSU patients who were uncontrolled on standard-of-care antihistamines and refractory to omalizumab with results providing additional supporting data.
The potential use of Dupixent in CSU is currently under clinical development, and the safety and efficacy have not been fully evaluated by any regulatory authority.
About the CSU Clinical Trial Program
The clinical trial program, known as LIBERTY-CUPID, includes Studies A and B, two Phase 3 randomized, double-blind, placebo-controlled trials evaluating the efficacy and safety of Dupixent in two different patient populations with uncontrolled CSU.
Study A evaluated Dupixent as an add-on therapy to standard-of-care H1 antihistamines compared to antihistamines alone in 138 patients with CSU aged 6 years and older who remained symptomatic despite antihistamine use and were not previously treated with omalizumab. Study B evaluated Dupixent in 108 patients with CSU aged 12 to 80 years who remained symptomatic despite standard-of-care treatment and were intolerant or incomplete responders to omalizumab.
In addition to CSU, Regeneron and Sanofi are also studying Dupixent in chronic inducible urticaria triggered by cold (LIBERTY-CINDU CUrIADS program) in an ongoing Phase 3 trial.
About Dupixent
Dupixent, which was invented using Regeneron's proprietary VelocImmune® technology, is a fully human monoclonal antibody that inhibits the signaling of the IL-4 and IL-13 pathways and is not an immunosuppressant. The Dupixent development program has shown significant clinical benefit and a decrease in type 2 inflammation in Phase 3 trials, establishing that IL-4 and IL-13 are key and central drivers of the type 2 inflammation that plays a major role in multiple related and often co-morbid diseases. These diseases include approved indications for Dupixent, such as atopic dermatitis, asthma, chronic rhinosinusitis with nasal polyposis (CRSwNP), prurigo nodularis and eosinophilic esophagitis (EoE).
Dupixent has received regulatory approvals in one or more countries around the world for use in certain patients with atopic dermatitis, asthma, CRSwNP, EoE or prurigo nodularis in different age populations. Dupixent is currently approved for one or more of these indications in more than 60 countries, including in Europe, the U.S. and Japan. More than 500,000 patients have been treated with Dupixent globally.
About Regeneron’s VelocImmune Technology
Regeneron’s VelocImmune technology utilizes a proprietary genetically engineered mouse platform endowed with a genetically humanized immune system to produce optimized fully human antibodies. When Regeneron’s President and Chief Scientific Officer George D. Yancopoulos was a graduate student with his mentor Frederick W. Alt in 1985, they were the first to envision making such a genetically humanized mouse, and Regeneron has spent decades inventing and developing VelocImmune and related VelociSuite® technologies. Dr. Yancopoulos and his team have used VelocImmune technology to create a substantial proportion of all original, FDA-approved or authorized fully human monoclonal antibodies currently available. This includes Dupixent, REGEN-COV® (casirivimab and imdevimab), Libtayo® (cemiplimab-rwlc), Praluent® (alirocumab), Kevzara® (sarilumab), Evkeeza® (evinacumab-dgnb) and Inmazeb® (atoltivimab, maftivimab, and odesivimab-ebgn).
Dupilumab Development Program
Dupilumab is being jointly developed by Regeneron and Sanofi under a global collaboration agreement. To date, dupilumab has been studied across more than 60 clinical trials involving more than 10,000 patients with various chronic diseases driven in part by type 2 inflammation.
In addition to the currently approved indications, Regeneron and Sanofi are studying dupilumab in a broad range of diseases driven by type 2 inflammation or other allergic processes in Phase 3 trials, including pediatric EoE, atopic hand and foot dermatitis, chronic inducible urticaria-cold, CSU, chronic pruritus of unknown origin, chronic obstructive pulmonary disease with evidence of type 2 inflammation, chronic rhinosinusitis without nasal polyposis, allergic fungal rhinosinusitis, allergic bronchopulmonary aspergillosis and bullous pemphigoid. These potential uses of dupilumab are currently under clinical investigation, and the safety and efficacy in these conditions have not been fully evaluated by any regulatory authority.
U.S. INDICATIONS
DUPIXENT is a prescription medicine used:
Please see accompanying full Prescribing Information including Patient Information.
About Regeneron
Regeneron is a leading biotechnology company that invents, develops and commercializes life-transforming medicines for people with serious diseases. Founded and led for 35 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to nine FDA-approved treatments and numerous product candidates in development, almost all of which were homegrown in our laboratories. Our medicines and pipeline are designed to help patients with eye diseases, allergic and inflammatory diseases, cancer, cardiovascular and metabolic diseases, pain, hematologic conditions, infectious diseases and rare diseases.
Regeneron is accelerating and improving the traditional drug development process through our proprietary VelociSuite® technologies, such as VelocImmune®, which uses unique genetically humanized mice to produce optimized fully human antibodies and bispecific antibodies, and through ambitious research initiatives such as the Regeneron Genetics Center®, which is conducting one of the largest genetics sequencing efforts in the world.
For more information, please visit www.Regeneron.com or follow @Regeneron on Twitter.
About Sanofi
We are an innovative global healthcare company, driven by one purpose: we chase the miracles of science to improve people's lives. Our team, across some 100 countries, is dedicated to transforming the practice of medicine by working to turn the impossible into the possible. We provide potentially life-changing treatment options and life-saving vaccine protection to millions of people globally, while putting sustainability and social responsibility at the center of our ambitions.
Sanofi is listed on EURONEXT: SAN and NASDAQ: SNY.
![]()
Source: Regeneron Pharmaceuticals, Inc.
Mar. 07, 2023 6:39 AM ET
Regeneron Pharmaceuticals, Inc. (REGN), SNY
By: Mamta Mayani, SA News Editor
03/03/2023CATEGORY:
Third authorized indication in Europe for Reblozyl, a first-in-class treatment for patients with diseases impacted by anemia
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) today announced that the European Commission (EC) has granted full Marketing Authorization for Reblozyl® (luspatercept), a first-in-class therapeutic option, for treatment in adult patients of anemia associated with non-transfusion-dependent (NTD) beta thalassemia. Reblozyl is currently approved in the European Union (EU), United States and Canada to address anemia associated with transfusion-dependent beta thalassemia and transfusion-dependent lower-risk myelodysplastic syndromes. The centralized Marketing Authorization approves use of Reblozyl in all EU member states, as well as Norway, Iceland and Liechtenstein.*
“Beta thalassemia is an inherited blood disorder that puts patients at significant risk for long-term clinical complications due to anemia, leaving a substantial need for treatment options, regardless of a patient’s dependence on blood transfusions. This announcement is welcome news for patients with non-transfusion-dependent beta thalassemia associated anemia across the EU who are seeking newer treatment options to reduce these burdens,” said Noah Berkowitz, M.D., Ph.D., senior vice president, Hematology Development, Bristol Myers Squibb. “Today’s approval represents the third indication for Reblozyl in Europe, and we look forward to continuing to evaluate this first-in-class therapeutic option across multiple diseases impacted by the burden of anemia in a broad clinical development program.”
The EC approval of Reblozyl was based on results from the Phase 2 BEYOND study, evaluating the efficacy and safety of Reblozyl versus placebo in 145 adults with NTD beta thalassemia. Patients were eligible to receive best supportive care, including red blood cell transfusions and iron-chelating agents.
Reblozyl is being developed and commercialized through a global collaboration with Merck following Merck’s acquisition of Acceleron Pharma, Inc. in November 2021.
*Centralized Marketing Authorization does not include approval in Great Britain (England, Scotland and Wales).
About BEYOND
BEYOND (NCT03342404) is a Phase 2, double-blind, randomized, placebo-controlled, multicenter study to determine the efficacy and safety of luspatercept-aamt (ACE-536) versus placebo in adults with non-transfusion-dependent beta thalassemia. The study is divided into the Screening Period, Double-blind Treatment Period (DBTP) and Post-Treatment Follow-up Period (PTFP) and randomized 145 subjects at a 2:1 ratio of Reblozyl versus placebo. All patients were eligible to receive best supportive care, which included red blood cell transfusions; iron-chelating agents; use of antibiotic, antiviral, and antifungal therapy; and/or nutritional support, as needed. The primary endpoint of the study is the proportion of subjects who have an increase from baseline ≥1.0 g/dL in mean of hemoglobin values over a continuous 12-week interval from Week 13 to Week 24 of treatment in the absence of transfusions. Key secondary endpoints include mean change in non-transfusion-dependent beta thalassemia-patient reported outcome (NTDT-PRO) Tiredness and Weakness (TW) domain score and baseline hemoglobin (Hb).
Results demonstrated 74 of 96 (77.1%) patients in the Reblozyl treatment arm achieved the study’s primary endpoint, ≥1.0 g/dL mean Hb increase from baseline, versus 0 of 49 (0%) patients in the placebo arm (P<0.0001).
In a key secondary endpoint of the study, 47 of 96 patients (49.0%) treated with Reblozyl achieved mean Hb increase of ≥1.5 g/dL compared to baseline from Week 37-48 in the absence of transfusions versus 0 patients (0%) in the placebo arm (P<0.0001). In the Reblozyl arm, 89.6% of patients remained transfusion free at weeks 1-24 versus 67.3% of patients in the placebo arm (P=0.0013). Improvements in patient-reported QoL outcomes (tiredness and weakness) were also observed to correlate with Hb increases.
Serious adverse reactions occurred in 11.5% of patients (n=11) who received Reblozyl. The most common adverse reactions occurring in ≥10% of patients treated with Reblozyl were bone pain (36%), headache (30%), arthralgia (29%), back pain (28%), prehypertension (23%), hypertension (20%), cough (18%), diarrhea (17%), influenza-like illness (17%), asthenia (13%), influenza (13%), insomnia (11%) and nausea (10%).
About Beta Thalassemia
Beta thalassemia is an inherited blood disorder caused by a genetic defect in hemoglobin. It is one of the most common autosomal recessive disorders, and the total annual incidence of symptomatic individuals is estimated at 1 in 100,000 people globally.1 The disease is associated with ineffective erythropoiesis, which results in the production of fewer and less healthy red blood cells (RBCs), often leading to severe anemia—a condition that can be debilitating and can lead to other complications for patients—as well as other serious health issues.2 Treatment options for anemia associated with beta thalassemia are limited, consisting mainly of frequent RBC transfusions that have the potential to contribute to iron overload, which can cause serious complications such as organ damage.1 Non-transfusion-dependent beta thalassemia is a term used to describe patients who do not require lifelong regular transfusions for survival, although they may experience a range of clinical complications and require occasional or even frequent transfusions, usually for defined periods of time.
About Reblozyl®
Reblozyl, a first-in-class therapeutic option, promotes late-stage red blood cell (RBC) maturation in animal models.1 Reblozyl is being developed and commercialized through a global collaboration with Merck following Merck’s acquisition of Acceleron Pharma, Inc. in November 2021. Reblozyl is currently approved in the U.S. for the treatment of:
Reblozyl is not indicated for use as a substitute for RBC transfusions in patients who require immediate correction of anemia.
Please see full Prescribing Information for REBLOZYL.
About Bristol Myers Squibb
Bristol Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information about Bristol Myers Squibb, visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
Source: Bristol Myers Squibb
Mar. 03, 2023 7:35 AM ET
Bristol-Myers Squibb Company (BMY)
By: Mamta Mayani, SA News Editor
February 27, 2023
NORTH CHICAGO, Ill., Feb. 27, 2023 /PRNewswire/ -- AbbVie (NYSE: ABBV) will present further analyses on SKYRIZI® (risankizumab) in Crohn's disease (CD) and RINVOQ® (upadacitinib) in ulcerative colitis (UC) as well as the investigational use of upadacitinib in CD at the 18th Congress of European Crohn's and Colitis Organisation (ECCO), to be held March 1-4 in Copenhagen, Denmark. AbbVie will present 24 abstracts, including four oral presentations, two digital oral presentations and 18 posters from a broad range of studies across its inflammatory bowel disease (IBD) portfolio.
"We are excited by the opportunity to present further analyses on the advancements of our expanding IBD portfolio at this year's 18th ECCO Congress," said Sofie Berg, Pharm.D, Ph.D., head of gastroenterology, global medical affairs, AbbVie. "Our data at ECCO underscore AbbVie's unwavering commitment to driving discoveries and delivering advancements for patients where expanded options are needed with the goal of positively impacting the treatment of those living with IBD."
Key data presentations will include:
"Despite continued advancements in the treatment of IBD, many patients are still negatively impacted by the burdens of living with their disease," said Silvio Danese, M.D. Ph.D. director of gastroenterology and endoscopy at IRCCS Ospedale San Raffaele and professor of gastroenterology at University Vita-Salute San Raffaele, Milan, Italy. "AbbVie's research at ECCO helps support their goal of providing a diverse portfolio of treatments and research that continues to advance the standard of care for IBD patients."
The full scientific program for the 18th ECCO Congress is available here.
SKYRIZI® (risankizumab) is part of a collaboration between Boehringer Ingelheim and AbbVie, with AbbVie leading development and commercialization globally.
About Ulcerative Colitis
Ulcerative colitis is a chronic, idiopathic, immune-mediated inflammatory bowel disease (IBD) of the large intestine that causes continuous mucosal inflammation extending, to a variable extent, from the rectum to the more proximal colon.1,2 The hallmark signs and symptoms of ulcerative colitis include rectal bleeding, abdominal pain, bloody diarrhea, tenesmus (a sense of pressure), urgency and fecal incontinence.1,3 The disease course of ulcerative colitis varies between patients and can range from quiescent disease to chronic refractory disease, which in some cases can lead to surgery or complications, including cancer or death.2,4 The severity of symptoms and unpredictability of disease course can lead to substantial burden and often disability among those living with the disease.5
About Crohn's Disease
Crohn's disease is a chronic, systemic disease that manifests as inflammation within the gastrointestinal (or digestive) tract, causing persistent diarrhea, abdominal pain and rectal bleeding.2,6,7 It is a progressive disease, meaning it gets worse over time.2,7 Because the signs and symptoms of Crohn's disease are unpredictable, it causes a significant burden on people living with the disease—not only physically, but also emotionally and economically.5
About RINVOQ® (upadacitinib)
Discovered and developed by AbbVie scientists, RINVOQ is a selective JAK inhibitor that is being studied in several immune-mediated inflammatory diseases. Based on enzymatic and cellular assays, RINVOQ demonstrated greater inhibitory potency for JAK-1 vs JAK-2, JAK-3, and TYK-2.8 The relevance of inhibition of specific JAK enzymes to therapeutic effectiveness and safety is not currently known.
In the U.S., RINVOQ 15 mg is approved for adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more TNF blockers; adults with active psoriatic arthritis who have had an inadequate response or intolerance to one or more TNF blockers; adults with active ankylosing spondylitis (AS) who have had an inadequate response or intolerance to one or more tumor necrosis factor (TNF) blockers and adults with active non-radiographic axial spondyloarthritis with objective signs of inflammation who have had an inadequate response or intolerance to TNF blocker therapy.8 RINVOQ 45 mg is approved for use in adult patients with moderately to severely active ulcerative colitis who have had an inadequate response or intolerance to one or more TNF blockers as an induction therapy once daily for 8 weeks. The recommended dose of RINVOQ for maintenance treatment is 15 mg once daily. A dosage of 30 mg once daily may be considered for patients with refractory, severe or extensive disease. Discontinue RINVOQ if an adequate response is not achieved with the 30 mg dose. Use the lowest effective dosage needed to maintain response. RINVOQ 15 mg once daily can also be initiated in adults and children 12 years of age and older weighing at least 40 kg with refractory, moderate to severe atopic dermatitis whose disease is not adequately controlled with other systemic drug products, including biologics or when use of those therapies is inadvisable. In these children and adults less than 65 years of age who do not achieve an adequate response, the dose may be increased to 30 mg once daily.
Phase 3 trials of RINVOQ in rheumatoid arthritis, atopic dermatitis, psoriatic arthritis, axial spondyloarthritis, Crohn's disease, ulcerative colitis, giant cell arteritis and Takayasu arteritis are ongoing.9-16
US Indications and Important Safety Information about RINVOQ® (upadacitinib)8
USES
RINVOQ is a prescription medicine used to treat:
It is not known if RINVOQ is safe and effective in children with juvenile idiopathic arthritis, psoriatic arthritis, ulcerative colitis, ankylosing spondylitis, or non-radiographic axial spondyloarthritis.
RINVOQ is safe and effective in children 12 years of age and older weighing at least 88 pounds (40 kg) with atopic dermatitis.
It is not known if RINVOQ is safe and effective in children under 12 years of age with atopic dermatitis.
About AbbVie
AbbVie's mission is to discover and deliver innovative medicines that solve serious health issues today and address the medical challenges of tomorrow. We strive to have a remarkable impact on people's lives across several key therapeutic areas: immunology, oncology, neuroscience, eye care, virology, women's health and gastroenterology, in addition to products and services across our Allergan Aesthetics portfolio. For more information about AbbVie, please visit us at www.abbvie.com. Follow @abbvie on Twitter, Facebook, LinkedIn or Instagram.
Feb. 27, 2023 5:48 AM ET
By: Dulan Lokuwithana, SA News Editor
March 3, 2023Download PDF
High risk patients eligible for Verzenio can now be identified solely based on nodal status, tumor size, and tumor grade, regardless of Ki-67 score
Approval supported by four-year data from the monarchE trial; Verzenio added to adjuvant endocrine therapy (ET) reduced the risk of recurrence by 35% compared to adjuvant ET alone
Verzenio remains the first and only CDK4/6 inhibitor approved in the adjuvant setting
INDIANAPOLIS, March 3, 2023 /PRNewswire/ -- Eli Lilly and Company (NYSE: LLY) today announced that the U.S. Food and Drug Administration (FDA) approved an expanded indication for Verzenio® (abemaciclib), in combination with endocrine therapy (ET), for the adjuvant treatment of adult patients with hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2-), node-positive, early breast cancer (EBC) at a high risk of recurrence. High risk patients eligible for Verzenio can now be identified solely based on nodal status, tumor size, and tumor grade (4+ positive nodes, or 1-3 positive nodes and at least one of the following: tumors that are ≥5 cm or Grade 3).1 This expanded adjuvant indication removes the Ki-67 score requirement for patient selection.
This label expansion is supported by four-year data from the Phase 3 monarchE trial of adjuvant Verzenio in combination with ET, which showed a deepened benefit in invasive disease-free survival (IDFS) beyond the two-year treatment course with adjuvant Verzenio. The absolute difference in IDFS between treatment groups increased over time. At four years, 85.5% of patients remained recurrence-free with Verzenio plus ET, compared to 78.6% with ET alone, an absolute difference in IDFS of 6.9%. At two years and at three years, the absolute differences between treatment groups were 3.1% and 5.0%, respectively; see Fig. 1 below. The addition of Verzenio to ET reduced the risk of recurrence by 35% compared to ET alone (HR=0.653 [95% CI: 0.567-0.753]). There were no new safety findings, and overall results are consistent with the well-established safety profile for Verzenio. These four-year monarchE data were presented at the 2022 San Antonio Breast Cancer Symposium and simultaneously published in The Lancet Oncology.2
The monarchE study enrolled 5,637 adults with high risk HR+, HER2-, node-positive EBC into two cohorts. Verzenio is now approved for use in the full Cohort 1 patient population, which comprised 91% of the study population. A statistically significant difference in IDFS was observed in the intent-to-treat (ITT) population, primarily due to patients in Cohort 1. As of the data cut-off date, while overall survival (OS) data remain immature across the entire study, an OS trend in favor of Verzenio was observed in the Cohort 1 population, but not the Cohort 2 population where more deaths were seen with Verzenio plus ET compared to ET alone (10/253 vs. 5/264). The "About the monarchE Study" section below provides more details on study design.
"Our goal in intensifying treatment for early breast cancer is to maintain remission and prevent the recurrence of cancer. The magnitude of benefit seen in the four-year data from the monarchE study reinforces my confidence in adjuvant Verzenio as the standard-of-care for high risk patients in this setting," said Erika P. Hamilton, M.D., medical oncologist, director of Breast and Gynecologic Cancer Research at Sarah Cannon Research Institute, and an investigator on the monarchE clinical trial. "The initial Verzenio FDA approval in early breast cancer was practice-changing and now, through this indication expansion, we have the potential to reduce the risk of breast cancer recurrence for many more patients, relying solely on commonly utilized clinicopathologic features to identify them."
More than 300,000 people are expected to be diagnosed with breast cancer in the U.S. in 2023.3 It is estimated that 90% of all breast cancers are detected at an early stage.4 Approximately 70% of all breast cancer cases are the HR+, HER2- subtype.5 Although the prognosis for HR+, HER2- EBC is generally favorable, high risk patients are three times more likely than those with low risk characteristics to experience recurrence – with the majority being incurable metastatic disease.6 These patients have an increased risk of recurrence during the first two years of endocrine therapy.
"This expanded approval will allow us to bring Verzenio to many more women and men with HR+, HER2-, high risk early breast cancer in the curative setting – before patients experience recurrence, potentially to incurable metastatic disease," said Jacob Van Naarden, chief executive officer of Loxo@Lilly. "The initial adjuvant approval for Verzenio changed the treatment paradigm, and the strength of the monarchE results supporting this approval underscores the role this differentiated CDK4/6 inhibitor can play in reducing the risk of recurrence in early breast cancer."
"This expanded approval for Verzenio is welcome news for our community," said Jean Sachs, chief executive officer of Living Beyond Breast Cancer. "A significant number of women and men have HR+, HER2- early breast cancer at high risk of returning. Making effective treatment options available is crucial to allowing people to make the best care decisions for themselves, together with their healthcare providers. We're pleased Verzenio will now be available to more people with this type of early breast cancer."
Concurrent with this expanded indication approval in EBC, the FDA has also broadened the indicated use of Verzenio in metastatic breast cancer (MBC) when used in combination with an aromatase inhibitor as initial endocrine-based therapy for the treatment of people with HR+, HER2- advanced or MBC. This updated MBC indication now includes all adult patients, with the expanded indication including pre-/perimenopausal women when used in combination with ovarian suppression. See below "Indications for Verzenio" for full details on indicated uses in HR+, HER2- advanced or metastatic breast cancer. Verzenio is available in tablet strengths of 50 mg, 100 mg, 150 mg, and 200 mg.
The labeling for Verzenio contains warnings and precautions for diarrhea, neutropenia, interstitial lung disease (ILD/pneumonitis), hepatotoxicity, venous thromboembolism, and embryo-fetal toxicity. Instruct patients at the first sign of loose stools to initiate antidiarrheal therapy, increase oral fluids, and notify their healthcare provider. Perform complete blood counts and liver function tests prior to the start of Verzenio treatment, every two weeks for the first two months, monthly for the next two months and as clinically indicated. Based on results, Verzenio may require dose modification. Monitor patients for signs and symptoms of thrombosis and pulmonary embolism and treat as medically appropriate. Advise patients of potential risk to a fetus and to use effective contraception.
See Important Safety Information below and full Prescribing Information for additional information.
Click here to view the early breast cancer infographic.
About the monarchE Study
monarchE was a global, randomized, open-label, two cohort, multicenter Phase 3 clinical trial that enrolled 5,637 adults with HR+, HER2-, node-positive EBC at high risk of recurrence. To be enrolled in Cohort 1 (n=5,120), which is the FDA-approved population, patients had to have 4+ positive nodes or 1-3 positive nodes and at least one of the following: tumors that were ≥5 cm or Grade 3. Patients enrolled in Cohort 2 could not have met the eligibility criteria for Cohort 1. To be enrolled in Cohort 2 (n=517), patients had to have 1-3 positive nodes and Ki-67 score ≥20%. Patients in each cohort were randomized 1:1 to receive either Verzenio 150 mg twice daily plus standard-of-care adjuvant ET (Cohort 1, n=2,555; Cohort 2, n=253) or standard-of-care adjuvant ET alone (Cohort 1, n=2,565; Cohort 2, n=264) for 2 years. ET continued for at least 5 years if deemed medically appropriate. The primary endpoint was IDFS. Consistent with expert guidelines, IDFS was defined as the length of time before breast cancer comes back, any new cancer develops, or death.
About Early Breast Cancer and Risk of Recurrence
It is estimated that 90% of all breast cancers are detected at an early stage.4 Approximately 70% of all breast cancer cases are the HR+, HER2- subtype.5 Although the prognosis for HR+, HER2- EBC is generally favorable, high risk patients are three times more likely than those with low risk characteristics to experience recurrence – with the majority being incurable metastatic disease.6 These patients have an increased risk of recurrence during the first two years of endocrine therapy.
Factors associated with high risk of recurrence in HR+, HER2- early breast cancer include: positive nodal status, the number of positive nodes, large tumor size (≥5 cm), and high tumor grade (Grade 3). Node-positive means that cancer cells from the tumor in the breast have been found in the lymph nodes near the breast. Although breast cancer is removed through surgery, the presence of cancer cells in the lymph nodes signifies that there is a higher chance of developing recurrence and distant metastatic disease.
About Breast Cancer
Breast cancer has surpassed lung cancer as the most commonly diagnosed cancer worldwide, according to GLOBOCAN. The estimated 2.3 million new cases indicate that 1 in every 8 cancers diagnosed in 2020 is breast cancer. With approximately 685,000 deaths in 2020, breast cancer is the fifth-leading cause of cancer death worldwide.7 In the U.S., it is estimated that there will be more than 300,000 new cases of breast cancer diagnosed in 2023. Breast cancer is the second leading cause of cancer death in women in the U.S.3
About Verzenio® (abemaciclib)
Verzenio® (abemaciclib) is a targeted treatment known as a CDK4/6 inhibitor. Verzenio is a nonchemotherapy oral tablet.
Verzenio works inside the cell to block CDK4/6 activity and help stop the growth of cancer cells so that they may eventually die (based on preclinical studies). Cyclin-dependent kinases (CDK)4/6 are activated by binding to D-cyclins. In estrogen receptor-positive (ER+) breast cancer cell lines, cyclin D1 and CDK4/6 promote phosphorylation of the retinoblastoma protein (Rb), cell cycle progression and cell proliferation.
In vitro, continuous exposure to Verzenio inhibited Rb phosphorylation and blocked progression from G1 to S phase of the cell cycle, resulting in senescence and apoptosis (cell death). Preclinically, Verzenio dosed daily without interruption resulted in reduction of tumor size. Inhibiting CDK4/6 in healthy cells can result in side effects, some of which may be serious. Clinical evidence also suggests that Verzenio crosses the blood-brain barrier. In patients with advanced cancer, including breast cancer, concentrations of Verzenio and its active metabolites (M2 and M20) in cerebrospinal fluid are comparable to unbound plasma concentrations.
Verzenio is Lilly's first solid oral dosage form to be made using a faster, more efficient process known as continuous manufacturing. Continuous manufacturing is a new and advanced type of manufacturing within the pharmaceutical industry, and Lilly is one of the first companies to use this technology.
INDICATIONS FOR VERZENIO®
VERZENIO® is a kinase inhibitor indicated:
About Lilly
Lilly unites caring with discovery to create medicines that make life better for people around the world. We've been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world's most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer's disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we're motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram and LinkedIn. P-LLY
PP-AL-US-3657 © Lilly USA, LLC 2023. ALL RIGHTS RESERVED.
Verzenio® is a registered trademark owned or licensed by Eli Lilly and Company, its subsidiaries, or affiliates.
![]()
View original content to download multimedia:https://www.prnewswire.com/news-releases/us-fda-broadens-indication-for-verzenio-abemaciclib-in-hr-her2--node-positive-high-risk-early-breast-cancer-301762459.html
SOURCE Eli Lilly and Company
Mar. 03, 2023 3:20 PM ET
By: Jonathan Block, SA News Editor1 Comment
March 1, 2023 6:45 am ET
In pivotal study, KEYTRUDA® (pembrolizumab) in combination with chemotherapy before surgery and continuing as a single agent after surgery showed a statistically significant improvement in EFS versus pre-operative chemotherapy
KEYTRUDA-based regimen also showed statistically significant improvements in key secondary endpoints of pathological complete response and major pathological response
FDA accepted application based on these data and set PDUFA date of October 16, 2023
RAHWAY, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside of the United States and Canada, announced today that the Phase 3 KEYNOTE-671 trial investigating KEYTRUDA, Merck’s anti-PD-1 therapy, met one of its dual primary endpoints, event-free survival (EFS), as a perioperative treatment regimen for patients with resectable stage II, IIIA or IIIB non-small cell lung cancer (NSCLC). A perioperative treatment regimen includes treatment before surgery (neoadjuvant) and continued after surgery (adjuvant). The trial will continue to evaluate the other dual primary endpoint of overall survival (OS).
At a prespecified interim analysis conducted by an independent Data Monitoring Committee, neoadjuvant KEYTRUDA plus chemotherapy followed by resection and adjuvant single-agent KEYTRUDA demonstrated a statistically significant and clinically meaningful improvement in EFS compared to neoadjuvant placebo plus chemotherapy followed by adjuvant placebo. Statistically significant improvements in the trial’s key secondary endpoints of pathological compete response (pCR) and major pathological response (mPR) were also demonstrated at this analysis. No new safety signals were observed.
Results will be presented at an upcoming medical meeting. The U.S. Food and Drug Administration (FDA) has accepted Merck’s new supplemental Biologics License Application (sBLA) based on these data for KEYTRUDA for the treatment of patients with resectable stage II, IIIA, or IIIB (T3-4N2) NSCLC in combination with platinum containing chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment. The FDA has set a Prescription Drug User Fee Act (PDUFA), or target action, date of October 16, 2023.
“Results from KEYNOTE-671 show that KEYTRUDA in combination with chemotherapy provided significant improvement in event-free survival, pathological complete response and major pathological response over chemotherapy alone as a perioperative treatment regimen for patients with resectable stage II, IIIA or IIIB non-small cell lung cancer,” said Dr. Eliav Barr, senior vice president, head of global clinical development and chief medical officer, Merck Research Laboratories. “By moving this KEYTRUDA-based regimen into earlier stages of non-small cell lung cancer, we may be able to significantly reduce the risk of recurrence for these patients. This study is an important milestone, and we look forward to sharing the detailed results with the medical community as soon as possible. We thank the patients and investigators for their important contributions to this study.”
Merck has an extensive clinical development program in lung cancer and is advancing multiple registration-enabling studies, with research directed at earlier stages of disease and novel combinations. Key studies in earlier stages of NSCLC include KEYNOTE-671, KEYNOTE-091, KEYNOTE-867, KEYLYNK-012 and KEYVIBE-006.
About KEYNOTE-671
KEYNOTE-671 is a randomized, double-blind Phase 3 trial (ClinicalTrials.gov, NCT03425643) evaluating neoadjuvant KEYTRUDA plus chemotherapy, followed by adjuvant KEYTRUDA as a single agent versus placebo plus neoadjuvant chemotherapy followed by adjuvant placebo in patients with resectable stage II, IIIA or IIIB (T3-4N2) NSCLC. The trial’s dual primary endpoints are EFS and OS. Key secondary endpoints include pCR and mPR. The study enrolled 786 patients who were randomly assigned (1:1) to receive either:
About lung cancer
Lung cancer is the leading cause of cancer death worldwide. In 2020 alone, there were more than 2.2 million new cases and 1.8 million deaths from lung cancer globally. Non-small cell lung cancer is the most common type of lung cancer, accounting for about 81% of all cases. In the U.S., the overall five-year survival rate for patients diagnosed with lung cancer is 25%, which is a 21% improvement over the last five years. Improved survival rates are due, in part, to earlier detection and screening, reduction in smoking, advances in diagnostic and surgical procedures, as well as the introduction of new therapies. Early detection and screening remain an important unmet need, as 44% of lung cancer cases are not found until they are advanced. Only 5.8% of people in the U.S. who are eligible were screened for lung cancer in 2021.
About Merck’s research in lung cancer
Merck is advancing research aimed at transforming the way lung cancer is treated, with a goal of improving outcomes for patients affected by this deadly disease. Through nearly 200 clinical trials evaluating more than 36,000 patients around the world, Merck is at the forefront of lung cancer research. In NSCLC, KEYTRUDA has five approved U.S. indications (see indications below) and is approved for advanced disease in more than 95 countries. Among Merck’s research efforts are trials focused on evaluating KEYTRUDA in earlier stages of lung cancer as well as identifying new combinations and coformulations with KEYTRUDA.
About Merck’s early-stage cancer clinical program
Finding cancer at an earlier stage may give patients a greater chance of long-term survival. Many cancers are considered most treatable and potentially curable in their earliest stage of disease. Building on the strong understanding of the role of KEYTRUDA in later-stage cancers, Merck is studying KEYTRUDA in earlier disease states, with approximately 20 ongoing registrational studies across multiple types of cancer.
About KEYTRUDA® (pembrolizumab) injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.
Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [tumor proportion score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
KEYTRUDA, as a single agent, is indicated as adjuvant treatment following resection and platinum-based chemotherapy for adult patients with stage IB (T2a ≥4 cm), II, or IIIA NSCLC.
See additional selected indications for KEYTRUDA in the U.S. after the Selected Important Safety Information
Pediatric Use
In KEYNOTE-051, 161 pediatric patients (62 pediatric patients aged 6 months to younger than 12 years and 99 pediatric patients aged 12 years to 17 years) were administered KEYTRUDA 2 mg/kg every 3 weeks. The median duration of exposure was 2.1 months (range: 1 day to 24 months).
Adverse reactions that occurred at a ≥10% higher rate in pediatric patients when compared to adults were pyrexia (33%), vomiting (30%), leukopenia (30%), upper respiratory tract infection (29%), neutropenia (26%), headache (25%), and Grade 3 anemia (17%).
Additional Indications for KEYTRUDA in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of adult and pediatric (12 years and older) patients with stage IIB, IIC, or III melanoma following complete resection.
Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [Combined Positive Score (CPS) ≥1] as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy. KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Carcinoma
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
Non-muscle Invasive Bladder Cancer
KEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with MSI-H central nervous system cancers have not been established.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC) as determined by an FDA-approved test.
Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or gastroesophageal junction (GEJ) (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
KEYTRUDA, in combination with chemotherapy, with or without bevacizumab, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
KEYTRUDA is indicated for the adjuvant treatment of patients with RCC at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions.
Endometrial Carcinoma
KEYTRUDA, as a single agent, is indicated for the treatment of patients with advanced endometrial carcinoma that is MSI-H or dMMR, as determined by an FDA-approved test, who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation.
Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
About the Merck Access Program for KEYTRUDA
At Merck, we are committed to supporting accessibility to our cancer medicines. Merck provides multiple programs to help appropriate patients who are prescribed KEYTRUDA have access to our anti-PD-1 therapy. The Merck Access Program provides reimbursement support for patients receiving KEYTRUDA, including information to help with out-of-pocket costs and co-pay assistance for eligible patients. More information is available by calling 855-257-3932 or visiting https://www.merckaccessprogram-keytruda.com/.
About Merck’s Patient Support Program for KEYTRUDA
Merck is committed to helping provide patients and their caregivers support throughout their treatment with KEYTRUDA. The KEY+YOU Patient Support Program provides a range of resources and support. For further information and to sign up, eligible patients may call 85-KEYTRUDA (855- 398-7832) or visit www.keytruda.com.
Merck’s focus on cancer
Our goal is to translate breakthrough science into innovative oncology medicines to help people with cancer worldwide. At Merck, the potential to bring new hope to people with cancer drives our purpose and supporting accessibility to our cancer medicines is our commitment. As part of our focus on cancer, Merck is committed to exploring the potential of immuno-oncology with one of the largest development programs in the industry across more than 30 tumor types. We also continue to strengthen our portfolio through strategic acquisitions and are prioritizing the development of several promising oncology candidates with the potential to improve the treatment of advanced cancers. For more information about our oncology clinical trials, visit www.merck.com/clinicaltrials.
About Merck
At Merck, known as MSD outside of the United States and Canada, we are unified around our purpose: We use the power of leading-edge science to save and improve lives around the world. For more than 130 years, we have brought hope to humanity through the development of important medicines and vaccines. We aspire to be the premier research-intensive biopharmaceutical company in the world – and today, we are at the forefront of research to deliver innovative health solutions that advance the prevention and treatment of diseases in people and animals. We foster a diverse and inclusive global workforce and operate responsibly every day to enable a safe, sustainable and healthy future for all people and communities. For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf .
Source: Merck & Co., Inc.
Mar. 01, 2023 7:26 AM ET
By: Dulan Lokuwithana, SA News Editor
Merck (NYSE:MRK) announced Wednesday that its blockbuster immunotherapy Keytruda reached one of the dual primary endpoints in a Phase 3 trial as perioperative therapy for certain lung cancer patients.
The trial named KEYNOTE-671 tested neoadjuvant KEYTRUDA (before surgery) plus chemotherapy, followed by adjuvant KEYTRUDA (after surgery) as a single agent against placebo plus neoadjuvant chemotherapy followed by adjuvant placebo.
02/28/2023CATEGORY:
The applications are based on results from the Phase 3 CheckMate -76K trial, in which Opdivo demonstrated a statistically significant and clinically meaningful benefit in recurrence-free survival
The U.S. Food and Drug Administration has assigned a target action date of October 13, 2023
PRINCETON, N.J.--(BUSINESS WIRE)--
U.S. Food and Drug Administration Accepts Bristol Myers Squibb’s Supplemental Biologics License Application and European Medicines Agency Validates Application for Opdivo (nivolumab) as an Adjuvant Treatment for Patients with Completely Resected Stage IIB or IIC Melanoma
Bristol Myers Squibb (NYSE: BMY) today announced that the U.S. Food and Drug Administration (FDA) has accepted the supplemental Biologics License Application (sBLA) and the European Medicines Agency (EMA) has validated the Type II Variation Marketing Authorization Application (MAA) for Opdivo® (nivolumab) as monotherapy in the adjuvant setting for the treatment of patients with completely resected stage IIB or IIC melanoma. In the U.S., the FDA has assigned a Prescription Drug User Fee Act (PDUFA) date of October 13, 2023. In Europe, the EMA’s validation of the application confirms the submission is complete and begins the start of the EMA’s centralized review process.
“Melanoma can be a devastating diagnosis, and patients with stage IIB or IIC melanoma tend to be at high risk of disease recurrence. Approximately one third of stage IIB and half of stage IIC patients experience recurrence within five years after surgery,” said Gina Fusaro, PhD, vice president, development program lead, Bristol Myers Squibb. “The data from the CheckMate -76K trial demonstrate the benefit that Opdivo can have for patients with this earlier stage of cancer. We look forward to working with the U.S. Food and Drug Administration and the European Medicines Agency to potentially offer a treatment option to patients with stage IIB or IIC melanoma that could help prevent recurrence.”
The submissions were based on safety and efficacy results from the pivotal Phase 3 CheckMate -76K trial, in which Opdivo demonstrated a statistically significant and clinically meaningful benefit in recurrence-free survival (RFS) versus placebo in patients with completely resected stage IIB or IIC melanoma. The safety profile of Opdivo was consistent with previously reported studies.
Results from CheckMate -76K were presented as late-breaking data during a plenary session at the Society for Melanoma Research (SMR) Annual Meeting in October 2022.
CheckMate -76K is part of BMS’ development program studying Opdivo and Opdivo-based combinations in earlier stages of cancer (adjuvant, neoadjuvant, and peri-operative), which currently spans seven tumor types. To date, Opdivo-based therapies have shown improved efficacy in the neoadjuvant or adjuvant treatment of four tumor types: non-small cell lung cancer (NSCLC), bladder cancer, esophageal/gastroesophageal junction cancer, and melanoma.
Bristol Myers Squibb thanks the patients and investigators involved in the CheckMate -76K trial.
About CheckMate -76K
CheckMate -76K is a randomized Phase 3, double-blind study evaluating adjuvant Opdivo (nivolumab) 480 mg Q4W for up to 12 months versus placebo in patients with completely resected stage IIB/C melanoma.
The primary endpoint of the trial is recurrence-free survival (RFS). Secondary endpoints of the trial include overall survival (OS), distant metastases-free survival (DMFS), progression-free survival on next-line therapy (PFS2), and safety endpoints.
About Melanoma
Melanoma is a form of skin cancer characterized by the uncontrolled growth of pigment-producing cells (melanocytes) located in the skin. Metastatic melanoma is the deadliest form of the disease and occurs when cancer spreads beyond the surface of the skin to other organs. In the United States, approximately 97,610 new diagnoses of melanoma and about 7,990 related deaths are estimated for 2023. Globally, the World Health Organization estimates that by 2035, melanoma incidence will reach 424,102, with 94,308 related deaths. Melanomas can be mostly treatable when caught in very early stages; however, survival rates can decrease as the disease progresses.
Bristol Myers Squibb: Creating a Better Future for People with Cancer
Bristol Myers Squibb is inspired by a single vision — transforming patients’ lives through science. The goal of the company’s cancer research is to deliver medicines that offer each patient a better, healthier life and to make cure a possibility. Building on a legacy across a broad range of cancers that have changed survival expectations for many, Bristol Myers Squibb researchers are exploring new frontiers in personalized medicine, and through innovative digital platforms, are turning data into insights that sharpen their focus. Deep scientific expertise, cutting-edge capabilities and discovery platforms enable the company to look at cancer from every angle. Cancer can have a relentless grasp on many parts of a patient’s life, and Bristol Myers Squibb is committed to taking actions to address all aspects of care, from diagnosis to survivorship. Because as a leader in cancer care, Bristol Myers Squibb is working to empower all people with cancer to have a better future.
About Opdivo
Opdivo is a programmed death-1 (PD-1) immune checkpoint inhibitor that is designed to uniquely harness the body’s own immune system to help restore anti-tumor immune response. By harnessing the body’s own immune system to fight cancer, Opdivo has become an important treatment option across multiple cancers.
Opdivo’s leading global development program is based on Bristol Myers Squibb’s scientific expertise in the field of Immuno-Oncology and includes a broad range of clinical trials across all phases, including Phase 3, in a variety of tumor types. To date, the Opdivo clinical development program has treated more than 35,000 patients. The Opdivo trials have contributed to gaining a deeper understanding of the potential role of biomarkers in patient care, particularly regarding how patients may benefit from Opdivo across the continuum of PD-L1 expression.
In July 2014, Opdivo was the first PD-1 immune checkpoint inhibitor to receive regulatory approval anywhere in the world. Opdivo is currently approved in more than 65 countries, including the United States, the European Union, Japan, and China. In October 2015, the Company’s Opdivo and Yervoy combination regimen was the first Immuno-Oncology combination to receive regulatory approval for the treatment of metastatic melanoma and is currently approved in more than 50 countries, including the United States and the European Union.
INDICATIONS
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of adult and pediatric patients 12 years of age or older with unresectable or metastatic melanoma.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adult and pediatric patients 12 years of age or older with unresectable or metastatic melanoma.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of adult and pediatric patients 12 years of age or older with melanoma with involvement of lymph nodes or metastatic disease who have undergone complete resection.
OPDIVO® (nivolumab), in combination with platinum-doublet chemotherapy, is indicated as neoadjuvant treatment of adult patients with resectable (tumors ≥4 cm or node positive) non-small cell lung cancer (NSCLC).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) whose tumors express PD-L1 (≥1%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab) and 2 cycles of platinum-doublet chemotherapy, is indicated for the first-line treatment of adult patients with metastatic or recurrent non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) with progression on or after platinum-based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving OPDIVO.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with unresectable malignant pleural mesothelioma (MPM).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with intermediate or poor risk advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab), in combination with cabozantinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with advanced renal cell carcinoma (RCC) who have received prior anti-angiogenic therapy.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with classical Hodgkin lymphoma (cHL) that has relapsed or progressed after autologous hematopoietic stem cell transplantation (HSCT) and brentuximab vedotin or after 3 or more lines of systemic therapy that includes autologous HSCT. This indication is approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN) with disease progression on or after platinum-based therapy.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
OPDIVO® (nivolumab), as a single agent, is indicated for the adjuvant treatment of adult patients with urothelial carcinoma (UC) who are at high risk of recurrence after undergoing radical resection of UC.
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of adult and pediatric (12 years and older) patients with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adults and pediatric patients 12 years and older with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adult patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with unresectable advanced, recurrent or metastatic esophageal squamous cell carcinoma (ESCC) after prior fluoropyrimidine- and platinum-based chemotherapy.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of completely resected esophageal or gastroesophageal junction cancer with residual pathologic disease in adult patients who have received neoadjuvant chemoradiotherapy (CRT).
OPDIVO® (nivolumab), in combination with fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of adult patients with unresectable advanced or metastatic esophageal squamous cell carcinoma (ESCC).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with unresectable advanced or metastatic esophageal squamous cell carcinoma (ESCC).
OPDIVO® (nivolumab), in combination with fluoropyrimidine- and platinum- containing chemotherapy, is indicated for the treatment of adult patients with advanced or metastatic gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma.
Please see US Full Prescribing Information for OPDIVO and YERVOY.
About the Bristol Myers Squibb and Ono Pharmaceutical Collaboration
In 2011, through a collaboration agreement with Ono Pharmaceutical Co., Bristol Myers Squibb expanded its territorial rights to develop and commercialize Opdivo globally, except in Japan, South Korea and Taiwan, where Ono had retained all rights to the compound at the time. On July 23, 2014, Ono and Bristol Myers Squibb further expanded the companies’ strategic collaboration agreement to jointly develop and commercialize multiple immunotherapies – as single agents and combination regimens – for patients with cancer in Japan, South Korea and Taiwan.
About Bristol Myers Squibb
Bristol Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information about Bristol Myers Squibb, visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
![]()
Download:Download original 99 KB 1505 x 211
Download thumbnail 5 KB 200 x 28
Download lowres 15 KB 480 x 67
Download square 7 KB 250 x 250
Feb. 28, 2023 8:08 AM ET
Bristol-Myers Squibb Company (BMY)
By: Ravikash, SA News Editor
PUBLISHED22 February 2023
AstraZeneca’s Imfinzi (durvalumab) and Imjudo (tremelimumab) immunotherapy combinations have been approved in the European Union (EU) for the treatment of advanced liver and lung cancers.
The approvals authorise Imfinzi in combination with Imjudo for the 1st-line treatment of adult patients with advanced or unresectable hepatocellular carcinoma (HCC) and Imfinzi in combination with Imjudo and platinum-based chemotherapy for the treatment of adult patients with metastatic (Stage IV) non-small cell lung cancer (NSCLC).
The two approvals by the European Commission follow positive recommendations by The Committee for Medicinal Products for Human Use of the European Medicines Agency in December 2022 and are based on positive results from the HIMALAYA Phase III trial, published in the New England Journal of Medicine Evidence and the POSEIDON Phase III trial, published in the Journal of Clinical Oncology.
Bruno Sangro, MD, PhD, Director of the Liver Unit and Professor of Internal Medicine at Clínica Universidad de Navarra, and a lead investigator in the HIMALAYA Phase III trial, said: “This approval in Europe is welcome news for eligible patients with advanced liver cancer, who face a poor prognosis and are in need of well-tolerated treatments that can meaningfully extend overall survival. In HIMALAYA, an estimated 31% of patients treated with this novel combination of tremelimumab with durvalumab were alive at three years, while only 20% of patients treated with sorafenib were still alive at the same duration of follow-up.”
Solange Peters, MD, PhD, Head of the Medical Oncology Service and Chair of Thoracic Oncology at Hospitalier Universitaire Vaudois, Lausanne, Switzerland, and principal investigator in the POSEIDON Phase III trial, said: “Patients with metastatic non-small cell lung cancer continue to need new therapies that can meaningfully extend survival, including for many patients whose disease does not respond to current therapies. The approval of tremelimumab added to durvalumab and chemotherapy means that patients in Europe with this devastating cancer now have a valuable new treatment approach with demonstrated long-term survival benefits.”
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “Lung cancer is one of the most prevalent and deadly cancers in Europe, and rates of liver cancer are rising steadily across the region. These approvals for Imfinzi and Imjudo bring critically needed, novel immunotherapy-based treatment regimens that will potentially deliver life-extending benefits for European patients with advanced liver and non-small cell lung cancers.”
Imjudo and Imfinzi approved in liver cancer
The approval for the treatment of HCC is based on results from the HIMALAYA Phase III trial, where the Single Tremelimumab Regular Interval Durvalumab (STRIDE) regimen, comprised of a single dose of the anti-CTLA-4 antibody Imjudo (300mg) combined with anti-PD-L1 antibody Imfinzi (1500mg dose) followed by Imfinzi every four weeks significantly reduced the risk of death by 22% versus sorafenib (hazard ratio [HR] 0.78; 95% confidence interval [CI], 0.66-0.92; p=0.0035). Median overall survival (OS) was 16.4 months versus 13.8 for sorafenib. An estimated 31% of patients treated with the combination were still alive after three years with 20% of patients treated with sorafenib still alive at the same duration of follow-up.
The safety profile of the combination of Imjudo added to Imfinzi were consistent with the known profiles of each medicine, and no new safety signals were identified.
Liver cancer is the third-leading cause of cancer death and the sixth most commonly diagnosed cancer worldwide.1,2 Approximately 87,000 Europeans were diagnosed with liver cancer in 2020, with 51% of patients at an advanced cancer stage at time of diagnosis. Rates of liver cancer continue to rise rapidly, with a 70% increase of liver cancer-related mortality in Europe from 1990-2019.3
Imfinzi and Imjudo approved in NSCLC
The approval for the treatment of metastatic NSCLC is based on results from the POSEIDON Phase III trial, which showed a limited course of five cycles of the anti-CTLA-4 antibody Imjudo added to Imfinzi plus four cycles of platinum-based chemotherapy significantly reduced the risk of death by 23% versus a range of chemotherapy options (HR 0.77; 95% CI, 0.65-0.92; p=0.00304). Median OS was 14.0 months versus 11.7 months for chemotherapy. An estimated 33% of patients were alive at two years versus 22% for chemotherapy. This treatment combination also reduced the risk of disease progression or death by 28% compared to chemotherapy alone (HR 0.72; 95% CI, 0.60-0.86; p=0.00031) with a median progression-free survival (PFS) of 6.2 months versus 4.8 months, respectively.
Updated results from the POSEIDON Phase III trial after approximately four years of follow-up presented at the European Society for Medical Oncology Congress 2022 demonstrated sustained survival benefit, reducing the risk of death by 25% compared to chemotherapy alone (HR 0.75; 95% CI, 0.63-0.88). Updated median OS was 14 months for the combination versus 11.7 months for chemotherapy alone. An estimated 25% of patients treated with the combination were alive at three years versus 13.6% for those treated with chemotherapy alone.
The safety profile for Imjudo plus Imfinzi and chemotherapy was consistent with the known profiles of each medicine, and no new safety signals were identified.
Metastatic lung cancer is the most advanced form of lung cancer.4,5 Approximately 40% of people with NSCLC have metastatic disease at the time of diagnosis.6 An estimated 5% of patients with metastatic NSCLC in Europe will survive five years after diagnosis.7-8
For the POSEIDON indication in the EU, Imjudo will be temporarily marketed under the name Tremelimumab AstraZeneca until the second half of 2023.
Notes
Liver cancer
About 75% of all primary liver cancers in adults are HCC.1 Between 80-90% of all patients with HCC also have cirrhosis.9 Chronic liver diseases are associated with inflammation that over time can lead to the development of HCC.9
More than half of patients are diagnosed at advanced stages of the disease, often when symptoms first appear.10 A critical unmet need exists for patients with HCC who face limited treatment options.10 The unique immune environment of liver cancer provides clear rationale for investigating medications that harness the power of the immune system to treat HCC.10
Stage IV NSCLC
Lung cancer is the leading cause of cancer-related death and is the second most common form of cancer globally, with more than two million patients diagnosed in 2020.11 Lung cancer is broadly split into NSCLC and small-cell lung cancer (SCLC), with 80-85% classified as NSCLC. Within NSCLC, patients are classified as squamous, representing 25-30% of patients, or non-squamous, in approximately 70-75% of patients.4, 12-13
HIMALAYA
HIMALAYA is a randomised, open-label, multicentre, global Phase III trial of Imfinzi monotherapy and a regimen comprising a single priming dose of Imjudo 300mg added to Imfinzi 1500mg followed by Imfinzi every four weeks versus sorafenib, a standard-of-care multi-kinase inhibitor.
The trial included a total of 1,324 randomised patients with unresectable, advanced HCC who had not been treated with prior systemic therapy and were not eligible for locoregional therapy (treatment localised to the liver and surrounding tissue).
The trial was conducted in 181 centres across 16 countries, including in the US, Canada, Europe, South America and Asia. The primary endpoint was OS for the combination versus sorafenib and key secondary endpoints included OS for Imfinzi versus sorafenib, objective response rate and PFS for the combination and for Imfinzi alone.
POSEIDON
POSEIDON is a randomised, open-label, multi-centre, global, Phase III trial of Imfinzi plus platinum-based chemotherapy or Imfinzi, tremelimumab and chemotherapy versus chemotherapy alone in the 1st-line treatment of 1,013 patients with metastatic NSCLC. The trial population included patients with either non-squamous or squamous disease and the full range of PD-L1 expression levels. POSEIDON excluded patients with certain epidermal growth factor receptor (EGFR) mutations or anaplastic lymphoma kinase (ALK) fusions.
In the experimental arms, patients were treated with a flat dose of either Imfinzi (1,500mg) or Imfinzi plus Imjudo (75mg) with up to four cycles of chemotherapy every three weeks before either Imfinzi maintenance once every four weeks or Imfinzi and a fifth dose of Imjudo given at week 16. In comparison, the control arm allowed up to six cycles of chemotherapy. Pemetrexed maintenance treatment was allowed in all arms in patients with non-squamous disease if given during the induction phase. Nearly all patients with non-squamous disease (95.5%) had pemetrexed and platinum, while the majority of patients with squamous disease receiving chemotherapy (88.3%) received gemcitabine and platinum.
Primary endpoints included PFS and OS for the Imfinzi plus chemotherapy arm. Key secondary endpoints included PFS and OS in the Imfinzi plus Imjudo and chemotherapy arm. As PFS endpoints were met for both experimental arms, the prespecified statistical analysis plan allowed for testing OS in the Imfinzi plus Imjudo and chemotherapy arm. The OS trend observed in the Imfinzi plus chemotherapy arm did not achieve statistical significance. The trial was conducted in more than 150 centres across 18 countries, including the US, Europe, South America, Asia and South Africa.
Imfinzi
Imfinzi (durvalumab) is a human monoclonal antibody that binds to the PD-L1 protein and blocks the interaction of PD-L1 with the PD-1 and CD80 proteins, countering the tumour's immune-evading tactics and releasing the inhibition of immune responses.
Imfinzi is the only approved immunotherapy in the curative-intent setting of unresectable, Stage III NSCLC in patients whose disease has not progressed after chemoradiotherapy and is the global standard of care in this setting based on the PACIFIC Phase III trial.
Imfinzi is also approved in the US, EU, Japan, China and many other countries around the world for the treatment of extensive-stage SCLC based on the CASPIAN Phase III trial. In an exploratory analysis in 2021, updated results from the CASPIAN trial showed Imfinzi plus chemotherapy tripled patient survival at three years versus chemotherapy alone.
Imfinzi is also approved in combination with Imjudo and chemotherapy in metastatic NSCLC in the US, EU and Japan; in combination with chemotherapy in locally advanced or metastatic biliary tract cancer (BTC) in the US, EU, Japan and several other countries; in combination with Imjudo in unresectable HCC in the US, EU and Japan; as monotherapy in unresectable HCC in Japan; and in previously treated patients with advanced bladder cancer in several countries.
Since the first approval in May 2017, more than 100,000 patients have been treated with Imfinzi.
As part of a broad development programme, Imfinzi is being tested as a single treatment and in combinations with other anti-cancer treatments for patients with SCLC, NSCLC, bladder cancer, several gastrointestinal (GI) cancers, ovarian cancer, endometrial cancer and other solid tumours.
Imjudo
Imjudo (tremelimumab) is a human monoclonal antibody that targets the activity of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). Imjudo blocks the activity of CTLA-4, contributing to T-cell activation, priming the immune response to cancer and fostering cancer cell death.
In addition to its approved indications in liver and lung cancers, Imjudo is being tested in combination with Imfinzi across multiple tumour types including locoregional HCC (EMERALD-3 trial), SCLC (ADRIATIC trial) and bladder cancer (VOLGA and NILE trials).
AstraZeneca in GI cancers
AstraZeneca has a broad development programme for the treatment of GI cancers across several medicines and a variety of tumour types and stages of disease. In 2020, GI cancers collectively represented approximately 5.1 million new cancer cases leading to approximately 3.6 million deaths.14
Within this programme, the Company is committed to improving outcomes in gastric, liver, biliary tract, oesophageal, pancreatic, and colorectal cancers.
In addition to its approved indications in biliary tract and liver cancers, Imfinzi is being assessed in combinations, including with Imjudo, in liver, oesophageal and gastric cancers in an extensive development programme spanning early to late-stage disease across settings.
Enhertu (trastuzumab deruxtecan), a HER2-directed antibody drug conjugate, is approved in the US, EU and several other countries for HER2-positive advanced gastric cancer and is being assessed in colorectal cancer. Enhertu is jointly developed and commercialised by AstraZeneca and Daiichi Sankyo.
Lynparza (olaparib), a first-in-class PARP inhibitor, is approved in the US, EU and several other countries for the treatment of BRCA-mutated metastatic pancreatic cancer. Lynparza is developed and commercialised in collaboration with MSD (Merck & Co., Inc. inside the US and Canada).
AstraZeneca in lung cancer
AstraZeneca is working to bring patients with lung cancer closer to cure through the detection and treatment of early-stage disease, while also pushing the boundaries of science to improve outcomes in the resistant and advanced settings. By defining new therapeutic targets and investigating innovative approaches, the Company aims to match medicines to the patients who can benefit most.
The Company's comprehensive portfolio includes leading lung cancer medicines and the next wave of innovations, including Tagrisso (osimertinib) and Iressa (gefitinib); Imfinzi (durvalumab) and Imjudo (tremelimumab); Enhertu (trastuzumab deruxtecan) and datopotamab deruxtecan in collaboration with Daiichi Sankyo; Orpathys (savolitinib) in collaboration with HUTCHMED; as well as a pipeline of potential new medicines and combinations across diverse mechanisms of action.
AstraZeneca is a founding member of the Lung Ambition Alliance, a global coalition working to accelerate innovation and deliver meaningful improvements for people with lung cancer, including and beyond treatment.
AstraZeneca in immuno-oncology (IO)
AstraZeneca is a pioneer in introducing the concept of immunotherapy into dedicated clinical areas of high unmet medical need. The Company has a comprehensive and diverse IO portfolio and pipeline anchored in immunotherapies designed to overcome evasion of the anti-tumour immune response and stimulate the body’s immune system to attack tumours.
AstraZeneca aims to reimagine cancer care and help transform outcomes for patients with Imfinzi as a single treatment and in combination with Imjudo as well as other novel immunotherapies and modalities. The Company is also exploring next-generation immunotherapies like bispecific antibodies and therapeutics that harness different aspects of immunity to target cancer.
AstraZeneca is boldly pursuing an innovative clinical strategy to bring IO-based therapies that deliver long-term survival to new settings across a wide range of cancer types. With an extensive clinical programme, the Company also champions the use of IO treatment in earlier disease stages, where there is the greatest potential for cure.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Feb. 22, 2023 4:27 AM ET
By: Ravikash, SA News Editor
February 24, 2023 at 7:07 AM ESTPDF Version
WILMINGTON, Del.--(BUSINESS WIRE)--Feb. 24, 2023-- Incyte (Nasdaq:INCY) today announced that the European Medicines Agency (EMA) Committee for Medicinal Products for Human Use (CHMP) has issued a positive opinion recommending the approval of ruxolitinib cream (Opzelura™) for the treatment of non-segmental vitiligo with facial involvement in adults and adolescents from 12 years of age.
“The positive CHMP opinion brings us one step closer to bringing ruxolitinib cream, the first ever treatment for repigmentation in non-segmental vitiligo, to patients and healthcare professionals in the European Union (EU),” said Steven Stein, M.D., Chief Medical Officer, Chief Medical Officer, Incyte. “With no centrally approved treatment option currently available in the EU, this positive opinion marks a significant milestone for the vitiligo community.”
The CHMP opinion recommending the approval of ruxolitinib cream was based on data from two pivotal Phase 3 clinical trials (TRuE-V1 and TRuE-V2) evaluating the safety and efficacy of ruxolitinib cream versus vehicle (non-medicated cream) in more than 600 people with non-segmental vitiligo, age 12 and older1. Results from the TRuE-V program, recently published in The New England Journal of Medicine, showed that treatment with ruxolitinib cream resulted in significant improvements in facial and total body repigmentation versus vehicle as shown by the number of patients reaching the facial and total body Vitiligo Area Scoring Index (F-VASI-T-VASI) endpoints at Week 24 compared to vehicle, with a higher proportion of patients responding at Week 521. The most common adverse reactions (incidence ≥ 1%) were application site acne, application site pruritus, nasopharyngitis, headache, urinary tract infection, application site erythema, and pyrexia2.
The CHMP’s opinion is now being reviewed by the European Commission, which has the authority to grant centralized marketing authorizations for medicinal products in the EU. When approved, this will be the first approved vitiligo therapy available in the EU indicated for the treatment of non-segmental vitiligo with facial involvement in adults and adolescents from 12 years of age.
“Given its complex pathogenesis and unpredictable progression, vitiligo can be very challenging for dermatologists to treat,” said Thierry Passeron M.D., Ph.D., Professor and Chair, Department of Dermatology, Université Côte d'Azur in Nice, France and one of the lead investigators of the TRUE-V trials. “I welcome today’s news and look forward to the potential approval of an effective therapy that can address repigmentation, providing a much-needed option for those patients living with vitiligo who are actively seeking treatment, as well as the clinical community dedicated to its treatment.”
About Vitiligo
Vitiligo is a chronic autoimmune disease characterized by depigmentation of skin that results in patchy loss of skin color from the progressive destruction of pigment-producing cells known as melanocytes. Overactivity of the JAK signaling pathway is believed to drive inflammation involved in the pathogenesis and progression of vitiligo. In Europe, approximately 1.5 million patients are diagnosed with vitiligo (0.2 to 0.8% of the population3,4), and its overall prevalence is estimated to be less than 1%, with the majority of patients (approximately 8 in 10) suffering from non-segmental vitiligo5. Vitiligo can occur at any age, although many patients with vitiligo will experience initial onset before the age of 306. Vitiligo not only impacts physical health but also places a heavy burden on quality of life including employment and psychosocial health such as depression.
About TRuE-V
The TRuE-V clinical trial program includes two Phase 3 studies, TRuE-V1 (NCT04052425) and TRuE-V2 (NCT04057573), evaluating the safety and efficacy of ruxolitinib cream in patients with vitiligo. Each study enrolled approximately 300 patients (age ≥12 years) who have been diagnosed with non-segmental vitiligo.
About Ruxolitinib Cream (Opzelura™)
Ruxolitinib cream (Opzelura™), a novel cream formulation of Incyte’s selective JAK1/JAK2 inhibitor ruxolitinib, approved by the U.S. Food & Drug Administration for the topical treatment of nonsegmental vitiligo in patients 12 years of age and older, is the first and only treatment for repigmentation approved for use in the United States. Opzelura is also approved in the U.S. for the topical short-term and non-continuous chronic treatment of mild to moderate atopic dermatitis (AD) in non-immunocompromised patients 12 years of age and older whose disease is not adequately controlled with topical prescription therapies, or when those therapies are not advisable. Use of Opzelura in combination with therapeutic biologics, other JAK inhibitors, or potent immunosuppressants, such as azathioprine or cyclosporine, is not recommended.
Incyte has worldwide rights for the development and commercialization of ruxolitinib cream, marketed in the United States as Opzelura. In April 2022, Incyte entered into a strategic alliance agreement with Maruho Co., Ltd. for the development, manufacturing and exclusive commercialization of ruxolitinib cream for treatment of autoimmune and inflammatory dermatology indications in Japan.
Opzelura is a trademark of Incyte.
About Incyte Dermatology
Incyte’s science-first approach and expertise in immunology has formed the foundation of the company. Today, we are building on this legacy as we discover and develop innovative dermatology treatments to bring solutions to patients in need.
Our research and development efforts in dermatology are initially focused on leveraging our knowledge of the JAK-STAT pathway. We are exploring the potential of JAK inhibition for a number of immune-mediated dermatologic conditions with a high unmet medical need, including atopic dermatitis, vitiligo and hidradenitis suppurativa.
To learn more, visit the Dermatology section of Incyte.com.
About Incyte
Incyte is a Wilmington, Delaware-based, global biopharmaceutical company focused on finding solutions for serious unmet medical needs through the discovery, development and commercialization of proprietary therapeutics. For additional information on Incyte, please visit Incyte.com and follow @Incyte.
View source version on businesswire.com: https://www.businesswire.com/news/home/20230223006046/en/
Source: Incyte Corporation
Feb. 24, 2023 7:52 AM ETIncyte Corporation (INCY)
By: Dulan Lokuwithana, SA News Editor
Incyte (NASDAQ:INCY) announced Friday that a group of medical experts in the EU recommended the marketing authorization of ruxolitinib cream for non-segmental vitiligo with facial involvement in those aged 12 years and above.
https://seekingalpha.com/news/3940592-incyte-wins-eu-nod-vitiligo-cream-opzelura
February 8, 2023 at 6:45 PM EST
ROP is a leading cause of childhood blindness worldwide
EYLEA now approved to treat five retinal conditions caused by ocular angiogenesis
TARRYTOWN, N.Y., Feb. 08, 2023 (GLOBE NEWSWIRE) -- Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) today announced that the U.S. Food and Drug Administration (FDA) has approved EYLEA® (aflibercept) Injection to treat preterm infants with retinopathy of prematurity (ROP). Following this first pediatric approval, EYLEA is now indicated to treat five retinal conditions caused by ocular angiogenesis.
“Retinopathy of prematurity is a leading cause of childhood blindness worldwide. Until now, the only FDA-approved treatment in common use was laser photocoagulation, a complex and lengthy procedure that permanently ablates retina tissue and is stressful not only for infant patients but also the family navigating a delicate time after a preterm birth,” said George D. Yancopoulos, M.D., Ph.D., President and Chief Scientific Officer of Regeneron, and a principal inventor of EYLEA. “For the first time, physicians will now have an FDA approved medication in EYLEA to treat this heartbreaking disease in these smallest of patients. We thank the investigators and the many families who participated in the clinical trials.”
Each year in the U.S., between 1,100 to 1,500 infants develop ROP that is severe enough to require medical treatment. This rare eye disease often impacts infants who are born before 31 weeks of pregnancy have been completed or who weigh less than 1,500 grams (3.3 lbs) pounds at birth. As retinal blood vessels are often only fully developed once an infant is full-term (~9 months of pregnancy), these infants are at risk of developing retinal blood vessels that are abnormal (retinal neovascularization) potentially leading to retinal detachment and irreversible vision loss. Mild cases of ROP may improve without treatment, but some cases require treatment to keep ROP from causing significant visual impairment and even blindness.
“With no existing FDA approved guidance for the treatment of retinopathy of prematurity with anti-VEGF therapies, there was a significant need for research to understand how best to treat the disease in a manner that puts patient safety first and preserves vision for a lifetime,” said Jeff Todd, Chief Executive Officer, Prevent Blindness. “Regeneron’s trials investigating EYLEA in retinopathy of prematurity have advanced our understanding of how to treat this disease and provided a needed evidence-based treatment option to potentially help preterm infants preserve their vision.”
About the Phase 3 Program in ROP
The FDA approval is supported by data from two randomized global Phase 3 trials – FIREFLEYE (N=113) and BUTTERFLEYE (N=120) – investigating EYLEA 0.4 mg versus laser photocoagulation (laser) in infants with ROP. In both trials, approximately 80% of EYLEA-treated infants achieved an absence of both active ROP and unfavorable structural outcomes at 52 weeks of age, which is better than would have been expected without treatment.
No new EYLEA safety signals were observed in either trial. Comparing EYLEA to laser, ocular adverse events (AEs) among patients occurred in 39% versus 37% in FIREFLEYE and 18% versus 26% in BUTTERFLEYE, with serious ocular AEs occurring in 8% for both groups in FIREFLEYE and 6.5% versus 11% in BUTTERFLEYE. AEs in both trials were consistent with infant prematurity or to the injection procedure, and with the AEs in similar ROP trials. The results of FIREFLEYE were published in Journal of the American Medical Association, and data from BUTTERFLEYE were presented at ROP Update 2022 meeting in the U.S.
Both trials were conducted pursuant to FDA Pediatric Written Request, and a Pediatric Exclusivity Determination was granted by FDA on October 12, 2022. This grant extends the period of U.S. market exclusivity for EYLEA by an additional six months through May 17, 2024.
EYLEA is being jointly developed by Regeneron and Bayer. The lead sponsors of the trials were Regeneron for BUTTERFLEYE and Bayer for FIREFLEYE. Bayer and Regeneron are collaborating on the global development of EYLEA. Regeneron maintains exclusive rights of EYLEA in the United States. Bayer has licensed the exclusive marketing rights outside the United States, where the companies share equally the profits from sales of EYLEA.
About EYLEA
EYLEA is a VEGF inhibitor formulated as an injection for the eye. It is designed to block the growth of new blood vessels and decrease the ability of fluid to pass through blood vessels (vascular permeability) in the eye by blocking VEGF-A and placental growth factor (PLGF), two growth factors involved in ocular angiogenesis. The EYLEA safety and efficacy profile is supported by a robust body of research that includes eight pivotal Phase 3 trials, more than 11 years of real-world experience and greater than 57 million EYLEA injections globally.
IMPORTANT SAFETY INFORMATION AND INDICATIONS FOR EYLEA
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.fda.gov/medwatch, or call 1-800-FDA-1088.
INDICATIONS
EYLEA® (aflibercept) Injection 2 mg (0.05 mL) is a prescription medicine approved for the treatment of patients with Wet Age-related Macular Degeneration (AMD), Macular Edema following Retinal Vein Occlusion (RVO), Diabetic Macular Edema (DME), Diabetic Retinopathy (DR), and Retinopathy of Prematurity (ROP).
Please see the full Prescribing Information for EYLEA.
About Regeneron
Regeneron (NASDAQ: REGN) is a leading biotechnology company that invents, develops and commercializes life-transforming medicines for people with serious diseases. Founded and led for 35 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to nine FDA-approved treatments and numerous product candidates in development, almost all of which were homegrown in our laboratories. Our medicines and pipeline are designed to help patients with eye diseases, allergic and inflammatory diseases, cancer, cardiovascular and metabolic diseases, pain, hematologic conditions, infectious diseases and rare diseases.
Regeneron is accelerating and improving the traditional drug development process through our proprietary VelociSuite® technologies, such as VelocImmune®, which uses unique genetically humanized mice to produce optimized fully human antibodies and bispecific antibodies, and through ambitious research initiatives such as the Regeneron Genetics Center®, which is conducting one of the largest genetics sequencing efforts in the world.
For more information, please visit www.Regeneron.com or follow @Regeneron on Twitter.
![]()
Source: Regeneron Pharmaceuticals, Inc.
Feb. 23, 2023 8:24 AM ET
Regeneron Pharmaceuticals, Inc. (REGN)BAYRY, BAYZF
By: Jonathan Block, SA News Editor
PUBLISHED22 February 2023
AstraZeneca’s tablet formulation of Calquence (acalabrutinib) has been approved in the European Union (EU) for the treatment of adult patients with chronic lymphocytic leukaemia (CLL).
The approval by the European Commission follows the positive opinion of the Committee for Medicinal Products for Human Use and is based on results from the ELEVATE-PLUS trials published in the American Society of Haematology journal, Blood.1
In the trials, results showed the Calquence capsule and tablet formulations are bioequivalent, indicating the same efficacy and safety profile can be expected when prescribed with the same dosing strength and schedule.1 The tablet can be taken with gastric acid-reducing agents, including proton pump inhibitors (PPIs), antacids and H2-receptor antagonists (H2RAs).1 The majority of observed adverse events (AEs) in these studies were mild with no new safety concerns identified.1
Paolo Ghia, MD, Director, Strategic Research Program on CLL, Università Vita-Salute San Raffaele in Milan, said: “Many patients with chronic lymphocytic leukaemia face multiple medical conditions that require daily treatment, including the use of acid-reducing agents for conditions such as gastro-oesophageal reflux. The tablet formulation allows for co-administration with these drugs, allowing more patients with chronic lymphocytic leukaemia to assume Calquence.”
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “The Calquence tablet formulation underscores our commitment to understanding the needs of the chronic lymphocytic leukaemia community and providing patient-oriented treatment solutions. Today’s approval offers physicians and patients in the EU more flexibility to determine the right treatment plan and enables more patients to potentially benefit from this medicine.”
Calquence is approved as a capsule formulation for CLL in the EU. It is also approved in the US as a capsule and tablet formulation for patients with CLL, small lymphocytic lymphoma (SLL) and relapsed or refractory mantle cell lymphoma (MCL). Additionally, Calquence is approved as a capsule formulation in many other countries worldwide. Indications may vary by market.
Notes
CLL
CLL is the most prevalent type of leukaemia in adults, with over 100,000 new cases globally in 2019.2 Although some people with CLL may not experience any symptoms at diagnosis, others may experience symptoms, such as weakness, fatigue, weight loss, chills, fever, night sweats, swollen lymph nodes and abdominal pain.3
In CLL, there is an accumulation of abnormal lymphocytes within the bone marrow and in blood and lymph nodes. As the number of abnormal cells increases, there is less room within the marrow for the production of normal white blood cells, red blood cells and platelets. This could result in anaemia, infection and bleeding.4 B-cell receptor signalling through BTK is one of the essential growth pathways for CLL.
ELEVATE-PLUS
ELEVATE-PLUS is comprised of three Phase I, open-label, single-dose, cross-over studies conducted in 116 healthy subjects. The trials established bioequivalence between acalabrutinib tablets (100mg) and acalabrutinib (100mg) capsules, evaluated the PPI effect of acalabrutinib tablets administered in the presence versus absence of PPI rabeprazole and investigated the effect of food by comparing acalabrutinib tablets administered with a high-fat diet versus fasted.1
Calquence
Calquence (acalabrutinib) is a next-generation, selective inhibitor of Bruton’s tyrosine kinase (BTK). Calquence binds covalently to BTK, thereby inhibiting its activity.5,6 In B cells, BTK signalling results in activation of pathways necessary for B-cell proliferation, trafficking, chemotaxis and adhesion.5
Calquence is available for prescribing in capsule and tablet formulations in the US and EU for the treatment of CLL. Capsules have restrictions in relation to use with gastric acid reducing agents.
Calquence capsules are approved for CLL in Japan, Canada, Australia and many other countries worldwide.
In the US and several other countries, Calquence capsules are also approved for the treatment of adult patients with MCL who have received at least one prior therapy. The US MCL indication is approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials. Calquence is not currently approved for the treatment of MCL in Europe or Japan.
As part of an extensive clinical development programme, AstraZeneca and Acerta Pharma are currently evaluating Calquence in more than 20 company-sponsored clinical trials. Calquence is being evaluated for the treatment of multiple B-cell blood cancers, including CLL, MCL, diffuse large B-cell lymphoma, Waldenström’s macroglobulinaemia, follicular lymphoma and marginal zone lymphoma.
AstraZeneca in haematology
AstraZeneca is pushing the boundaries of science to redefine care in haematology. We have expanded our commitment to patients with haematologic conditions, not only in oncology but also in rare diseases with the acquisition of Alexion, allowing us to reach more patients with high unmet needs. By applying our deep understanding of blood cancers, leveraging our strength in solid tumour oncology and delivering on Alexion’s pioneering legacy in complement science to provide innovative medicines for rare diseases, we are pursuing the end-to-end development of novel therapies designed to target underlying drivers of disease.
By targeting haematologic conditions with high unmet medical needs, we aim to deliver innovative medicines and approaches to improve patient outcomes. Our goal is to help transform the lives of patients living with malignant, rare and other related haematologic diseases, shaped by insights from patients, caregivers and physicians to have the most meaningful impact.
AstraZeneca in Oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients. The Company’s focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development and commercialisation of prescription medicines in Oncology, Rare Diseases and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Feb. 22, 2023 4:44 AM ET
By: Ravikash, SA News Editor
February 17, 2023
– Oral Presentati on Highlights Trodelvy Efficacy of 13.5 Months Overall Survival in Pati ents with Platinum-Ineligible Metastatic UC After Checkpoint Inhibitor Therapy –
– Trodelvy Demonstrated 12.8 Months Overall Survival in Pa tients with Metastatic UC Whose Disease Progressed Rapidly Following Platinum-Based Chemotherapy –
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced new and updated positive results from three cohorts of the Phase 2 TROPHY-U-01 study of Trodelvy® (sacituzumab govitecan-hziy) for the treatment of metastatic urothelial cancer (mUC). These data demonstrate that Trodelvy produced both rapid and durable responses for patients across a range of hard-to-treat types of mUC including platinum-ineligible and rapidly progressing, post-platinum mUC. These findings will be featured in both an oral session (abstract #520) and in poster presentations (abstract #518, #526) during the 2023 American Society of Clinical Oncology Genitourinary Cancers Symposium (ASCO-GU) Annual Meeting February 16-18.
“The TROPHY-U-01 data show consistent benefit of Trodelvy across multiple types of metastatic urothelial cancer, including the most difficult-to-treat and, often times, frail patients where treatment options are still scarce,” said Bill Grossman, MD, PhD, Senior Vice President, Therapeutic Area Head, Gilead Oncology. “Trodelvy has the potential to become a cornerstone treatment in metastatic urothelial cancer, and we are excited about the expected results from the ongoing Phase 3 TROPiCS-04 study that may serve to convert our U.S. accelerated approval to full approval for Trodelvy to treat patients with locally advanced or metastatic urothelial cancer following a platinum-containing chemotherapy and PD-1/PD-L1 inhibitor.”
Longer-term follow-up across Cohorts 1, 2, and 3 of TROPHY-U-01 provides an increasing body of evidence supporting the potential benefit of treating mUC with Trodelvy across clinically relevant, hard-to-treat patient populations.
In April 2021, the U.S. FDA granted accelerated approval of Trodelvy for use in adult patients with locally advanced or mUC who have previously received a platinum-containing chemotherapy and either a PD-1 or PD-L1 inhibitor. This approval is based on ORR and DOR established in Cohort 1.
Trodelvy has not been approved by any regulatory agency for the treatment of platinum-ineligible patients with mUC who progressed after prior CPI therapy, or in combination with pembrolizumab in patients with mUC who progressed after platinum-based therapy. Its safety and efficacy have not been established for these indications.
Trodelvy has a Boxed Warning for severe or life-threatening neutropenia and severe diarrhea; please see below for additional Important Safety Information.
About Metastatic Urothelial Cancer
Urothelial Cancer (UC) is the most common type of bladder cancer and occurs when the urothelial cells that line the inside of the bladder and other parts of the urinary tract grow unusually or uncontrollably. An estimated 83,000 Americans will be diagnosed with bladder cancer in 20231, and almost 90% of those diagnoses will be UC2. In total, 30% of cases are considered advanced or metastatic disease.3 Despite advancements in treating mUC, long-term survival remains low.
About the TROPHY U-01 Study
The Phase 2 TROPHY-U-01 trial is an ongoing, international, multi-center, open-label, multi-cohort, single-arm study evaluating Trodelvy monotherapy or combination therapy in patients with mUC after progression on a platinum-based regimen and anti-PD-1/PD-L1-based immunotherapy.
Cohort 1 is assessing Trodelvy after progression on platinum-based chemotherapy and checkpoint inhibitor (CPI) therapy. Cohorts 2, 3, 4 and 5 of the study are ongoing. Cohort 2 is assessing Trodelvy monotherapy in platinum-ineligible patients after progression on anti-PD-1/PD-L1-based immunotherapy. Cohort 3 is assessing Trodelvy in patients with rapidly progressing, mUC who progressed after platinum-based therapy. Cohorts 4 and 5 are assessing Trodelvy combination therapy in patients with treatment naive mUC, with those in Cohort 4 receiving cisplatin and those in Cohort 5 receiving cisplatin and avelumab, respectively, in addition to Trodelvy.
More information about TROPHY is available at https://clinicaltrials.gov/ct2/show/NCT03547973.
About Trodelvy
Trodelvy® (sacituzumab govitecan-hziy) is a first-in-class Trop-2 directed antibody-drug conjugate. Trop-2 is a cell surface antigen highly expressed in multiple tumor types, including in more than 90% of breast and bladder cancers. Trodelvy is intentionally designed with a proprietary hydrolyzable linker attached to SN-38, a topoisomerase I inhibitor payload. This unique combination delivers potent activity to both Trop-2 expressing cells and the microenvironment.
Trodelvy is approved in more than 40 countries, with multiple additional regulatory reviews underway worldwide, for the treatment of adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (TNBC) who have received two or more prior systemic therapies, at least one of them for metastatic disease.
Trodelvy is also approved in the U.S. to treat certain patients with pre-treated HR+/HER2- metastatic breast cancer and has an accelerated approval for treatment of certain patients with second-line metastatic urothelial cancer; see below for full indication statements.
Trodelvy is also being developed for potential investigational use in other TNBC, HR+/HER2- and mUC populations, as well as a range of tumor types where Trop-2 is highly expressed, including metastatic non-small cell lung cancer (NSCLC), metastatic small cell lung cancer (SCLC), head and neck cancer, and endometrial cancer.
U.S. Indications for Trodelvy
In the United States, Trodelvy is indicated for the treatment of adult patients with:
Please see full Prescribing Information , including BOXED WARNING.
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer. Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California.
Source: Gilead Sciences, Inc.
Feb. 17, 2023 9:44 AM ET
Gilead Sciences, Inc. (GILD)MRK
By: Dulan Lokuwithana, SA News Editor
February 18, 2023
OSAKA, Japan and CAMBRIDGE, Massachusetts,
February 18, 2023 – Takeda (TSE:4502/NYSE:TAK) today announced late-breaking data from the Phase 3 GRAPHITE study presented at the 2023 Tandem Meetings, demonstrating vedolizumab achieved a statistically significant and clinically meaningful improvement in lower gastrointestinal (GI) aGvHD-free survival by Day 180 after allo-HSCT with no relevant differences in safety profile versus placebo.1 Intestinal aGvHD is a serious complication characterized by inflammation of the GI tract which can affect patients undergoing allo-HSCT, a common treatment for blood cancers.
“Lower GI aGvHD represents a critical unmet need in patients undergoing allo-HSCT.” said Yi-Bin Chen, MD, Director, Hematopoietic Cell Transplant & Cell Therapy Program, Mass General Cancer Center. “I’m excited that this study can contribute to the understanding of this common and life-threatening complication in stem cell transplantation.”
Vedolizumab is not currently indicated for use in aGvHD.
The Phase 3, randomized, double-blind, placebo-controlled, multicenter GRAPHITE study evaluated the efficacy and safety of vedolizumab as prophylaxis for intestinal aGvHD in patients undergoing allo-HSCT from unrelated donors for the treatment of hematological malignancies. The study met its primary endpoint, with vedolizumab achieving a statistically significant improvement in intestinal aGvHD-free survival versus placebo by Day 180 after allo-HSCT (85.5% of patients in the vedolizumab arm versus 70.9% in the placebo arm [HR=0.45; 95% CI: 0.27, 0.73; p<0.001]).1 Statistically significant superiority of vedolizumab over placebo was also demonstrated for intestinal aGvHD-free and relapse-free survival by Day +180 (HR= 0.56, 95% CI: 0.37, 0.86; p = 0.0043), and for Grade C-D aGvHD-free (with any organ involvement) survival at Day +180 (HR: 0.59, 95% CI: 0.39, 0.91; p = 0.0204).1 In addition, no relevant differences in safety profile between the vedolizumab and placebo arms were observed, and no new safety signals were identified. Treatment-related adverse events were reported in 24.8% versus 28.4%, and treatment related serious adverse events in 8.5% versus 6.5% of patients treated with placebo versus vedolizumab, respectively. The most common adverse events of special interest were hypersensitivity reactions (placebo 82.4%, vedolizumab 79.3%), serious infections (placebo 67.3%, vedolizumab 74.0%), and liver injury (placebo 41.8%, vedolizumab 40.2%).1
“These results have advanced our understanding of vedolizumab, currently indicated for IBD, in another critical GI inflammatory condition.” said Chinwe Ukomadu, M.D., Ph.D., Head, Gastroenterology Therapeutic Area Unit at Takeda. “Our Phase 3 study in the prevention of lower GI aGvHD is the latest example of Takeda’s commitment to advancing the science of vedolizumab, furthering the understanding of its mechanism of action, and exploring new ways to help patients.”
Intestinal aGvHD can occur after stem cell transplantation when the immune cells of the donor (the graft) consider the recipient’s body (the host) as foreign and attack the organs and tissue.2 Intestinal aGvHD results in the majority of morbidity and mortality associated with GvHD. Effective prevention of aGvHD, especially with lower intestinal involvement, has been an important treatment goal for physicians when patients are undergoing allo-HSCT.3
GRAPHITE (vedolizumab-3035) is a randomized, double-blind, placebo controlled study designed to evaluate the use of vedolizumab as prophylaxis of intestinal aGvHD in participants who receive allo-HSCT as treatment for a hematologic malignancy or myeloproliferative disorder from an unrelated donor.1 The study enrolled 333 patients who were randomly assigned in a 1:1 ratio to one of two treatment groups receiving either an intravenous infusion of vedolizumab 300 mg or placebo on days -1 (before allo-HSCT), and on days +13, +41, +69, +97, +125, and +153 following allo-HSCT, alongside a background GvHD prophylaxis regimen. The overall time of participation in the study was 12 months.4
Vedolizumab is a gut-selective anti-lymphocyte therapy and is approved in both intravenous (IV) and subcutaneous (SC) formulations (approvals vary by market; vedolizumab is not currently approved in the SC formulation in the U.S.).5,6 It is a humanized monoclonal antibody designed to specifically antagonize the alpha4beta7 integrin, inhibiting the binding of alpha4beta7 integrin to intestinal mucosal addressin cell adhesion molecule 1 (MAdCAM-1), but not vascular cell adhesion molecule 1 (VCAM-1).7 MAdCAM-1 is preferentially expressed on blood vessels and lymph nodes of the gastrointestinal tract.8 The alpha4beta7 integrin is expressed on a subset of circulating white blood cells.7 These cells have been shown to play a role in mediating the inflammatory process in ulcerative colitis (UC) and Crohn’s disease (CD).7,9,10 By inhibiting alpha4beta7 integrin, vedolizumab may limit the ability of certain white blood cells to infiltrate gut tissues.7
Vedolizumab is approved for the treatment of adult patients with moderately to severely active UC and CD, who have had an inadequate response with, lost response to, or were intolerant to either conventional therapy or a tumor necrosis factor-alpha (TNFα)-antagonist.5,6 Vedolizumab IV has been granted marketing authorization in over 70 countries, including the United States and European Union, with more than 1,000,000 patient years of exposure to date.11 Vedolizumab SC has been granted marketing authorization in the European Union and over 50 countries.
Vedolizumab is indicated for the treatment of adult patients with moderately to severely active ulcerative colitis who have had an inadequate response with, lost response to, or were intolerant to either conventional therapy or a tumor necrosis factor-alpha (TNFα) antagonist.
Vedolizumab is indicated for the treatment of adult patients with moderately to severely active Crohn’s disease who have had an inadequate response with, lost response to, or were intolerant to either conventional therapy or a tumor necrosis factor-alpha (TNFα) antagonist.
Vedolizumab IV is indicated in the EU for the treatment of adult patients with moderately to severely active chronic pouchitis, who have undergone proctocolectomy and ileal pouch-anal anastomosis (IPAA) for ulcerative colitis (UC) and have had an inadequate response with or lost response to antibiotic therapy.
Please consult with your local regulatory agency for approved labeling in your country.
For EU audiences, please see the Summary of Product Characteristics (SmPC) for ENTYVIO®.
For U.S. audiences, please see the full Prescribing Information, including Medication Guide for ENTYVIO® IV.
Takeda is a global, values-based, RD-driven biopharmaceutical leader headquartered in Japan, committed to discover and deliver life-transforming treatments, guided by our commitment to patients, our people and the planet. Takeda focuses its RD efforts on four therapeutic areas: Oncology, Rare Genetics and Hematology, Neuroscience, and Gastroenterology (GI), with expertise in immune and inflammatory diseases. We also make targeted RD investments in Plasma-Derived Therapies and Vaccines. We are focusing on developing highly innovative medicines that contribute to making a difference in people’s lives by advancing the frontier of new treatment options and leveraging our enhanced collaborative RD engine and capabilities to create a robust, modality-diverse pipeline. Our employees are committed to improving quality of life for patients and to working with our partners in health care in approximately 80 countries and regions. For more information, visit .
Feb. 20, 2023 5:18 AM ET
Takeda Pharmaceutical Company Limited (TAK)
By: Ravikash, SA News Editor
Takeda Pharmaceutical's (NYSE:TAK) drug vedolizumab met the main goal of a phase 3 trial to prevent intestinal acute graft-versus-host disease (aGvHD) in patients undergoing allogeneic hematopoietic stem cell transplantation (allo-HSCT).

02/17/2023 CATEGORY:
Three-year median follow-up data demonstrate significantly improved disease-free survival, non-urothelial tract recurrence-free survival, distant metastasis-free survival and second progression-free survival with adjuvant Opdivo compared to placebo
Randomized patients who received Opdivo after radical surgery remained disease-free more than twice as long vs. placebo; patients whose tumor cells express PD-L1 ≥1% remained disease-free more than six times as long vs. placebo
Updated results from the Phase 3 CheckMate -274 trial will be presented in a late-breaking oral presentation at ASCO GU 2023
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) today announced three-year follow-up results from the Phase 3 CheckMate -274 trial, demonstrating significant sustained clinical benefits with Opdivo (nivolumab) for the adjuvant treatment of patients with surgically resected, high-risk muscle-invasive urothelial carcinoma. With a median follow-up of 36.1 months (31.6 months minimum), adjuvant Opdivo continued to show improved disease-free survival (DFS), non-urothelial tract recurrence-free survival (NUTRFS), distant metastasis-free survival (DMFS) and second progression-free survival (PFS2) compared to placebo across all-randomized patients and in patients whose tumor cells express PD-L1 ≥1%. These updated results will be featured in a late-breaking oral presentation at the American Society of Clinical Oncology (ASCO) 2023 Genitourinary Cancers Symposium from February 16-18, 2023.
“Patients with muscle-invasive urothelial carcinoma face a high chance of recurrence due to micrometastatic disease, especially within the first three years after surgical removal of the bladder or kidney. The three-year results from CheckMate -274 show a stable decrease in the risk of disease with adjuvant nivolumab with longer follow-up,” said Matthew D. Galsky,* M.D., Professor of Medicine, Director of Genitourinary Medical Oncology, Associate Director for Translational Research, and Co-Director of the Center of Excellence for Bladder Cancer at The Tisch Cancer Institute and the Icahn School of Medicine at Mount Sinai. “Nivolumab remains the only immunotherapy, as well as the only medical treatment in general, to decrease the risk of urothelial cancer recurrence after radical surgery in patients who received chemotherapy prior to surgery or who are ineligible for chemotherapy. The results of this trial have changed the way that urothelial cancer is treated.”
With three years of follow-up in the CheckMate -274 trial:
“We strive to provide hope for patients by offering safe and effective options that may help prevent recurrence and improve long-term outcomes. This is why earlier stages of cancer is an important research area for Bristol Myers Squibb across multiple tumor types, including hard-to-treat cancers with high unmet needs like urothelial carcinoma,” said Dana Walker, M.D., M.S.C.E., vice president, development program lead, genitourinary cancers, Bristol Myers Squibb. “The durable follow-up results from CheckMate -274 continue to fuel our excitement toward our ongoing research in earlier stages and its potential to change outcomes for patients. We look forward to closely following the CheckMate -274 trial, which is ongoing to assess additional key secondary endpoints, including overall survival to which we currently remain blinded.”
Bristol Myers Squibb thanks the patients and investigators involved in the CheckMate -274 clinical trial.
About CheckMate -274
CheckMate -274 is a Phase 3 randomized, double-blind, multi-center study evaluating Opdivo compared to placebo in patients with muscle-invasive urothelial carcinoma at a high risk of recurrence after radical resection. A total of 709 patients were randomized 1:1 to receive Opdivo 240 mg or placebo every two weeks for up to one year. The primary endpoints of the trial are disease-free survival (DFS) in all randomized patients (i.e., the intention-to-treat population) and in the subset of patients whose tumor cells express PD-L1 ≥1%. Key secondary endpoints include overall survival (OS), non-urothelial tract recurrence-free survival (NUTRFS) and disease-specific survival (DSS). Key exploratory endpoints include distant metastasis-free survival (DMFS), quality of life (QoL) and second progression-free survival (PFS2).
About Urothelial Carcinoma
Bladder cancer is the 10th most common cancer in the world, with more than 573,000 new cases diagnosed annually. Urothelial carcinoma, which most frequently begins in the cells that line the inside of the bladder, accounts for approximately 90% of bladder cancer cases. In addition to the bladder, urothelial carcinoma can occur in other parts of the urinary tract, including the ureters and renal pelvis. The majority of urothelial carcinomas are diagnosed at an early stage, but rates of recurrence and disease progression are high. Approximately 50% of patients who undergo radical surgery will experience disease recurrence, especially within the first three years after surgical removal of the bladder or kidney. For patients whose disease recurs as metastatic cancer, the prognosis is poor, with a median overall survival of approximately 12 to 14 months when treated with systemic therapy.
Bristol Myers Squibb: Creating a Better Future for People with Cancer
Bristol Myers Squibb is inspired by a single vision — transforming patients’ lives through science. The goal of the company’s cancer research is to deliver medicines that offer each patient a better, healthier life and to make cure a possibility. Building on a legacy across a broad range of cancers that have changed survival expectations for many, Bristol Myers Squibb researchers are exploring new frontiers in personalized medicine, and through innovative digital platforms, are turning data into insights that sharpen their focus. Deep scientific expertise, cutting-edge capabilities and discovery platforms enable the company to look at cancer from every angle. Cancer can have a relentless grasp on many parts of a patient’s life, and Bristol Myers Squibb is committed to taking actions to address all aspects of care, from diagnosis to survivorship. Because as a leader in cancer care, Bristol Myers Squibb is working to empower all people with cancer to have a better future.
About Opdivo
Opdivo is a programmed death-1 (PD-1) immune checkpoint inhibitor that is designed to uniquely harness the body’s own immune system to help restore anti-tumor immune response. By harnessing the body’s own immune system to fight cancer, Opdivo has become an important treatment option across multiple cancers.
Opdivo’s leading global development program is based on Bristol Myers Squibb’s scientific expertise in the field of Immuno-Oncology and includes a broad range of clinical trials across all phases, including Phase 3, in a variety of tumor types. To date, the Opdivo clinical development program has treated more than 35,000 patients. The Opdivo trials have contributed to gaining a deeper understanding of the potential role of biomarkers in patient care, particularly regarding how patients may benefit from Opdivo across the continuum of PD-L1 expression.
In July 2014, Opdivo was the first PD-1 immune checkpoint inhibitor to receive regulatory approval anywhere in the world. Opdivo is currently approved in more than 65 countries, including the United States, the European Union, Japan and China. In September 2015, the Company’s Opdivo and Yervoy combination regimen was the first Immuno-Oncology combination to receive regulatory approval for the treatment of metastatic melanoma and is currently approved in more than 50 countries, including the United States and the European Union.
INDICATIONS
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of adult and pediatric (12 years and older) patients with unresectable or metastatic melanoma.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adult and pediatric (12 years and older) patients with unresectable or metastatic melanoma.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of adult and pediatric (12 years and older) patients with melanoma with involvement of lymph nodes or metastatic disease who have undergone complete resection.
OPDIVO® (nivolumab), in combination with platinum-doublet chemotherapy, is indicated as neoadjuvant treatment of adult patients with resectable (tumors ≥4 cm or node positive) non-small cell lung cancer (NSCLC).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) whose tumors express PD-L1 (≥1%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab) and 2 cycles of platinum-doublet chemotherapy, is indicated for the first-line treatment of adult patients with metastatic or recurrent non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) with progression on or after platinum-based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving OPDIVO.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with unresectable malignant pleural mesothelioma (MPM).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with intermediate or poor risk advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab), in combination with cabozantinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with advanced renal cell carcinoma (RCC) who have received prior anti-angiogenic therapy.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with classical Hodgkin lymphoma (cHL) that has relapsed or progressed after autologous hematopoietic stem cell transplantation (HSCT) and brentuximab vedotin or after 3 or more lines of systemic therapy that includes autologous HSCT. This indication is approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN) with disease progression on or after platinum-based therapy.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
OPDIVO® (nivolumab), as a single agent, is indicated for the adjuvant treatment of adult patients with urothelial carcinoma (UC) who are at high risk of recurrence after undergoing radical resection of UC.
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of adult and pediatric (12 years and older) patients with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adult and pediatric (12 years and older) patients with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adult patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with unresectable advanced, recurrent or metastatic esophageal squamous cell carcinoma (ESCC) after prior fluoropyrimidine- and platinum-based chemotherapy.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of completely resected esophageal or gastroesophageal junction cancer with residual pathologic disease in adult patients who have received neoadjuvant chemoradiotherapy (CRT).
OPDIVO® (nivolumab), in combination with fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of adult patients with unresectable advanced or metastatic esophageal squamous cell carcinoma (ESCC).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with unresectable advanced or metastatic esophageal squamous cell carcinoma (ESCC).
OPDIVO® (nivolumab), in combination with fluoropyrimidine- and platinum- containing chemotherapy, is indicated for the treatment of adult patients with advanced or metastatic gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma.
Please see US Full Prescribing Information for OPDIVO and YERVOY.
About Bristol Myers Squibb
Bristol Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information about Bristol Myers Squibb, visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
For more information, please see U.S. Full Prescribing Information and Medication Guide for OPDIVO,
and U.S. Full Prescribing Information and Medication Guide for YERVOY, or talk to your healthcare team.
Information provided on this website is not a substitute for talking with your healthcare professional.
Your healthcare professional is the best source of information about your disease.
All individuals depicted are models used for illustrative purposes only.
Feb. 17, 2023 11:01 AM ET
Bristol-Myers Squibb Company (BMY)
By: Jonathan Block, SA News Editor
Feb 21, 2023
– PDUFA Date Set for October 8, 2023 –
CAMBRIDGE, Mass.--(BUSINESS WIRE)--Feb. 21, 2023-- Alnylam Pharmaceuticals, Inc. (Nasdaq: ALNY), the leading RNAi therapeutics company, today announced that the U.S. Food and Drug Administration (FDA) has accepted for filing the Company’s supplemental New Drug Application (sNDA) for patisiran, an investigational RNAi therapeutic in development for the treatment of the cardiomyopathy of transthyretin-mediated (ATTR) amyloidosis. The FDA has set an action date of October 8, 2023 under the Prescription Drug User Fee Act (PDUFA). In their file acceptance letter, the FDA stated that they have not identified any review issues. The Agency also noted that they are planning to hold an advisory committee meeting to discuss the application. Patisiran is the established name for ONPATTRO®, which is currently approved by the U.S. FDA for the treatment of the polyneuropathy of hereditary ATTR amyloidosis in adults.
“ATTR amyloidosis with cardiomyopathy is an increasingly recognized cause of heart failure for which there are limited treatment options. The FDA’s acceptance of our sNDA for patisiran is a positive step forward as we work to bring patients with ATTR amyloidosis with cardiomyopathy a new treatment option that addresses the underlying cause of disease and has the potential to meaningfully improve functional capacity and quality of life,” said Rena N. Denoncourt, Vice President, TTR Franchise Lead. “The acceptance also marks another important milestone as we continue to build an industry-leading franchise for the treatment of ATTR amyloidosis.”
The application to the FDA was based on positive results from APOLLO-B, a randomized, double-blind, placebo-controlled, multicenter, global Phase 3 study that demonstrated favorable effects of patisiran on both functional capacity and quality of life in patients with ATTR amyloidosis with cardiomyopathy relative to placebo at 12 months. The majority of adverse events were mild or moderate in severity, and the overall safety profile in APOLLO-B was consistent with prior clinical trials and post-marketing experience with ONPATTRO. The 12-month results from the study were presented at the 18th International Symposium on Amyloidosis (ISA) on September 8, 2022 and at the Heart Failure Society of America’s Annual Scientific Meeting on September 30, 2022.
ONPATTRO Indication and Important Safety Information
Indication
ONPATTRO is indicated for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults.
About ONPATTRO® (patisiran)
ONPATTRO (patisiran) is an RNAi therapeutic that is approved in the United States and Canada for the treatment of the polyneuropathy of hATTR amyloidosis in adults. ONPATTRO is also approved in the European Union, Switzerland and Brazil for the treatment of hATTR amyloidosis in adults with Stage 1 or Stage 2 polyneuropathy, and in Japan for the treatment of hATTR amyloidosis with polyneuropathy. ONPATTRO is an intravenously administered RNAi therapeutic targeting transthyretin (TTR). It is designed to target and silence TTR messenger RNA, thereby reducing the production of TTR protein before it is made. Reducing the pathogenic protein leads to a reduction in amyloid deposits in tissues.
About ATTR Amyloidosis
Transthyretin-mediated (ATTR) amyloidosis is a rare, rapidly progressive, debilitating disease caused by misfolded transthyretin (TTR) proteins which accumulate as amyloid fibrils in multiple tissues including the nerves, heart, and gastrointestinal (GI) tract. There are two different types of ATTR amyloidosis – Hereditary ATTR (hATTR) amyloidosis, caused by a TTR gene variant, and Wild-type ATTR (wtATTR) amyloidosis, which occurs without a TTR gene variant. hATTR amyloidosis affects approximately 50,000 people worldwide, while wtATTR amyloidosis is estimated to impact 200,000 – 300,000 people worldwide.
About LNP Technology
Alnylam has licenses to Arbutus Biopharma LNP intellectual property for use in RNAi therapeutic products using LNP technology.
About RNAi
RNAi (RNA interference) is a natural cellular process of gene silencing that represents one of the most promising and rapidly advancing frontiers in biology and drug development today. Its discovery has been heralded as "a major scientific breakthrough that happens once every decade or so," and was recognized with the award of the 2006 Nobel Prize for Physiology or Medicine. By harnessing the natural biological process of RNAi occurring in our cells, a new class of medicines known as RNAi therapeutics is now a reality. Small interfering RNA (siRNA), the molecules that mediate RNAi and comprise Alnylam's RNAi therapeutic platform, function upstream of today’s medicines by potently silencing messenger RNA (mRNA) – the genetic precursors – that encode for disease-causing or disease pathway proteins, thus preventing them from being made. This is a revolutionary approach with the potential to transform the care of patients with genetic and other diseases.
About Alnylam Pharmaceuticals
Alnylam Pharmaceuticals (Nasdaq: ALNY) has led the translation of RNA interference (RNAi) into a whole new class of innovative medicines with the potential to transform the lives of people afflicted with rare and prevalent diseases with unmet need. Based on Nobel Prize-winning science, RNAi therapeutics represent a powerful, clinically validated approach yielding transformative medicines. Since its founding 20 years ago, Alnylam has led the RNAi Revolution and continues to deliver on a bold vision to turn scientific possibility into reality. Alnylam’s commercial RNAi therapeutic products are ONPATTRO® (patisiran), AMVUTTRA® (vutrisiran), GIVLAARI® (givosiran), OXLUMO® (lumasiran), and Leqvio® (inclisiran), which is being developed and commercialized by Alnylam’s partner, Novartis. Alnylam has a deep pipeline of investigational medicines, including multiple product candidates that are in late-stage development. Alnylam is executing on its “Alnylam P5x25” strategy to deliver transformative medicines in both rare and common diseases benefiting patients around the world through sustainable innovation and exceptional financial performance, resulting in a leading biotech profile. Alnylam is headquartered in Cambridge, MA. For more information about our people, science and pipeline, please visit www.alnylam.com and engage with us on Twitter at @Alnylam, on LinkedIn, or on Instagram.
View source version on businesswire.com: https://www.businesswire.com/news/home/20230221005335/en/
Alnylam Pharmaceuticals, Inc.
Source: Alnylam Pharmaceuticals, Inc.
Feb. 21, 2023 9:50 AM ET
Alnylam Pharmaceuticals, Inc. (ALNY)
By: Ravikash, SA News Editor
February 17, 2023 6:45 am ET
FDA also accepts a separate supplemental application to extend prophylaxis with PREVYMIS to 200 days in certain HSCT recipients
RAHWAY, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside of the United States and Canada, today announced the U.S. Food and Drug Administration (FDA) has accepted for review two supplemental new drug applications (sNDA) for PREVYMIS™ (letermovir). The FDA granted priority review for the sNDA for PREVYMIS for prophylaxis of cytomegalovirus (CMV) disease in adult kidney transplant recipients at high risk (D+/R-); the Prescription Drug User Fee Act (PDUFA), or target action date, is June 5, 2023. The FDA grants priority review to medicines and vaccines that, if approved, would provide a significant improvement in the safety or effectiveness of the treatment or prevention of a serious condition. A second sNDA to extend use of PREVYMIS from 100 days to 200 days in adults receiving an allogeneic hematopoietic stem cell transplant (HSCT) who are at risk for late CMV infection and disease was also accepted for review, with a PDUFA date of Sept. 7, 2023.
“Certain high-risk individuals who develop CMV infection following receipt of a kidney transplant are at increased risk for transplant failure and death. PREVYMIS has the potential to be an important new option with a favorable safety profile for patients at risk for CMV infection following a kidney transplant,” said Dr. Nicholas Kartsonis, senior vice president, vaccines and infectious diseases, Global Clinical Development, Merck Research Laboratories. “We look forward to the FDA’s review of our filings for PREVYMIS.”
PREVYMIS is a first-in-class antiviral agent that was approved by the U.S. FDA in 2017 and is indicated for prophylaxis of CMV infection and disease in adult CMV-seropositive recipients [R+] of an allogeneic hematopoietic stem cell transplant (HSCT).
Data supporting the sNDA for CMV prophylaxis in kidney transplant recipients
The sNDA for use of PREVYMIS for CMV prophylaxis in kidney transplant recipients is supported by a Phase 3, randomized, double-blind clinical trial (NCT03443869) that demonstrated non-inferior efficacy and a more favorable safety profile for PREVYMIS compared to valganciclovir, the current standard of care for CMV prophylaxis in kidney transplant recipients. Data from the trial were presented during a late-breaking oral session at the IDWeek Annual Meeting in October 2022.
Key safety results
In the study, PREVYMIS had a more favorable safety profile compared to valganciclovir, with fewer drug-related adverse events (AEs) and study drug discontinuations due to adverse events reported in the PREVYMIS group compared to the valganciclovir group. The incidence of leukopenia (decrease in leukocytes, or white blood cells) and neutropenia (decrease in neutrophils, the most common type of white blood cell) was lower with PREVYMIS than with valganciclovir:
Additional safety information from the trial:
Data supporting the sNDA for 200 days of prophylaxis in HSCT patients
The sNDA for 200 days of prophylaxis with PREVYMIS is supported by a Phase 3, randomized, double-blind, placebo-controlled trial (NCT03930615) that evaluated the safety and efficacy of extending prophylaxis with PREVYMIS from 100 to 200 days, compared to placebo, in CMV-seropositive recipients [R+] at high risk for clinically significant CMV infection following an HSCT. This trial met its primary endpoint and prophylaxis with PREVYMIS extended to 200 days was superior to placebo in reducing the rate of clinically significant CMV infection from Week 14 through Week 28 post-HSCT in allogeneic R+ HSCT recipients. PREVYMIS had an adverse event profile similar to placebo and was well-tolerated. These data will be presented on Saturday, Feb. 18, at the 2023 Tandem Meetings / Transplantation & Cellular Therapy Meetings of ASTCT™ (American Society for Transplantation and Cellular Therapy) and CIBMTR® (Center for International Blood & Marrow Transplant Research).
About PREVYMIS (letermovir)
PREVYMIS is the only drug approved in the United States for prophylaxis of CMV infection and disease in adults who are CMV-seropositive and have received an allogeneic HSCT. PREVYMIS is also approved in more than 60 countries outside of the United States, including EU member states, Canada, Japan and China. PREVYMIS is a first-in-class non-nucleoside CMV inhibitor (3,4 dihydro-quinazolines) and inhibits viral replication by specifically targeting the viral terminase complex. Cross resistance is not likely with drugs outside of this class. PREVYMIS is fully active against viral populations with substitutions conferring resistance to CMV DNA polymerase inhibitors. These DNA polymerase inhibitors are fully active against viral populations with substitutions conferring resistance to PREVYMIS. PREVYMIS has no activity against other viruses.
Selected Safety Information about PREVYMIS (letermovir)
PREVYMIS is contraindicated in patients receiving pimozide or ergot alkaloids. Increased pimozide concentrations may lead to QT prolongation and torsades de pointes. Increased ergot alkaloids concentrations may lead to ergotism. PREVYMIS is contraindicated with pitavastatin and simvastatin when co-administered with cyclosporine. Significantly increased pitavastatin or simvastatin concentrations may lead to myopathy or rhabdomyolysis.
The concomitant use of PREVYMIS and certain drugs may result in potentially significant drug interactions, some of which may lead to adverse reactions (PREVYMIS or concomitant drugs) or reduced therapeutic effect of PREVYMIS or the concomitant drug.
The rate of adverse events in the first 100 days following HSCT occurring in at least 10% of transplant recipients treated with PREVYMIS and at a frequency at least 2% greater than placebo were nausea (27% vs 23%), diarrhea (26% vs 24%), vomiting (19% vs 14%), peripheral edema (14% vs 9%), cough (14% vs 10%), headache (14% vs 9%), fatigue (13% vs 11%), and abdominal pain (12% vs 9%).
Hypersensitivity reaction, with associated moderate dyspnea, occurred in one subject following the first infusion of IV PREVYMIS after switching from oral PREVYMIS, leading to treatment discontinuation.
If PREVYMIS is co-administered with cyclosporine, the dosage of PREVYMIS should be decreased to 240 mg once daily.
Co-administration of PREVYMIS may alter the plasma concentrations of other drugs and other drugs may alter the plasma concentrations of PREVYMIS. Consult the full Prescribing Information prior to and during treatment for potential drug interactions.
Closely monitor serum creatinine levels in patients with CLcr less than 50 mL/min using PREVYMIS injection.
PREVYMIS is not recommended for patients with severe (Child-Pugh Class C) hepatic impairment.
The safety and efficacy of PREVYMIS in patients below 18 years of age have not been established.
For patients with CLcr greater than 10 mL/min (by Cockcroft-Gault equation), no dosage adjustment of PREVYMIS is required based on renal impairment. The safety of PREVYMIS in patients with end-stage renal disease (CLcr less than 10 mL/min), including patients on dialysis, is unknown.
About Merck
At Merck, known as MSD outside of the United States and Canada, we are unified around our purpose: We use the power of leading-edge science to save and improve lives around the world. For more than 130 years, we have brought hope to humanity through the development of important medicines and vaccines. We aspire to be the premier research-intensive biopharmaceutical company in the world – and today, we are at the forefront of research to deliver innovative health solutions that advance the prevention and treatment of diseases in people and animals. We foster a diverse and inclusive global workforce and operate responsibly every day to enable a safe, sustainable and healthy future for all people and communities. For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Please see Prescribing Information for PREVYMIS at https://www.merck.com/product/usa/pi_circulars/p/prevymis/prevymis_pi.pdf and Patient Information/Medication Guide for PREVYMIS at https://www.merck.com/product/usa/pi_circulars/p/prevymis/prevymis_ppi.pdf.
Source: Merck & Co., Inc.
Feb. 17, 2023 7:22 AM ET
By: Dulan Lokuwithana, SA News Editor
02/13/2023 CATEGORY: Corporate/Financial News
Data to be featured at ASCO GU 2023 demonstrate continued overall survival, progression-free survival and objective response rate benefits with Opdivo in combination with CABOMETYX compared to sunitinib, regardless of IMDC risk score
These three-year data – with a median follow-up of 44 months – from CheckMate -9ER represent the longest reported follow-up in any Phase 3 trial with an immunotherapy-tyrosine kinase inhibitor regimen in this population
In an exploratory biomarker analysis, median progression-free survival and overall survival were improved with the combination of Opdivo and CABOMETYX regardless of PD-L1 status
PRINCETON, N.J. & ALAMEDA, Calif.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) and Exelixis, Inc. (NASDAQ: EXEL) today announced three-year (36.5 months minimum; 44.0 months median) follow-up results from the Phase 3 CheckMate -9ER trial, demonstrating sustained survival and response rate benefits with the combination of Opdivo® (nivolumab) and CABOMETYX® (cabozantinib) versus sunitinib in the first-line treatment of advanced renal cell carcinoma (RCC). Additionally, a biomarker analysis showed that improvements in median progression-free survival (PFS) and overall survival (OS) were sustained with the combination of Opdivo and CABOMETYX regardless of PD-L1 status. These updated results will be featured in one oral and one poster presentation at the American Society of Clinical Oncology (ASCO) 2023 Genitourinary Cancers Symposium from February 16-18, 2023.
“Despite the progress made through science and medicine, there remains a need for treatment options that can durably extend survival for patients with metastatic renal cell carcinoma, especially for those classified as higher risk,” said Mauricio Burotto, M.D., medical director, Bradford Hill Clinical Research Center, Santiago, Chile. “With these updated results from CheckMate -9ER, we’ve now seen nivolumab in combination with cabozantinib durably extend survival and sustain response benefits compared to sunitinib for over three years, regardless of patients’ risk classification. These results reinforce the importance of this immunotherapy-tyrosine kinase inhibitor regimen for patients and its potential to help change survival expectations for patients with this challenging cancer.”
Abstract #603: Nivolumab plus cabozantinib vs sunitinib for first-line treatment of advanced renal cell carcinoma (aRCC): 3-year follow-up from the phase 3 CheckMate -9ER trial (Burotto, et. al.)
With a median follow-up of 44.0 months (36.5 months minimum), Opdivo in combination with CABOMETYX (n=323) continued to show superior OS, PFS, objective response rate (ORR), duration of response (DoR) and complete response (CR) rates versus sunitinib (n=328). No new safety signals were seen with extended follow-up. Within the full study population:
Results were also assessed by the following International Metastatic Renal Cell Carcinoma Database Consortium (IMDC) risk scores: favorable, intermediate, intermediate/poor and poor. Benefits were seen with Opdivo in combination with CABOMETYX across all efficacy measures (OS, PFS, ORR and CR), regardless of IMDC risk.
Abstract #608: Biomarker analysis from the phase 3 CheckMate 9ER trial of nivolumab + cabozantinib v sunitinib for advanced renal cell carcinoma (aRCC) (Choueiri, et. al.)
In an exploratory post-hoc analysis from CheckMate -9ER with 44.0 months median follow-up, improvements were observed in both median PFS and OS with Opdivo in combination with CABOMETYX regardless of PD-L1 status. PFS and OS were evaluated by tumor PD-L1 expression (<1% or ≥1%), CD8% (low, medium, high by tertiles), and CD8 topology phenotype (cold, excluded, inflamed), and assessed for association using Kaplan-Meier (KM) methods with log-rank test (PD-L1 and CD8), and Cox proportional hazard (Cox PH) models (CD8). Biomarkers previously found to be predictive of anti-PD-L1 + anti-VEGF outcomes, including established gene expression signatures, were not necessarily predictive of efficacy with anti-PD-1 + anti-VEGF targeted therapy, suggesting that key determinants of response to anti-PD-1 vs. anti-PD-L1 therapies may differ.
“We have a strong heritage of bringing immunotherapy-based combinations to patients with advanced renal cell carcinoma and transforming patient outcomes with Opdivo-based combinations across several cancer types,” said Dana Walker, M.D., M.S.C.E., vice president, development program lead, genitourinary cancers, Bristol Myers Squibb. “The updated data from CheckMate -9ER further reinforce the use of Opdivo in combination with CABOMETYX for the first-line treatment of patients with advanced renal cell carcinoma. We remain committed to collaboration across the scientific community and our ongoing exploration of combination approaches with our proven cancer agents in the pursuit of solutions that may help improve long-term outcomes for as many patients as possible.”
“With forty-four months median follow-up, the compelling survival benefit further reinforces the value of the CABOMETYX and Opdivo combination regimen as a first-line option for patients with advanced kidney cancer,” said Vicki L. Goodman, M.D., Executive Vice President, Product Development & Medical Affairs, and Chief Medical Officer, Exelixis. “We are pleased to share these long-term findings at ASCO GU and remain steadfast in our longstanding commitment to improving outcomes for patients living with advanced kidney cancer.”
Bristol Myers Squibb and Exelixis thank the patients and investigators involved in the CheckMate -9ER clinical trial.
About CheckMate -9ER
CheckMate -9ER is an open-label, randomized, multi-national Phase 3 trial evaluating patients with previously untreated advanced or metastatic renal cell carcinoma (RCC). A total of 651 patients (23% favorable risk, 58% intermediate risk, 20% poor risk; 25% PD-L1≥1%) were randomized to receive Opdivo plus CABOMETYX (n=323) vs. sunitinib (n=328). The primary endpoint is progression-free survival (PFS). Secondary endpoints include overall survival (OS) and objective response rate (ORR). The primary efficacy analysis is comparing the doublet combination vs. sunitinib in all randomized patients. The trial is sponsored by Bristol Myers Squibb and Ono Pharmaceutical Co. and co-funded by Exelixis, Inc., Ipsen Pharma SAS and Takeda Pharmaceutical Company Limited.
About Renal Cell Carcinoma
Renal cell carcinoma (RCC) is the most common type of kidney cancer in adults, accounting for more than 431,000 new cases and 179,000 deaths worldwide each year. RCC is approximately twice as common in men as in women, with the highest rates of the disease in North America and Europe. At diagnosis, up to 30% of patients present with advanced or metastatic RCC.
Bristol Myers Squibb: Creating a Better Future for People with Cancer
Bristol Myers Squibb is inspired by a single vision — transforming patients’ lives through science. The goal of the company’s cancer research is to deliver medicines that offer each patient a better, healthier life and to make cure a possibility. Building on a legacy across a broad range of cancers that have changed survival expectations for many, Bristol Myers Squibb researchers are exploring new frontiers in personalized medicine, and through innovative digital platforms, are turning data into insights that sharpen their focus. Deep scientific expertise, cutting-edge capabilities and discovery platforms enable the company to look at cancer from every angle. Cancer can have a relentless grasp on many parts of a patient’s life, and Bristol Myers Squibb is committed to taking actions to address all aspects of care, from diagnosis to survivorship. Because as a leader in cancer care, Bristol Myers Squibb is working to empower all people with cancer to have a better future.
About Opdivo
Opdivo is a programmed death-1 (PD-1) immune checkpoint inhibitor that is designed to uniquely harness the body’s own immune system to help restore anti-tumor immune response. By harnessing the body’s own immune system to fight cancer, Opdivo has become an important treatment option across multiple cancers.
Opdivo’s leading global development program is based on Bristol Myers Squibb’s scientific expertise in the field of Immuno-Oncology and includes a broad range of clinical trials across all phases, including Phase 3, in a variety of tumor types. To date, the Opdivo clinical development program has treated more than 35,000 patients. The Opdivo trials have contributed to gaining a deeper understanding of the potential role of biomarkers in patient care, particularly regarding how patients may benefit from Opdivo across the continuum of PD-L1 expression.
In July 2014, Opdivo was the first PD-1 immune checkpoint inhibitor to receive regulatory approval anywhere in the world. Opdivo is currently approved in more than 65 countries, including the United States, the European Union, Japan and China. In September 2015, the Company’s Opdivo and Yervoy combination regimen was the first Immuno-Oncology combination to receive regulatory approval for the treatment of metastatic melanoma and is currently approved in more than 50 countries, including the United States and the European Union.
OPDIVO INDICATIONS
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of adult patients with unresectable or metastatic melanoma.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adult patients with unresectable or metastatic melanoma.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of adult patients with melanoma with involvement of lymph nodes or metastatic disease who have undergone complete resection.
OPDIVO® (nivolumab), in combination with platinum-doublet chemotherapy, is indicated as neoadjuvant treatment of adult patients with resectable (tumors ≥4 cm or node positive) non-small cell lung cancer (NSCLC).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) whose tumors express PD-L1 (≥1%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab) and 2 cycles of platinum-doublet chemotherapy, is indicated for the first-line treatment of adult patients with metastatic or recurrent non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) with progression on or after platinum-based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving OPDIVO.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with unresectable malignant pleural mesothelioma (MPM).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with intermediate or poor risk advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab), in combination with cabozantinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with advanced renal cell carcinoma (RCC) who have received prior anti-angiogenic therapy.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with classical Hodgkin lymphoma (cHL) that has relapsed or progressed after autologous hematopoietic stem cell transplantation (HSCT) and brentuximab vedotin or after 3 or more lines of systemic therapy that includes autologous HSCT. This indication is approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN) with disease progression on or after platinum-based therapy.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
OPDIVO® (nivolumab), as a single agent, is indicated for the adjuvant treatment of adult patients with urothelial carcinoma (UC) who are at high risk of recurrence after undergoing radical resection of UC.
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of adult and pediatric (12 years and older) patients with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adults and pediatric patients 12 years and older with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adult patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with unresectable advanced, recurrent or metastatic esophageal squamous cell carcinoma (ESCC) after prior fluoropyrimidine- and platinum-based chemotherapy.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of completely resected esophageal or gastroesophageal junction cancer with residual pathologic disease in adult patients who have received neoadjuvant chemoradiotherapy (CRT).
OPDIVO® (nivolumab), in combination with fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of adult patients with unresectable advanced or metastatic esophageal squamous cell carcinoma (ESCC).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with unresectable advanced or metastatic esophageal squamous cell carcinoma (ESCC).
OPDIVO® (nivolumab), in combination with fluoropyrimidine- and platinum- containing chemotherapy, is indicated for the treatment of adult patients with advanced or metastatic gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma.
About CABOMETYX® (cabozantinib)
In the U.S., CABOMETYX tablets are approved for the treatment of patients with advanced renal cell carcinoma (RCC); for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib; for patients with advanced RCC as a first-line treatment in combination with nivolumab; and for adult and pediatric patients 12 years of age and older with locally advanced or metastatic differentiated thyroid cancer that has progressed following prior VEGFR-targeted therapy and who are radioactive iodine-refractory or ineligible. CABOMETYX tablets have also received regulatory approvals in the European Union and additional countries and regions worldwide. In 2016, Exelixis granted Ipsen exclusive rights for the commercialization and further clinical development of cabozantinib outside of the U.S. and Japan. In 2017, Exelixis granted exclusive rights to Takeda for the commercialization and further clinical development of cabozantinib for all future indications in Japan. Exelixis holds the exclusive rights to develop and commercialize cabozantinib in the U.S.
Please see accompanying full Prescribing Information https://www.cabometyx.com/downloads/CABOMETYXUSPI.pdf
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.FDA.gov/medwatch or call 1-800-FDA-1088.
About the Bristol Myers Squibb and Ono Pharmaceutical Collaboration
In 2011, through a collaboration agreement with Ono Pharmaceutical Co., Bristol Myers Squibb expanded its territorial rights to develop and commercialize Opdivo globally, except in Japan, South Korea and Taiwan, where Ono had retained all rights to the compound at the time. On July 23, 2014, Ono and Bristol Myers Squibb further expanded the companies’ strategic collaboration agreement to jointly develop and commercialize multiple immunotherapies – as single agents and combination regimens – for patients with cancer in Japan, South Korea and Taiwan.
About Bristol Myers Squibb
Bristol Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information about Bristol Myers Squibb, visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
About Exelixis
Exelixis is a globally ambitious oncology company innovating next-generation medicines and regimens at the forefront of cancer care. Powered by bi-coastal centers of discovery and development excellence, we are rapidly evolving our product portfolio to target an expanding range of tumor types and indications with our clinically differentiated pipeline of small molecules, antibody-drug conjugates and other biotherapeutics. This comprehensive approach harnesses decades of robust investment in our science and partnerships to advance our investigational programs and extend the impact of our flagship commercial product, CABOMETYX® (cabozantinib). Exelixis is driven by a bold scientific pursuit to create transformational treatments that give more patients hope for the future. For information about the company and its mission to help cancer patients recover stronger and live longer, visit www.exelixis.com, follow @ExelixisInc on Twitter, like Exelixis, Inc. on Facebook and follow Exelixis on LinkedIn.
Source: Bristol Myers Squibb and Exelixis, Inc.
Feb. 14, 2023 5:59 AM ET
Bristol-Myers Squibb Company (BMY), EXELPFE, IPSEF, IPSEY
By: Ravikash, SA News Editor2 Comments
Three year data from a phase 3 trial showed sustained survival and response rate benefits with Bristol Myers Squibb's (NYSE:BMY) Opdivo and Exelixis and Ipsen's (OTCPK:IPSEY) (OTCPK:IPSEF) Cabometyx compared to Pfizer's (PFE) Sutent (sunitinib) as first-line treatment of advanced renal cell carcinoma (RCC).
Bristol-Myers Squibb Company (BMY), EXELPFE, IPSEF, IPSEY
02/10/2023 CATEGORY:
Abecma more than tripled progression-free survival (13.3 months vs. 4.4 months) compared with standard regimens for triple-class exposed multiple myeloma
Safety results were consistent with the well-established and predictable safety profile of Abecma
Abecma is the first and only CAR T cell therapy to demonstrate superiority over standard regimens in triple-class exposed relapsed and refractory multiple myeloma in a randomized, controlled Phase 3 trial
Data published in The New England Journal of Medicine and presented at the EBMT and the European Hematology Association’s 5th European CAR T-cell Meeting
PRINCETON, N.J., & CAMBRIDGE, Mass.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) and 2seventy bio, Inc. (Nasdaq: TSVT) today announced the first publication and presentation of positive results from KarMMa-3, a pivotal Phase 3, open-label, global, randomized, controlled study evaluating Abecma (idecabtagene vicleucel) compared with standard combination regimens in adults with relapsed and refractory multiple myeloma after two to four prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody, and who were refractory to their last regimen. Data from KarMMa-3 are being published in The New England Journal of Medicine and simultaneously presented at the EBMT and the European Hematology Association’s (EHA) 5th European CAR T-cell Meeting on Friday, February 10 in an oral presentation during the Best Abstract Session.
At a median follow up of 18.6 months, treatment with Abecma (n=254)demonstrated a clinically meaningful and statistically significant improvement in the primary endpoint of progression-free survival (PFS) compared with standard regimens (n=132), with a median PFS of 13.3 months (95% CI: 11.8-16.1) vs. 4.4 months (95% CI: 3.4-5.9), respectively (HR:0.49; p<0.0001). This represents a 51% reduction in risk of disease progression or death with Abecma. Based on results from KarMMa-3, Abecma is the first and only chimeric antigen receptor (CAR) T cell therapy to demonstrate superiority over standard regimens in triple-class exposed relapsed and refractory multiple myeloma in a randomized, controlled Phase 3 trial.
“In earlier lines of treatment for multiple myeloma, regimens consisting of immunomodulatory agents, proteasome inhibitors, and anti-CD38 monoclonal antibodies are often used to help manage the disease. This shift in the treatment paradigm leaves many patients who are triple-class exposed with relapsed and refractory disease and in need of new treatment options sooner,” said Paula Rodriguez-Otero, M.D., Ph.D., Department of Hematology, Clinica Universidad de Navarra, Pamplona, Spain. “Results from the KarMMa-3 study with Abecma clearly demonstrate the benefit of earlier use of a CAR T cell therapy in providing the longest progression-free survival for patients with relapsed and refractory multiple myeloma compared to current standard regimens for these patients.”
“With Abecma, our first-in-class anti-BCMA CAR T cell therapy, we sought to deliver a personalized therapy that provides durable outcomes with a single infusion to advance the multiple myeloma treatment paradigm for patients,” said Anne Kerber, senior vice president, Cell Therapy Development, Bristol Myers Squibb. “This represents the third New England Journal of Medicine publication for Abecma, showing the clear clinical benefit of using Abecma across lines of therapy for patients with triple-class exposed relapsed and refractory multiple myeloma to provide the best chance for lasting disease control.”
Results for the key secondary endpoint of overall response rate also met statistical significance with the majority of patients (71%) treated with Abecma achieving a response, and 39% achieving a complete response or stringent complete response. In comparison, less than half of patients (41%) who received standard regimens achieved a response, with 5% experiencing a complete response or stringent complete response (p<0.0001). Responses with Abecma were durable with a median duration of 14.8 months (95% CI: 12.0-18.6) compared with 9.7 months (95% CI: 5.4-16.3) for standard regimens. Clinical benefit with Abecma was consistently observed across difficult-to-treat subgroups.
“For relapsing triple-class exposed multiple myeloma patients, median progression-free survival is just 4.6 months and there is no established standard treatment approach that provides durable responses,” said Sergio Giralt, M.D., Division of Hematologic Malignancies, Memorial Sloan Kettering Cancer Center. “In this study, we are seeing efficacy among a population with historically difficult-to-treat disease, with a significant improvement in progression-free survival and deep and lasting responses. These results from KarMMa-3 introduce the potential for this anti-BCMA CAR T cell therapy to become a standard of care earlier in the treatment course for relapsed and refractory multiple myeloma.”
Abecma exhibited a consistent and generally predictable safety profile, including no new safety signals, with mostly low-grade occurrences of cytokine release syndrome (CRS) and neurotoxicity. In patients treated with Abecma, 88% experienced any grade CRS, with Grade 3/4 events occurring in 4% of patients. Two patients (1%) experienced a Grade 5 CRS event. Median time to onset of CRS was 1 day (range: 1-14) and median duration of CRS was 3.5 days (range: 1-51). Any grade neurotoxicity occurred in 15% of patients, with Grade 3/4 neurotoxicity occurring in 3% of patients, and no Grade 5 events reported. Median time to onset of neurotoxicity was 3 days (range: 1-317) and median duration of neurotoxicity was 2 days (range: 1-37).
“The KarMMa-3 study is the first with a BCMA-directed CAR T therapy to demonstrate superiority versus standard regimens for patients with relapsed and refractory multiple myeloma, illustrating the potential of Abecma to change the standard of care of triple-class exposed multiple myeloma in early lines,” said Steve Bernstein, M.D., chief medical officer, 2seventy bio. “We are pleased to present and have these data published to build on the compelling efficacy profile of Abecma demonstrating significant improvement in progression-free survival and we look forward to working with regulatory authorities to make Abecma available to more myeloma patients who could benefit from this important treatment option.”
Bristol Myers Squibb and 2seventy bio intend to include these data in a planned supplemental Biologics License Application submission to the U.S. Food and Drug Administration (FDA) in 2023. Abecma is the first-in-class B-cell maturation antigen (BCMA)-directed CAR T cell immunotherapy approved by the FDA for the treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody. Please see the Important Safety Information section below, including Boxed WARNINGS for Abecma regarding CRS, neurologic toxicities, Hemophagocytic Lymphohistiocytosis/Macrophage Activation Syndrome and Prolonged Cytopenia. Abecma is also approved in the European Union, Switzerland, Japan, Canada, the United Kingdom and Israel for adult patients with triple-class exposed relapsed or refractory multiple myeloma after three to four or more prior lines of therapy.
Memorial Sloan Kettering Cancer Center disclosures: Dr. Giralt and Memorial Sloan Kettering Cancer Center have financial interests associated with the research described in this release.
About KarMMa-3
KarMMa-3 (NCT03651128) is a pivotal, Phase 3, open-label, global, randomized, controlled trial evaluating Abecma compared to standard regimens in patients with relapsed and refractory multiple myeloma who have received two to four prior lines of treatment, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody, and were refractory to the last treatment regimen. Patients were randomized to receive Abecma or standard regimens that consisted of combinations that included daratumumab, pomalidomide, and dexamethasone (DPd), daratumumab, bortezomib, and dexamethasone (DVd), ixazomib, lenalidomide, and dexamethasone (IRd), carfilzomib and dexamethasone (Kd) or elotuzumab, pomalidomide and dexamethasone (EPd) chosen based on their most recent treatment regimen and investigator discretion. The primary endpoint evaluated in this study is progression-free survival, defined as time from randomization to the first documentation of progressive disease or death due to any cause, whichever occurs first. Key secondary endpoints include overall response rate and overall survival.
About Abecma
Abecma recognizes and binds to BCMA on the surface of multiple myeloma cells leading to CAR T cell proliferation, cytokine secretion, and subsequent cytolytic killing of BCMA-expressing cells. Abecma is being jointly developed and commercialized in the U.S. as part of a Co-Development, Co-Promotion, and Profit Share Agreement between Bristol Myers Squibb and 2seventy bio.
The companies’ broad clinical development program for Abecma includes clinical studies (KarMMa-2, KarMMa-3, KarMMa-9) in earlier lines of treatment for patients with multiple myeloma. For more information visit clinicaltrials.gov.
Please see full Prescribing Information, including Boxed WARNINGS and Medication Guide.
Bristol Myers Squibb: Creating a Better Future for People with Cancer
Bristol Myers Squibb is inspired by a single vision—transforming patients’ lives through science. The goal of the company’s cancer research is to deliver medicines that offer each patient a better, healthier life and to make cure a possibility. Building on a legacy across a broad range of cancers that have changed survival expectations for many, Bristol Myers Squibb researchers are exploring new frontiers in personalized medicine, and through innovative digital platforms, are turning data into insights that sharpen their focus. Deep scientific expertise, cutting-edge capabilities and discovery platforms enable the company to look at cancer from every angle. Cancer can have a relentless grasp on many parts of a patient’s life, and Bristol Myers Squibb is committed to taking actions to address all aspects of care, from diagnosis to survivorship. Because as a leader in cancer care, Bristol Myers Squibb is working to empower all people with cancer to have a better future.
Learn more about the science behind cell therapy and ongoing research at Bristol Myers Squibb here.
About Bristol Myers Squibb
Bristol Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information about Bristol Myers Squibb, visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
About 2seventy bio
Our name, 2seventy bio, reflects why we do what we do - TIME. Cancer rips time away, and our goal is to work at the maximum speed of translating human thought into action – 270 miles per hour – to give the people we serve more time. We are building the leading immuno-oncology cell therapy company, focused on discovering and developing new therapies that truly disrupt the cancer treatment landscape With a deep understanding of the human body’s immune response to tumor cells and how to translate cell therapies into practice, we’re applying this knowledge to deliver next generation cellular therapies that focus on a broad range of hematologic malignancies, including the first FDA-approved CAR T cell therapy for multiple myeloma, as well as solid tumors. Our research and development is focused on delivering therapies that are designed with the goal to “think” smarter and faster than the disease. Importantly, we remain focused on accomplishing these goals by staying genuine and authentic to our “why” and keeping our people and culture top of mind every day.
For more information, visit www.2seventybio.com.
Follow 2seventy bio on social media: Twitter and LinkedIn.
2seventy bio is a trademark of 2seventy bio, Inc.
Source: Bristol Myers Squibb
Feb. 10, 2023 11:46 AM ET
Bristol-Myers Squibb Company (BMY), TSVTJNJ, LEGN
By: Dulan Lokuwithana, SA News Editor
Bristol Myers Squibb (NYSE:BMY) and 2seventy bio (NASDAQ:TSVT) on Friday announced data from a pivotal Phase 3 trial for BCMA-directed CAR T cell therapy Abecma in bone marrow cancer multiple myeloma (MM).
Bristol-Myers Squibb Company (BMY), TSVT
https://seekingalpha.com/symbol/BMY/
https://www.2seventybio.com/our-science

10 February 2023
Issued: London, UK For media and investors only
GSK plc (LSE/NYSE: GSK) today reports that the US Food and Drug Administration (FDA) granted full approval for Jemperli (dostarlimab-gxly) for the treatment of adult patients with mismatch repair-deficient (dMMR) recurrent or advanced endometrial cancer, as determined by a US FDA-approved test, that has progressed on or following a prior platinum-containing regimen in any setting and are not candidates for curative surgery or radiation.
Hesham Abdullah, Senior Vice President, Global Head of Oncology Development, GSK, said: “This US regulatory action confirms our confidence in Jemperli as an important treatment option for patients with dMMR recurrent or advanced endometrial cancer. We continue to unlock the potential of Jemperli as the backbone for our immuno-oncology development programs to address the unmet needs of patients, including earlier lines of endometrial cancer and other solid tumors.”
In April 2021, Jemperli received accelerated approval for the treatment of adult patients with dMMR recurrent or advanced endometrial cancer that had progressed on or following prior treatment with a platinum-containing regimen.
This approval is based on additional data collected from the A1 expansion cohort of the ongoing GARNET trial, a phase I, multicenter, open-label, single-arm study of Jemperli monotherapy in patients with advanced or recurrent solid tumors. Cohort A1 evaluated the efficacy of Jemperli in 141 patients with dMMR advanced or recurrent endometrial cancer that has progressed on or following prior treatment with a platinum-containing regimen. The major efficacy outcome measures were overall response rate (ORR) and duration of response (DOR) as assessed by a blinded independent central review according to RECIST v1.1. Confirmed ORR was 45.4% (95% CI: 37.0, 54.0), with a 15.6% complete response rate and a 29.8% partial response rate. Median DOR was not reached (range: 1.2+, 52.8+ months), measured from the time of first response, with 85.9% of patients having duration ≥12 months and 54.7% of patients having duration ≥24 months. Median follow-up for duration of response was 27.9 months.
Treatment-related adverse events were consistent with previous analyses for cohort A1. The most common adverse reactions (≥20%) were fatigue/asthenia, anemia, rash, nausea, diarrhea, constipation, and vomiting. The most common Grade 3 or 4 adverse reactions (≥2%) were anemia, increased transaminases, urinary tract infection, fatigue/asthenia, and diarrhea.
About endometrial cancer
Endometrial cancer is found in the inner lining of the uterus, known as the endometrium. Endometrial cancer is the most common gynecologic cancer globally, with approximately 417,000 new cases reported each year worldwide[1], and incidence rates are expected to rise by almost 40% by 2040.[2][3] Approximately 15-20% of patients with endometrial cancer will be diagnosed with advanced disease at the time of diagnosis.[4]
About GARNET
The ongoing GARNET phase I trial is evaluating Jemperli as monotherapy in patients with advanced solid tumors. Part 2B of the study includes five expansion cohorts: dMMR/MSI-H endometrial cancer (cohort A1), mismatch repair proficient/microsatellite stable (MMRp/MSS) endometrial cancer (cohort A2), non-small cell lung cancer (cohort E), dMMR/MSI-H non-endometrial or POLE-mut solid tumor basket cohort (cohort F), and platinum-resistant ovarian cancer without BRCA mutations (cohort G). GARNET continues to enroll patients.
In cohort A1, patients received 500mg of Jemperli as an intravenous infusion once every three weeks (Q3W) for four doses, followed by 1,000mg once every six weeks until disease progression, discontinuation or withdrawal.
About Jemperli (dostarlimab-gxly)
Jemperli is a programmed death receptor-1 (PD-1)-blocking antibody that binds to the PD-1 receptor and blocks its interaction with the PD-1 ligands PD-L1 and PD-L2.[5] GSK’s ambition is for Jemperli to become the backbone of the Company’s ongoing immuno-oncology-based research and development program when used alone and in combination with standard of care and future novel cancer therapies, particularly for patients who currently have limited treatment options. Jemperli is being investigated in registrational enabling studies as monotherapy and as part of combination regimens, including in patients with recurrent or primary advanced endometrial cancer, patients with Stage III or IV non-mucinous epithelial ovarian cancer, and patients with other advanced solid tumors or metastatic cancers.
Jemperli was discovered by AnaptysBio, Inc. and licensed to TESARO, Inc., under a collaboration and exclusive license agreement signed in March 2014. The collaboration has resulted in three monospecific antibody therapies that have progressed into the clinic. These are: Jemperli (GSK4057190), a PD-1 antagonist; cobolimab, (GSK4069889), a TIM-3 antagonist; and GSK4074386, a LAG-3 antagonist. GSK is responsible for the ongoing research, development, commercialization, and manufacturing of each of these medicines under the agreement.
Indications and Important Safety Information for JEMPERLI (dostarlimab-gxly)
JEMPERLI is indicated for the treatment of adult patients with mismatch repair deficient (dMMR) recurrent or advanced:
Please see the full US Prescribing Information.
GSK in oncology
GSK is committed to maximizing patient survival through transformational medicines. GSK’s pipeline is focused on immuno-oncology, tumor cell targeting therapies and synthetic lethality. Our goal is to achieve a sustainable flow of new treatments based on a diversified portfolio of investigational medicines utilizing modalities such as small molecules, antibodies, and antibody-drug conjugates, either alone or in combination.
About GSK
GSK is a global biopharma company with a purpose to unite science, technology, and talent to get ahead of disease together. Find out more at us.gsk.com/en-us/company.
Feb. 10, 2023 4:35 AM ET
By: Ravikash, SA News Editor
The U.S. Food and Drug Administration (FDA) granted full approval to GSK's (NYSE:GSK) Jemperli converting it from accelerated approval to treat certain patients with a type of endometrial cancer.
February 09, 2023
-- In Three-Year Follow-up in Adults with Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia, Tecartus Shows High Rates of Durable Response (CR+CRi 71%) and a Median Overall Survival of 26 Months --
SANTA MONICA, Calif.--(BUSINESS WIRE)-- Kite, a Gilead Company (Nasdaq: GILD), today announced the three-year follow-up results from the pivotal ZUMA-3 study of the CAR T-cell therapy Tecartus® (brexucabtagene autoleucel). Results from the analysis showed a median overall survival (OS) of 26 months and demonstrated that responses remained durable in adults with relapsed/refractory B-cell acute lymphoblastic leukemia (R/R B-ALL) with a consistent safety profile observed since the two-year analysis. These findings were presented today during a poster session at the 5th European CAR T-cell Meeting, taking place in Rotterdam, the Netherlands.
“For adult patients living with ALL, there is a need for therapeutic options that provide long-term responses,” said Bijal Shah, MD, ZUMA-3 investigator and medical oncologist, Moffitt Cancer Center, Tampa, Florida. “The continued durable response and significant improvement in survival indicated by these new data can potentially establish a new standard of care for adult patients living with this aggressive form of leukemia.”
In the Phase 2 treated patient cohort (n=55) the median follow-up was 38.8 months (range, 32.7-44.6). The OS rate at 36.0 months was 47.1% (95% CI, 32.7-60.2), with a median OS of 26.0 months among all treated Phase 2 patients (n=55) and 38.9 months in patients with complete remission (CR) or complete remission with incomplete hematologic recovery (CRi; n=39). Overall CR rate (CR + CRi), CR, and subsequent allogeneic stem cell transplant (alloSCT) rates remained unchanged since the prior data cut at 71%, 56%, and 20%, respectively. Median (95% CI) relapse-free survival (RFS) censored and not censored at subsequent alloSCT were both 11.6 (2.7-20.5) and 11.7 months (2.8-20.5), respectively.
For patients treated at the pivotal dose in both Phase 1 and 2 (n=78), the median follow-up at data cutoff was 41.6 months (range, 32.7-70.3). Median (95% CI) DOR censored and not censored at subsequent alloSCT was 18.6 (9.6-24.1) and 20.0 (10.3-24.1) months, respectively. Median (95% CI) RFS were both 11.7 (6.1-20.5) months. At data cutoff, 36% of patients (28) were still alive with a median OS of 25.6 months (95% CI, 16.2-47.0) in all treated patients (n=78). The proportion of pooled Phase 1 and 2 patients with Grade ≥3 AEs that were deemed treatment-related was unchanged since the prior data cut. No Grade 5 AEs occurred since the prior data cut off.
“We are encouraged by the sustained benefit that a single one-time treatment of Tecartus continues to provide for patients living with this difficult-to-treat blood cancer,” said Frank Neumann, MD, PhD, SVP, Kite’s Global Head of Clinical Development. “Our hope is that these results, along with our commitment to long-term research of Tecartus, will continue to provide clarity to physicians on optimal treatment methods for these patients living with this rare disease who have suffered historically poor outcomes.”
In this trial, longer follow-up of the pivotal analysis and outcomes of a larger pooled analysis of Phase 1 and 2 patients who received the pivotal dose of Tecartus were reported. Most patients in the analysis were heavily pre-treated, with a median of two prior therapies, and 47% had received three or more prior therapies.
About ZUMA-3
ZUMA-3 is an ongoing international multicenter (US, Canada, Europe), single arm, open label, registrational Phase 1/2 study of Tecartus in adult patients (≥18 years old) with ALL whose disease is refractory to or has relapsed following standard systemic therapy or hematopoietic stem cell transplantation. The primary endpoint is the rate of overall complete remission or complete remission with incomplete hematological recovery by central assessment. Duration of remission and relapse-free survival, overall survival, minimal residual disease (MRD) negativity rate, and alloSCT rate were assessed as secondary endpoints.
About Tecartus
Please see full FDA Prescribing Information, including BOXED WARNING and Medication Guide.
Tecartus is a CD19-directed genetically modified autologous T cell immunotherapy indicated for the treatment of:
This indication is approved under accelerated approval based on overall response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
About Kite
Kite, a Gilead Company, is a global biopharmaceutical company based in Santa Monica, California, focused on cell therapy to treat and potentially cure cancer. As the global cell therapy leader, Kite has treated more patients with CAR T-cell therapy than any other company. Kite has the largest in-house cell therapy manufacturing network in the world, spanning process development, vector manufacturing, clinical trial production and commercial product manufacturing. For more information on Kite, please visit www.kitepharma.com.
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer. Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California. Gilead Sciences acquired Kite in 2017.
U.S. Prescribing Information for Tecartus including BOXED WARNING, is available at www.kitepharma.com and www.gilead.com.
Kite, the Kite logo, Tecartus and GILEAD are trademarks of Gilead Sciences, Inc. or its related companies.
For more information on Kite, please visit the company’s website at www.kitepharma.com or call Gilead Public Affairs at 1-800-GILEAD-5 or 1-650-574-3000. Follow Kite on social media on Twitter (@KitePharma) and LinkedIn.
Source: Gilead Sciences, Inc.
Feb. 09, 2023 11:01 AM ET
By: Jonathan Block, SA News Editor2 Comments
PUBLISHED7 February 2023
Forxiga (dapagliflozin) has been approved in the European Union to extend the indication for heart failure (HF) with reduced ejection fraction (HFrEF) to cover patients across the full spectrum of left ventricular ejection fraction (LVEF), including HF with mildly reduced and preserved ejection fraction (HFmrEF, HFpEF).
The approval by the European Commission follows the positive opinion of the Committee for Medicinal Products for Human Use in December 2022 and was based on the positive results from the DELIVER Phase III trial1. Results from the prespecified pooled analysis of DELIVER and DAPA-HF Phase III trials also established Forxiga as the first HF medication to demonstrate mortality benefit across the full ejection fraction range2.
Mene Pangalos, Executive Vice President, BioPharmaceuticals R&D, AstraZeneca, said: “This broader indication for Forxiga for the treatment of symptomatic chronic heart failure across the full ejection fraction range will help more patients to benefit from this well-tolerated and guideline-directed treatment. We are redefining treatment of cardiorenal diseases with Forxiga’s demonstration of life-saving benefits, underscoring AstraZeneca’s commitment to provide innovative solutions that can help address the complexities of heart failure across the spectrum of the disease.”
HF is a chronic, long-term condition that worsens over time3 and affects about 15 million people in Europe4. Approximately half of HF patients die within five years of diagnosis5 and patients with HFmrEF and HFpEF are not only at greater risk of death and hospitalisations but experience an especially high burden of symptoms and physical limitations, and a poor quality of life6.
HFmrEF and HFpEF are also severely underdiagnosed as signs and symptoms are often nonspecific and overlapping with other clinical conditions7. These conditions are frequently complicated by multiple interrelated diseases, specifically coronary heart disease, obesity, diabetes, long-standing hypertension, and chronic kidney disease (CKD), highlighting the importance of risk management for patients with this complex syndrome7.
Forxiga (known as Farxiga in the US) is approved for the treatment of patients with type-2 diabetes (T2D), HFrEF and CKD in more than 100 countries around the world including the US, the EU, China and Japan. It has most recently received regulatory approvals in Great Britain, Japan and Turkey to extend the HF indication to include patients across the full spectrum of LVEF. The HF indication extension application is currently under review in the US and other countries.
Notes
HF
HF is a chronic, long-term condition that worsens over time3. It affects nearly 64 million people globally8 and is associated with substantial morbidity and mortality5. Chronic HF is the leading cause of hospitalisation for those over the age of 65 and represents a significant clinical and economic burden9. There are several types of HF often defined by LVEF, a measurement of the percentage of blood leaving the heart each time it contracts, including: HFrEF (LVEF less than or equal to 40%), HFmrEF (LVEF 41-49%) and HFpEF (LVEF greater than or equal to 50%)7. Approximately half of all HF patients have HFmrEF or HFpEF, with few therapeutic options available7,10.
DAPA-HF
DAPA-HF (Dapagliflozin And Prevention of Adverse-outcomes in Heart Failure) was an international, multi-centre, parallel-group, randomised, double-blinded Phase III trial in 4,744 patients with HFrEF, with and without T2D), designed to evaluate the effect of Forxiga 10mg, compared with placebo, given once daily in addition to standard of care (SoC). The primary composite endpoint was time to the first occurrence of a worsening HF event (hospitalisation or equivalent event, i.e. an urgent HF visit), or cardiovascular (CV) death. The median duration of follow-up was 18.2 months. Key secondary endpoints included the total number of hospitalisations for HF (hHF) (including repeat admissions) and CV deaths, change from baseline to 8 months in the total symptom score on the Kansas City Cardiomyopathy Questionnaire (KCCQ)11.
DELIVER
DELIVER was an international, randomised, double-blind, parallel-group, placebo-controlled, event-driven Phase III trial designed to evaluate the efficacy of Forxiga, compared with placebo, in the treatment of HF patients with LVEF greater than 40%, with or without T2D. Forxiga was given once daily in addition to background therapy (regional SoC for all comorbidities, including diabetes and hypertension, with the exception of concomitant use of SGLT2 inhibitor)12. DELIVER is the largest clinical trial to date in HF patients with LVEF above 40%, with 6,263 randomised patients12.
The primary composite endpoint was the time to first occurrence of CV death, hHF or an urgent HF visit. Key secondary endpoints include the total number of HF events (hHF or urgent HF visit) and CV death, change from baseline in the total symptom score of the KCCQ at eight months, time to the occurrence of CV death and time to the occurrence of death from any cause12.
Forxiga
Forxiga (dapagliflozin) is a first-in-class, oral, once-daily SGLT2 inhibitor. Research has shown Forxiga’s efficacy in preventing and delaying cardiorenal disease, while also protecting the organs – important findings given the underlying links between the heart, kidneys and pancreas11,13,14. Damage to one of these organs can cause the other organs to fail, contributing to leading causes of death worldwide, including T2D, HF and CKD8,15-17.
Forxiga is approved in adults and children aged 10 years and above for the treatment of insufficiently controlled T2D mellitus as an adjunct to diet and exercise. Forxiga is also approved for the treatment of HFrEF in adults and the treatment of CKD in adults based on the findings of the DAPA-HF and DAPA-CKD Phase III trials.
AstraZeneca in CVRM
Cardiovascular, Renal and Metabolism (CVRM), part of BioPharmaceuticals, forms one of AstraZeneca’s main disease areas and is a key growth driver for the Company. By following the science to understand more clearly the underlying links between the heart, kidneys and pancreas, AstraZeneca is investing in a portfolio of medicines for organ protection and improving outcomes by slowing disease progression, reducing risks and tackling co-morbidities. The Company’s ambition is to modify or halt the natural course of CVRM diseases and potentially regenerate organs and restore function, by continuing to deliver transformative science that improves treatment practices and CV health for millions of patients worldwide.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Feb. 07, 2023 4:25 AM ET
By: Ravikash, SA News Editor
The European Commission (EC) approved AstraZeneca (NASDAQ:AZN) Forxiga for expanded use to treat heart failure (HF) with reduced ejection fraction (HFrEF) to cover patients across the full spectrum of left ventricular ejection fraction (LVEF), including HF with mildly reduced and preserved ejection fraction (HFmrEF, HFpEF).

February 8, 2023 at 6:45 PM EST
ROP is a leading cause of childhood blindness worldwide
EYLEA now approved to treat five retinal conditions caused by ocular angiogenesis
TARRYTOWN, N.Y., Feb. 08, 2023 (GLOBE NEWSWIRE) -- Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) today announced that the U.S. Food and Drug Administration (FDA) has approved EYLEA® (aflibercept) Injection to treat preterm infants with retinopathy of prematurity (ROP). Following this first pediatric approval, EYLEA is now indicated to treat five retinal conditions caused by ocular angiogenesis.
“Retinopathy of prematurity is a leading cause of childhood blindness worldwide. Until now, the only FDA-approved treatment in common use was laser photocoagulation, a complex and lengthy procedure that permanently ablates retina tissue and is stressful not only for infant patients but also the family navigating a delicate time after a preterm birth,” said George D. Yancopoulos, M.D., Ph.D., President and Chief Scientific Officer of Regeneron, and a principal inventor of EYLEA. “For the first time, physicians will now have an FDA approved medication in EYLEA to treat this heartbreaking disease in these smallest of patients. We thank the investigators and the many families who participated in the clinical trials.”
Each year in the U.S., between 1,100 to 1,500 infants develop ROP that is severe enough to require medical treatment. This rare eye disease often impacts infants who are born before 31 weeks of pregnancy have been completed or who weigh less than 1,500 grams (3.3 lbs) pounds at birth. As retinal blood vessels are often only fully developed once an infant is full-term (~9 months of pregnancy), these infants are at risk of developing retinal blood vessels that are abnormal (retinal neovascularization) potentially leading to retinal detachment and irreversible vision loss. Mild cases of ROP may improve without treatment, but some cases require treatment to keep ROP from causing significant visual impairment and even blindness.
“With no existing FDA approved guidance for the treatment of retinopathy of prematurity with anti-VEGF therapies, there was a significant need for research to understand how best to treat the disease in a manner that puts patient safety first and preserves vision for a lifetime,” said Jeff Todd, Chief Executive Officer, Prevent Blindness. “Regeneron’s trials investigating EYLEA in retinopathy of prematurity have advanced our understanding of how to treat this disease and provided a needed evidence-based treatment option to potentially help preterm infants preserve their vision.”
About the Phase 3 Program in ROP
The FDA approval is supported by data from two randomized global Phase 3 trials – FIREFLEYE (N=113) and BUTTERFLEYE (N=120) – investigating EYLEA 0.4 mg versus laser photocoagulation (laser) in infants with ROP. In both trials, approximately 80% of EYLEA-treated infants achieved an absence of both active ROP and unfavorable structural outcomes at 52 weeks of age, which is better than would have been expected without treatment.
No new EYLEA safety signals were observed in either trial. Comparing EYLEA to laser, ocular adverse events (AEs) among patients occurred in 39% versus 37% in FIREFLEYE and 18% versus 26% in BUTTERFLEYE, with serious ocular AEs occurring in 8% for both groups in FIREFLEYE and 6.5% versus 11% in BUTTERFLEYE. AEs in both trials were consistent with infant prematurity or to the injection procedure, and with the AEs in similar ROP trials. The results of FIREFLEYE were published in Journal of the American Medical Association, and data from BUTTERFLEYE were presented at ROP Update 2022 meeting in the U.S.
Both trials were conducted pursuant to FDA Pediatric Written Request, and a Pediatric Exclusivity Determination was granted by FDA on October 12, 2022. This grant extends the period of U.S. market exclusivity for EYLEA by an additional six months through May 17, 2024.
EYLEA is being jointly developed by Regeneron and Bayer. The lead sponsors of the trials were Regeneron for BUTTERFLEYE and Bayer for FIREFLEYE. Bayer and Regeneron are collaborating on the global development of EYLEA. Regeneron maintains exclusive rights of EYLEA in the United States. Bayer has licensed the exclusive marketing rights outside the United States, where the companies share equally the profits from sales of EYLEA.
About EYLEA
EYLEA is a VEGF inhibitor formulated as an injection for the eye. It is designed to block the growth of new blood vessels and decrease the ability of fluid to pass through blood vessels (vascular permeability) in the eye by blocking VEGF-A and placental growth factor (PLGF), two growth factors involved in ocular angiogenesis. The EYLEA safety and efficacy profile is supported by a robust body of research that includes eight pivotal Phase 3 trials, more than 11 years of real-world experience and greater than 57 million EYLEA injections globally.
INDICATIONS
EYLEA® (aflibercept) Injection 2 mg (0.05 mL) is a prescription medicine approved for the treatment of patients with Wet Age-related Macular Degeneration (AMD), Macular Edema following Retinal Vein Occlusion (RVO), Diabetic Macular Edema (DME), Diabetic Retinopathy (DR), and Retinopathy of Prematurity (ROP).
Please see the full Prescribing Information for EYLEA.
About Regeneron
Regeneron (NASDAQ: REGN) is a leading biotechnology company that invents, develops and commercializes life-transforming medicines for people with serious diseases. Founded and led for 35 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to nine FDA-approved treatments and numerous product candidates in development, almost all of which were homegrown in our laboratories. Our medicines and pipeline are designed to help patients with eye diseases, allergic and inflammatory diseases, cancer, cardiovascular and metabolic diseases, pain, hematologic conditions, infectious diseases and rare diseases.
Regeneron is accelerating and improving the traditional drug development process through our proprietary VelociSuite® technologies, such as VelocImmune®, which uses unique genetically humanized mice to produce optimized fully human antibodies and bispecific antibodies, and through ambitious research initiatives such as the Regeneron Genetics Center®, which is conducting one of the largest genetics sequencing efforts in the world.
For more information, please visit www.Regeneron.com or follow @Regeneron on Twitter.
![]()
Source: Regeneron Pharmaceuticals, Inc.
Feb. 09, 2023 7:47 AM ET
Regeneron Pharmaceuticals, Inc. (REGN), BAYZF, BAYRY
By: Dulan Lokuwithana, SA News Editor
February 03, 2023
-- First Trop-2 Directed ADC to Demonstrate Overall Survival Benefit in HR+/HER2- Metastatic Breast Cancer Patients who had Received Prior Endocrine-based Therapy and at Least Two Chemotherapies --
-- Trodelvy has Now Improved Survival in both Pre-Treated HR+/HER2- Metastatic Breast Cancer and in Second-Line Metastatic Triple-Negative Breast Cancer --
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced the U.S. Food and Drug Administration (FDA) has approved Trodelvy® (sacituzumab govitecan-hziy) for the treatment of adult patients with unresectable locally advanced or metastatic hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative (IHC 0, IHC 1+ or IHC 2+/ISH–) breast cancer who have received endocrine-based therapy and at least two additional systemic therapies in the metastatic setting. The approval is based on statistically significant and clinically meaningful progression-free survival and overall survival data from the Phase 3 TROPiCS-02 study. Trodelvy is now also recommended as a Category 1, preferred treatment for metastatic HR+/HER2- breast cancer by the National Comprehensive Cancer Network® (NCCN®) as defined in the Clinical Practice Guidelines in Oncology (NCCN Guidelines®)i.
![]()
(Graphic: Business Wire)
“Despite decades of advances, people living with pre-treated HR+/HER2- metastatic breast cancer need new treatment options. Nearly all people with this type of breast cancer will eventually develop resistance to endocrine-based therapies and progress on available chemotherapies,” said Hope S. Rugo, MD, Professor of Medicine and Director, Breast Oncology and Clinical Trials Education at the UCSF Helen Diller Family Comprehensive Cancer Center, U.S. and principal investigator of the TROPiCS-02 study. “This approval is significant for the breast cancer community. We have had limited options to offer patients after endocrine-based therapy and chemotherapy, and to see a clinically meaningful survival benefit of more than three months with a quality of life benefit for these women is exceptional.”
In the TROPiCS-02 study, Trodelvy demonstrated a statistically significant and clinically meaningful overall survival (OS) benefit of 3.2 months versus comparator single-agent chemotherapy (treatment of physician’s choice; TPC) (median OS: 14.4 months vs. 11.2 months; hazard ratio [HR]=0.79; 95% CI: 0.65-0.96; p=0.02). Trodelvy also demonstrated a 34% reduction in risk of disease progression or death (median PFS: 5.5 versus 4.0 months; HR: 0.66; 95% CI: 0.53-0.83; p=0.0003). Three times as many people treated with Trodelvy were progression free at one year versus those treated with chemotherapy (21% versus 7%). In a post-hoc analysis, data demonstrated Trodelvy’s efficacy across HER2-low and IHC0 status in pre-treated metastatic breast cancer patients in the TROPiCS-02 trial.
Trodelvy also significantly improved additional secondary endpoint measures, including objective response rate and time to deterioration (TTD) assessed by the Global Health Status/Quality of Life and Fatigue scale per EORTC-QLQ-C30. No statistically significant difference in TTD in Pain Scale was observed.
“The FDA approval is an important step forward for both women and men living with metastatic breast cancer, especially for those individuals whose tumor is no longer responding to endocrine-based therapies and who are facing a poor prognosis,” said Laura Carfang, Executive Director, SurvivingBreastCancer.org. “We need to combat this terrible disease, and all options that potentially slow its progress and extend life for those living with metastatic breast cancer are welcomed.”
“We are pleased that Trodelvy could now provide new hope for people living with pre-treated HR+/HER2- metastatic breast cancer, building on the transformative role that Trodelvy is already playing for people with metastatic triple-negative breast cancer,” said Daniel O'Day, Chairman and Chief Executive Officer, Gilead Sciences. “We thank the physicians, patients and their families who put their trust in the TROPiCS-02 study and helped make this milestone possible.”
The safety profile for Trodelvy was consistent with prior studies, with no new safety signals identified in this patient population. In TROPiCS-02 the most frequent serious adverse reactions (>1%) were diarrhea (5%), febrile neutropenia (4%), neutropenia (3%), abdominal pain, colitis, neutropenic colitis, pneumonia, and vomiting (each 2%). The most common Grade 3-4 lab abnormalities (incidence ≥25%) in the TROPiCS-02 study were reduced neutrophils and leukocytes. No patients treated with Trodelvy experienced interstitial lung disease.
This review by the FDA was conducted under Project Orbis and granted Priority Review.
The European Medicines Agency has also validated a Type II Variation Marketing Authorization Application for Trodelvy in HR+/HER2- metastatic breast cancer.
Trodelvy has a Boxed Warning for severe or life-threatening neutropenia and severe diarrhea; please see below for additional Important Safety Information.
About the TROPiCS-02 Study
The TROPiCS-02 study is a global, multicenter, open-label, Phase 3 study, randomized 1:1 to evaluate Trodelvy versus physicians’ choice of chemotherapy (eribulin, capecitabine, gemcitabine, or vinorelbine) in 543 patients with HR+/HER2- metastatic breast cancer who were previously treated with endocrine therapy, CDK4/6 inhibitor and two to four lines of chemotherapy for metastatic disease. The primary endpoint is progression-free survival per Response Evaluation Criteria in Solid Tumors (RECIST 1.1) as assessed by blinded independent central review (BICR) for participants treated with Trodelvy compared to those treated with chemotherapy. Secondary endpoints include overall survival, overall response rate, clinical benefit rate and duration of response, as well as assessment of safety and tolerability and quality of life measures. In the study, HER2 negativity was defined per American Society of Clinical Oncology (ASCO) and the College of American Pathologists (CAP) criteria as immunohistochemistry (IHC) score of 0, IHC 1+ or IHC 2+ with a negative in-situ hybridization (ISH) test. More information about TROPiCS-02 is available at https://clinicaltrials.gov/ct2/show/NCT03901339.
About Trodelvy
Trodelvy® (sacituzumab govitecan-hziy) is a first-in-class Trop-2 directed antibody-drug conjugate. Trop-2 is a cell surface antigen highly expressed in multiple tumor types, including in more than 90% of breast and bladder cancers. Trodelvy is intentionally designed with a proprietary hydrolyzable linker attached to SN-38, a topoisomerase I inhibitor payload. This unique combination delivers potent activity to both Trop-2 expressing cells and the microenvironment.
Trodelvy is approved in more than 40 countries, with multiple additional regulatory reviews underway worldwide, for the treatment of adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (TNBC) who have received two or more prior systemic therapies, at least one of them for metastatic disease.
Trodelvy is also approved in the U.S. to treat certain patients with pre-treated HR+/HER2- metastatic breast cancer and has an accelerated approval for treatment of certain patients with second-line metastatic urothelial cancer; see below for full indication statements.
Trodelvy is also being developed for potential investigational use in other TNBC, HR+/HER2- and metastatic UC populations, as well as a range of tumor types where Trop-2 is highly expressed, including metastatic non-small cell lung cancer (NSCLC), metastatic small cell lung cancer (SCLC), head and neck cancer, and endometrial cancer.
U.S. Indications for Trodelvy
In the United States, Trodelvy is indicated for the treatment of adult patients with:
Please see full Prescribing Information , including BOXED WARNING.
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer. Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California.
U.S. Prescribing Information for Trodelvy including BOXED WARNING, is available at www.gilead.com .
Trodelvy, Gilead and the Gilead logo are trademarks of Gilead Sciences, Inc., or its related companies.
For more information about Gilead, please visit the company’s website at www.gilead.com , follow Gilead on Twitter (@GileadSciences) or call Gilead Public Affairs at 1-800-GILEAD-5 or 1-650-574-3000.
Feb. 03, 2023 10:56 AM ET
Gilead Sciences, Inc. (GILD)AZN, DSKYF, DSNKY
By: Dulan Lokuwithana, SA News Editor
Gilead Sciences (NASDAQ:GILD) announced Friday that the FDA approved its antibody-drug conjugate Trodelvy as a late-line option for certain adult patients with HR+/HER2- metastatic breast cancer.
https://seekingalpha.com/news/3931880-gilead-wins-fda-nod-trodelvy-new-breast-cancer-indication
February 3, 2023 6:45 am ET
KEYTRUDA plus chemotherapy significantly improved PFS versus standard of care chemotherapy alone as first-line treatment for patients with stage III-IV or recurrent endometrial carcinoma regardless of mismatch repair status
RAHWAY, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside of the United States and Canada, today announced that the Phase 3 NRG-GY018 trial evaluating KEYTRUDA, Merck’s anti-PD-1 therapy, in combination with standard of care chemotherapy (carboplatin and paclitaxel) met its primary endpoint of progression-free survival (PFS) for the treatment of patients with stage III-IV or recurrent endometrial carcinoma regardless of mismatch repair status. At a pre-specified interim analysis review conducted by an independent Data Monitoring Committee, KEYTRUDA in combination with chemotherapy then continued as single agent every six weeks for up to 14 cycles demonstrated a statistically significant and clinically meaningful improvement in PFS compared with chemotherapy alone in these patients whose endometrial carcinoma was either mismatch repair proficient (pMMR) or mismatch repair deficient (dMMR).
The safety profile of KEYTRUDA in this trial was consistent with that observed in previously reported studies; no new safety signals were identified. Results will be presented at an upcoming medical meeting and discussed with regulatory authorities.
“Patients with advanced stage or recurrent endometrial cancer, the most common type of gynecologic cancer in the U.S., face a poor prognosis with limited treatment options. This is particularly notable in patients who progress after prior platinum-based adjuvant therapy with disease not amenable to curative surgery or radiation,” said Dr. Ramez Eskander, principal investigator and gynecologic oncologist, University of California, San Diego. “In this study, pembrolizumab in combination with carboplatin and paclitaxel resulted in a statistically significant and clinically meaningful improvement in progression-free survival in both the dMMR and pMMR study populations. We look forward to presenting these exciting findings at an upcoming scientific congress.”
“In certain patients with advanced endometrial cancer who have progressed following prior systemic therapy and are not candidates for surgery or radiation, KEYTRUDA has become an important treatment option, both as monotherapy and in combination,” said Dr. Eliav Barr, senior vice president, head of global clinical development and chief medical officer, Merck Research Laboratories. “These latest results in the first-line setting are very encouraging and show the potential of KEYTRUDA plus chemotherapy for patients with stage III to IV or recurrent disease regardless of mismatch repair status. We thank our collaborators for their partnership on this study, and we are grateful to the patients and investigators for their participation.”
This trial was sponsored by the U.S. National Cancer Institute (NCI), part of the National Institutes of Health. NRG Oncology designed and led the trial with funding from the NCI and participation from all the National Clinical Trials Network (NCTN) Groups. Merck provided funding and support through a Cooperative Research and Development Agreement (CRADA) between Merck and NCI.
Merck has a comprehensive clinical development program in endometrial cancer. In the U.S., KEYTRUDA has two approved indications in endometrial cancer: in combination with LENVIMA® (lenvatinib), for the treatment of patients with advanced endometrial carcinoma that is pMMR, as determined by an FDA-approved test, or not microsatellite instability-high (MSI-H), who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation; and as a single agent, for the treatment of patients with advanced endometrial carcinoma that is MSI-H or dMMR, as determined by an FDA-approved test, who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation.
Additionally, Merck is evaluating KEYTRUDA in first-line advanced endometrial cancer both as monotherapy (KEYNOTE-C93/ENGOT-en15/GOG-3064) and in combination with LENVIMA (LEAP-001/ENGOT-en9), as well as in the adjuvant setting (KEYNOTE-B21/ENGOT-en11/GOG-3053).
About NRG-GY018
NRG-GY018 is a randomized, blinded, placebo-controlled Phase 3 trial (ClinicalTrials.gov, NCT03914612) evaluating KEYTRUDA in combination with standard of care chemotherapy (paclitaxel and carboplatin) versus placebo plus standard of care chemotherapy alone for the treatment of measurable stage III, IVA, IVB or recurrent endometrial cancer in pMMR and dMMR cohorts. The primary endpoint is PFS, and secondary endpoints include overall survival, objective response rate, duration of response and safety. The trial enrolled 819 patients who were randomized to receive KEYTRUDA plus chemotherapy every three weeks for approximately six cycles followed by KEYTRUDA as a single agent every six weeks for up to 14 cycles, or placebo plus chemotherapy. Enrolled patients were required to have MMR testing prior to randomization; approximately 70% of patients were pMMR, and approximately 30% were dMMR.
About endometrial carcinoma
Endometrial carcinoma begins in the inner lining of the uterus, which is known as the endometrium, and is the most common type of cancer in the uterus. This disease remains the only gynecologic malignancy with a rising incidence and mortality. In the U.S., it is estimated there will be approximately 66,000 new cases of uterine body cancer and approximately 13,000 deaths from the disease in 2023. Globally, endometrial cancer is the sixth most common cancer in women and 15th most common cancer overall.
About KEYTRUDA® (pembrolizumab) injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.
Endometrial Carcinoma
KEYTRUDA, in combination with LENVIMA, is indicated for the treatment of patients with advanced endometrial carcinoma that is mismatch repair proficient (pMMR), as determined by an FDA-approved test, or not microsatellite instability-high (MSI-H), who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with advanced endometrial carcinoma that is MSI-H or dMMR, as determined by an FDA-approved test, who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation.
See additional selected KEYTRUDA indications in the U.S. after the Selected Important Safety Information.
Additional Selected KEYTRUDA Indications in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of adult and pediatric (12 years and older) patients with stage IIB, IIC, or III melanoma following complete resection.
Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [tumor proportion score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
KEYTRUDA, as a single agent, is indicated as adjuvant treatment following resection and platinum-based chemotherapy for adult patients with Stage IB (T2a ≥4 cm), II, or IIIA NSCLC.
Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [Combined Positive Score (CPS) ≥1] as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy.
KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Carcinoma
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
Non-muscle Invasive Bladder Cancer
KEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with MSI-H central nervous system cancers have not been established.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC) as determined by an FDA-approved test.
Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or gastroesophageal junction (GEJ) (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
KEYTRUDA, in combination with chemotherapy, with or without bevacizumab, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
KEYTRUDA, in combination with lenvatinib, is indicated for the first-line treatment of adult patients with advanced RCC.
KEYTRUDA is indicated for the adjuvant treatment of patients with RCC at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions.
Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf .
Merck’s focus on cancer
Our goal is to translate breakthrough science into innovative oncology medicines to help people with cancer worldwide. At Merck, the potential to bring new hope to people with cancer drives our purpose and supporting accessibility to our cancer medicines is our commitment. As part of our focus on cancer, Merck is committed to exploring the potential of immuno-oncology with one of the largest development programs in the industry across more than 30 tumor types. We also continue to strengthen our portfolio through strategic acquisitions and are prioritizing the development of several promising oncology candidates with the potential to improve the treatment of advanced cancers. For more information about our oncology clinical trials, visit www.merck.com/clinicaltrials.
About Merck
At Merck, known as MSD outside of the United States and Canada, we are unified around our purpose: We use the power of leading-edge science to save and improve lives around the world. For more than 130 years, we have brought hope to humanity through the development of important medicines and vaccines. We aspire to be the premier research-intensive biopharmaceutical company in the world – and today, we are at the forefront of research to deliver innovative health solutions that advance the prevention and treatment of diseases in people and animals. We foster a diverse and inclusive global workforce and operate responsibly every day to enable a safe, sustainable and healthy future for all people and communities. For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Source: Merck & Co., Inc.
Feb. 03, 2023 7:15 AM ET
By: Ravikash, SA News Editor1 Comment
Merck's (NYSE:MRK) blockbuster drug Keytruda, with chemotherapy, met the main goal of progression free survival (PFS) in patients with a type of cancer of the uterus in a phase 3 trial.
The study, dubbed NRG-GY018, evaluated Keytruda (pembrolizumab), in combination with standard of care chemotherapy (carboplatin and paclitaxel) to treat patients with stage 3-4 or recurrent endometrial carcinoma regardless of mismatch repair status.
January 30, 2023 at 1:00 AM EST
Approximately 60% of patients aged 12 years and older treated with Dupixent 300 mg weekly in the pivotal trial experienced histological disease remission; patients also significantly improved their ability to swallow compared to placebo
Dupixent is now an option for the approximately 50,000 adults and adolescents living with severe uncontrolled eosinophilic esophagitis in the European Union (EU)
Dupixent now approved to treat five diseases with underlying type 2 inflammation in the EU
TARRYTOWN, N.Y. and PARIS, Jan. 30, 2023 (GLOBE NEWSWIRE) -- Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) and Sanofi today announced that the European Commission (EC) expanded the marketing authorization for Dupixent® (dupilumab) in the European Union (EU) to treat eosinophilic esophagitis (EoE) in adults and adolescents 12 years and older, weighing at least 40 kg, who are inadequately controlled by, are intolerant to, or who are not candidates for conventional medicinal therapy. EoE is a chronic, progressive inflammatory disease that damages the esophagus and prevents it from working properly. With this approval, Dupixent is the first and only targeted medicine specifically indicated to treat EoE in Europe and the U.S.
“This latest approval establishes Dupixent as the only targeted medicine specifically indicated for eosinophilic esophagitis in the European Union. Dupixent is also the only biologic shown in pivotal trials to help patients achieve histological remission, reduce difficulty swallowing and improve health-related quality of life – all of which are crucial to reducing the burden of this debilitating disease,” said George D. Yancopoulos, M.D., Ph.D., President and Chief Scientific Officer at Regeneron, and a principal inventor of Dupixent. “Since its first approval, Dupixent has redefined the treatment of certain chronic diseases with underlying type 2 inflammation and is now indicated for five conditions in the European Union. We remain committed to investigating Dupixent’s potential in additional diseases in which IL-4 and IL-13 may play a key role.”
“The impact of EoE on a patient’s daily life cannot be overstated – the narrowing and scarring of the esophagus can make something as simple as eating a painful and distressing experience, and may lead to choking and food impaction,” said Naimish Patel, M.D., Head of Global Development, Immunology and Inflammation at Sanofi. “With this latest approval for Dupixent, adults and adolescents in the EU suffering from the chronic and often debilitating symptoms of EoE now have the first and only targeted treatment option clinically proven to reduce both esophageal inflammation and damage, as well as improve swallowing ability, pain and health-related quality of life.”
The EC decision is supported by 52-week data from a Phase 3 trial consisting of three parts (Part A, B and C). Part A and Part B investigated Dupixent 300 mg weekly (Part A n=42; Part B n=80) compared to placebo (Part A n=39; Part B n=79) for 24 weeks. Part C (n=188) observed patients who had continued on or switched to Dupixent from Parts A and B for an additional 28 weeks.
Dupixent patients in Parts A and B, respectively, experienced:
Histological disease remission, swallowing improvement and reduction in abnormal endoscopic findings were consistent with the overall population in patients who were uncontrolled, or not responsive to or not eligible for swallowed topical corticosteroids. Longer term efficacy in Part C was similar to results observed in Parts A and B.
The safety results of the trial were generally consistent with the known safety profile of Dupixent in its approved indications. The most common side effects across indications were injection site reactions, conjunctivitis, conjunctivitis allergic, arthralgia, oral herpes and eosinophilia. Adverse events more commonly observed in EoE patients treated with Dupixent (n=122) compared to placebo (n=117) included infections (32% vs. 25%). An additional adverse reaction of injection site bruising was reported in the EoE trial. The safety profile through 52 weeks was generally consistent with the safety profile observed at 24 weeks.
About the Dupixent Eosinophilic Esophagitis Trial
The three-part Phase 3 randomized, double-blind, placebo-controlled trial evaluated the efficacy and safety of Dupixent in patients aged 12 years and older with EoE. All patients had previously not responded to proton pump inhibitors, and, across Parts A and B, 74% of patients were previously treated with swallowed topical corticosteroids.
At 24 weeks, the co-primary endpoints in Parts A and B assessed patient-reported measures of difficulty swallowing (change from baseline in the DSQ on a 0-84 scale) and esophageal inflammation (proportion of patients achieving histological disease remission, defined as peak esophageal intraepithelial eosinophil count of ≤6 eos/hpf).
Additional endpoints included abnormal endoscopic findings (EoE Endoscopic Reference Score [EoE-EREFS] on a 0-18 scale), swallowing-related pain (DSQ pain score), health-related quality of life (EoE Impact Questionnaire [EoE-IQ]) and frequency of other non-dysphagia symptoms (EoE Symptom Questionnaire [EoE-SQ]).
About Dupixent
Dupixent is an injection administered under the skin (subcutaneous injection) at different injection sites. In the EU for adolescents and adults with EoE, Dupixent is administered at 300 mg every week. It is available as both a pre-filled pen and pre-filled syringe at the 300 mg dose. Dupixent is intended for use under the guidance of a healthcare professional and can be given in a clinic or at home by self-administration after training by a healthcare professional.
Dupixent, which was invented using Regeneron's proprietary VelocImmune® technology, is a fully human monoclonal antibody that inhibits the signaling of the interleukin-4 (IL-4) and interleukin-13 (IL-13) pathways and is not an immunosuppressant. The Dupixent development program has shown significant clinical benefit and a decrease in type 2 inflammation in Phase 3 trials, establishing that IL-4 and IL-13 are key and central drivers of the type 2 inflammation that plays a major role in multiple related and often co-morbid diseases. These diseases include approved indications for Dupixent, such as atopic dermatitis, asthma, chronic rhinosinusitis with nasal polyposis (CRSwNP), prurigo nodularis and EoE.
Dupixent has received regulatory approvals in one or more countries around the world for use in certain patients with atopic dermatitis, asthma, CRSwNP, EoE or prurigo nodularis in different age populations. Dupixent is currently approved for one or more of these indications in more than 60 countries, including in Europe, the U.S. and Japan. More than 500,000 patients have been treated with Dupixent globally.
About Regeneron's VelocImmune Technology
Regeneron's VelocImmune technology utilizes a proprietary genetically engineered mouse platform endowed with a genetically humanized immune system to produce optimized fully human antibodies. When Regeneron's co-Founder, President and Chief Scientific Officer George D. Yancopoulos was a graduate student with his mentor Frederick W. Alt in 1985, they were the first to envision making such a genetically humanized mouse, and Regeneron has spent decades inventing and developing VelocImmune and related VelociSuite® technologies. Dr. Yancopoulos and his team have used VelocImmune technology to create a substantial proportion of all original, FDA-approved or authorized fully human monoclonal antibodies. This includes REGEN-COV® (casirivimab and imdevimab), Dupixent, Libtayo® (cemiplimab-rwlc), Praluent® (alirocumab), Kevzara® (sarilumab), Evkeeza® (evinacumab-dgnb) and Inmazeb® (atoltivimab, maftivimab and odesivimab-ebgn).
Dupilumab Development Program
Dupilumab is being jointly developed by Regeneron and Sanofi under a global collaboration agreement. To date, dupilumab has been studied across more than 60 clinical trials involving more than 10,000 patients with various chronic diseases driven in part by type 2 inflammation.
In addition to the currently approved indications, Regeneron and Sanofi are studying dupilumab in a broad range of diseases driven by type 2 inflammation or other allergic processes in Phase 3 trials, including pediatric EoE, hand and foot atopic dermatitis, chronic inducible urticaria-cold, chronic spontaneous urticaria, chronic pruritus of unknown origin, chronic obstructive pulmonary disease with evidence of type 2 inflammation, chronic rhinosinusitis without nasal polyposis, allergic fungal rhinosinusitis, allergic bronchopulmonary aspergillosis, and bullous pemphigoid. These potential uses of dupilumab are currently under clinical investigation, and the safety and efficacy in these conditions have not been fully evaluated by any regulatory authority.
U.S. INDICATIONS
DUPIXENT is a prescription medicine used:
Please see accompanying full Prescribing Information including Patient Information.
About Regeneron
Regeneron (NASDAQ: REGN) is a leading biotechnology company that invents, develops and commercializes life-transforming medicines for people with serious diseases. Founded and led for 35 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to nine FDA-approved treatments and numerous product candidates in development, almost all of which were homegrown in our laboratories. Our medicines and pipeline are designed to help patients with eye diseases, allergic and inflammatory diseases, cancer, cardiovascular and metabolic diseases, pain, hematologic conditions, infectious diseases and rare diseases.
Regeneron is accelerating and improving the traditional drug development process through our proprietary VelociSuite® technologies, such as VelocImmune®, which uses unique genetically humanized mice to produce optimized fully human antibodies and bispecific antibodies, and through ambitious research initiatives such as the Regeneron Genetics Center, which is conducting one of the largest genetics sequencing efforts in the world.
For more information, please visit www.Regeneron.com or follow @Regeneron on Twitter.
About Sanofi
We are an innovative global healthcare company, driven by one purpose: we chase the miracles of science to improve people’s lives. Our team, across some 100 countries, is dedicated to transforming the practice of medicine by working to turn the impossible into the possible. We provide potentially life-changing treatment options and life-saving vaccine protection to millions of people globally, while putting sustainability and social responsibility at the center of our ambitions.
Sanofi is listed on EURONEXT: SAN and NASDAQ: SNY.
![]()
Source: Regeneron Pharmaceuticals, Inc.
Jan. 30, 2023 4:26 AM ET
By: Ravikash, SA News Editor
The European Commission (EC) approved Sanofi (NASDAQ:SNY) and Regeneron Pharmaceuticals' (NASDAQ:REGN) Dupixent to treat eosinophilic esophagitis (EoE) in patients 12 years and older, weighing at least 40 kg, who are inadequately controlled by, are intolerant to, or who are not eligible for conventional medicinal therapy.
PUBLISHED26 January 2023 26 January 2023 07:00 GMT
AstraZeneca and Daiichi Sankyo’s Enhertu (trastuzumab deruxtecan) has been approved in the European Union (EU) as monotherapy for the treatment of adult patients with unresectable or metastatic HER2-low (IHC 1+ or IHC 2+/ISH-) breast cancer who have received prior chemotherapy in the metastatic setting or developed disease recurrence during or within six months of completing adjuvant chemotherapy.
Enhertu is a specifically engineered HER2-directed antibody drug conjugate (ADC) being jointly developed and commercialised by AstraZeneca and Daiichi Sankyo.
The approval by the European Commission follows the positive opinion of the Committee for Medicinal Products for Human Use and is based on results from the DESTINY-Breast04 Phase III trial, which were presented at the American Society of Clinical Oncology 2022 Annual Meeting and published in The New England Journal of Medicine.1
In the trial, Enhertu significantly reduced the risk of disease progression or death by 50% versus physician’s choice of chemotherapy (based on a hazard ratio [HR] of 0.50; 95% confidence interval [CI]: 0.40-0.63; p<0.0001) in patients with HER2-low metastatic breast cancer with hormone receptor (HR)-positive or HR-negative disease. A median progression-free survival (PFS) of 9.9 months was seen with Enhertu versus 5.1 months in those treated with chemotherapy, as assessed by blinded independent central review (BICR). A 36% reduction in the risk of death was seen with Enhertu compared to chemotherapy (hazard ratio of 0.64; 95% CI: 0.49-0.84; p=0.001) with a median overall survival (OS) of 23.4 months versus 16.8 months.
Javier Cortés, MD, PhD, Head, International Breast Cancer Center, Barcelona, Spain, said: “The European approval of Enhertu in the HER2-low metastatic breast cancer population marks the first time we will have the opportunity to treat patients with lower levels of HER2 expression with a HER2-directed therapy. Enhertu has shown a significant improvement in outcomes compared to chemotherapy for these patients, reinforcing its potential to become a new standard of care.”
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “Historically, patients with breast cancer who have tumours with low levels of HER2 expression have been classified as HER2-negative, giving them limited treatment options beyond chemotherapy. This approval reinforces the important role Enhertu may have for patients with HER2-low disease and highlights the need to evolve the way breast cancer is treated to improve patient outcomes.”
Ken Keller, Global Head of Oncology Business, and President and CEO, Daiichi Sankyo, Inc, said: “The approval of Enhertu in HER2-low metastatic breast cancer represents a significant clinical advance for patients in Europe with both HR-positive and HR-negative disease who previously have had limited treatment options in the late-line setting. This milestone also supports our vision to bring Enhertu to more patients across the HER2 spectrum, which requires a change to the breast cancer classification system that has been guiding treatment for more than two decades.”
The safety profile observed in patients treated with Enhertu in the DESTINY-Breast04 trial was consistent with that seen in other trials of Enhertu in breast cancer with no new safety signals identified.
Financial considerations
Following EU approval, an amount of $150m is due from AstraZeneca to Daiichi Sankyo as a milestone payment for this HER2-low breast cancer post-chemotherapy indication. The milestone payment will be capitalised as an addition to the upfront payment made by AstraZeneca to Daiichi Sankyo in 2019 and subsequent capitalised milestones.
Sales of Enhertu in most EU territories are recognised by Daiichi Sankyo. AstraZeneca reports its share of gross profit margin from Enhertu sales in those territories as collaboration revenue in the Company’s financial statements. AstraZeneca will record product sales in respect of sales made in territories where AstraZeneca is the selling party.
Further details on the financial arrangements were set out in the March 2019 announcement of the collaboration.
DESTINY-Breast04
DESTINY-Breast04 is a global, randomised, open-label, Phase III trial evaluating the efficacy and safety of Enhertu (5.4mg/kg) versus physician’s choice of chemotherapy (capecitabine, eribulin, gemcitabine, paclitaxel or nab-paclitaxel) in patients with HR-positive or HR-negative, HER2-low unresectable and/or metastatic breast cancer previously treated with one or two prior lines of chemotherapy. Patients were randomised 2:1 to receive either Enhertu or chemotherapy.
The primary endpoint of DESTINY-Breast04 is PFS in patients with HR-positive disease based on BICR. Key secondary endpoints include PFS based on BICR in all randomised patients (HR-positive and HR-negative disease), OS in patients with HR-positive disease and OS in all randomised patients (HR-positive and HR-negative disease). Other secondary endpoints include PFS based on investigator assessment, objective response rate based on BICR and on investigator assessment, duration of response based on BICR and safety.
DESTINY-Breast04 enrolled 557 patients at multiple sites in Asia, Europe and North America. For more information about the trial, visit ClinicalTrials.gov.
Enhertu
Enhertu is a HER2-directed ADC. Designed using Daiichi Sankyo’s proprietary DXd ADC technology, Enhertu is the lead ADC in the oncology portfolio of Daiichi Sankyo and the most advanced programme in AstraZeneca’s ADC scientific platform. Enhertu consists of a HER2 monoclonal antibody attached to a topoisomerase I inhibitor payload, an exatecan derivative, via a stable tetrapeptide-based cleavable linker.
Enhertu (5.4mg/kg) is approved in more than 40 countries for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received a (or one or more) prior anti-HER2-based regimen, either in the metastatic setting or in the neoadjuvant or adjuvant setting, and have developed disease recurrence during or within six months of completing therapy based on the results from the DESTINY-Breast03 trial.
Enhertu (5.4mg/kg) is approved in more than 30 countries for the treatment of adult patients with unresectable or metastatic HER2-low (IHC 1+ or IHC 2+/ ISH-) breast cancer who have received a prior systemic therapy in the metastatic setting or developed disease recurrence during or within six months of completing adjuvant chemotherapy based on the results of the DESTINY-Breast04 trial.
Enhertu (5.4mg/kg) is approved under accelerated approval in the US for the treatment of adult patients with unresectable or metastatic non-small cell lung cancer whose tumours have activating HER2 (ERBB2) mutations, as detected by an FDA-approved test, and who have received a prior systemic therapy based on the results from the DESTINY-Lung02 trial. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
Enhertu (6.4mg/kg) is approved in more than 30 countries for the treatment of adult patients with locally advanced or metastatic HER2-positive gastric or gastroesophageal junction adenocarcinoma who have received a prior trastuzumab-based regimen based on the results from the DESTINY-Gastric01 trial and/or DESTINY-Gastric02 trials.
Enhertu development programme
A comprehensive global development programme is underway evaluating the efficacy and safety of Enhertu monotherapy across multiple HER2-targetable cancers including breast, gastric, lung and colorectal cancers. Trials in combination with other anticancer treatments, such as immunotherapy, are also underway.
Daiichi Sankyo collaboration
Daiichi Sankyo Company, Limited (TSE: 4568) [referred to as Daiichi Sankyo] and AstraZeneca entered into a global collaboration to jointly develop and commercialise Enhertu (a HER2-directed ADC) in March 2019, and datopotamab deruxtecan (DS-1062; a TROP2-directed ADC) in July 2020, except in Japan where Daiichi Sankyo maintains exclusive rights. Daiichi Sankyo is responsible for the manufacturing and supply of Enhertu and datopotamab deruxtecan.
AstraZeneca in breast cancer
Driven by a growing understanding of breast cancer biology, AstraZeneca is starting to challenge, and redefine, the current clinical paradigm for how breast cancer is classified and treated to deliver even more effective treatments to patients in need – with the bold ambition to one day eliminate breast cancer as a cause of death.
AstraZeneca has a comprehensive portfolio of approved and promising compounds in development that leverage different mechanisms of action to address the biologically diverse breast cancer tumour environment.
With Enhertu (trastuzumab deruxtecan), a HER2-directed ADC, AstraZeneca and Daiichi Sankyo are aiming to improve outcomes in previously treated HER2-positive and HER2-low metastatic breast cancer and are exploring its potential in earlier lines of treatment and in new breast cancer settings.
In HR-positive breast cancer, AstraZeneca continues to improve outcomes with foundational medicines Faslodex (fulvestrant) and Zoladex (goserelin) and aims to reshape the HR-positive space with next-generation SERD and potential new medicine camizestrant as well as a potential first-in-class AKT kinase inhibitor, capivasertib. AstraZeneca is also collaborating with Daiichi Sankyo to explore the potential of TROP2-directed ADC, datopotamab deruxtecan, in this setting.
PARP inhibitor Lynparza (olaparib) is a targeted treatment option that has been studied in early and metastatic breast cancer with an inherited BRCA mutation. AstraZeneca with MSD (Merck & Co., Inc. in the US and Canada) continue to research Lynparza in these settings and to explore its potential in earlier disease.
To bring much-needed treatment options to patients with triple-negative breast cancer, an aggressive form of breast cancer, AstraZeneca is evaluating the potential of datopotamab deruxtecan alone and in combination with immunotherapy Imfinzi (durvalumab), capivasertib in combination with chemotherapy, and Imfinzi in combination with other oncology medicines, including Lynparza and Enhertu.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Jan. 26, 2023 4:36 AM ET
AstraZeneca PLC (AZN), DSKYF, DSNKY
By: Ravikash, SA News Editor
Now Offers Patients the Choice of Administration at Home or in a Doctor's Office
THOUSAND OAKS, Calif., Feb. 2, 2023 /PRNewswire/ -- Amgen (NASDAQ:AMGN) and AstraZeneca today announced the U.S. Food and Drug Administration (FDA) has approved TEZSPIRE® (tezepelumab-ekko) for self-administration in a pre-filled, single-use pen for patients aged 12 years and older with severe asthma.1 First approved by the FDA in December 2021, TEZSPIRE is the only biologic approved for severe asthma with no phenotype (e.g., eosinophilic or allergic) or biomarker limitation within its approved label.2-9
"People with severe asthma will now have the flexibility to administer TEZSPIRE at home or continue to receive their medicine in their doctor's office," said Murdo Gordon, executive vice president of Global Commercial Operations at Amgen. "This approval reinforces our continued efforts to improve accessibility to TEZSPIRE, a first-in-class medicine proven to consistently and significantly reduce exacerbations across a broad population of people with severe asthma."
The approval by the FDA was based on results from the PATHFINDER clinical trial program, which included results from the PATH-BRIDGE Phase 1 trial and the PATH-HOME trial Phase 3 trial.10 The majority (92%) of healthcare providers, patients and caregivers were able to successfully administer TEZSPIRE both in the clinic and at home throughout the PATH-HOME trial. The improvements in asthma control and the safety profile of TEZSPIRE observed in the PATH-HOME trial were consistent with previous clinical trials.10
"Severe asthma continues to be a very complex condition to manage, so we welcome the TEZSPIRE pre-filled pen as an option that will empower patients and healthcare providers with increased choice," said Kenneth Mendez, president and chief executive officer of the Asthma and Allergy Foundation of America. "We believe self-administration alternatives can play an important role in patients' lives and address unmet needs for those living with severe asthma."
The most common adverse reactions (incidence ≥3% and more common than placebo) of TEZSPIRE are pharyngitis, arthralgia, and back pain.1
TEZSPIRE self-administration and the TEZSPIRE pre-filled pen are also approved in the European Union (EU) and are under regulatory review in several other countries around the world. TEZSPIRE is currently approved for the treatment of severe asthma in the U.S., EU, Japan and other countries.
TEZSPIRE® (tezepelumab-ekko) U.S. Indication
TEZSPIRE is indicated for the add-on maintenance treatment of adult and pediatric patients aged 12 years and older with severe asthma.
TEZSPIRE is not indicated for the relief of acute bronchospasm or status asthmaticus.
TEZSPIRE® (tezepelumab-ekko) Important Safety Information
CONTRAINDICATIONS
Known hypersensitivity to tezepelumab-ekko or excipients.
Please see the full Prescribing Information including Patient Information and Instructions for Use.
About TEZSPIRE® (tezepelumab-ekko)
TEZSPIRE is a first-in-class human monoclonal antibody that works on the primary source of inflammation: the airway epithelium, which is the first point of contact for viruses, allergens, pollutants and other environmental insults. Specifically, TEZSPIRE targets and blocks TSLP, a key epithelial cytokine that sits at the top of multiple inflammatory cascades and initiates an overreactive immune response to allergic, eosinophilic and other types of airway inflammation associated with severe asthma.11,12 TSLP is released in response to multiple triggers associated with asthma exacerbations, including allergens, viruses and other airborne particles.11,12
Expression of TSLP is increased in the airways of patients with asthma and has been correlated with disease severity.11,13 Blocking TSLP may prevent the release of pro-inflammatory cytokines by immune cells, resulting in the prevention of asthma exacerbations and improved asthma control.11,13 By working at the top of the cascade, TEZSPIRE helps stop inflammation at the source and has the potential to treat a broad population of severe asthma patients.11,13
TEZSPIRE is also in development for other potential indications including chronic obstructive pulmonary disease, chronic rhinosinusitis with nasal polyps, chronic spontaneous urticaria and eosinophilic esophagitis (EoE). In October 2021, tezepelumab was granted Orphan Drug Designation by the FDA for the treatment of EoE.
About the Amgen and AstraZeneca Collaboration
In 2020, Amgen and AstraZeneca updated the 2012 collaboration agreement for TEZSPIRE. Both companies will continue to share costs and profits equally after payment by AstraZeneca of a mid-single-digit royalty to Amgen. AstraZeneca continues to lead development and Amgen continues to lead manufacturing. All aspects of the collaboration are under the oversight of joint governing bodies. Under the amended agreement, Amgen and AstraZeneca will jointly commercialize TEZSPIRE in North America. Amgen will record product sales in the U.S., with AstraZeneca recording its share of U.S. profits as Collaboration Revenue. Outside of the U.S., AstraZeneca will record product sales, with Amgen recording profit share as Other/Collaboration revenue.
About Amgen
Amgen is committed to unlocking the potential of biology for patients suffering from serious illnesses by discovering, developing, manufacturing and delivering innovative human therapeutics. This approach begins by using tools like advanced human genetics to unravel the complexities of disease and understand the fundamentals of human biology.
Amgen focuses on areas of high unmet medical need and leverages its expertise to strive for solutions that improve health outcomes and dramatically improve people's lives. A biotechnology pioneer since 1980, Amgen has grown to be one of the world's leading independent biotechnology companies, has reached millions of patients around the world and is developing a pipeline of medicines with breakaway potential.
Amgen is one of the 30 companies that comprise the Dow Jones Industrial Average and is also part of the Nasdaq-100 index. In 2022, Amgen was named one of the "World's Best Employers" by Forbes and one of "America's 100 Most Sustainable Companies" by Barron's.
For more information, visit Amgen.com and follow us on Twitter, LinkedIn, Instagram, TikTok and YouTube.
![]()
View original content to download multimedia:https://www.prnewswire.com/news-releases/tezspire-approved-for-self-administration-in-the-us-with-a-new-pre-filled-pen-301736900.html
SOURCE Amgen
Feb. 02, 2023 9:42 AM ET
By: Ravikash, SA News Editor
European Commission approves label expansion of Roche’s Hemlibra to include people with moderate haemophilia A in the EUFebruary 01, 2023
Basel, 01 February 2023 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the European Commission approved the expansion of the Hemlibra® (emicizumab) European Union (EU) marketing authorisation. The label will now include the routine prophylaxis of bleeding episodes in people with haemophilia A (congenital factor VIII deficiency) without factor VIII inhibitors, who have moderate disease (FVIII ≥1% and ≤ 5%) with a severe bleeding phenotype.3 Haemophilia A affects around 900,000 people worldwide,4,5 approximately 14% of whom have a moderate form of the disease.6
“We welcome the European Commission's decision to approve Hemlibra also for people with moderate haemophilia A in the EU, since even moderate disease can produce bleeds that cause irreversible joint damage and impact quality of life,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “As its benefit expands to broader populations, we remain committed to determining how Hemlibra might help even more people with haemophilia A to live a bleed-free life.”
While the treatment and care of people with severe haemophilia A are well-established, there is less information and guidance on prophylaxis for moderate haemophilia A.7 Additionally, the severity of haemophilia A, traditionally measured by an individual’s factor VIII levels, is not always reflective of bleeding behaviour. Many people with moderate haemophilia A may not be receiving prophylactic treatments and could have a worsened clinical burden. Approximately 85% of people with moderate haemophilia A have bleeds within a given year and one in three have long-term joint problems, many of which require surgery and impact quality of life.1,7,8
This approval is based on the results of the phase III HAVEN 6 trial, in which Hemlibra demonstrated effective bleed control and a favourable safety profile in people with non-severe haemophilia A without factor VIII inhibitors, where prophylaxis was clinically indicated.2 The decision was also based on real world data. This label expansion will provide an effective and convenient prophylactic treatment option with a favourable safety profile for people in the EU with moderate haemophilia A with a severe bleeding phenotype.
Hemlibra is approved as a prophylactic treatment option for people with haemophilia A with factor VIII inhibitors in more than 110 countries and for people without factor VIII inhibitors in more than 100 countries worldwide. It has been studied in one of the largest clinical trial programmes in people with haemophilia A with and without factor VIII inhibitors, including eight phase III studies.
About HAVEN 6
HAVEN 6 is a phase III clinical study designed to evaluate the safety, efficacy, pharmacokinetics and pharmacodynamics of Hemlibra in people with non-severe haemophilia A without factor VIII inhibitors. Data from the primary analysis of HAVEN 6 was presented at the 30th International Society on Thrombosis and Haemostasis (ISTH) Annual Congress, on 11 July 2022.2 The analysis included data from 72 participants, including three women, 51 (70.8%) of whom had moderate haemophilia A without factor VIII inhibitors. The data indicate that Hemlibra has effective bleed control and a favourable safety profile in people with non-severe haemophilia A without factor VIII inhibitors, with no new safety signals identified. Hemlibra achieved clinically meaningful bleed control, with 66.7% of participants experiencing no bleeds that required treatment, 81.9% experiencing no spontaneous bleeds that required treatment, and 88.9% experiencing no joint bleeds that required treatment. Model-based annualised bleed rates (ABR) remained low throughout the evaluation period at 0.9 (95% CI: 0.55-1.52).2
About Hemlibra® (emicizumab)
Hemlibra is a bispecific factor IXa- and factor X-directed antibody. It is designed to bring together factor IXa and factor X, proteins involved in the natural coagulation cascade, and restore the blood clotting process for people with haemophilia A. Hemlibra is a prophylactic (preventative) treatment that can be administered by an injection of a ready-to-use solution under the skin (subcutaneously) once-weekly, every two weeks, or every four weeks (after an initial once-weekly dose for the first four weeks). Hemlibra was created by Chugai Pharmaceutical Co., Ltd. and is being co-developed globally by Chugai, Roche and Genentech. It is marketed in the United States by Genentech as Hemlibra (emicizumab-kxwh), with kxwh as the suffix designated in accordance with Nonproprietary Naming of Biological Products Guidance for Industry issued by the U.S. Food and Drug Administration.9
About Roche in haematology
Roche has been developing medicines for people with malignant and non-malignant blood diseases for more than 20 years; our experience and knowledge in this therapeutic area runs deep. Today, we are investing more than ever in our effort to bring innovative treatment options to patients across a wide range of haematological diseases. Our approved medicines include MabThera®/Rituxan® (rituximab), Gazyva®/Gazyvaro® (obinutuzumab), Polivy® (polatuzumab vedotin), Venclexta®/Venclyxto® (venetoclax) in collaboration with AbbVie, Hemlibra® (emicizumab) and Lunsumio® (mosunetuzumab). Our pipeline of investigational haematology medicines includes T-cell engaging bispecific antibodies glofitamab, targeting both CD20 and CD3, and cevostamab, targeting both FcRH5 and CD3; Tecentriq® (atezolizumab), a monoclonal antibody designed to bind with PD-L1 and crovalimab, an anti-C5 antibody engineered to optimise complement inhibition. Our scientific expertise, combined with the breadth of our portfolio and pipeline, also provides a unique opportunity to develop combination regimens that aim to improve the lives of patients even further.
About Roche
Founded in 1896 in Basel, Switzerland, as one of the first industrial manufacturers of branded medicines, Roche has grown into the world’s largest biotechnology company and the global leader in in-vitro diagnostics. The company pursues scientific excellence to discover and develop medicines and diagnostics for improving and saving the lives of people around the world. We are a pioneer in personalised healthcare and want to further transform how healthcare is delivered to have an even greater impact. To provide the best care for each person we partner with many stakeholders and combine our strengths in Diagnostics and Pharma with data insights from the clinical practice.
In recognising our endeavour to pursue a long-term perspective in all we do, Roche has been named one of the most sustainable companies in the pharmaceuticals industry by the Dow Jones Sustainability Indices for the thirteenth consecutive year. This distinction also reflects our efforts to improve access to healthcare together with local partners in every country we work.
Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan.
For more information, please visit www.roche.com.
Feb. 02, 2023 5:21 AM ET
Roche Holding AG (RHHBY), RHHBF
By: Ravikash, SA News Editor
January 25, 2023 6:45 am ET
RAHWAY, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside of the United States and Canada, today announced positive results from the Phase 3 KEYNOTE-966 trial. In the final analysis of this trial, KEYTRUDA, Merck’s anti-PD-1 therapy, in combination with standard of care chemotherapy (gemcitabine and cisplatin) demonstrated a statistically significant and clinically meaningful improvement in overall survival (OS) versus chemotherapy alone for the first-line treatment of patients with advanced or unresectable biliary tract cancer (BTC). The safety profile of KEYTRUDA in this trial was consistent with that observed in previously reported studies. Results will be presented at an upcoming medical meeting and will be submitted to regulatory authorities.
“Biliary tract cancer is typically diagnosed at an advanced stage, and these patients face a poor prognosis, with five-year survival rates estimated to be approximately 5% to 15%,” said Dr. Eliav Barr, senior vice president, head of global clinical development and chief medical officer, Merck Research Laboratories. “We are very encouraged by these overall survival results that show the potential benefit of KEYTRUDA in addition to chemotherapy for biliary cancer patients who are in urgent need of new treatment options.”
Merck has an extensive clinical development program evaluating KEYTRUDA in gastrointestinal cancers and is continuing to study KEYTRUDA for multiple uses in gastric, hepatobiliary, esophageal, pancreatic and colorectal cancers.
About KEYNOTE-966
KEYNOTE-966 is a randomized, double-blind Phase 3 trial (ClinicalTrials.gov, NCT04003636) evaluating KEYTRUDA in combination with gemcitabine and cisplatin compared to placebo plus gemcitabine and cisplatin for the first-line treatment of advanced and/or unresectable BTC. The primary endpoint was OS, and the secondary endpoints included progression-free survival, objective response rate, duration of response and safety. The trial enrolled 1,069 patients who were randomized to receive KEYTRUDA (200 mg every three weeks for up to approximately two years) plus gemcitabine and cisplatin, or placebo plus gemcitabine and cisplatin.
About Biliary Tract Cancer (BTC)
Biliary tract cancer is a group of rare and highly aggressive cancers in the gallbladder and bile ducts. Biliary tract cancer is the second most common type of primary liver cancer after hepatocellular carcinoma, accounting for 15% of all liver cancers. It is estimated there are approximately 211,000 new cases of BTC diagnosed and 174,000 deaths from the disease each year globally. Biliary tract cancer is most frequently diagnosed in patients between 50 to 70 years old, and 70% of BTC patients are diagnosed at an advanced stage. Patients diagnosed with BTC face a very poor prognosis, as the five-year survival rate is estimated to be between 5% and 15%.
About KEYTRUDA® (pembrolizumab) injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of adult and pediatric (12 years and older) patients with stage IIB, IIC, or III melanoma following complete resection.
Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [tumor proportion score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [Combined Positive Score (CPS) ≥1] as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy.
KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Carcinoma
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
Non-muscle Invasive Bladder Cancer
KEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with MSI-H central nervous system cancers have not been established.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC) as determined by an FDA-approved test.
Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or gastroesophageal junction (GEJ) (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
KEYTRUDA, in combination with chemotherapy, with or without bevacizumab, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
KEYTRUDA is indicated for the adjuvant treatment of patients with RCC at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions.
Endometrial Carcinoma
KEYTRUDA, as a single agent, is indicated for the treatment of patients with advanced endometrial carcinoma that is MSI-H or dMMR, as determined by an FDA-approved test, who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation.
Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
About Merck
At Merck, known as MSD outside of the United States and Canada, we are unified around our purpose: We use the power of leading-edge science to save and improve lives around the world. For more than 130 years, we have brought hope to humanity through the development of important medicines and vaccines. We aspire to be the premier research-intensive biopharmaceutical company in the world – and today, we are at the forefront of research to deliver innovative health solutions that advance the prevention and treatment of diseases in people and animals. We foster a diverse and inclusive global workforce and operate responsibly every day to enable a safe, sustainable and healthy future for all people and communities. For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf .
Source: Merck & Co., Inc.
Jan. 25, 2023 7:50 AM ET
By: Ravikash, SA News Editor
Merck's (NYSE:MRK) Keytruda, with standard of care chemotherapy, met the main goal of overall survival (OS) in patients with advanced or unresectable biliary tract cancer (BTC) in a phase 3 trial.
January 27, 2023 6:45 am ET
Approval based on KEYNOTE-091 trial, which demonstrated a clinically meaningful improvement in disease-free survival with KEYTRUDA in these patients following surgical resection and platinum-based chemotherapy versus placebo
Approval marks fifth indication for KEYTRUDA-based regimens in NSCLC and 34th indication for KEYTRUDA in the US
RAHWAY, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside of the United States and Canada, announced today that the U.S. Food and Drug Administration (FDA) has approved KEYTRUDA, Merck’s anti-PD-1 therapy, as a single agent, for adjuvant treatment following surgical resection and platinum-based chemotherapy for adult patients with stage IB (T2a ≥4 centimeters [cm]), II, or IIIA non-small cell lung cancer (NSCLC).
Jan 27, 2023 United States
Study unblinded following recommendation of Independent Data Monitoring Committee
RARITAN, New Jersey, January 27, 2023 – The Janssen Pharmaceutical Companies of Johnson & Johnson today announced that the Phase 3 CARTITUDE-4 study evaluating CARVYKTI® (ciltacabtagene autoleucel; cilta-cel) versus pomalidomide, bortezomib, and dexamethasone (PVd) or daratumumab, pomalidomide, and dexamethasone (DPd) for the treatment of patients with relapsed and lenalidomide-refractory multiple myeloma met its primary endpoint of significant improvement in progression-free survival (PFS) at the first pre-specified interim analysis. As a result of meeting the primary endpoint, the Independent Data Monitoring Committee recommended the unblinding of the study.
“The CARTITUDE-4 study represents the first Phase 3 program in our comprehensive clinical development strategy for CARVYKTI, and further demonstrates our commitment to advance the treatment of patients with relapsed/refractory multiple myeloma,” said Jordan Schecter, M.D., Vice President, Clinical Development Cellular Therapy Program, Janssen Research & Development, LLC. “We look forward to the presentation of the data from the CARTITUDE-4 study at a future medical meeting.”
CARTITUDE-4 (NCT04181827) is the first randomized Phase 3 study evaluating the efficacy and safety of CARVYKTI®. The study compares CARVYKTI® with standard of care treatments PVd or DPd in adult patients with relapsed and lenalidomide-refractory multiple myeloma who received one to three prior lines of therapy. The primary endpoint of the study is PFS; safety, overall survival, minimal residual disease negative rate and overall response rate are secondary endpoints. Patients enrolled in the CARTITUDE-4 study will continue to be followed for primary, secondary and exploratory endpoints for the duration of the study.
Results from the CARTITUDE-4 study will be presented at an upcoming scientific congress and shared with health authorities in planned submission applications.
About CARVYKTI® (ciltacabtagene autoleucel)
CARVYKTI® (cilta-cel) received U.S. Food and Drug Administration approval in February 2022 for the treatment of adults with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.[1] In May 2022, the European Commission granted conditional marketing authorization of CARVYKTI® (cilta-cel) for the treatment of adults with relapsed and refractory multiple myeloma who have received at least three prior therapies, including an immunomodulatory agent, a proteasome inhibitor and an anti-CD38 antibody, and have demonstrated disease progression on the last therapy. In September 2022, Japan’s Ministry of Health, Labour and Welfare (MHLW) approved CARVYKTI® (cilta-cel) for the treatment of adults with relapsed or refractory multiple myeloma in patients that have no history of CAR-positive T cell infusion therapy targeting BCMA and who have received three or more lines of therapies, including an immunomodulatory agent, a proteasome inhibitor and an anti-CD38 monoclonal antibody, and in whom multiple myeloma has not responded to or has relapsed following the most recent therapy.
CARVYKTI® is a BCMA-directed, genetically modified autologous T-cell immunotherapy, which involves reprogramming a patient’s own T-cells with a transgene encoding chimeric antigen receptor (CAR) that directs the CAR positive T-cells to eliminate cells that express BCMA. BCMA is primarily expressed on the surface of malignant multiple myeloma B-lineage cells, as well as late-stage B cells and plasma cells. The CARVYKTI® CAR protein features two BCMA-targeting single domains designed to confer high avidity against human BCMA. Upon binding to BCMA-expressing cells, the CAR promotes T-cell activation, expansion, and elimination of target cells.
In December 2017, Janssen Biotech, Inc. entered into an exclusive worldwide license and collaboration agreement with Legend Biotech USA, Inc. to develop and commercialize CARVYKTI®.
For more information, visit www.CARVYKTI.com.
CARVYKTI® INDICATIONS AND USAGE
CARVYKTI® (ciltacabtagene autoleucel) is a B-cell maturation antigen (BCMA)-directed genetically modified autologous T cell immunotherapy indicated for the treatment of adult patients with relapsed or refractory multiple myeloma, after four or more prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.
Further information is available at www.CARVYKTIrems.com or 1-844-672-0067.
Please read full Prescribing Information including Boxed Warning for CARVYKTI®.
About the Janssen Pharmaceutical Companies of Johnson & Johnson
At Janssen, we're creating a future where disease is a thing of the past. We're the Pharmaceutical Companies of Johnson & Johnson, working tirelessly to make that future a reality for patients everywhere by fighting sickness with science, improving access with ingenuity, and healing hopelessness with heart. We focus on areas of medicine where we can make the biggest difference: Cardiovascular, Metabolism & Retina; Immunology; Infectious Diseases & Vaccines; Neuroscience; Oncology; and Pulmonary Hypertension.
Learn more at www.janssen.com. Follow us at @JanssenUS and @JanssenGlobal. Janssen Research & Development, LLC and Janssen Biotech, Inc. are part of the Janssen Pharmaceutical Companies of Johnson & Johnson.
Jan. 27, 2023 9:09 AM ET
Legend Biotech Corporation (LEGN), JNJ
By: Dulan Lokuwithana, SA News Editor
dulaglutide
On 26 January 2023, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion recommending a change to the terms of the marketing authorisation for the medicinal product Trulicity. The marketing authorisation holder for this medicinal product is Eli Lilly Nederland B.V.
The CHMP adopted an extension to the existing indication to include children from 10 years of age. For information, the full indication will be as follows:1
Type 2 Diabetes Mellitus
Trulicity is indicated for the treatment of adultspatients 10 years and above with insufficiently controlled type 2 diabetes mellitus as an adjunct to diet and exercise
For study results with respect to combinations, effects on glycaemic control and cardiovascular events, and the populations studied, see sections 4.4, 4.5 and 5.1.
Detailed recommendations for the use of this product will be described in the updated summary of product characteristics (SmPC), which will be published in the revised European public assessment report (EPAR), and will be available in all official European Union languages after a decision on this change to the marketing authorisation has been granted by the European Commission.
1 New text in bold, removed text as strikethrough
Jan. 27, 2023 7:14 AM ET
By: Ravikash, SA News Editor1 Comment

January 20, 2023Download PDF
The supplemental New Drug Application is based on results from the landmark EMPA-KIDNEY phase III trial, which showed Jardiance® (empagliflozin) tablets significantly reduced the risk of kidney disease progression* or cardiovascular death in adults with CKD by 28% (absolute risk reduction [ARR]: 3.8%) compared with placebo, both on top of standard of care.
RIDGEFIELD, Conn. and INDIANAPOLIS, Jan. 20, 2023 /PRNewswire/ -- The U.S. Food and Drug Administration (FDA) has accepted a supplemental New Drug Application (sNDA) for Jardiance® (empagliflozin) tablets, which is being investigated as a potential treatment to reduce the risk of kidney disease progression and cardiovascular death in adults with chronic kidney disease (CKD), Boehringer Ingelheim and Eli Lilly and Company (NYSE: LLY) announced.
"There is a significant need for additional therapies that reduce the risk of kidney disease progression and hospitalizations in adults with CKD," said Mohamed Eid, M.D., M.P.H., M.H.A., vice president, Clinical Development & Medical Affairs, Cardio-Renal-Metabolism & Respiratory Medicine, Boehringer Ingelheim Pharmaceuticals, Inc. "This application acceptance is an important step forward for the approximately 37 million people in the U.S. living with CKD."
The sNDA is based on results from the landmark EMPA-KIDNEY phase III trial, in which Jardiance significantly reduced the risk of kidney disease progression* or cardiovascular death in adults with CKD by 28% (ARR: 3.8%) compared with placebo, both on top of standard of care. Results were presented during the American Society of Nephrology (ASN)'s Kidney Week 2022 and simultaneously published in The New England Journal of Medicine. EMPA-KIDNEY is the first SGLT2 inhibitor CKD trial to show a significant reduction in risk of hospitalization for any cause, with a 14% relative risk reduction with Jardiance versus placebo (24.8 vs. 29.2 events/100 patient-years, respectively), both on top of standard of care, in a pre-specified key secondary endpoint. Reductions in other key secondary endpoints of hospitalization for heart failure or cardiovascular death or all-cause death were not statistically significant. Hospitalizations account for 35% to 55% of total healthcare costs for people with CKD in the U.S.
EMPA-KIDNEY, the largest and broadest dedicated SGLT2 inhibitor trial in CKD to date, provides additional data for patients commonly seen in clinical practice. The trial enrolled 6,609 participants, including people without diabetes (56%), those with various underlying causes of CKD and those across the spectrum of eGFR and urine albumin-creatinine ratio (measures of kidney function and excess albumin in the urine, respectively). Overall, the safety data in EMPA-KIDNEY were consistent with the previously known safety profile of Jardiance.
Initially approved in 2014, Jardiance is a once-daily tablet used along with diet and exercise to lower blood sugar in adults with type 2 diabetes; and to reduce the risk of cardiovascular death in adults with type 2 diabetes and known cardiovascular disease. Jardiance is also indicated to reduce the risk of cardiovascular death and hospitalization for heart failure in adults with heart failure. Jardiance is not for patients with type 1 diabetes, or to improve glycemic control in adults with type 2 diabetes with an eGFR <30 mL/min/1.73 m2. Jardiance is contraindicated in people with hypersensitivity to empagliflozin or any of the excipients in Jardiance, and in patients on dialysis. Please see additional Important Safety Information below.
"This marks another exciting milestone for Jardiance, potentially extending its ability to positively impact the approximately one billion people diagnosed with a cardio, renal or metabolic condition," said Jeff Emmick, M.D., Ph.D., vice president, Product Development, Lilly. "We look forward to working with the FDA during the review process and eagerly await a decision later this year on the indication for CKD, which doubles a person's risk for hospitalization."
In March 2020, the FDA granted Fast Track designation to the clinical investigation of Jardiance to reduce the risk of kidney disease progression and cardiovascular death in adults with CKD. According to the FDA, Fast Track designation is designed to facilitate the development of drugs and expedite treatments that may address serious conditions and fill an unmet medical need. Jardiance is not indicated for the treatment of CKD.
*Kidney disease progression: Defined as end-stage kidney disease (the initiation of maintenance dialysis or receipt of a kidney transplant), a sustained decline in estimated glomerular filtration rate (eGFR) to below 10 mL/min/1.73 m2, kidney death or a sustained decline of at least 40% in eGFR from randomization).
About EMPA-KIDNEY: The study of heart and kidney protection with Jardiance
EMPA-KIDNEY (NCT03594110) is a multinational, randomized, double-blind, placebo-controlled clinical trial, designed to evaluate the effect of Jardiance on kidney disease progression and cardiovascular mortality risk. The primary outcome is defined as time to a first event of either cardiovascular death or kidney disease progression, defined as end-stage kidney disease (the need for kidney replacement therapy such as dialysis or kidney transplantation), a sustained decline in eGFR to <10 mL/min/1.73 m2, kidney death or a sustained decline of ≥40 percent in eGFR from randomization. Key secondary outcomes include cardiovascular death or hospitalization for heart failure, all-cause hospitalization and all-cause mortality. EMPA-KIDNEY includes 6,609 adults randomized from eight countries with established CKD both with and without diabetes, as well as with and without albuminuria, receiving either Jardiance 10 mg or placebo, on top of current standard of care.
What is JARDIANCE?
JARDIANCE is a prescription medicine used to:
JARDIANCE is not for people with type 1 diabetes. It may increase their risk of diabetic ketoacidosis (increased ketones in the blood or urine).
JARDIANCE is not for use to lower blood sugar in adults with type 2 diabetes who have severe kidney problems, because it may not work.
https://www.jardiance.com/type-2-diabetes/
For more information, please see Prescribing Information and Medication Guide.
Boehringer Ingelheim and Eli Lilly and Company
In January 2011, Boehringer Ingelheim and Eli Lilly and Company announced an Alliance that centers on compounds representing several of the largest diabetes treatment classes. Depending on geographies, the companies either co-promote or separately promote the respective molecules each contributing to the Alliance. The Alliance leverages the strengths of two of the world's leading pharmaceutical companies to focus on patient needs. By joining forces, the companies demonstrate their commitment, not only to the care of people with diabetes, but also to investigating the potential to address areas of unmet medical need.
About Boehringer Ingelheim
Boehringer Ingelheim is working on breakthrough therapies that improve the lives of humans and animals. As a leading research-driven biopharmaceutical company, the company creates value through innovation in areas of high unmet medical need. Founded in 1885 and family-owned ever since, Boehringer Ingelheim takes a long-term perspective. Around 52,000 employees serve more than 130 markets in the three business areas, Human Pharma, Animal Health, and Biopharmaceutical Contract Manufacturing. Learn more at www.boehringer-ingelheim.us.
About Lilly
Lilly unites caring with discovery to create medicines that make life better for people around the world. We've been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world's most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer's disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we're motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram and LinkedIn.
This press release contains forward-looking statements (as that term is defined in the Private Securities Litigation Reform Act of 1995) about Jardiance® as a treatment for adults with type 2 diabetes, to reduce the risk of cardiovascular death in adults with type 2 diabetes and known cardiovascular disease, and to reduce the risk of cardiovascular death and hospitalization for heart failure in adults with heart failure, and as a potential treatment for adults with cardio-kidney-metabolic conditions and reflects Lilly's current beliefs and expectations. However, as with any pharmaceutical product, there are substantial risks and uncertainties in the process of drug research, development and commercialization. Among other things, there can be no guarantee that planned or ongoing studies will be completed as planned, that future study results will be consistent with the results to date or that Jardiance® will receive additional regulatory approvals. For a further discussion of these and other risks and uncertainties that could cause actual results to differ from Lilly's expectations, please see Lilly's most recent Forms 10-K and 10-Q filed with the U.S. Securities and Exchange Commission. Lilly undertakes no duty to update forward-looking statements.
Jardiance®, EMPEROR-Reduced®, EMPEROR-Preserved®, EMPA-REG OUTCOME® and EMPACT-MI® are registered trademarks of Boehringer Ingelheim.
![]()
![]()
View original content to download multimedia:https://www.prnewswire.com/news-releases/us-fda-accepts-supplemental-new-drug-application-for-jardiance-for-adults-with-chronic-kidney-disease-301726873.html
SOURCE Eli Lilly and Company
Jan. 20, 2023 10:02 AM ET
By: Jonathan Block, SA News Editor
Jan 19, 2023 7:00 AM
MHRA marketing authorizations follow recent European Commission marketing authorizations
BRUKINSA is the only treatment authorized for MZL in Great Britain
CAMBRIDGE, Mass. & BASEL, Switzerland & BEIJING--(BUSINESS WIRE)-- BeiGene (NASDAQ: BGNE; HKEX: 06160; SSE: 688235), a global biotechnology company, today announced that the Medicines and Healthcare products Regulatory Agency (MHRA) has granted marketing authorizations for BRUKINSA (zanubrutinib) in Great Britain for both the treatment of adult patients with chronic lymphocytic leukemia (CLL) and the treatment of adult patients with marginal zone lymphoma (MZL) who have received at least one prior anti-CD20-based therapy.
“BRUKINSA is a highly selective BTK inhibitor that has demonstrated clinically meaningful improvements over the first generation of BTK inhibitor in relapsed CLL,” said Dr Renata Walewska, Department of Hematology, University Hospitals Dorset, Bournemouth, UK. “The authorization of BRUKINSA for MZL and CLL in Great Britain is a significant step forward for eligible patients and their physicians as there is no targeted treatment currently authorized for MZL patients other than chemoimmunotherapy, and it represents an alternative to current BTKi treatments for patients with CLL.”
The MHRA authorization for CLL is based on two global Phase 3 clinical trials: SEQUOIA (NCT03336333)1, comparing BRUKINSA against bendamustine plus rituximab (BR) in patients with previously untreated CLL, and ALPINE (NCT03734016)2, comparing BRUKINSA® against IMBRUVICA® (ibrutinib) in patients with relapsed/refractory (R/R) CLL.
The MHRA authorization for MZL is based on results from the multicenter, global, single-arm, open-label, Phase 2 MAGNOLIA3 trial in patients with R/R MZL who received at least one anti-CD-20 based regimen.
“As a BTK inhibitor designed to maximize BTK occupancy and minimize off-target binding, we believe BRUKINSA presents a very promising treatment option for eligible patients with MZL and CLL,” said Mehrdad Mobasher, M.D., M.P.H., Chief Medical Officer, Hematology at BeiGene.
“As we strive for delivering cancer medicines more quickly to more patients around the world, we are pleased with the progress we’ve made in bringing BRUKINSA to more eligible patients with hematological malignancies in Great Britain following these authorizations,” added Dr. Robert Mulrooney, General Manager, UK & Ireland at BeiGene.
Earlier this year, the National Institute for Health and Care Excellence (NICE), recommended BRUKINSA for the treatment of Waldenström’s macroglobulinemia (WM) in adults who have had at least one treatment, only if bendamustine plus rituximab is also suitable. BRUKINSA has also been recommended by the Scottish Medicines Consortium for the treatment of adult patients with WM who have received at least one prior therapy, or in first-line treatment for patients unsuitable for chemoimmunotherapy.
BRUKINSA is currently authorized in the European Union, and Northern Ireland, as per rulings set out in the Northern Ireland Protocol,for the treatment of adult patients with WM who have received at least one prior therapy or as the first-line treatment for patients unsuitable for chemoimmunotherapy; for the treatment of adult patients with CLL and for adult patients with MZL who have received at least one prior anti-CD20-based therapy.
About BRUKINSA
BRUKINSA is a small-molecule inhibitor of Bruton’s tyrosine kinase (BTK) discovered by BeiGene scientists that is currently being evaluated globally in a broad clinical program as a monotherapy and in combination with other therapies to treat various B-cell malignancies. BRUKINSA was specifically designed to deliver targeted and sustained inhibition of the BTK protein by optimizing bioavailability, half-life, and selectivity. With differentiated pharmacokinetics compared to other approved BTK inhibitors, BRUKINSA® has been demonstrated to inhibit the proliferation of malignant B cells within a number of disease-relevant tissues.
BRUKINSA is supported by a broad clinical program which includes more than 4,700 subjects in 35 trials in more than 30 geographies. To date, BRUKINSA is approved in over 60 markets, including the United States, China, the European Union, Great Britain, Canada, Australia, South Korea, Iceland, Norway and Switzerland.
About BeiGene
BeiGene is a global biotechnology company that is developing and commercializing innovative and affordable oncology medicines to improve treatment outcomes and access for far more patients worldwide. With a broad portfolio, we are expediting development of our diverse pipeline of novel therapeutics through our internal capabilities and collaborations. We are committed to radically improving access to medicines for far more patients who need them. Our growing global team of more than 9,000 colleagues spans five continents, with administrative offices in Cambridge, U.S.; Basel, Switzerland and Beijing, China. To learn more about BeiGene, please visit www.beigene.com and follow us on Twitter at @BeiGeneGlobal.
View source version on businesswire.com: https://www.businesswire.com/news/home/20230119005082/en/
Source: BeiGene
Jan. 19, 2023 7:48 AM ET
By: Ravikash, SA News Editor
PUBLISHED13 January 2023 13 January 2023 07:00 GMT
AstraZeneca’s Tezspire (tezepelumab) has received a positive opinion from the European Medicine Agency’s Committee for Medicinal Products for Human Use (CHMP) for self-administration in a pre-filled, single-use pen for patients aged 12 years and older with severe asthma. The CHMP opinion can be implemented without the need for a European Commission decision due to the nature of the Type-II label variation.
The approval for self-administration was based on results from the PATHFINDER clinical trial programme, which included results from the PATH-BRIDGE Phase I trial and the PATH-HOME Phase III trial.1,2 The majority (92%) of healthcare providers, patients and caregivers were able to successfully administer Tezspire both in the clinic and at home throughout the PATH-HOME trial.2 The improvements in asthma control and the safety profile of Tezspire observed in the PATH-HOME trial were consistent with previous clinical trials.2 Tezspire is the only biologic approved for severe asthma with no phenotype (e.g. eosinophilic or allergic) or biomarker limitation within its approved label.3
Professor Ian Pavord, Professor of Respiratory Medicine at the University of Oxford and Honorary Consultant Physician at the Oxford University Hospitals, said: “Severe asthma continues to have a debilitating impact for people living with the disease. I believe the approval of the Tezspire pre-filled pen will be welcome news for physicians and patients in Europe as it offers increased choice and greater flexibility when administering this important medicine.”
Mene Pangalos, Executive Vice President, BioPharmaceuticals R&D, AstraZeneca, said: “Tezspire is the first and only biologic approved in Europe for patients with severe asthma with no phenotype or biomarker limitation. With the approval of the Tezspire pre-filled pen, we can give patients in Europe greater flexibility and support physicians in treating a broad population of severe asthma patients.”
AstraZeneca anticipates a regulatory decision by the US Food and Drug Administration (FDA) on self-administration and the new pre-filled pen in the first half of 2023. Tezspire is currently approved for the treatment of severe asthma in the US, EU, Japan and other countries.4-6
Notes
Tezspire pre-filled pen
Tezspire will be available as a fixed-dose 210mg subcutaneous injection via a pre-filled, single-use auto-injector (the Tezspire pre-filled pen), in addition to the pre-filled, single-use syringe (Tezspire pre-filled syringe). Both are administered every four weeks.
The Tezspire pre-filled pen enables patients and caregivers to self-administer the medicine at home or in clinic via a simple process. The device is fitted with a safety guard and viewing window and has audible clicks at the start and end of the injection to guide patients.
Clinical trials
PATH-HOME was a Phase III multi-centre, open-label, parallel-group trial designed to assess patient, caregiver and healthcare provider-reported functionality and performance of a single-use, pre-filled syringe (PFS) or auto-injector (AI) with a fixed 210mg dose of Tezspire administered subcutaneously every four weeks in a clinic and in an at-home setting in 216 patients aged 12 years and older with severe asthma.2
The majority (92%) of healthcare providers, patients and caregivers were able to successfully administer the Tezspire pre-filled pen both in the clinic and at home throughout the trial.2 At-home administration of the Tezspire pre-filled pen at weeks 12 and 16 was successful in 97% of the patients or caregivers (102/105).2 The trial also demonstrated for the first time that adolescents can successfully administer Tezspire using the two devices.2 The very low proportion of device malfunctions (0.9% of PFS and 0.8% of AIs) provides support that the instructions for use provided to healthcare providers, patients and caregivers is adequate for successful subcutaneous administration of Tezspire both in the clinic and at home.2
PATH-BRIDGE was a single-centre, randomised, open-label, parallel-group Phase I trial in healthy people to compare the pharmacokinetic (PK) exposure following a single 210mg dose of Tezspire by using a vial-and-syringe (V-S), PFS or pre-filled AI device.1 Tezspire PK exposure was comparable following subcutaneous administration via V-S, PFS or AI.1 In addition, injection site-pain was low in severity and injection-site reactions were uncommon in all device groups.1
In addition to PATH-BRIDGE and PATH-HOME, the PATHFINDER clinical trial programme included the pivotal NAVIGATOR Phase III trial in which Tezspire demonstrated superiority across every primary and key secondary endpoint in patients with severe asthma, compared to placebo, when added to standard therapy.7
NAVIGATOR was the first Phase III trial to show benefit in severe asthma irrespective of eosinophils by targeting thymic stromal lymphopoietin (TSLP).7 These results support the FDA Breakthrough Therapy Designation granted to Tezspire in September 2018 for patients with severe asthma, without an eosinophilic phenotype. In July 2021, Tezspire was the first and only biologic to be granted Priority Review in the US for the treatment of asthma by the FDA.
Tezspire
Tezspire (tezepelumab) is being developed by AstraZeneca in collaboration with Amgen as a first-in-class human monoclonal antibody that inhibits the action of TSLP, a key epithelial cytokine that sits at the top of multiple inflammatory cascades and is critical in the initiation and persistence of allergic, eosinophilic and other types of airway inflammation associated with severe asthma, including airway hyperresponsiveness.8,9 TSLP is released in response to multiple triggers associated with asthma exacerbations, including allergens, viruses and other airborne particles.8,9 Expression of TSLP is increased in the airways of patients with asthma and has been correlated with disease severity.8,9 Blocking TSLP may prevent the release of pro-inflammatory cytokines by immune cells, resulting in the prevention of asthma exacerbations and improved asthma control.7,8,10 Tezspire acts at the top of the inflammation cascade and has the potential to help address a broad population of severe asthma patients irrespective of biomarker levels.7,8
Tezspire is approved in the US, EU, Japan and other countries for the treatment of severe asthma.4-6 Tezspire is also in development for other potential indications including chronic obstructive pulmonary disease (COPD), chronic rhinosinusitis with nasal polyps, chronic spontaneous urticaria and eosinophilic esophagitis (EoE). In October 2021, tezepelumab was granted Orphan Drug Designation by the FDA for the treatment of EoE.
Amgen collaboration
In 2020, Amgen and AstraZeneca updated a 2012 collaboration agreement for Tezspire. Both companies will continue to share costs and profits equally after payment by AstraZeneca of a mid single-digit inventor royalty to Amgen. AstraZeneca continues to lead development and Amgen continues to lead manufacturing. All aspects of the collaboration are under the oversight of joint governing bodies. Under the amended agreement, Amgen and AstraZeneca will jointly commercialise Tezspire in North America. Amgen will record product sales in the US, with AZ recording its share of US profits as Collaboration Revenue. Outside of the US, AstraZeneca will record product sales, with Amgen recording profit share as Other/Collaboration revenue.
AstraZeneca in Respiratory & Immunology
Respiratory & Immunology, part of BioPharmaceuticals, is one of AstraZeneca’s main disease areas and is a key growth driver for the Company.
AstraZeneca is an established leader in respiratory care with a 50-year heritage. The Company aims to transform the treatment of asthma and COPD by focusing on earlier biology-led treatment, eliminating preventable asthma attacks, and removing COPD as a top-three leading cause of death. The Company’s early respiratory research is focused on emerging science involving immune mechanisms, lung damage and abnormal cell-repair processes in disease and neuronal dysfunction.
With common pathways and underlying disease drivers across respiratory and immunology, AstraZeneca is following the science from chronic lung diseases to immunology-driven disease areas. The Company’s growing presence in immunology is focused on five mid- to late-stage franchises with multi-disease potential, in areas including rheumatology (including systemic lupus erythematosus), dermatology, gastroenterology, and systemic eosinophilic-driven diseases. AstraZeneca’s ambition in Respiratory & Immunology is to achieve disease modification and durable remission for millions of patients worldwide.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Jan. 13, 2023 4:33 AM ET
By: Ravikash, SA News Editor
A panel of the European Medicine Agency (EMA) issued a positive opinion on a self-administration option of Amgen (NASDAQ:AMGN) and AstraZeneca's (NASDAQ:AZN) asthma therapy Tezspire.
01/19/2023
– First FDA-approved treatment in HER2-positive metastatic colorectal cancer –
– Combination regimen is approved for use in the second-line treatment setting and beyond –
BOTHELL, Wash.--(BUSINESS WIRE)-- Seagen Inc. (Nasdaq: SGEN) today announced that the U.S. Food and Drug Administration (FDA) has granted accelerated approval to TUKYSA® (tucatinib) in combination with trastuzumab for adult patients with RASwild-type, HER2-positive unresectable or metastatic colorectal cancer that has progressed following treatment with fluoropyrimidine-, oxaliplatin- and irinotecan-based chemotherapy. TUKYSA is approved under the FDA’s Accelerated Approval Program based on tumor response rate and durability of response from the phase 2 MOUNTAINEER clinical trial. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials. This is the first FDA-approved treatment in HER2-positive metastatic colorectal cancer. The FDA previously granted Breakthrough Therapy Designation and Priority Review for TUKYSA in this setting.
“Historically, patients with HER2-positive metastatic colorectal cancer who have progressed following frontline therapy have had poor outcomes,” said John Strickler, M.D., associate professor of medicine, Duke University Medical Center, and lead investigator for the MOUNTAINEER trial. “The FDA approval of a chemotherapy-free combination regimen that specifically targets HER2 is great news for these patients.”
“Biomarker testing is bringing new hope to people living with some types of colorectal cancer by opening the door to targeted treatments like TUKYSA for those who have RAS wild-type, HER2-positive disease,” said Michael Sapienza, CEO, Colorectal Cancer Alliance. “It is critical that physicians and patients understand the importance of comprehensive biomarker testing at diagnosis because it can inform treatment decisions and help improve outcomes.”
Results from the MOUNTAINEER trial showed a 38% overall response rate (ORR) (95% Confidence Interval [CI]: 28, 49) per blinded independent central review (BICR) in the patients who received TUKYSA in combination with trastuzumab (N=84 with a median age of 55.0 years [range: 24 to 77]). Complete responses were observed in 3.6% of patients (n=3), and partial responses were observed in 35% of patients (n=29). The median duration of response (DOR) per BICR was 12.4 months (95% CI: 8.5, 20.5). At study entry, 64% and 70% of these patients had liver or lung metastases, respectively.
The Prescribing Information for TUKYSA includes warnings and precautions for diarrhea, hepatotoxicity and embryo-fetal toxicity, some of which may be severe or fatal. In MOUNTAINEER, serious adverse reactions occurred in 22% of patients; the most common (in ≥2% of patients) were intestinal obstruction (7%), urinary tract infection (3.5%), pneumonia, abdominal pain and rectal perforation (2.3% each). The most common adverse reactions (≥20%) in patients treated with TUKYSA and trastuzumab were diarrhea, fatigue, rash, nausea, abdominal pain, infusion-related reactions and pyrexia. Adverse reactions leading to permanent discontinuation of TUKYSA occurred in 6% of patients; the most common (in ≥2% of patients) was increased alanine aminotransferase (ALT) (2.3%). Please see additional Important Safety Information below.
“The accelerated approval of TUKYSA for RAS wild-type, HER2-positive metastatic colorectal cancer expands TUKYSA-based therapy to patients across two distinct types of cancer,” said Marjorie Green, M.D., Senior Vice President and Head of Late-Stage Development, Seagen. “We believe the efficacy and safety profile of the TUKYSA and trastuzumab-based regimen further establishes its role as an important backbone of dual HER2 inhibition in the treatment of adult patients with certain HER2-expressing breast and colorectal cancers.”
The FDA’s Accelerated Approval Program allows for approval of a medicine based on a surrogate endpoint that is reasonably likely to predict clinical benefit if the medicine fills an unmet medical need for a serious condition. A global, randomized phase 3 clinical trial (MOUNTAINEER-03) is ongoing and will compare TUKYSA in combination with trastuzumab and mFOLFOX6 with standard of care, which is intended to serve as a confirmatory trial and potentially support future global regulatory submissions. Merck, known as MSD outside of the U.S. and Canada, is commercializing TUKYSA in regions outside of the U.S., Canada and Europe and plans to discuss results from the MOUNTAINEER trial with certain health authorities as it continues to accelerate the filing of TUKYSA in its territories.
About Colorectal Cancer
In the U.S., approximately 153,000 people will be diagnosed with colorectal cancer in 2023, and the rate of the disease is increasing in younger adults.1 Approximately 22% of U.S. patients with colorectal cancer are diagnosed at the advanced stage.2 Human epidermal growth factor receptor 2 (HER2) is overexpressed in 3-5% of patients with metastatic colorectal cancer and approximately 10% of patients with RASwild-type metastatic colorectal cancer.3 In 2023, colorectal cancer is anticipated to lead to about 52,550 deaths in the U.S., where it is the third-leading cause of cancer-related deaths.1
About MOUNTAINEER
MOUNTAINEER is a U.S. and European open-label, multicenter phase 2 clinical trial of TUKYSA in combination with trastuzumab that evaluated 84 patients with HER2-positive, RASwild-type, unresectable or metastatic colorectal cancer following previous standard-of-care therapies. Patients evaluated in MOUNTAINEER had not received prior anti-HER2 therapy. Patients received TUKYSA (300 mg) twice per day orally in combination with trastuzumab intravenously (8 mg/kg loading dose, then 6 mg/kg every three weeks thereafter) until disease progression or unacceptable toxicity. The major efficacy outcome measures were overall response rate (ORR) and duration of response (DOR) as assessed by blinded independent central review (BICR) according to RECIST (Response Evaluation Criteria in Solid Tumors) version 1.1.
About TUKYSA (tucatinib)
TUKYSA (tucatinib) tablets (50 | 150 mg tablets) is an oral medicine that is a tyrosine kinase inhibitor of the HER2 protein. In vitro (in lab studies), TUKYSA inhibited phosphorylation of HER2 and HER3, resulting in inhibition of downstream MAPK and AKT signaling and cell growth (proliferation), and showed anti-tumor activity in HER2-expressing tumor cells. In vivo (in living organisms), TUKYSA inhibited the growth of HER2-expressing tumors. The combination of TUKYSA and the anti-HER2 antibody trastuzumab showed increased anti-tumor activity in vitro and in vivo compared to either medicine alone.
TUKYSA was approved by the U.S. FDA in April 2020 in combination with trastuzumab and capecitabine for treatment of adult patients with advanced unresectable or metastatic HER2-positive breastcancer, including patients with brain metastases, who have received one or more prior anti-HER2-based regimens in the metastatic setting. In January 2023, TUKYSA received accelerated approval by the U.S. FDA in combination with trastuzumab for adult patients with RASwild-type, HER2-positive unresectable or metastatic colorectal cancer that has progressed following treatment with fluoropyrimidine-, oxaliplatin- and irinotecan-based chemotherapy. Merck, known as MSD outside the U.S. and Canada, has exclusive rights to commercialize TUKYSA in Asia, the Middle East and Latin America and other regions outside of the U.S., Canada and Europe.
About Seagen
Seagen is a global biotechnology company that discovers, develops and commercializes transformative cancer medicines to make a meaningful difference in people’s lives. Seagen is headquartered in the Seattle, Washington area, and has locations in California, Canada, Switzerland and the European Union. For more information on the company’s marketed products and robust pipeline, visit www.seagen.com and follow @SeagenGlobal on Twitter.
Jan. 19, 2023 2:37 PM ET
Seagen Inc. (SGEN), MRKRHHBY, RHHBF
By: Dulan Lokuwithana, SA News Editor1 Comment
Roche announces the European Commission approval of Xofluza for the treatment and prevention of influenza in children aged one year and above
January 12, 2023
Basel, 12 January 2023 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the European Commission (EC) has approved Xofluza® (baloxavir marboxil) in children aged one year and above for the treatment of uncomplicated influenza and for post-exposure prophylaxis of influenza. Post-exposure prophylaxis aims to prevent influenza in individuals following contact with someone infected with the influenza virus. The Commission’s Decision is based on the results of the phase III miniSTONE-2 and BLOCKSTONE studies.1,2 This approval marks the first single-dose, oral influenza medicine approved in Europe for children, and now means that Xofluza can be used to treat and prevent uncomplicated influenza in children aged one year and above, and in adolescents and adults.3
“We are delighted that the European Commission has approved Xofluza for children aged one year and above, as influenza can be particularly dangerous for younger children,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “We are hopeful that Xofluza’s convenient single-dose oral regimen will help children recover quickly, as well as reduce the societal burden of influenza.”
Influenza is a serious infectious disease and represents a significant threat to public health.4-6 Rates of influenza in Europe are rising and it is expected that the virus will infect around one in four children each year.6,7 In addition to being at higher risk of infection, children also play a significant role in the spread of influenza from one person to another.6,8 In studies of influenza transmission in households, the risk of infection from others in the household was as high as 38%.9 Prophylactic treatment with Xofluza, which stops viral replication faster than oseltamivir, may help to limit the spread of influenza and therefore reduce the impact and burden of influenza on society.2,10,11
The approval is based on the results of the phase III miniSTONE-2 and BLOCKSTONE studies.1,2 The miniSTONE-2 study met its primary endpoint of safety and showed that Xofluza reduced the length of time that influenza was released from the body by more than two days compared with oseltamivir (viral replication; median time of 24.2 hours versus 75.8 hours, respectively). Xofluza was well tolerated with no new safety signals identified.1
In the BLOCKSTONE study Xofluza showed a statistically significant prophylactic effect after a single oral dose, by reducing the risk of people developing influenza after exposure to an infected household member by 86% versus placebo (1.9% in participants treated with Xofluza and 13.6% in the placebo group).2
Xofluza was first approved for use in Europe in 2021 for the treatment of uncomplicated influenza and for post-exposure prophylaxis of influenza in adults and adolescents aged 12 years and above.12 Xofluza is now approved in more than 70 countries for the treatment of influenza types A and B.13 Roche is continuing to investigate Xofluza in phase III trials in children under the age of one (NCT03653364), as well as to assess its potential to reduce transmission of influenza from an infected person to household members (NCT03969212).14,15
About miniSTONE-21
miniSTONE-2 was a phase III, multicentre, randomised, double-blind study that evaluated the safety, pharmacokinetics and efficacy of a single-dose of Xofluza® (baloxavir marboxil) compared with oseltamivir in otherwise healthy children aged one to less than 12 years with influenza infection and displaying influenza symptoms for no more than 48 hours (temperature of 38°C or over, and one or more respiratory symptoms).
Participants enrolled in the study were recruited in parallel into two cohorts: children aged five to less than 12 years and children aged one to less than five years. Participants were randomly assigned to receive a single-dose of Xofluza or oseltamivir twice a day over five days (dosing according to body weight).
Time to alleviation of influenza signs and symptoms were comparable between Xofluza and oseltamivir. The median time to alleviation of signs and symptoms in influenza-infected participants was 138 hours (95% CI: 117, 163) and 150 hours (95% CI: 115, 166) for those who received Xofluza or oseltamivir, respectively. Xofluza reduced the length of time that influenza was released from the body by more than two days compared with oseltamivir (viral replication; median time of 24.2 hours versus 75.8 hours, respectively). Xofluza was well tolerated with no new safety signals identified.
About BLOCKSTONE2
BLOCKSTONE was a phase III, double-blind, multicentre, randomised, placebo-controlled, post-exposure prophylaxis study that evaluated single-dose Xofluza® (baloxavir marboxil) compared with placebo in household members (adults and children), who were living with someone with influenza, confirmed by a rapid influenza diagnostic test (the ‘index patient’).
Participants enrolled in the study were household members of someone who had been diagnosed with influenza. The participants were randomised to receive a single-dose of Xofluza (dose according to body weight) or placebo as a preventive measure against developing influenza.
Xofluza showed a statistically significant prophylactic effect after a single oral dose, by reducing the risk of people developing influenza after exposure to an infected household member by 86% versus placebo. The proportion of households, who developed laboratory-confirmed clinical influenza was 1.9% in participants treated with Xofluza and 13.6% in the placebo-treated group.
Xofluza was well tolerated in this study and no new safety signals were identified. The study was conducted in Japan by Shionogi & Co., Ltd.
About Xofluza® (baloxavir marboxil)
Xofluza is a first-in-class, single-dose oral medicine with an innovative mechanism of action that has demonstrated efficacy in a wide range of influenza viruses, including in vitro activity against oseltamivir-resistant strains and avian strains (H7N9, H5N1) in non-clinical studies.10,16,17 Xofluza is the first in a class of antivirals designed to inhibit the cap-dependent endonuclease protein, which is essential for viral replication.10
Xofluza is approved in more than 70 countries for the treatment of influenza types A and B. In Europe, Xofluza is now approved for the treatment of uncomplicated influenza and for post-exposure prophylaxis of influenza in children aged one year and above, and in adolescents and adults. Xofluza is the first innovation in mechanism of action for an influenza antiviral approved by the European Commission in almost 20 years.18
Robust clinical evidence has demonstrated the benefit of Xofluza in several populations (otherwise-healthy, high-risk, children and post-exposure prophylaxis in individuals aged one year and above).1,2,10,19 Xofluza is being further studied in a phase III development programme, including in children under the age of one (NCT03653364) as well as to assess its potential to reduce transmission of influenza from an infected person to healthy people (NCT03969212).14,15
Xofluza was discovered by Shionogi & Co., Ltd. and is being further developed and commercialised globally in collaboration with the Roche Group (which includes Genentech in the US) and Shionogi & Co., Ltd. Under the terms of this agreement, Roche holds worldwide rights to Xofluza excluding Japan and Taiwan, which will be retained exclusively by Shionogi & Co., Ltd.
About Roche in influenza
Influenza is a serious infectious disease and represents a significant threat to public health. Seasonal epidemics result in three to five million cases of severe disease, millions of hospitalisations and up to 650,000 deaths globally every year.4 Roche has a long heritage in developing medicines that contribute to public health. We are committed to bringing innovation in the field of infectious diseases, including influenza. Tamiflu® (oseltamivir) has made a significant difference both to the treatment of seasonal influenza as well as in the management of recent pandemics, and we are proud to have brought this innovative medicine to patients. Although vaccines are an important first line of defence in preventing influenza, there is a need for new medical options for prevention (prophylaxis) and treatment. Roche is committed to addressing the unmet need in this area through its agreement with Shionogi & Co., Ltd. to develop and commercialise Xofluza® (baloxavir marboxil).
About Roche
Founded in 1896 in Basel, Switzerland, as one of the first industrial manufacturers of branded medicines, Roche has grown into the world’s largest biotechnology company and the global leader in in-vitro diagnostics. The company pursues scientific excellence to discover and develop medicines and diagnostics for improving and saving the lives of people around the world. We are a pioneer in personalised healthcare and want to further transform how healthcare is delivered to have an even greater impact. To provide the best care for each person we partner with many stakeholders and combine our strengths in Diagnostics and Pharma with data insights from the clinical practice.
In recognising our endeavor to pursue a long-term perspective in all we do, Roche has been named one of the most sustainable companies in the pharmaceuticals industry by the Dow Jones Sustainability Indices for the thirteenth consecutive year. This distinction also reflects our efforts to improve access to healthcare together with local partners in every country we work.
Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan.
For more information, please visit www.roche.com.
All trademarks used or mentioned in this release are protected by law.
Jan. 12, 2023 4:27 AM ET
Roche Holding AG (RHHBY), RHHBF
By: Ravikash, SA News Editor
PUBLISHED5 January 2023 5 January 2023 07:00 GMT
AstraZeneca’s Biologics License Application (BLA) for nirsevimab has been accepted for review by the US Food and Drug Administration (FDA) for the prevention of respiratory syncytial virus (RSV) lower respiratory tract disease in newborns and infants entering or during their first RSV season, and for children up to 24 months of age who remain vulnerable to severe RSV disease through their second RSV season.
Nirsevimab is being developed and commercialised by AstraZeneca in collaboration with Sanofi and is the first single-dose preventative option for the broad infant population, including those born healthy, at term or preterm, or with specific health conditions. The FDA has indicated it will work to expedite its review. The Prescription Drug User Fee Act date, the FDA target action date for its decision, is in the third quarter of 2023. If approved at that time, nirsevimab will be available in the US for the 2023/2024 RSV season.
RSV is a very contagious virus that can lead to serious respiratory illness, according to the Centers for Disease Control and Prevention.1 In the US, RSV is the leading cause of hospitalisation for babies under one.2 Any infant can be hospitalised in their first RSV season and about 75% of infants hospitalised for RSV in the US were born at term with no underlying conditions.3-5
Dr William Muller, Associate Professor, Pediatrics, Northwestern University Feinberg School of Medicine and Scientific Director, Clinical and Community Trials, Ann & Robert H. Lurie Children’s Hospital of Chicago, Illinois, US, said: “A substantial burden of disease from RSV affects infants, families, and healthcare providers every year. Effective interventions to prevent RSV are a critical need. This year in the US, we’ve seen first-hand how frightening the impact of this respiratory disease is on our patients and how stressful it is on the healthcare system, highlighting the urgency of addressing this problem.”
Iskra Reic, Executive Vice President, Vaccines and Immune Therapies, AstraZeneca, said: “This decision brings us a step closer to delivering a first-in-class preventative option for a broad infant population in the US. If approved, we believe nirsevimab may transform the medical community’s approach to respiratory syncytial virus prevention in infants and we are committed to working with the FDA to support completion of the review as quickly as possible.”
The BLA was based on results from the comprehensive nirsevimab clinical development programme, including the MELODY Phase III (primary cohort and all subjects), MEDLEY Phase II/III (first and second RSV season), and Phase IIb trials.6-11 Data from the MELODY trial was published in the New England Journal of Medicine in March 2022 and demonstrated a reduction in the incidence of medically-attended lower respiratory tract infections (LRTI) caused by RSV by 74.5% (95% CI 49.6, 87.1; p<0.001) vs. placebo through day 151 (a typical RSV season) with a single dose.6 Nirsevimab also demonstrated a comparable safety and tolerability profile to Synagis (palivizumab) in the MEDLEY Phase II/III trial, with occurrence of treatment emergent adverse events (TEAEs) or treatment emergent serious adverse events (TESAEs) similar between groups.10-11
In November 2022, nirsevimab was granted marketing authorisation in the European Union for the prevention of RSV lower respiratory tract disease in newborns and infants during their first RSV season, under the name Beyfortus.12 Additional global regulatory submissions are underway.
Notes
Nirsevimab
Nirsevimab is a single dose long-acting antibody, developed and commercialised in partnership by AstraZeneca and Sanofi using AstraZeneca’s YTE technology. It is designed to protect infants entering or during their first RSV season and for children up to 24 months of age who remain vulnerable to severe RSV disease through their second RSV season.
Nirsevimab has been developed to offer newborns and infants direct RSV protection via an antibody to help prevent LRTI caused by RSV. Monoclonal antibodies do not require the activation of the immune system to help offer timely, rapid and direct protection against disease.14
Nirsevimab has been granted regulatory and other designations to facilitate expedited development by several major regulatory agencies around the world. These include Breakthrough Therapy Designation by the China Center for Drug Evaluation under the National Medical Products Administration; Breakthrough Therapy Designation from the US Food and Drug Administration; access granted to the European Medicines Agency (EMA PRIority Medicines (PRIME) scheme; and named “a medicine for prioritized development” under the Project for Drug Selection to Promote New Drug Development in Pediatrics by the Japan Agency for Medical Research and Development (AMED). In November 2022, Beyfortus was approved by the European Commission and by the UK Medicines and Healthcare products Regulatory Agency (MHRA).12-13
Pivotal clinical trials
The Phase IIb study was a randomised, placebo-controlled trial designed to measure the efficacy of nirsevimab against medically attended LRTI through 150 days postdose. Healthy preterm infants of 29–35 weeks’ gestation were randomised (2:1) to receive a single 50mg intramuscular injection of nirsevimab or placebo.8-9
The dosing regimen was recommended based on further exploration of the Phase IIb data.8 The subsequent Phase III study, MELODY applied the recommended dosing regimen.6-7
The MELODY Phase III study was a randomised, placebo-controlled trial conducted across 21 countries designed to determine efficacy of nirsevimab against medically attended LRTI due to RSV confirmed by reverse transcriptase polymerase chain reaction testing through 150 days after dosing, versus placebo, in healthy late preterm and term infants (35 weeks gestational age or greater) entering their first RSV season.6-7
MEDLEY was a Phase II/III, randomised, double-blind, Synagis-controlled trial with the primary objective of assessing safety and tolerability for nirsevimab in preterm infants and infants with congenital heart disease (CHD) and/or chronic lung disease of prematurity (CLD) eligible to receive Synagis.10-11 Between July 2019 and May 2021 approximately 918 infants entering their first RSV season were randomised to receive a single 50mg (in infants weighing <5kg) or 100mg (in infants weighing ≥5kg) intramuscular injection of nirsevimab or Synagis. Safety was assessed by monitoring the occurrence of TEAEs and TESAEs through 360 days post-dose.10-11 Serum levels of nirsevimab following dosing (on day 151) in this trial were comparable with those observed in the MELODY Phase III trial, indicating similar protection in this population to that in the healthy term and late preterm infants is likely.11 Data was published in the New England Journal of Medicine (NEJM) in March 2022.
The safety profile of nirsevimab was similar to Synagis in the MEDLEY Phase II/III and consistent with the safety profile in term and preterm infants studied in the MELODY Phase III trial.6-7,10-11 While uncommon, the most reported adverse reactions were: rash 14 days post-dose, (the majority of which were mild to moderate); pyrexia (fever) within 7 days post-dose; non-serious injection site reactions within 7 days post-dose.
The results of MELODY, MEDLEY Phase II/III and the Phase IIb trials demonstrate that nirsevimab helps protect infants during their first RSV season against RSV disease with a single dose.6-11,15-16 This broad infant population includes preterm, healthy late preterm and term infants, as well as infants with specific conditions.
These trials formed the basis of regulatory submissions which began in 2022.
Results from the first year MELODY Phase III trial
The primary endpoint of the MELODY Phase III trial was met, reducing the incidence of medically attended LRTI, such as bronchiolitis or pneumonia, caused by RSV by 74.5% (95% CI 49.6, 87.1; P<0.001) compared to placebo. Infants were randomised (2:1) to receive a single 50mg (in infants weighing <5kg) or 100mg (in infants weighing ≥5kg) intramuscular injection of nirsevimab or placebo. Between July 2019 and March 2020, 1,490 infants were randomised to receive either nirsevimab or placebo at the RSV season start.6-7 Data was published in NEJM in March 2022.
Sanofi Alliance
In March 2017, AstraZeneca and Sanofi announced an agreement to develop and commercialise nirsevimab. Under the terms of the agreement, AstraZeneca leads all development and manufacturing activities, and Sanofi leads commercialisation activities and records revenue. Under the terms of the global agreement, Sanofi made an upfront payment of €120m, has paid a development milestone of €30m and will pay up to a further €465m upon achievement of certain development and sales-related milestones. The two companies share costs and profits. Revenue from the agreement is reported as Collaboration Revenue in the Company’s financial statements.
Sobi agreement
Related, in November 2018, AstraZeneca agreed to sell US commercial rights for Synagis (palivizumab) to Swedish Orphan Biovitrum AB (publ) (Sobi) in addition to the right to participate in payments that may be received by AstraZeneca from the US profits or losses for nirsevimab. Under the agreement AstraZeneca received upfront consideration and also received non-contingent payments for nirsevimab during 2019-2021. AstraZeneca is also entitled to receive certain milestone payments for nirsevimab, including a $175m cash payment following the date on which the Biologics License Application (BLA) for nirsevimab was accepted for filing by the FDA and a $90m cash payment following the date on which BLA approval in the US occurs. AstraZeneca will continue to manufacture and supply nirsevimab globally and is entitled to an additional royalty from Sobi if profits from nirsevimab in the US exceed a pre-specified level.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Jan. 05, 2023 2:40 PM ET
By: Jonathan Block, SA News Editor
[Ad hoc announcement pursuant to Art. 53 LR] Roche’s Tecentriq plus Avastin is the first treatment combination to reduce the risk of cancer returning in people with certain types of early-stage liver cancer in a Phase III trial January 19, 2023
Basel, 19 January 2023 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the Phase III IMbrave050 study met its primary endpoint of recurrence-free survival (RFS) at the prespecified interim analysis. The study is evaluating Tecentriq® (atezolizumab) in combination with Avastin® (bevacizumab) as adjuvant treatment following surgery for people with early-stage hepatocellular carcinoma (HCC) at high risk of disease recurrence. The Tecentriq combination showed a statistically significant improvement in RFS in the intention-to-treat population of HCC patients who have an increased risk of recurrence following resection or ablation with curative intent, compared with active surveillance.
Overall survival data were immature at the time of interim analysis and follow-up will continue to the next analysis. Safety for Tecentriq and Avastin was consistent with the known safety profile of each therapeutic agent and with the underlying disease. Results from the IMbrave050 study will be discussed with health authorities, including the US Food and Drug Administration and the European Medicines Agency, and presented at an upcoming medical meeting.
“Today, more than 70% of people with early-stage HCC may have their cancer return after surgery, which is associated with poorer prognosis and shorter survival. IMbrave050 is the first Phase III study to show that a cancer immunotherapy combination reduced the risk of disease returning in people with this type of HCC,” said Levi Garraway, M.D., Ph.D., Chief Medical Officer and Head of Global Product Development. “We are excited by the clinical benefit that this adjuvant Tecentriq combination may bring to people with early liver cancer and look forward to seeing more mature data to further confirm the benefit.”
With liver cancer incidence increasing and mortality rates rising globally, effective treatments are urgently required.1-4 Roche is working in partnership with the community and leveraging its diagnostic and pharmaceutical expertise to develop solutions for patients that address unmet needs at each stage of liver cancer. IMbrave050 is part of Roche’s overall commitment to drive fundamental treatment change and improve outcomes for people living with liver cancer. In unresectable HCC (uHCC), for example, Tecentriq plus Avastin was the first treatment in over a decade to significantly improve overall survival over the existing standard of care, based on data from the IMbrave150 study. The Tecentriq combination quickly became a standard of care in uHCC and is clearly defined as a preferred front-line treatment in multiple international clinical guidelines.
Roche has an extensive development programme for Tecentriq, including multiple ongoing and planned Phase III studies across different lung, genitourinary, skin, breast, gastrointestinal, gynaecological, and head and neck cancers. This includes studies evaluating Tecentriq both alone and in combination with other medicines, as well as studies in metastatic, adjuvant and neoadjuvant settings across various tumour types.
About the IMbrave050 study
IMbrave050 is a Phase III global, multicentre, open-label, randomised study evaluating the efficacy and safety of adjuvant Tecentriq plus Avastin, compared with active surveillance, in people with HCC at high risk of recurrence (determined by the size and number of cancerous lesions and the histopathology results, if available) after surgical resection or ablation with curative intent.
The study randomised 662 people with a ratio of 1:1 to receive either Tecentriq (1,200 mg every three weeks) plus Avastin (15 mg/kg every three weeks) for a maximum of 12 months, or no intervention with active surveillance. The primary endpoint is independent review facility-assessed RFS. Key secondary endpoints include overall survival, RFS as determined by the investigator and RFS in patients with PD-L1-positive disease.
About Tecentriq
Tecentriq is a cancer immunotherapy approved for some of the most aggressive and difficult-to-treat forms of cancer. Tecentriq was the first cancer immunotherapy approved for the treatment of a certain type of early-stage non-small cell lung cancer (NSCLC), small cell lung cancer (SCLC) and HCC. Tecentriq is also approved in countries around the world, either alone or in combination with targeted therapies and/or chemotherapies, for various forms of metastatic NSCLC, certain types of metastatic urothelial cancer, PD-L1-positive metastatic triple-negative breast cancer and BRAF V600 mutation-positive advanced melanoma.
Tecentriq is a monoclonal antibody designed to bind with a protein called programmed death ligand-1 (PD-L1), which is expressed on tumour cells and tumour-infiltrating immune cells, blocking its interactions with both PD-1 and B7.1 receptors. By inhibiting PD-L1, Tecentriq may enable the activation of T-cells. Tecentriq is a cancer immunotherapy that has the potential to be used as a foundational combination partner with other immunotherapies, targeted medicines and various chemotherapies across a broad range of cancers. In addition to intravenous infusion, the formulation of Tecentriq is also being investigated as a subcutaneous injection to help address the growing burden of cancer treatment for patients and healthcare systems.
https://www.avastin.com/hcp.html
https://www.avastin.com/patient/mcrc.html
About Roche
Founded in 1896 in Basel, Switzerland, as one of the first industrial manufacturers of branded medicines, Roche has grown into the world’s largest biotechnology company and the global leader in in-vitro diagnostics. The company pursues scientific excellence to discover and develop medicines and diagnostics for improving and saving the lives of people around the world. We are a pioneer in personalised healthcare and want to further transform how healthcare is delivered to have an even greater impact. To provide the best care for each person we partner with many stakeholders and combine our strengths in Diagnostics and Pharma with data insights from the clinical practice.
In recognising our endeavor to pursue a long-term perspective in all we do, Roche has been named one of the most sustainable companies in the pharmaceuticals industry by the Dow Jones Sustainability Indices for the thirteenth consecutive year. This distinction also reflects our efforts to improve access to healthcare together with local partners in every country we work.
Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan.
For more information, please visit www.roche.com.
All trademarks used or mentioned in this release are protected by law.
Jan. 19, 2023 4:55 AM ET
Roche Holding AG (RHHBY), RHHBF
By: Ravikash, SA News Editor
Roche (OTCQX:RHHBY) (OTCQX:RHHBF) said Tecentriq in combination with Avastin met the main goal of recurrence-free survival (RFS) in a phase 3 trial in patients with a type of liver cancer.
Roche Holding AG (RHHBY), RHHBF
JANUARY 6, 2023 • NEWS RELEASE
Accelerated Approval is based on Phase 2 data showing a reduction in amyloid-beta plaques in early AD patients treated with LEQEMBI™
Treatment with LEQEMBI should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in clinical trials
TOKYO and CAMBRIDGE, Mass., Jan. 6, 2023 /PRNewswire/ -- Eisai Co., Ltd. (Headquarters: Tokyo, CEO: Haruo Naito, "Eisai") and Biogen Inc. (Nasdaq: BIIB, Corporate headquarters: Cambridge, Massachusetts, CEO: Christopher A. Viehbacher, "Biogen") announced today that under the Accelerated Approval Pathway the U.S. Food and Drug Administration (FDA) has approved lecanemab-irmb (Brand Name in the U.S.: LEQEMBI™) 100 mg/mL injection for intravenous use, a humanized immunoglobulin gamma 1 (IgG1) monoclonal antibody directed against aggregated soluble ("protofibril")* and insoluble forms of amyloid beta (Aβ) for the treatment of Alzheimer's disease (AD). The approval is based on Phase 2 data that demonstrated that LEQEMBI reduced the accumulation of Aβ plaque in the brain, a defining feature of AD. Using the recently published data from the large global confirmatory Phase 3 clinical trial, Clarity AD, Eisai will work quickly to file a Supplemental Biologics License Application (sBLA) to the FDA for approval under the traditional pathway.
INDICATION
LEQEMBI is indicated for the treatment of Alzheimer's disease. Treatment with LEQEMBI should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in clinical trials. There are no safety or effectiveness data on initiating treatment at earlier or later stages of the disease than were studied. This indication is approved under accelerated approval based on reduction in amyloid beta plaques observed in patients treated with LEQEMBI. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial.
INDICATION
LEQEMBI is indicated for the treatment of Alzheimer's disease. Treatment with LEQEMBI should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in clinical trials. There are no safety or effectiveness data on initiating treatment at earlier or later stages of the disease than were studied. This indication is approved under accelerated approval based on reduction in amyloid beta plaques observed in patients treated with LEQEMBI. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial.
Please see full Prescribing Information.
1. About LEQEMBITM (lecanemab-irmb)
LEQEMBITM (lecanemab-irmb) is a humanized immunoglobulin gamma 1 (IgG1) monoclonal antibody directed against aggregated soluble (protofibril) and insoluble forms of amyloid-beta (Aβ). LEQEMBI is indicated for the treatment of Alzheimer's disease (AD) in the U.S. This indication is approved under accelerated approval based on reduction in Aβ plaques observed in patients treated with LEQEMBI. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial.
LEQEMBI is the result of a strategic research alliance between Eisai and BioArctic. Eisai has been initiated submission of data for BLA to the National Medical Products Administration (NMPA) of China in December 2022. Eisai plans to file for marketing authorization applications of lecanemab in Japan and Europe by the end of Eisai's FY2022.
Since July 2020 Eisai's Phase 3 clinical study (AHEAD 3-45) for individuals with preclinical AD, meaning they are clinically normal and have intermediate or elevated levels of amyloid in their brains, is ongoing. AHEAD 3-45 is conducted as a public-private partnership between the Alzheimer's Clinical Trial Consortium that provides the infrastructure for academic clinical trials in AD and related dementias in the U.S, funded by the National Institute on Aging, part of the National Institutes of Health, Eisai and Biogen.
Since January 2022, the Tau NexGen clinical study for Dominantly Inherited AD (DIAD), that is conducted by Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU), led by Washington University School of Medicine in St. Louis, is ongoing. Eisai has completed a LEQEMBI subcutaneous bioavailability study, and subcutaneous dosing is currently being evaluated in the Clarity AD (Study 301) OLE.
2. About Amyloid-Related Imaging Abnormalities (ARIA)
ARIA is an important adverse event of amyloid-lowering therapies that is critical to monitor and manage during treatment. ARIA is most commonly seen as temporary swelling/effusion (ARIA-E) in areas of the brain that usually resolves over time. Some people may also have small spots of bleeding in or on the surface of the brain (ARIA-H) with the swelling. Although most people with ARIA-E do not have symptoms, some people may have symptoms such as headache, confusion, dizziness, vision changes and nausea.
3. About the Collaboration between Eisai and Biogen for AD
Eisai and Biogen have been collaborating on the joint development and commercialization of AD treatments since 2014. Eisai serves as the lead of LEQEMBI development and regulatory submissions globally with both companies co-commercializing and co-promoting the product and Eisai having final decision-making authority.
4. About the Collaboration between Eisai and BioArctic for AD
Since 2005, Eisai and BioArctic have had a long-term collaboration regarding the development and commercialization of AD treatments. Eisai obtained the global rights to study, develop, manufacture and market LEQEMBI for the treatment of AD pursuant to an agreement with BioArctic in December 2007. The development and commercialization agreement on the antibody LEQEMBI back-up was signed in May 2015.
5. About Eisai Co., Ltd.
Eisai's Corporate Concept is "to give first thought to patients and people in the daily living domain, and to increase the benefits that health care provides." Under this Concept (also known as human health care (hhc) Concept), we aim to effectively achieve social good in the form of relieving anxiety over health and reducing health disparities. With a global network of R&D facilities, manufacturing sites and marketing subsidiaries, we strive to create and deliver innovative products to target diseases with high unmet medical needs, with a particular focus in our strategic areas of Neurology and Oncology.
In addition, we demonstrate our commitment to the elimination of neglected tropical diseases (NTDs), which is a target (3.3) of the United Nations Sustainable Development Goals (SDGs), by working on various activities together with global partners.
For more information about Eisai, please visit www.eisai.com (for global headquarters: Eisai Co., Ltd.), and connect with us on Twitter @Eisai_SDGs.
6. About Biogen
As pioneers in neuroscience, Biogen discovers, develops and delivers worldwide innovative therapies for people living with serious neurological diseases as well as related therapeutic adjacencies. One of the world's first global biotechnology companies, Biogen was founded in 1978 by Charles Weissmann, Heinz Schaller, Sir Kenneth Murray, and Nobel Prize winners Walter Gilbert and Phillip Sharp. Today, Biogen has a leading portfolio of medicines to treat multiple sclerosis, has introduced the first approved treatment for spinal muscular atrophy, and developed the first approved treatment to address a defining pathology of Alzheimer's disease. Biogen is also commercializing biosimilars and focusing on advancing one of the industry's most diversified pipelines in neuroscience that will transform the standard of care for patients in several areas of high unmet need.
The company routinely posts information that may be important to investors on its website at www.biogen.com. To learn more, please visit www.biogen.com and Follow Biogen on social media – Twitter, LinkedIn, Facebook, YouTube.
View original content to download multimedia:https://www.prnewswire.com/news-releases/fda-approves-leqembi-lecanemab-irmb-under-the-accelerated-approval-pathway-for-the-treatment-of-alzheimers-disease-301715691.html
SOURCE Eisai Inc.
Jan. 06, 2023 2:17 PM ET
Eisai Co., Ltd. (ESALF), ESALY, BIIB
By: Jonathan Block, SA News Editor23 Comments
tang90246
NOTE: Updated with information about Leqembi availability and price.
The US FDA on Friday approved Biogen (NASDAQ:BIIB) and Eisai's (OTCPK:ESALY) Alzheimer's therapy lecanemab, which will be marketed as Leqembi.
Eisai Co., Ltd. (ESALF), ESALY, BIIB
Jan. 09, 2023 7:01 AM ET
MONTRÉAL, Jan. 9, 2023 /CNW Telbec/ - AbbVie (NYSE: ABBV) announced today that Health Canada has approved QULIPTA (atogepant) for the prevention of episodic migraine (< 15 migraine days per month) in adults.1 QULIPTA, the first and only oral calcitonin gene-related peptide (CGRP) receptor antagonist (gepant) preventive treatment, marks the latest innovation in AbbVie's migraine portfolio to support Canadians impacted by migraine.

"Migraine is one of the leading causes of disability in Canada and impacts a person's ability to function and perform their daily routines," says Dr. Elizabeth Leroux, Founder and Chair, Migraine Canada. "People with migraine face incredible challenges managing a debilitating neurological condition that others cannot see or truly understand. Migraine Canada welcomes the approval of QULIPTA for Canadians who will now have an opportunity to experience a new treatment that will help prevent and manage episodic migraine appropriately and take control of their migraine attacks before they even start."
The approval is supported by data from a robust clinical program evaluating the efficacy, safety and tolerability of QULIPTA in nearly 2,000 patients who experienced 4 to 14 migraine days per month. QULIPTA demonstrated statistically significant, clinically meaningful, rapid and continuous reductions in mean monthly migraine days among adults with episodic migraine compared to placebo across the 12-week treatment period with significant reductions seen in weeks 1-4. 1
"AbbVie is committed to bringing new innovative treatments to address the needs of Canadians living with migraine," says Tracey Ramsay, Vice President and General Manager, AbbVie Canada. "With the approval of QULIPTA for the preventive treatment of episodic migraine, AbbVie has expanded on our support to those living with migraine given the legacy of BOTOX® for the treatment of chronic migraine, and recently approved UBRELVY® for acute treatment of migraine. AbbVie is proud to partner with the migraine community to advance treatment and care for people living with this neurological disease."
About Migraine
Migraine is a complex neurological disease with recurrent attacks that lasts 4-72 hours. It can be defined by symptoms such as moderate to severe pain intensity, nausea, vomiting, photophobia and phonophobia.2 An estimated 2.7 million Canadians are reported diagnosed with migraine, however the number of people living with migraine is much higher.3 Episodic migraine is characterized as having less than 15 headache days per month, while 15 headache days or more per month, is considered chronic. 4
About QULIPTA (atogepant)
QULIPTA (atogepant) is the first and only oral calcitonin gene-related peptide (CGRP) receptor antagonist (gepant) specifically developed for the preventive treatment of episodic migraine. QULIPTA is an orally administered, small molecule, selective calcitonin gene-related peptide (CGRP) receptor antagonist that blocks the binding of the CGRP to its receptor. CGRP is a neuropeptide that may play a role in migraine pathophysiology.1
For important safety information, please consult the QULIPTA™ Product Monograph at www.abbvie.ca.
About AbbVie
AbbVie's mission is to discover and deliver innovative medicines that solve serious health issues today and address the medical challenges of tomorrow. We strive to have a remarkable impact on people's lives across several key therapeutic areas: immunology, oncology, neuroscience, eye care, virology, women's health and gastroenterology, in addition to products and services across its Allergan Aesthetics portfolio. For more information about AbbVie, please visit us at www.abbvie.ca. Follow AbbVie Canada on Twitter, on Instagram or find us on LinkedIn.
For more information on AbbVie's complete migraine portfolio, please visit www.abbvie.ca.
SOURCE AbbVie Canada
January 03, 2023
– Application Based on Statistically Significant and Clinically Meaningful Overall Survival and Progression-Free Survival Results from the Phase 3 TROPiCS-02 Study –
– Supplemental Biologics License Application Already Under Priority Review by the U.S. FDA –
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the European Medicines Agency (EMA) has validated a Type II variation Marketing Authorization Application (MAA) for Trodelvy® (sacituzumab govitecan-hziy) for the treatment of adult patients with unresectable or metastatic hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative (IHC 0, IHC 1+ or IHC 2+/ISH–) breast cancer who have received endocrine-based therapy and at least two additional systemic therapies in the metastatic setting.
“At Gilead Oncology our ambition is to transform care for people with cancer,” said Bill Grossman, MD, PhD, Senior Vice President, Therapeutic Area Head, Gilead Oncology. “Trodelvy is already moving us towards this ambition and changing the standard of care in second-line metastatic triple-negative breast cancer across the EU. The validation of our Marketing Authorization Application in pre-treated HR+/HER2- metastatic breast cancer marks an important step forward to potentially making Trodelvy available to even more patients with severely limited treatment options.”
This Marketing Authorization Application is based on data from the registrational Phase 3 TROPiCS-02 study, which met its primary endpoint of progression-free survival (PFS) and key secondary endpoint of overall survival (OS) versus comparator chemotherapies (treatment of physician’s choice (TPC) of chemotherapy). PFS data were published in the Journal of Clinical Oncology,and OS data were recently presented at ESMO Congress 2022.
The safety profile for Trodelvy in TROPiCS-02 was consistent with prior studies, and no new safety signals were identified in this population.
In October 2022, the U.S. Food and Drug Administration (FDA) accepted for priority review the supplemental Biologics License Application (sBLA) for Trodelvy for the treatment of adult patients with unresectable locally advanced or metastatic HR+/HER2-negative (IHC 0, IHC 1+ or IHC 2+/ISH–) breast cancer who have received endocrine-based therapy and at least two additional systemic therapies in the metastatic setting. The Prescription Drug User Fee Act (PDUFA) target action date is currently set for February 2023.
Trodelvy has not been approved by any regulatory agency for the treatment of HR+/HER2- metastatic breast cancer. Its safety and efficacy have not been established for this indication.
Trodelvy has a Boxed Warning for severe or life-threatening neutropenia and severe diarrhea; please see below for additional Important Safety Information.
About the TROPiCS-02 Study
The TROPiCS-02 study is a global, multicenter, open-label, Phase 3 study, randomized 1:1 to evaluate Trodelvy versus physicians’ choice of chemotherapy (eribulin, capecitabine, gemcitabine, or vinorelbine) in 543 patients with HR+/HER2- metastatic breast cancer who were previously treated with endocrine therapy, CDK4/6 inhibitors and two to four lines of chemotherapy for metastatic disease. The primary endpoint is progression-free survival per Response Evaluation Criteria in Solid Tumors (RECIST 1.1) as assessed by blinded independent central review (BICR) for participants treated with Trodelvy compared to those treated with chemotherapy. Secondary endpoints include overall survival, overall response rate, clinical benefit rate and duration of response, as well as assessment of safety and tolerability and quality of life measures. In the study, HER2 negativity was defined per American Society of Clinical Oncology (ASCO) and the College of American Pathologists (CAP) criteria as immunohistochemistry (IHC) score of 0, IHC 1+ or IHC 2+ with a negative in-situ hybridization (ISH) test. More information about TROPiCS-02 is available at https://clinicaltrials.gov/ct2/show/NCT03901339.
About Trodelvy
Trodelvy® (sacituzumab govitecan-hziy) is a first-in-class Trop-2 directed antibody-drug conjugate. Trop-2 is a cell surface antigen highly expressed in multiple tumor types, including in more than 90% of breast and bladder cancers. Trodelvy is intentionally designed with a proprietary hydrolyzable linker attached to SN-38, a topoisomerase I inhibitor payload. This unique combination delivers potent activity to both Trop-2 expressing cells and the microenvironment.
Trodelvy is approved in more than 40 countries, with multiple additional regulatory reviews underway worldwide, for the treatment of adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (TNBC) who have received two or more prior systemic therapies, at least one of them for metastatic disease. Trodelvy is also approved in the U.S. under the accelerated approval pathway for the treatment of adult patients with locally advanced or metastatic urothelial cancer (UC) who have previously received a platinum-containing chemotherapy and either programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor.
Trodelvy is also being developed for potential investigational use in other TNBC and metastatic UC populations, as well as a range of tumor types where Trop-2 is highly expressed, including HR+/HER2- metastatic breast cancer, metastatic non-small cell lung cancer (NSCLC), metastatic small cell lung cancer (SCLC), head and neck cancer, and endometrial cancer.
U.S. Indications for Trodelvy
In the United States, Trodelvy is indicated for the treatment of:
View source version on businesswire.com: https://www.businesswire.com/news/home/20230102005076/en/
Source: Gilead Sciences, Inc.
Jan. 03, 2023 10:38 AM ET
By: Anuron Mitra, SA News Editor
Gilead Sciences (NASDAQ:GILD) on Tuesday said Europe's drug regulator had approved a change to its marketing authorization application for breast cancer treatment Trodelvy.
https://www.trodelvy.com/bladder-cancer
January 02, 2023 04:45 AM Eastern Standard Time
SOMERSET, N.J.--(BUSINESS WIRE)--Legend Biotech Corporation (NASDAQ: LEGN) (Legend Biotech), a global biotechnology company developing, manufacturing and commercializing novel therapies to treat life-threatening diseases, announced today that China’s National Medical Products Administration (NMPA) has formally accepted its New Drug Application (NDA) for ciltacabtagene autoleucel (cilta-cel).
This submission is based on data from the confirmatory Phase 2 clinical study CARTIFAN-1 (NCT03758417) conducted in China, which evaluated the efficacy and safety of cilta-cel in adult patients with relapsed or refractory multiple myeloma who have received 3 or more prior lines of therapy, including a proteasome inhibitor and immunomodulatory drug.
Saijuan Chen, M.D., hematologist and molecular geneticist, Academician of the Chinese Academy of Engineering, and principal investigator of the CARTIFAN-1 clinical trial, said: “Incidence and mortality rates of multiple myeloma have recently increased in China, and the disease remains incurable. As a result, there is a huge unmet medical need for new treatment options. The data from the CARTIFAN-1 study showed that cilta-cel provided deep and durable responses in patients with relapsed or refractory multiple myeloma. We hope this drug becomes available to eligible patients as soon as possible.”
Ying Huang, Ph.D., Chief Executive Officer of Legend Biotech, said: “Meeting medical needs and serving patients around the world has always been the goal of Legend Biotech’s innovative research and development. Cilta-cel has been approved for marketing in the United States and Japan and has received conditional marketing authorization in Europe. We look forward to the possibility of providing a new treatment option for appropriate patients with relapsed and refractory multiple myeloma in China.”
###
About Ciltacabtagene autoleucel (cilta-cel)
Ciltacabtagene autoleucel is a BCMA-directed, genetically modified autologous T-cell immunotherapy, which involves reprogramming a patient’s own T-cells with a transgene encoding a chimeric antigen receptor (CAR) that identifies and eliminates cells that express BCMA. BCMA is primarily expressed on the surface of malignant multiple myeloma B-lineage cells, as well as late-stage B-cells and plasma cells. The cilta-cel CAR protein features two BCMA-targeting single domain antibodies designed to confer high avidity against human BCMA. Upon binding to BCMA-expressing cells, the CAR promotes T-cell activation, expansion, and elimination of target cells.1
In December 2017, Legend Biotech Corporation entered into an exclusive worldwide license and collaboration agreement with Janssen Biotech, Inc. (Janssen) to develop and commercialize cilta-cel.
In February 2022, cilta-cel was approved by the U.S. Food and Drug Administration (FDA) under the brand name CARVYKTI® for the treatment of adults with relapsed or refractory multiple myeloma.2 In May 2022, the European Commission (EC) granted conditional marketing authorization of CARVYKTI® for the treatment of adults with relapsed and refractory multiple myeloma.3 In September 2022, Japan’s Ministry of Health, Labour and Welfare (MHLW) approved CARVYKTI®.4 Cilta-cel was granted Breakthrough Therapy Designation in the U.S. in December 2019 and in China in August 2020. In addition, cilta-cel received a PRIority MEdicines (PRIME) designation from the European Commission in April 2019. Cilta-cel also received Orphan Drug Designation from the U.S. FDA in February 2019, from the European Commission in February 2020, and from the Pharmaceuticals and Medicinal Devices Agency (PMDA) in Japan in June 2020. In March 2022, the European Medicines Agency’s Committee for Orphan Medicinal Products recommended by consensus that the orphan designation for cilta-cel be maintained on the basis of clinical data demonstrating improved and sustained complete response rates following treatment.5
About CARTIFAN-1
CARTIFAN-1 (NCT03758417)6 is a Phase 2 open-label, confirmatory trial evaluating the efficacy and safety of cilta-cel in Chinese patients with relapsed/refractory multiple myeloma who have received at least three prior lines of treatments including a proteasome inhibitor and immunomodulatory drug. The primary endpoint is overall response rate.
CARVYKTI® U.S. Important Safety Information
INDICATIONS AND USAGE
CARVYKTI® (ciltacabtagene autoleucel) is a B-cell maturation antigen (BCMA)-directed genetically modified autologous T cell immunotherapy indicated for the treatment of adult patients with relapsed or refractory multiple myeloma, after four or more prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.
Please read full Prescribing Information including Boxed Warning for CARVYKTI®.
About Legend Biotech
Legend Biotech is a global biotechnology company dedicated to treating, and one day curing, life-threatening diseases. Headquartered in Somerset, New Jersey, we are developing advanced cell therapies across a diverse array of technology platforms, including autologous and allogeneic chimeric antigen receptor T-cell and natural killer (NK) cell-based immunotherapy. From our three R&D sites around the world, we apply these innovative technologies to pursue the discovery of cutting-edge therapeutics for patients worldwide.
Learn more at www.legendbiotech.com and follow us on Twitter and LinkedIn.
Jan. 03, 2023 4:24 AM ET
Legend Biotech Corporation (LEGN), JNJ
By: Ravikash, SA News Editor
Dec 30, 2022 9:00 AM
Submission seeks marketing authorization for first-line treatment of unresectable or metastatic hepatocellular carcinoma
CAMBRIDGE, Mass. & BASEL, Switzerland & BEIJING--(BUSINESS WIRE)-- BeiGene (NASDAQ: BGNE; HKEX: 06160; SSE: 688235), a global biotechnology company announced that the Center for Drug Evaluation (CDE) of the China National Medical Products Administration (NMPA) has accepted a supplemental biologics license application (sBLA) for tislelizumab in patients with first-line unresectable or metastatic hepatocellular carcinoma (HCC).
Hepatocellular carcinoma is the most common type of primary liver cancer worldwide and is associated with a very poor prognosis.1 New cases and deaths due to HCC in China account for half of the global numbers and the 5-year survival rate for patients with HCC in China is only 14%.2
“While the incidence of HCC is increasing in China, the treatment landscape has not advanced accordingly; survival benefits with newer treatments are modest and multi-kinase inhibitors have sub-optimal tolerability,” said Lai Wang, Ph.D., Global Head of R&D at BeiGene. “We believe the evidence from our rigorously conducted global clinical development program for tislelizumab in HCC support the efficacy and favorable tolerability profile and look forward to working with NMPA on this submission and bringing a new treatment option to patients with HCC in China.”
The sBLA is supported by data from the RATIONALE 301 clinical trial (NCT03412773) that enrolled 674 patients from research centers across Asia, Europe, and the United States. RATIONALE 301 results were presented as a late-breaking oral presentation at the 2022 European Society for Medical Oncology (ESMO) Congress in Paris.
Tislelizumab was approved by the China NMPA as a treatment for nine indications, including conditional approval ‘for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with at least one systemic therapy’. Additional tislelizumab’s sBLAs under review at CDE include: combination with chemotherapy as a first-line treatment for patients with advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumors express PD-L1; combination with chemotherapy as first-line treatment in patients with unresectable locally advanced, recurrent or metastatic esophageal squamous cell carcinoma. Tislelizumab is not approved for use outside of China.
About Tislelizumab
Tislelizumab is a humanized IgG4 anti-PD-1 monoclonal antibody specifically designed to minimize binding to Fc-gamma (Fcγ) receptors on macrophages, helping to aid the body’s immune cells to detect and fight tumors. In pre-clinical studies, binding to Fcγ receptors on macrophages has been shown to compromise the anti-tumor activity of PD-1 antibodies through activation of antibody-dependent macrophage-mediated killing of T effector cells.
Tislelizumab is the first investigational medicine from BeiGene’s immuno-oncology biologics program and is being evaluated in solid tumor and hematologic malignancies, as monotherapy and in combination.
The global tislelizumab clinical development program includes more than 11,500 subjects enrolled to-date in 21 registration-enabling trials, from more than 30 countries and regions.
https://www.beigene.com/our-science-and-medicines/tislelizumab/
About RATIONALE 301
RATIONALE 301 (NCT03412773) is a global, Phase 3, randomized, open-label study of tislelizumab compared with sorafenib as a first-line treatment in adult patients with unresectable HCC. The primary endpoint of the study is non-inferiority of Overall Survival between the two treatment groups. The key secondary endpoint is Overall Response Rate, as assessed by Blinded Independent Review Committee (BIRC) per RECIST v1.1. Other secondary endpoints include other efficacy assessments such as Progression Free Survival, Duration of Response, and Time to Progression per BIRC, as well as measures of health-related quality of life, and safety and tolerability.
About BeiGene
BeiGene is a global biotechnology company that is developing and commercializing innovative and affordable oncology medicines to improve treatment outcomes and access for far more patients worldwide. With a broad portfolio, we are expediting development of our diverse pipeline of novel therapeutics through our internal capabilities and collaborations. We are committed to radically improving access to medicines for far more patients who need them. Our growing global team of more than 9,000 colleagues spans five continents, with administrative offices in Cambridge, U.S., & Basel, Switzerland & Beijing, China. To learn more about BeiGene, please visit www.beigene.com and follow us on Twitter at @BeiGeneGlobal.
View source version on businesswire.com: https://www.businesswire.com/news/home/20221230005150/en/
Source: BeiGene
Dec. 30, 2022 9:37 AM ET
By: Anuron Mitra, SA News Editor
PUBLISHED28 December 2022 28 December 2022 07:00 GMT
AstraZeneca’s immunotherapies Imfinzi (durvalumab) and Imjudo (tremelimumab) have been approved in Japan for the treatment of three cancer types: advanced liver, biliary tract and lung.
The approvals authorise Imfinzi in combination with Imjudo for the treatment of adult patients with unresectable hepatocellular carcinoma (HCC) and for the treatment of adult patients with unresectable, advanced or recurrent non-small cell lung cancer (NSCLC) in combination with chemotherapy. Imfinzi was also authorised for the treatment of adult patients with unresectable HCC as monotherapy and for the treatment of adult patients with curatively unresectable biliary tract cancer (BTC) in combination with chemotherapy (gemcitabine plus cisplatin).
The concurrent approvals by the Japanese Ministry of Health, Labour, and Welfare are based on positive results from the HIMALAYA and TOPAZ-1 Phase III trials, each published in the New England Journal of Medicine Evidence and the POSEIDON Phase III trial, published in the Journal of Clinical Oncology.
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “Japan has one of the highest rates of diagnosis for liver and biliary tract cancers in the world, and lung cancer remains the country’s leading cause of cancer death. With these approvals for Imfinzi and Imjudo, patients in Japan can now be treated with novel immunotherapy-based treatment regimens that have demonstrated significant survival benefits across three complex cancers with poor prognoses.”
Imfinzi and Imjudo approved in liver cancer
The approval of Imfinzi in combination with Imjudo for the treatment of unresectable HCC brings the first dual immunotherapy treatment regimen to patients in Japan. The approval is based on results from the HIMALAYA Phase III trial, in which a single dose of the anti-CTLA-4 antibody Imjudo 300mg added to the anti-PD-L1 antibody Imfinzi 1500mg followed by Imfinzi every four weeks (STRIDE regimen: Single Tremelimumab Regular Interval Durvalumab) significantly reduced the risk of death versus sorafenib. The addition of Imjudo to Imfinzi did not increase severe liver toxicity, and no bleeding risk was observed.
HIMALAYA also served as the basis for the approval of Imfinzi monotherapy in the same disease setting. In HIMALAYA, Imfinzi demonstrated non-inferior overall survival (OS) compared to sorafenib, and an improved tolerability profile versus sorafenib.
The safety profiles of the combination of Imjudo added to Imfinzi and for Imfinzi alone were consistent with the known profiles of each medicine, and no new safety signals were identified.
Liver cancer is the fifth-leading cause of cancer death in Japan and the sixth most commonly diagnosed cancer worldwide.1,2 HCC is the most common type of liver cancer with 80% of cases occurring in the Asia-Pacific region.3,4 In Japan, approximately 45,000 people are diagnosed with HCC and 32,000 die of the disease each year.5,6
Imfinzi and Imjudo approved in NSCLC
The approval of Imfinzi and Imjudo plus chemotherapy for the treatment of unresectable, advanced or recurrent NSCLC is based on results from the POSEIDON Phase III trial, which showed a limited course of five cycles of the anti-CTLA-4 antibody Imjudo added to Imfinzi plus four cycles of platinum-based chemotherapy significantly reduced the risk of death versus a range of chemotherapy options.
The safety profile for Imjudo plus Imfinzi and chemotherapy was consistent with the known profiles of each medicine, and no new safety signals were identified.
In Japan, lung cancer is the most commonly diagnosed cancer, with more than 138,000 patients diagnosed in 2020.1 The prognosis for patients with metastatic NSCLC in Japan is particularly poor, as less than 20% will live beyond three years after diagnosis without treatment.7
Regulatory applications for Imfinzi and Imjudo are currently under review in the EU and several other countries based on the HIMALAYA, TOPAZ-1 and POSEIDON results.
Imfinzi plus chemotherapy approved in biliary tract cancer
The approval of Imfinzi plus chemotherapy brings the first immunotherapy regimen to patients with curatively unresectable BTC in Japan after more than a decade of limited innovation. The approval is based on results from an interim analysis of the TOPAZ-1 Phase III trial, which showed that Imfinzi plus chemotherapy significantly reduced the risk of death compared to chemotherapy alone. Imfinzi plus chemotherapy was generally well tolerated and did not increase the discontinuation rate due to adverse events compared to chemotherapy alone.
BTC is a group of rare and aggressive cancers that occur in the bile ducts and gallbladder.8,9 In 2021, approximately 23,300 people in Japan were diagnosed with BTC, which is the sixth-leading cause of cancer-related deaths for women and seventh-leading cause of cancer-related death for men in Japan.7 Patients face a poor prognosis, with approximately 19% to 31% of patients with BTC surviving five years.7
Notes
HIMALAYA
HIMALAYA was a randomised, open-label, multicentre, global Phase III trial of Imfinzi monotherapy and the STRIDE regimen comprising a single priming dose of Imjudo 300mg added to Imfinzi 1500mg followed by Imfinzi every four weeks versus sorafenib, a standard-of-care multi-kinase inhibitor.
The trial included a total of 1,324 patients with unresectable, advanced HCC who had not been treated with prior systemic therapy and were not eligible for locoregional therapy (treatment localised to the liver and surrounding tissue).
The trial was conducted in 181 centres across 16 countries, including in the US, Canada, Europe, South America and Asia. The primary endpoint was OS for the combination versus sorafenib and key secondary endpoints included OS for Imfinzi versus sorafenib, objective response rate (ORR) and progression-free survival (PFS) for the combination and for Imfinzi alone.
In HIMALAYA, the STRIDE regimen reduced the risk of death by 22% versus sorafenib (hazard ratio [HR] 0.78; 95% confidence interval [CI], 0.66-0.92; p= 0.0035). An estimated 31% of patients treated with the combination were still alive after three years, with 20% of patients treated with sorafenib still alive at the same duration of follow-up. The safety profiles of the combination of Imjudo added to Imfinzi and for Imfinzi alone were consistent with the known profiles of each medicine, and no new safety signals were identified.
POSEIDON
The POSEIDON trial was a randomised, open-label, multi-centre, global, Phase III trial of Imfinzi plus platinum-based chemotherapy, or Imfinzi, Imjudo and chemotherapy, versus chemotherapy alone in the 1st-line treatment of 1,013 patients with metastatic NSCLC. The trial population included patients with either non-squamous or squamous disease, and the full range of PD-L1 expression levels. POSEIDON excluded patients with certain epidermal growth factor receptor (EGFR) mutations or anaplastic lymphoma kinase (ALK) fusions.
In the experimental arms, patients were treated with a flat dose of either Imfinzi (1,500mg) or Imfinzi plus Imjudo (75mg) with up to four cycles of chemotherapy every three weeks before either Imfinzi maintenance once every four weeks or Imfinzi and a fifth dose of Imjudo given at week 16. In comparison, the control arm allowed up to six cycles of chemotherapy. Pemetrexed maintenance treatment was allowed in all arms in patients with non-squamous disease if given during the induction phase. Nearly all patients with non-squamous disease (95.5%) had pemetrexed and platinum, while the majority of patients with squamous disease receiving chemotherapy (88.3%) received gemcitabine and platinum.
Primary endpoints included PFS and OS for the Imfinzi plus chemotherapy arm. Key secondary endpoints included PFS and OS in the Imfinzi plus Imjudo and chemotherapy arm. As both PFS endpoints were met for Imfinzi plus chemotherapy and Imfinzi, Imjudo and chemotherapy, the prespecified statistical analysis plan allowed for testing OS in the Imfinzi plus Imjudo and chemotherapy arm. The trial was conducted in more than 150 centres across 18 countries, including the US, Europe, South America, Asia and South Africa.
Patients treated with a limited course of five cycles of the anti-CTLA-4 antibody Imjudo added to Imfinzi plus four cycles of platinum-based chemotherapy experienced a 23% reduction in the risk of death versus a range of chemotherapy options (HR 0.77; 95% CI, 0.65-0.92; p=0.00304). An estimated 33% of patients were alive at two years versus 22% for chemotherapy. This treatment combination also reduced the risk of disease progression or death by 28% compared to chemotherapy alone (HR 0.72; 95% CI, 0.60-0.86; p=0.00031).
Updated results after approximately four years of follow-up presented at the European Society for Medical Oncology Congress 2022 demonstrated sustained survival benefit, improving overall survival (OS) by 25% compared to chemotherapy alone (HR 0.75; 95% CI, 0.63-0.88). An estimated 25% of patients treated with the combination were alive at three years versus 13.6% for those treated with chemotherapy alone. The safety profile for Imjudo plus Imfinzi and chemotherapy was consistent with the known profiles of each medicine, and no new safety signals were identified.
TOPAZ-1
TOPAZ-1 was a randomised, double-blind, placebo controlled, multicentre, global Phase III trial of Imfinzi in combination with chemotherapy (gemcitabine plus cisplatin) versus placebo in combination with chemotherapy as a 1st-line treatment in 685 patients with unresectable advanced or metastatic BTC including intrahepatic and extrahepatic cholangiocarcinoma, and gallbladder cancer. Patients with ampullary carcinoma were excluded.
The primary endpoint was OS and key secondary endpoints included PFS, ORR and safety. The trial was conducted in 105 centres across 17 countries including in the US, Europe, South America and several countries in Asia including South Korea, Thailand, Japan and China.
At the interim analysis, Imfinzi plus chemotherapy reduced the risk of death by 20% versus chemotherapy alone (based on a hazard ratio [HR] of 0.80; 95% confidence interval [CI] 0.66-0.97; p=0.021). Updated results from TOPAZ-1 after an additional 6.5 months of follow-up showed a 24% reduction in the risk of death versus chemotherapy alone (HR 0.76; 95% CI, 0.64-0.91), with more than two times as many patients treated with Imfinzi plus chemotherapy estimated to be alive at two years versus chemotherapy alone (23.6% versus 11.5%). Updated median overall survival (OS) was 12.9 months versus 11.3 with chemotherapy. Imfinzi plus chemotherapy was generally well tolerated, with no new safety signals observed, and did not increase the discontinuation rate due to adverse events (AEs) compared to chemotherapy alone.
Imfinzi
Imfinzi (durvalumab) is a human monoclonal antibody that binds to the PD-L1 protein and blocks the interaction of PD-L1 with the PD-1 and CD80 proteins, countering the tumour's immune-evading tactics and releasing the inhibition of immune responses.
Imfinzi is the only approved immunotherapy in the curative-intent setting of unresectable, Stage III NSCLC in patients whose disease has not progressed after chemoradiotherapy and is the global standard of care in this setting based on the PACIFIC Phase III trial.
Imfinzi is also approved in the US, EU, Japan, China and many other countries around the world for the treatment of extensive-stage small cell lung cancer (ES-SCLC) based on the CASPIAN Phase III trial. In 2021, updated results from the CASPIAN trial showed Imfinzi plus chemotherapy tripled patient survival at three years versus chemotherapy alone.
Imfinzi is also approved in combination with Imjudo and chemotherapy in metastatic non-small cell lung cancer in the US and Japan; in combination with chemotherapy in locally advanced or metastatic BTC in the US, Japan and several other countries; in combination with Imjudo in unresectable HCC in the US and Japan; as monotherapy in unresectable HCC in Japan; and in previously treated patients with advanced bladder cancer in several countries.
Since the first approval in May 2017, more than 100,000 patients have been treated with Imfinzi.
As part of a broad development programme, Imfinzi is being tested as a single treatment and in combinations with other anti-cancer treatments for patients with SCLC, NSCLC, bladder cancer, several gastrointestinal (GI) cancers, ovarian cancer, endometrial cancer and other solid tumours.
Imjudo
Imjudo (tremelimumab) is a human monoclonal antibody that targets the activity of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). Imjudo blocks the activity of CTLA-4, contributing to T-cell activation, priming the immune response to cancer and fostering cancer cell death.
In addition to its approved indications in liver and lung cancers, Imjudo is being tested in combination with Imfinzi across multiple tumour types including locoregional HCC (EMERALD-3), SCLC (ADRIATIC) and bladder cancer (VOLGA and NILE).
AstraZeneca in GI cancers
AstraZeneca has a broad development programme for the treatment of GI cancers across several medicines and a variety of tumour types and stages of disease. In 2020, GI cancers collectively represented approximately 5.1 million new cancer cases leading to approximately 3.6 million deaths.10
Within this programme, the Company is committed to improving outcomes in gastric, liver, BTC, oesophageal, pancreatic, and colorectal cancers.
Imfinzi is approved in the US and several other countries in combination with chemotherapy (gemcitabine plus cisplatin) for advanced biliary tract cancer and in the US and Japan in combination with Imjudo in unresectable HCC. Imfinzi is also approved as a monotherapy in unresectable HCC in Japan. Imfinzi is being assessed in combinations, including with Imjudo, in liver, oesophageal and gastric cancers in an extensive development programme spanning early to late-stage disease across settings.
Enhertu (trastuzumab deruxtecan), a HER2-directed antibody drug conjugate, is approved in the US and several other countries for HER2-positive advanced gastric cancer and is being assessed in colorectal cancer. Enhertu is jointly developed and commercialised by AstraZeneca and Daiichi Sankyo.
Lynparza (olaparib), a first-in-class PARP inhibitor, is approved in the US and several other countries for the treatment of BRCA-mutated metastatic pancreatic cancer. Lynparza is developed and commercialised in collaboration with MSD (Merck & Co., Inc. inside the US and Canada).
AstraZeneca in lung cancer
AstraZeneca is working to bring patients with lung cancer closer to cure through the detection and treatment of early-stage disease, while also pushing the boundaries of science to improve outcomes in the resistant and advanced settings. By defining new therapeutic targets and investigating innovative approaches, the Company aims to match medicines to the patients who can benefit most.
The Company's comprehensive portfolio includes leading lung cancer medicines and the next wave of innovations, including Tagrisso (osimertinib) and Iressa (gefitinib); Imfinzi and Imjudo; Enhertu and datopotamab deruxtecan in collaboration with Daiichi Sankyo; Orpathys (savolitinib) in collaboration with HUTCHMED; as well as a pipeline of potential new medicines and combinations across diverse mechanisms of action.
AstraZeneca is a founding member of the Lung Ambition Alliance, a global coalition working to accelerate innovation and deliver meaningful improvements for people with lung cancer, including and beyond treatment.
AstraZeneca in immuno-oncology (IO)
AstraZeneca is a pioneer in introducing the concept of immunotherapy into dedicated clinical areas of high unmet medical need. The Company has a comprehensive and diverse IO portfolio and pipeline anchored in immunotherapies designed to overcome evasion of the anti-tumour immune response and stimulate the body’s immune system to attack tumours.
AstraZeneca aims to reimagine cancer care and help transform outcomes for patients with Imfinzi as a single treatment and in combination with Imjudo as well as other novel immunotherapies and modalities. The Company is also exploring next-generation immunotherapies like bispecific antibodies and therapeutics that harness different aspects of immunity to target cancer.
AstraZeneca is boldly pursuing an innovative clinical strategy to bring IO-based therapies that deliver long-term survival to new settings across a wide range of cancer types. With an extensive clinical programme, the Company also champions the use of IO treatment in earlier disease stages, where there is the greatest potential for cure.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Dec. 28, 2022 4:40 AM ET
AstraZeneca PLC (AZN) By: Ravikash, SA News Editor
Japan's Ministry of Health, Labour, and Welfare approved AstraZeneca's (NASDAQ:AZN) immunotherapies Imfinzi and Imjudo to treat certain types of liver and lung cancers and Imfinzi to treat biliary tract and liver cancers.
PUBLISHED28 December 2022
AstraZeneca’s Calquence (acalabrutinib), a selective Bruton’s tyrosine kinase (BTK) inhibitor, has been approved in Japan for the treatment of adult patients with treatment-naïve chronic lymphocytic leukaemia (CLL) (including small lymphocytic lymphoma [SLL]). Calquence was previously approved in Japan for the treatment of adults with relapsed or refractory CLL.
The approval by the Japanese Ministry of Health, Labour and Welfare (MHLW) was based on positive results from two clinical trials, including the ELEVATE-TN Phase III trial in adults with treatment-naïve CLL. This trial showed that Calquence combined with obinutuzumab or as monotherapy demonstrated a significantly improved progression-free survival (PFS) when compared with the chemotherapy-based combination of chlorambucil and obinutuzumab.1,2 Data from the interim analysis of ELEVATE-TN was published in The Lancet in 2020.2 Additionally, a Phase I trial in treatment-naïve Japanese patients with CLL was also submitted to MHLW supporting the approval, with the trial showing an overall response rate of 88.9% (95% CI: 63.2, 98.8%) for Calquence alone and 100% (95% CI: 66.4, 100%) for Calquence combined with obinutuzumab.
CLL is the most prevalent type of adult leukaemia across the globe but is considered a rare disease in Japan and East Asia, with fewer than one person newly diagnosed per 100,000 persons per year across Japan.3-5
Koji Izutsu, MD, PhD, Department Head, Department of Hematology, National Cancer Center Hospital, Tokyo, Japan, said: “Results from ELEVATE-TN and our local Japanese trial confirm that Calquence provides a significant improvement in progression-free survival compared with chemotherapy-based combination of chlorambucil and obinutuzumab for patients with treatment-naïve chronic lymphocytic leukaemia. Today’s approval marks great progress for physicians and patients in Japan, as they can now be treated with Calquence earlier in their treatment journey.”
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “The approval of Calquence in Japan for those with treatment-naïve chronic lymphocytic leukaemia now offers more patients a next-generation Bruton’s tyrosine kinase inhibitor that has proven longer-term efficacy and tolerability compared to standards of care. With this approval, people living with chronic lymphocytic leukaemia in Japan can now potentially benefit from our medicine in an earlier setting.”
Updated results of the ELEVATE-TN Phase III trial after a median follow-up of approximately five years were presented earlier this year. These results showed that Calquence maintained a statistically significant PFS benefit versus chlorambucil plus obinutuzumab, and a safety and tolerability profile consistent with the known profile for Calquence. At a median follow-up of 58.2 months, Calquence plus obinutuzumab reduced the risk of disease progression or death by 89% (based on a hazard ratio [HR] of 0.11, 95% confidence interval [CI] 0.07-0.16) and as a monotherapy by 79% (based on a HR of 0.21, 95% CI 0.15-0.30), compared with chlorambucil plus obinutuzumab.1
Calquence is approved for the treatment of CLL and SLL in the US and is approved for the treatment of CLL in the EU and in several other countries worldwide in the treatment-naïve and relapsed or refractory settings. Calquence is also approved in the US and several other countries for the treatment of adult patients with mantle cell lymphoma (MCL) who have received at least one prior therapy. Calquence is not currently approved for the treatment of MCL in Japan or the EU.
Notes
CLL
CLL is the most prevalent type of leukaemia in adults, with over 100,000 new cases globally in 2019, but is considered a rare disease in Japan and East Asia, with fewer than one person newly diagnosed per 100,000 persons per year across Japan.3-5 Although some people with CLL may not experience any symptoms at diagnosis, others may experience symptoms, such as weakness, fatigue, weight loss, chills, fever, night sweats, swollen lymph nodes and abdominal pain.6
In CLL, there is an accumulation of abnormal lymphocytes within the bone marrow. As the number of abnormal cells increases, there is less room within the marrow for the production of normal white blood cells, red blood cells and platelets.7 This could result in anaemia, infection and bleeding. B-cell receptor signalling through BTK is one of the essential growth pathways for CLL.
ELEVATE-TN
ELEVATE-TN (ACE-CL-007) is a randomised, multicentre, open-label Phase III trial evaluating the safety and efficacy of Calquence alone or in combination with obinutuzumab versus chlorambucil in combination with obinutuzumab in previously untreated patients with CLL. In the trial, 535 patients were randomised (1:1:1) into three arms. Patients in the first arm received chlorambucil in combination with obinutuzumab. Patients in the second arm received Calquence (100mg twice daily until disease progression) in combination with obinutuzumab. Patients in the third arm received Calquence monotherapy (100mg twice daily until disease progression).8
The primary endpoint was PFS in the Calquence and obinutuzumab arm compared to the chlorambucil and obinutuzumab arm, assessed by an independent review committee (IRC), and a key secondary endpoint was IRC-assessed PFS in the Calquence monotherapy arm compared to the chlorambucil and obinutuzumab arm. Other secondary endpoints included overall response rate, time to next treatment, overall survival and investigator assessed PFS. After interim analysis, assessments were by investigator only.8
Initial results from the ELEVATE-TN Phase III trial were presented in December 2019 at the American Society of Hematology Annual Meeting and Exhibition. The trial met its primary endpoint (IRC-assessed PFS with Calquence plus obinutuzumab versus chlorambucil plus obinutuzumab) at the data cut-off for the interim analysis after a median follow-up of 28.3 months. The findings, along with previously reported data from the ASCEND Phase III trial in relapsed or refractory CLL, supported the approvals of Calquence by the US FDA and the Australian Therapeutic Goods Administration for the treatment of adult patients with CLL or SLL and by the European Medicines Agency (EMA) and Health Canada for CLL.
Calquence
Calquence (acalabrutinib) is a next-generation, selective inhibitor of BTK. Calquence binds covalently to BTK, thereby inhibiting its activity.9 In B cells, BTK signalling results in activation of pathways necessary for B-cell proliferation, trafficking, chemotaxis and adhesion.
Calquence is approved for the treatment of CLL and SLL in the US, approved for CLL in the EU and many other countries worldwide and approved in Japan for relapsed or refractory CLL and SLL.
In the US and several other countries, Calquence is also approved for the treatment of adult patients with MCL who have received at least one prior therapy. The US MCL indication is approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials. Calquence is not currently approved for the treatment of MCL in Europe or Japan.
As part of an extensive clinical development programme, AstraZeneca and Acerta Pharma are currently evaluating Calquence in more than 20 company-sponsored clinical trials. Calquence is being evaluated for the treatment of multiple B-cell blood cancers, including CLL, MCL, diffuse large B-cell lymphoma, Waldenström’s macroglobulinaemia, marginal zone lymphoma and other haematologic malignancies.
https://www.calquencehcp.com/mcl.html
AstraZeneca in haematology
AstraZeneca is pushing the boundaries of science to redefine care in haematology. We have expanded our commitment to patients with haematologic conditions, not only in oncology but also in rare diseases with the acquisition of Alexion, allowing us to reach more patients with high unmet needs. By applying our deep understanding of blood cancers, leveraging our strength in solid tumour oncology and delivering on Alexion’s pioneering legacy in complement science to provide innovative medicines for rare diseases, we are pursuing the end-to-end development of novel therapies designed to target underlying drivers of disease.
By targeting haematologic conditions with high unmet medical needs, we aim to deliver innovative medicines and approaches to improve patient outcomes. Our goal is to help transform the lives of patients living with malignant, rare and other related haematologic diseases, shaped by insights from patients, caregivers and physicians to have the most meaningful impact.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company’s focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Dec. 28, 2022 4:57 AM ET
By: Ravikash, SA News Editor
PUBLISHED21 December 2022 21 December 2022 07:00 GMT
AstraZeneca’s Imfinzi (durvalumab) has been approved in the European Union (EU) for the 1st-line treatment of adult patients with unresectable or metastatic biliary tract cancer (BTC) in combination with chemotherapy (gemcitabine plus cisplatin).
The approval by the European Commission was based on the primary results from the TOPAZ-1 Phase III trial published in the New England Journal of Medicine Evidence, and on the updated results presented at the European Society for Medical Oncology Congress 2022. The approval follows the recommendation by The Committee for Medicinal Products for Human Use of the European Medicines Agency in November 2022.
At the interim analysis, Imfinzi plus chemotherapy reduced the risk of death by 20% versus chemotherapy alone (based on a hazard ratio [HR] of 0.80; 95% confidence interval [CI] 0.66-0.97; p=0.021). Updated results from TOPAZ-1 after an additional 6.5 months of follow-up showed a 24% reduction in the risk of death versus chemotherapy alone (HR 0.76; 95% CI, 0.64-0.91), with more than two times as many patients treated with Imfinzi plus chemotherapy estimated to be alive at two years versus chemotherapy alone (23.6% versus 11.5%). Updated median overall survival (OS) was 12.9 months versus 11.3 with chemotherapy.
BTC is a group of rare and aggressive cancers that occur in the bile ducts (cholangiocarcinoma) and gallbladder.1,2 There are approximately 211,000 new patients diagnosed with gallbladder and biliary tract cancer each year, and about 40,000 of these occur across Europe.3 These patients have a poor prognosis, with approximately 5% to 15% of patients with BTC surviving five years.4
Juan W. Valle, MD, Professor of Medical Oncology at the University of Manchester and The Christie NHS Foundation Trust, UK, and a lead investigator in the TOPAZ-1 Phase III trial, said: “Today’s approval marks an important shift in the treatment of this aggressive and often overlooked disease and a significant improvement compared to standard of care for these patients. After waiting over a decade for new therapeutic options, biliary tract cancer patients in the EU will now have the opportunity to benefit from an immunotherapy-based treatment for the first time.”
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “With this approval, Imfinzi plus chemotherapy becomes the only immunotherapy-based treatment option available to patients in the EU with advanced biliary tract cancer. This approval underscores our commitment to transform survival outcomes while addressing the high unmet need for new and improved treatments for patients with hepatobiliary cancers.”
Imfinzi plus chemotherapy was generally well tolerated, with no new safety signals observed, and did not increase the discontinuation rate due to adverse events (AEs) compared to chemotherapy alone. Grade 3 or 4 treatment-related AEs were experienced by 60.9% of patients treated with Imfinzi plus chemotherapy, and by 63.5% of patients treated with chemotherapy alone.
Imfinzi plus chemotherapy is approved in the US and other countries for the treatment of adults with locally advanced or metastatic BTC. Regulatory applications are also currently under review in Japan and several other countries based on the TOPAZ-1 results.
TOPAZ-1
TOPAZ-1 was a randomised, double-blind, placebo controlled, multicentre, global Phase III trial of Imfinzi in combination with chemotherapy (gemcitabine plus cisplatin) versus placebo in combination with chemotherapy as a 1st-line treatment in 685 patients with unresectable advanced or metastatic BTC including intrahepatic and extrahepatic cholangiocarcinoma, and gallbladder cancer. Patients with ampullary carcinoma were excluded.
The primary endpoint was overall survival and key secondary endpoints included progression-free survival, objective response rate and safety. The trial was conducted in 105 centres across 17 countries including in the US, Europe, South America and several countries in Asia including South Korea, Thailand, Japan and China.
Imfinzi
Imfinzi (durvalumab) is a human monoclonal antibody that binds to the PD-L1 protein and blocks the interaction of PD-L1 with the PD-1 and CD80 proteins, countering the tumour’s immune-evading tactics and releasing the inhibition of immune responses.
Imfinzi is the only approved immunotherapy in unresectable or metastatic BTC, unresectable hepatocellular carcinoma [in combination with Imjudo (tremelimumab)], and the curative-intent setting of unresectable, Stage III non-small cell lung cancer (NSCLC) in patients whose disease has not progressed after chemoradiotherapy. It is the global standard of care in this setting based on the PACIFIC Phase III trial.
Imfinzi is also approved in the US, EU, Japan, China and many other countries around the world for the treatment of extensive-stage small cell lung cancer (ES-SCLC) based on the CASPIAN Phase III trial. In 2021, updated results from the CASPIAN trial showed Imfinzi plus chemotherapy tripled patient survival at three years versus chemotherapy alone.
Imfinzi is also approved in the US for the treatment of adult patients with Stage IV (metastatic) NSCLC in combination with Imjudo and chemotherapy.
Imfinzi is also approved for previously treated patients with advanced bladder cancer in several countries.
As part of a broad development programme, Imfinzi is being tested as a single treatment and in combinations with other anti-cancer treatments for patients with SCLC, NSCLC, bladder cancer, several GI cancers, ovarian cancer, endometrial cancer, and other solid tumours.
AstraZeneca in GI cancers
AstraZeneca has a broad development programme for the treatment of GI cancers across several medicines and a variety of tumour types and stages of disease. In 2020, GI cancers collectively represented approximately 5.1 million new cancer cases leading to approximately 3.6 million deaths.7
Within this programme, the Company is committed to improving outcomes in gastric, liver, biliary tract, oesophageal, pancreatic, and colorectal cancers.
Imfinzi is approved in the US in combination with chemotherapy (gemcitabine plus cisplatin) for advanced BTC and in combination with Imjudo in unresectable hepatocellular carcinoma. Imfinzi is being assessed in combinations, including with Imjudo in liver, oesophageal and gastric cancers in an extensive development programme spanning early to late-stage disease across settings.
Enhertu (trastuzumab deruxtecan), a HER2-directed antibody drug conjugate, is approved in HER2-positive advanced gastric cancer and is being assessed in colorectal cancer. Enhertu is jointly developed and commercialised by AstraZeneca and Daiichi Sankyo.
Lynparza (olaparib), a first-in-class PARP inhibitor, is approved in BRCA-mutated metastatic pancreatic cancer. Lynparza is developed and commercialised in collaboration with MSD (Merck & Co., Inc. inside the US and Canada).
AstraZeneca in immuno-oncology (IO)
AstraZeneca is a pioneer in introducing the concept of immunotherapy into dedicated clinical areas of high unmet medical need. The Company has a comprehensive and diverse IO portfolio and pipeline anchored in immunotherapies designed to overcome evasion of the anti-tumour immune response and stimulate the body’s immune system to attack tumours.
AstraZeneca aims to reimagine cancer care and help transform outcomes for patients with Imfinzi as a single treatment and in combination with Imjudo as well as other novel immunotherapies and modalities. The Company is also exploring next-generation immunotherapies like bispecific antibodies and therapeutics that harness different aspects of immunity to target cancer.
AstraZeneca is boldly pursuing an innovative clinical strategy to bring IO-based therapies that deliver long-term survival to new settings across a wide range of cancer types. With an extensive clinical programme, the Company also champions the use of IO treatment in earlier disease stages, where there is the greatest potential for cure.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Dec. 21, 2022 4:50 AM ET
By: Ravikash, SA News Editor
December 22, 2022
– Based on Landmark ZUMA-7 Study, Patients with LBCL Treated with Yescarta in Second-Line Achieved Four-Fold Greater Improvement in Event-Free Survival of 8.3 Months vs. Two Months for Standard of Care (SOC)
– In ZUMA-7, Patients Treated with Yescarta Were 2.5 Times More Likely than SOC to be Alive at Two Years Without Disease Progression or Need for Additional Treatments –
SANTA MONICA, Calif. & TOKYO--(BUSINESS WIRE)-- Kite Pharma, Inc., a Gilead Company, (hereafter, Kite) (NASDAQ: GILD) and Daiichi Sankyo Co., Ltd. (hereafter, Daiichi Sankyo) (TSE: 4568) today jointly announced that the Japan Ministry of Health, Labour and Welfare (MHLW) has approved Yescarta (axicabtagene ciloleucel), a chimeric antigen receptor (CAR) T-cell therapy, for the initial treatment of patients with relapsed/refractory large B-cell lymphoma ( R/R LBCL): diffuse large B-cell lymphoma, primary mediastinal large B-cell lymphoma, transformed follicular lymphoma, and high-grade B-cell lymphoma. Yescarta should be used only in patients who have not received prior transfusion of CAR T-cells targeted at CD19 antigen.
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20221222005384/en/
The Standard of Care (SOC) to treat LBCL patients has historically been a multi-step process that starts with chemoimmunotherapy, followed by high-dose chemotherapy (HDT) and then ends with a stem cell transplant (ASCT). Although approximately 60% of newly diagnosed LBCL patients will respond to the initial treatment with chemotherapy, 40% will relapse or will not respond and need second-line treatment. Yescarta is now approved for the initial treatment of R/R LBCL patients in Japan.
“We are very proud of this additional Yescarta approval in Japan,” said Christi Shaw, CEO of Kite. “As Japan has the second-largest number of people diagnosed with non-Hodgkin lymphoma globally, this approval marks an important step in bringing this innovative therapy earlier to more patients.”
“We are glad to have achieved this important milestone in order to provide an innovative treatment option to more patients with large B-cell lymphoma in Japan,” said Wataru Takasaki, PhD, Executive Officer, Head of R&D Division in Japan, Daiichi Sankyo. “It also further demonstrates the expertise of our Daiichi Sankyo R&D organization as we had successfully expanded the indication for this CAR-T modality.”
The approval of Yescarta for the initial treatment of R/R LBCL patients in Japan is based on clinical data, including the results of the global pivotal trial conducted by Kite (ZUMA-7). ZUMA-7 is the largest and longest trial of a CAR T-cell therapy versus SOC in this patient population. Results from the study demonstrated that at a median follow-up of two years, Yescarta-treated patients had a four-fold greater improvement in the primary endpoint of event-free survival (EFS; hazard ratio 0.40; 95% CI: 0.31-0.51, P<0.0001) over the current SOC (8.3 months vs 2.0 months). Additionally, Yescarta demonstrated a 2.5 fold increase in patients who were alive at two years without disease progression or need for additional cancer treatment vs SOC (41% v 16%).
This indication was first approved by the U.S. FDA in April 2022 followed by the European Commission in October 2022.
On December 7, 2022, Daiichi Sankyo and Kite jointly announced that the Marketing Authorization for Yescarta will be transferred to Gilead Sciences K.K., the Japan subsidiary of Gilead Sciences, Inc., in 2023. The Kite Cell Therapy Business Unit at Gilead Sciences K.K. will manage the sales and promotion activities of the product in Japan after the Marketing Authorization transfer.
Kite’s manufacturing facility in El Segundo, California, U.S., has been approved by Japanese regulatory authorities to commence manufacturing Yescarta for the Japan market in 2023.
About Zuma-7
ZUMA-7 is an ongoing, randomized, open-label, global, multicenter (US, Australia, Canada, Europe, Israel) Phase 3 study of 359 patients at 77 centers, evaluating the safety and efficacy of a single-infusion of Yescarta versus current SOC for second-line therapy (platinum-based salvage combination chemotherapy regimen followed by high-dose chemotherapy and autologous stem cell transplant in those who respond to salvage chemotherapy) in adult patients with relapsed or refractory LBCL within 12 months of first-line therapy. The primary endpoint is event free survival (EFS). Key secondary endpoints include objective response rate (ORR) and overall survival (OS). Additional secondary endpoints include patient reported outcomes and safety.
About YESCARTA®
Yescarta (axicabtagene ciloleucel) is a CAR T-cell therapy directed against CD19 (a cell membrane protein), which harnesses a patient’s own immune system to fight cancer. Axicabtagene ciloleucel is made by removing a patient’s T cells from their blood and engineering them in the lab to express chimeric antigen receptors so that they can recognize and destroy cancer cells when they are infused back to the patient’s body. The CAR T-cell therapy is manufactured specifically for each patient and administered only once. Axicabtagene ciloleucel received Orphan Drug Designation from the Japan MHLW in 2018 for the treatment of diffuse large B-cell lymphoma, primary mediastinal B-cell lymphoma, transformed follicular lymphoma and high-grade B-cell lymphoma. Yescarta was approved in Japan for the treatment of patients with relapsed or refractory large B-cell lymphomas, a type of non-Hodgkin lymphoma, in January 2021.
https://www.yescarta.com/lbcl/
Please see full U.S. Prescribing Information, including BOXED WARNING and Medication Guide.
Yescarta® is a CD19-directed genetically modified autologous T cell immunotherapy indicated for the treatment of:
Limitations of Use: Yescarta is not indicated for the treatment of patients with primary central nervous system lymphoma. (1.1)
For the full European Prescribing Information, please visit: : https://www.ema.europa.eu/en/medicines/human/EPAR/yescarta.
About Daiichi Sankyo
Daiichi Sankyo is dedicated to creating new modalities and innovative medicines by leveraging our world-class science and technology for our purpose “to contribute to the enrichment of quality of life around the world.” In addition to our current portfolio of medicines for cancer and cardiovascular disease, Daiichi Sankyo is primarily focused on developing novel therapies for people with cancer as well as other diseases with high unmet medical needs. With more than 100 years of scientific expertise and a presence in more than 20 countries, Daiichi Sankyo and its 16,000 employees around the world draw upon a rich legacy of innovation to realize our 2030 Vision to become an “Innovative Global Healthcare Company Contributing to the Sustainable Development of Society.” For more information, please visit: www.daiichisankyo.com.
About Kite
Kite, a Gilead Company, is a global biopharmaceutical company based in Santa Monica, California, focused on cell therapy to treat and potentially cure cancer. As the global cell therapy leader, Kite has treated more patients with CAR T-cell therapy than any other company. Kite has the largest in-house cell therapy manufacturing network in the world, spanning process development, vector manufacturing, clinical trial production and commercial product manufacturing. For more information on Kite, please visit www.kitepharma.com.
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer. Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California. Gilead Sciences acquired Kite in 2017.
U.S. Prescribing Information for Yescarta including BOXED WARNING, is available at www.kitepharma.com and www.gilead.com .
Kite, the Kite logo, Yescarta, and GILEAD are trademarks of Gilead Sciences, Inc. or its related companies.
View source version on businesswire.com: https://www.businesswire.com/news/home/20221222005384/en/
Source: Gilead Sciences, Inc.
Dec. 22, 2022 4:59 PM ET
Gilead Sciences, Inc. (GILD), DSKYF, DSNKY
By: Anuron Mitra, SA News Editor
December 23, 2022 at 8:30 AM EST Back
TARRYTOWN, N.Y., Dec. 23, 2022 /PRNewswire/ -- Regeneron Pharmaceuticals (NASDAQ: REGN) today announced that the Ministry of Health, Labor and Welfare (MHLW) in Japan has granted marketing and manufacturing authorization for Libtayo® (cemiplimab) as monotherapy to treat patients with advanced or recurrent cervical cancer whose disease progressed after chemotherapy.
"In recent years, the incidence of cervical cancer has increased in Japan, with the prognosis for advanced stage disease remaining poor and treatment options limited," said Israel Lowy, M.D., Ph.D., Senior Vice President, Translational and Clinical Sciences, Oncology at Regeneron. "With this approval, Libtayo becomes the first single-agent immunotherapy approved in Japan for the treatment of advanced cervical cancer."
The MHLW approval is based on positive data from the international, multicenter Phase 3 EMPOWER-Cervical 1 trial, conducted in collaboration with NRG Oncology-Japan, the GOG Foundation, Inc., and the European Network for Gynaecological Oncological Trial (ENGOT) groups. The trial evaluated Libtayo compared to an investigator's choice of chemotherapy and enrolled 608 patients across 14 countries, including Japan, irrespective of PD-L1 expression status or histology. In March 2021, the trial was stopped early based on the highly significant effect of Libtayo on overall survival among squamous cell carcinoma patients following a unanimous recommendation by the Independent Data Monitoring Committee.
"With Libtayo, Japan now has an approved treatment option that has demonstrated significant survival benefits in this cervical cancer patient population compared to chemotherapy irrespective of PD-L1 expression – a milestone no other PD-1 inhibitor has achieved in a Phase 3 trial," said Prof. Kosei Hasegawa, Gynecologic Oncology, Saitama Medical University International medical Center, a trial investigator and a member of NRG Oncology-Japan. "We are proud to have contributed to bringing this new treatment option to women living with this difficult-to-treat cancer in Japan.
Cervical cancer is the fourth leading cause of cancer death in women worldwide and is most frequently diagnosed in women between the ages of 35 and 44. Almost all cases of cervical cancer are caused by human papillomavirus (HPV) infection. It is estimated that approximately 600,000 new cases of cervical cancer are diagnosed and 350,000 deaths from cervical cancer occur worldwide each year.
In November 2022, Libtayo was approved by the European Commission for the treatment of adult patients with recurrent or metastatic cervical cancer and disease progression on or after platinum-based chemotherapy.
About Libtayo
Libtayo is a fully human monoclonal antibody targeting the immune checkpoint receptor PD-1 on T cells and was invented using Regeneron's proprietary VelocImmune® technology. By binding to PD-1, Libtayo has been shown to block cancer cells from using the PD-1 pathway to suppress T-cell activation. In the U.S. and other countries Libtayo is indicated in certain patients with advanced basal cell carcinoma (BCC), advanced CSCC and advanced non-small cell lung cancer (NSCLC), as well as in advanced cervical cancer in Japan, the European Union, Canada and Brazil.
Libtayo was jointly developed by Sanofi and Regeneron under a global collaboration agreement. As of July 1, 2022, Regeneron is responsible for the development and marketing of Libtayo globally. In Japan, Libtayo is currently marketed by Sanofi on Regeneron's behalf over the course of a defined transition period.
The extensive clinical program for Libtayo is focused on difficult-to-treat cancers. Libtayo is currently being investigated in trials as a monotherapy, as well as in combination with either conventional or novel therapeutic approaches for other solid tumors and blood cancers. These potential uses are investigational, and their safety and efficacy have not been evaluated by any regulatory authority.
U.S. FDA-approved Indications
Libtayo is a prescription medicine used to treat:
-- People with a type of skin cancer called CSCC that has spread or cannot be cured by surgery or radiation.
-- People with a type of skin cancer called BCC:
-- Adults with a type of lung cancer called NSCLC:
It is not known if Libtayo is safe and effective in children.
Please see full Prescribing Information, including Medication Guide.
About Regeneron in Oncology
At Regeneron, we're applying more than three decades of scientific innovation with the goal of developing paradigm-changing therapies for patients with cancer. Our oncology portfolio is built around two foundational approaches – our approved PD-1 inhibitor Libtayo and investigational bispecific antibodies – which are being evaluated both as monotherapies and in combination with emerging therapeutic modalities. Together, they provide us with unique combinatorial flexibility to develop potentially synergistic treatments for a wide range of solid tumors and blood cancers.
If you are interested in learning more about our clinical trials, please contact us (clinicaltrials@regeneron.com or 844-734-6643) or visit our clinical trials website.
About Regeneron's VelocImmune Technology
Regeneron's VelocImmune technology utilizes a proprietary genetically engineered mouse platform endowed with a genetically humanized immune system to produce optimized fully human antibodies. When Regeneron's co-Founder, President and Chief Scientific Officer George D. Yancopoulos was a graduate student with his mentor Frederick W. Alt in 1985, they were the first to envision making such a genetically humanized mouse, and Regeneron has spent decades inventing and developing VelocImmune and related VelociSuite® technologies. Dr. Yancopoulos and his team have used VelocImmune technology to create approximately one in five of all original, FDA-approved or authorized fully human monoclonal antibodies. This includes REGEN-COV® (casirivimab and imdevimab), Dupixent® (dupilumab), Libtayo, Praluent® (alirocumab), Kevzara® (sarilumab), Evkeeza® (evinacumab-dgnb) and Inmazeb™ (atoltivimab, maftivimab and odesivimab-ebgn).
About Regeneron
Regeneron is a leading biotechnology company that invents, develops and commercializes life-transforming medicines for people with serious diseases. Founded and led for nearly 35 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to nine FDA-approved treatments and numerous product candidates in development, almost all of which were homegrown in our laboratories. Our medicines and pipeline are designed to help patients with eye diseases, allergic and inflammatory diseases, cancer, cardiovascular and metabolic diseases, pain, hematologic conditions, infectious diseases and rare diseases.
Regeneron is accelerating and improving the traditional drug development process through our proprietary VelociSuite technologies, such as VelocImmune, which uses unique genetically humanized mice to produce optimized fully human antibodies and bispecific antibodies, and through ambitious research initiatives such as the Regeneron Genetics Center®, which is conducting one of the largest genetics sequencing efforts in the world.
For more information, please visit www.Regeneron.com or follow @Regeneron on Twitter.
View original content:https://www.prnewswire.com/news-releases/libtayo-cemiplimab-approved-in-japan-for-advanced-or-recurrent-cervical-cancer-301709636.html
SOURCE Regeneron Pharmaceuticals, Inc.
Dec. 23, 2022 9:01 AM ET
Regeneron Pharmaceuticals, Inc. (REGN)
By: Jonathan Block, SA News Editor
12/22/2022
– Sunlenca is the First and Only Approved Capsid Inhibitor-Based HIV Treatment Option –
– New Drug Application Approval Based on High Rates of Sustained Virologic Suppression in the CAPELLA Trial –
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced that Sunlenca® (lenacapavir), in combination with other antiretroviral(s) (ARV), has been granted approval by the U.S. Food and Drug Administration (FDA) for the treatment of HIV-1 infection in heavily treatment-experienced (HTE) adults with multi-drug resistant (MDR) HIV-1 infection. Sunlenca has a multi-stage mechanism of action distinguishable from other currently approved classes of antiviral agents and no known cross resistance exhibited in vitro to other existing drug classes. Sunlenca offers a new, twice-yearly treatment option for adults with HIV that is not adequately controlled by their current treatment regimen.
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20221221005541/en/

Sunlenca Packaging (Photo: Business Wire)
“An effective antiretroviral regimen can be devised for most people living with the virus; however, some people living with HIV no longer have durable viral suppression due to resistance to multiple classes of antiretroviral therapies,” said Sorana Segal-Maurer, MD, Director of the Dr. James J. Rahal Jr. Division of Infectious Diseases at NewYork-Presbyterian Queens, Professor of Clinical Medicine at Weill Cornell Medicine and the Site Principal Investigator for the CAPELLA trial. “The availability of new classes of antiretroviral drugs is critical for heavily treatment-experienced people with multi-drug resistant HIV. Following today’s decision from the FDA, lenacapavir helps to fill a critical unmet need for people with complex prior treatment histories and offers physicians a long-awaited twice-yearly option for these patients who otherwise have limited therapy choices."
Despite the significant advances in ARV therapy, there remain numerous critical and pressing unmet needs for people living with HIV. This is particularly true for individuals who are heavily treatment experienced – which account for an estimated 2% of adults living with HIV who are on treatment globally –- and are unable to maintain virologic suppression due to resistance, intolerance or safety considerations. This type of complexity further increases the chance of treatment failure, underscoring the need for new treatment options that are active against resistant variants of the virus with a novel mechanism of action.
Lenacapavir is a breakthrough innovation with the potential to be a preferred and versatile foundational long-acting agent due to its therapeutic potency and a range of dosing frequencies and routes of administration. Lenacapavir is being developed as a foundation for Gilead’s future HIV therapies with the goal of offering several long-acting options that help address individual needs and preferences that may help optimize outcomes and reduce burden of care. Lenacapavir is being studied in multiple ongoing early and late-stage development programs and has the potential to offer a diverse set of person-centric options for treatment and prevention that could uniquely fit into the lives of people living with HIV and people who would benefit from pre-exposure prophylaxis (PrEP). The use of lenacapavir for HIV prevention is investigational and the safety and efficacy of lenacapavir for this use have not been established.
“This news is an important milestone in the work to help end the HIV epidemic as Sunlenca is now the only FDA-approved twice-yearly treatment for people with multi-drug resistant HIV,” said Daniel O’Day, Chairman and Chief Executive Officer, Gilead Sciences. “Gilead scientists have developed a unique and potent antiretroviral medicine in Sunlenca with the potential for flexible dosing options. Our goal is to deliver multiple long-acting options for treatment and prevention that are tailored to the needs of people living with HIV and people who could benefit from PrEP medicines.”
The FDA approval for Sunlenca is supported by data from the Phase 2/3 CAPELLA trial, which evaluated lenacapavir in combination with an optimized background regimen in people with multi-drug resistant HIV-1 who are heavily treatment experienced. CAPELLA participants had undergone previous treatment with a median of nine antiretroviral medications. In this patient population with significant unmet medical need, 83% (n=30/36) of participants randomly allocated to receive lenacapavir in addition to an optimized background regimen achieved an undetectable viral load (<50 copies/mL) at Week 52. Additionally, these participants achieved a mean increase in CD4 count of 82 cells/µL. These data were presented at the 29th Conference on Retroviruses and Opportunistic Infections (virtual CROI 2022).
Sunlenca was reviewed and approved as a medication with FDA Breakthrough Therapy Designation, which is intended to expedite the development and review of new drugs which may demonstrate substantial improvement over available therapy. In May 2019, the FDA granted Breakthrough Therapy Designation for the development of lenacapavir for the treatment of HIV-1 infection in heavily treatment-experienced patients with multi-drug resistance in combination with other antiretroviral drugs.
Lenacapavir, alone or in combination, is not approved by any regulatory authority outside of the United States, United Kingdom, Canada or the European Union for any use. The European Marketing Authorization applies to all 27 member states of the European Union, as well as Norway, Iceland and Liechtenstein.
Additional regulatory filings and decisions by regulatory authorities are anticipated to continue in 2023.
The use of lenacapavir for HIV prevention is investigational and the safety and efficacy of lenacapavir for this use have not been established. Gilead is studying the safety and efficacy of lenacapavir for HIV prevention in multiple ongoing clinical studies.
Please see below for the U.S. Indication and Important Safety Information for Sunlenca.
There is no cure for HIV or AIDS.
Sorana Segal-Maurer, MD, is a paid consultant for Gilead Sciences, Inc.
About CAPELLA (NCT04150068)
CAPELLA is a Phase 2/3, double-blinded, placebo-controlled global multicenter study designed to evaluate the antiviral activity of lenacapavir administered every six months as a subcutaneous injection in heavily treatment-experienced people with multi-drug resistant HIV-1 infection. CAPELLA includes men and women with HIV-1 and is being conducted at research centers in North America, Europe and Asia.
In CAPELLA, 36 participants with multi-class HIV-1 drug resistance and a detectable viral load while on a failing regimen were randomly allocated to receive oral lenacapavir or placebo in a 2:1 ratio for 14 days, in addition to continuing their failing regimen (functional monotherapy). An additional 36 participants were enrolled in a separate treatment cohort. Both cohorts are part of the ongoing maintenance period of the study evaluating the safety and efficacy of subcutaneous lenacapavir administered every six months in combination with an optimized background regimen. The primary endpoint was the proportion of participants randomly allocated to receive lenacapavir or placebo for 14 days, in addition to continuing their failing regimen, achieving ≥0.5 log10 copies/mL reduction from baseline in HIV-1 RNA at the end of the functional monotherapy period. The study found that 88% of participants receiving lenacapavir (n=21/24) experienced at least a 0.5 log10 reduction in HIV-1 viral load by the end of 14 days of functional monotherapy as compared with 17% of those receiving placebo (n=2/12).
Following the 14-day functional monotherapy period, participants randomly allocated to receive lenacapavir or placebo, in addition to continuing their failing regimen, started open-label lenacapavir and an optimized background regimen, while those enrolled in a separate treatment cohort received open-label lenacapavir and an optimized background regimen on Day 1. This ongoing maintenance period is evaluating the additional study endpoints of safety and efficacy of subcutaneous lenacapavir administered every six months in combination with an optimized background regimen.
The New England Journal of Medicine published the primary outcome results of the CAPELLA study in its May 11, 2022 issue - Capsid Inhibition with Lenacapavir in Multidrug-Resistant HIV-1 Infection.
For further information, please see https://clinicaltrials.gov/ct2/show/NCT04150068.
About Sunlenca ®
Sunlenca (300 mg tablet and 463.5 mg/1.5 mL injection) is a first-in-class, long-acting HIV capsid inhibitor approved in the United States, the United Kingdom, Canada and the European Union, for the treatment of HIV infection, in combination with other antiretroviral(s), in people with multi-drug resistant HIV who are heavily treatment-experienced. Sunlenca tablets are approved for oral loading during initiation of Sunlenca treatment, prior to or at the time of the first long-acting lenacapavir injection depending on initiation option. The multi-stage mechanism of action of Sunlenca is distinguishable from other currently approved classes of antiviral agents and is designed to provide a new avenue for the development of a long-acting treatment option for individuals with multi-drug resistant HIV whose virus no longer effectively responds to therapy. While most antivirals act on just one stage of viral replication, Sunlenca is designed to inhibit HIV at multiple stages of its lifecycle and has no known cross resistance exhibited in vitro to other existing drug classes. Sunlenca is the only HIV treatment option administered twice-yearly.
U.S. Indication for Sunlenca
Sunlenca, a human immunodeficiency virus type 1 (HIV-1) capsid inhibitor, in combination with other antiretroviral(s), is indicated for the treatment of HIV-1 infection in heavily treatment-experienced adults with multidrug resistant HIV-1 infection failing their current antiretroviral regimen due to resistance, intolerance, or safety considerations.
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer.
For 35 years, Gilead has been a leading innovator in the field of HIV, driving advances in treatment, prevention and cure research. Gilead researchers have developed 12 HIV medications, including the first single-tablet regimen to treat HIV, the first antiretroviral for pre-exposure prophylaxis (PrEP) to reduce the risk of acquiring HIV infection, and the first, long-acting injectable HIV treatment medication administered twice-yearly. Our advances in medical research have helped to transform HIV into a preventable, chronic condition for millions of people.
Gilead is committed to continued scientific innovation to provide solutions for the evolving needs of people affected by HIV around the world. Through partnerships and collaborations, the company also aims to improve education, expand access and address barriers to care, with the goal of ending the HIV epidemic for everyone, everywhere. Gilead was recognized as the number one philanthropic funder of HIV-related programs in a report released by Funders Concerned About AIDS.
Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California.
U.S. Prescribing Information for Sunlenca is available at www.gilead.com .
Sunlenca, Gilead and the Gilead logo are registered trademarks of Gilead Sciences, Inc., or its related companies.
For more information about Gilead, please visit the company’s website at www.gilead.com , follow Gilead on Twitter ( @Gilead Sciences ) or call Gilead Public Affairs at 1-800-GILEAD-5 or 1-650-574-3000.
View source version on businesswire.com: https://www.businesswire.com/news/home/20221221005541/en/
Source: Gilead Sciences, Inc.
Dec. 22, 2022 11:05 AM ET
By: Dulan Lokuwithana, SA News Editor
December 21, 2022 6:45 am ET
First PARP inhibitor and new hormonal agent combination approved for these patients in Europe
RAHWAY, N.J.--(BUSINESS WIRE)-- AstraZeneca and Merck (NYSE: MRK), known as MSD outside of the United States and Canada, today announced that LYNPARZA has been approved in the European Union (EU) in combination with abiraterone and prednisone or prednisolone for the treatment of adult patients with metastatic castration-resistant prostate cancer (mCRPC) in whom chemotherapy is not clinically indicated.
This approval by the European Commission follows the positive recommendation from the Committee for Medicinal Products for Human Use received in November this year and was based on the Phase 3 PROpel trial, results of which were published in NEJM Evidence in June 2022.
In PROpel, LYNPARZA in combination with abiraterone and prednisone or prednisolone reduced the risk of disease progression or death by 34% (HR=0.66 [95% CI, 0.54-0.81]; p<0.0001) versus placebo plus abiraterone and prednisone or prednisolone, based on investigator assessment. Median radiographic progression-free survival (rPFS) was 24.8 months for the LYNPARZA plus abiraterone arm (95% CI, 20.5, 27.6) versus 16.6 months for the placebo plus abiraterone arm (95% CI, 13.9, 19.2). A planned sensitivity analysis by blinded independent central review was consistent with the investigator-based analysis, with a median rPFS of 27.6 months for the LYNPARZA plus abiraterone arm compared to 16.4 months for the placebo plus abiraterone arm.
Interim results for the key secondary efficacy endpoint of overall survival (OS) did not reach statistical significance, with an event rate of 37.1% in the LYNPARZA plus abiraterone arm versus 43.1% in the placebo plus abiraterone arm at the time of the analysis (HR=0.83 [95% CI, 0.66-1.03]).
The safety and tolerability of LYNPARZA in combination with abiraterone and prednisone or prednisolone was generally consistent with that of the individual medicines. Approximately 16% of patients who received LYNPARZA in combination with abiraterone and prednisone or prednisolone discontinued treatment due to an adverse event (AE). As previously reported, based on an analysis from the PROpel trial presented earlier this year at the American Society of Clinical Oncology Genitourinary Cancers Symposium, the most common AEs (≥20%) were anemia (46%), fatigue (37%) and nausea (28%). Grade ≥3 AEs were anemia (15%), hypertension (4%), urinary tract infection (2%), fatigue (2%), decreased appetite (1%), vomiting (1%), back pain (1%), diarrhea (1%) and nausea (0.3%).
Prostate cancer is the most commonly diagnosed cancer in men in Europe, with an estimated 473,000 cases and 108,000 deaths in 2020. Approximately 10-20% of patients with prostate cancer are estimated to develop castration-resistant prostate cancer (CRPC) within five years, with at least 84% of these patients presenting with metastases at the time of CRPC diagnosis. Patients diagnosed with advanced prostate cancer have a particularly poor prognosis, with a five-year relative survival rate of about 30%, compared to patients diagnosed with earlier stages of the disease, with a five-year relative survival rate of more than 99%.
Noel Clarke, urological surgeon and professor of urological oncology at Manchester’s Christie/Salford Royal Hospitals and Manchester University, a senior investigator of the PROpel trial, said, “The results of the PROpel Phase 3 trial of olaparib in combination with abiraterone as a first-line treatment show that this therapeutic combination can provide significant clinical benefit to patients with mCRPC. Patients with this condition in the EU will now, for the first time, have the opportunity to benefit from this new treatment combination.”
Dave Fredrickson, executive vice president, oncology business unit, AstraZeneca, said, “Many patients with mCRPC are only able to receive one line of active therapy, as the disease can progress quickly. LYNPARZA in combination with abiraterone has been shown to reduce the risk of disease progression by 34% versus the standard of care treatment in the PROpel trial. Moreover, the combination of LYNPARZA with abiraterone as a first-line treatment expands the use of LYNPARZA to a broader group of mCRPC patients than those treated with LYNPARZA alone in the second-line setting in the PROfound trial. Today’s approval marks a significant advance toward addressing the unmet need of patients with mCRPC in the EU.”
Dr. Eliav Barr, senior vice president, head of global clinical development and chief medical officer, Merck Research Laboratories, said, “Merck is committed to developing new treatment options for patients with mCRPC, a complex disease that urgently needs more therapies. This approval by the European Commission marks another step toward delivering on that commitment, and we look forward to extending the benefits of LYNPARZA to more patients with mCRPC in the EU.”
Use of LYNPARZAin combination with abiraterone and prednisone or prednisolone is currently under review by the U.S. FDA for the treatment of adult patients with mCRPC. LYNPARZA is approved in the U.S. as monotherapy for patients with homologous recombination repair (HRR) gene-mutated mCRPC (BRCA-mutated and other HRR gene mutations) who have progressed following prior treatment with enzalutamide or abiraterone and in the EU, Japan and China for patients with BRCA-mutated mCRPC who have progressed following prior therapy that included a new hormonal agent (NHA). These approvals were based on the data from the Phase 3 PROfound trial.
About PROpel
PROpel is a randomized, double-blind Phase 3 trial testing the efficacy, safety and tolerability of LYNPARZA versus placebo when given in addition to abiraterone and prednisone or prednisolone in patients with mCRPC who had not received prior chemotherapy or NHAs in the mCRPC setting. The major efficacy outcome was rPFS as assessed by investigator per RECIST v1.1 and Prostate Cancer Working Group (bone) criteria. OS was an additional efficacy outcome measure.
INDICATIONS
LYNPARZA is a poly (ADP-ribose) polymerase (PARP) inhibitor indicated:
First-Line Maintenance BRCAm Advanced Ovarian Cancer
For the maintenance treatment of adult patients with deleterious or suspected deleterious germline or somatic BRCA-mutated (gBRCAm or sBRCAm) advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in complete or partial response to first-line platinum-based chemotherapy. Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
First-Line Maintenance HRD-Positive Advanced Ovarian Cancer in Combination with Bevacizumab
In combination with bevacizumab for the maintenance treatment of adult patients with advanced epithelial ovarian, fallopian tube or primary peritoneal cancer who are in complete or partial response to first-line platinum-based chemotherapy and whose cancer is associated with homologous recombination deficiency (HRD)-positive status defined by either:
Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
Maintenance Recurrent Ovarian Cancer
For the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in complete or partial response to platinum-based chemotherapy.
Adjuvant Treatment of gBRCAm, HER2-Negative, High-Risk Early Breast Cancer
For the adjuvant treatment of adult patients with deleterious or suspected deleterious gBRCAm, human epidermal growth factor receptor 2 (HER2)-negative high-risk early breast cancer who have been treated with neoadjuvant or adjuvant chemotherapy. Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
gBRCAm, HER2-Negative Metastatic Breast Cancer
For the treatment of adult patients with deleterious or suspected deleterious gBRCAm, human epidermal growth factor receptor 2 (HER2)-negative metastatic breast cancer who have been treated with chemotherapy in the neoadjuvant, adjuvant, or metastatic setting. Patients with hormone receptor (HR)-positive breast cancer should have been treated with a prior endocrine therapy or be considered inappropriate for endocrine therapy. Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
First-Line Maintenance gBRCAm Metastatic Pancreatic Cancer
For the maintenance treatment of adult patients with deleterious or suspected deleterious gBRCAm metastatic pancreatic adenocarcinoma whose disease has not progressed on at least 16 weeks of a first-line platinum-based chemotherapy regimen. Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
HRR Gene-mutated Metastatic Castration-Resistant Prostate Cancer
For the treatment of adult patients with deleterious or suspected deleterious germline or somatic homologous recombination repair (HRR) gene-mutated metastatic castration-resistant prostate cancer (mCRPC) who have progressed following prior treatment with enzalutamide or abiraterone. Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
Please see complete Prescribing Information , including Medication Guide .
Financial considerations
Under the oncology collaboration with AstraZeneca and following this new approval for LYNPARZA, AstraZeneca will receive a $105 million payment from Merck.
About LYNPARZA® (olaparib)
LYNPARZAis a first-in-class PARP inhibitor and the first targeted treatment to potentially exploit DNA damage response (DDR) pathway deficiencies, such as BRCA mutations, to preferentially kill cancer cells. Inhibition of PARP with LYNPARZA leads to the trapping of PARP bound to DNA single-strand breaks, stalling of replication forks, their collapse and the generation of DNA double-strand breaks and cancer cell death. LYNPARZA is being tested in a range of tumor types with defects and dependencies in the DDR.
LYNPARZA, which is being jointly developed and commercialized by AstraZeneca and Merck, has a broad clinical trial development program, and AstraZeneca and Merck are working together to understand how it may affect multiple PARP-dependent tumors as a monotherapy and in combination across multiple cancer types.
About the AstraZeneca and Merck strategic oncology collaboration
In July 2017, AstraZeneca and Merck, known as MSD outside of the United States and Canada, announced a global strategic oncology collaboration to co-develop and co-commercialize certain oncology products including LYNPARZA, the world’s first PARP inhibitor, for multiple cancer types. Working together, the companies will develop these products in combination with other potential new medicines and as monotherapies. Independently, the companies will develop these oncology productsin combination with their respective PD-L1 and PD-1 medicines.
Merck’s focus on cancer
Our goal is to translate breakthrough science into innovative oncology medicines to help people with cancer worldwide. At Merck, the potential to bring new hope to people with cancer drives our purpose and supporting accessibility to our cancer medicines is our commitment. As part of our focus on cancer, Merck is committed to exploring the potential of immuno-oncology with one of the largest development programs in the industry across more than 30 tumor types. We also continue to strengthen our portfolio through strategic acquisitions and are prioritizing the development of several promising oncology candidates with the potential to improve the treatment of advanced cancers. For more information about our oncology clinical trials, visit www.merck.com/clinicaltrials.
About Merck
At Merck, known as MSD outside of the United States and Canada, we are unified around our purpose: We use the power of leading-edge science to save and improve lives around the world. For more than 130 years, we have brought hope to humanity through the development of important medicines and vaccines. We aspire to be the premier research-intensive biopharmaceutical company in the world – and today, we are at the forefront of research to deliver innovative health solutions that advance the prevention and treatment of diseases in people and animals. We foster a diverse and inclusive global workforce and operate responsibly every day to enable a safe, sustainable and healthy future for all people and communities. For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Source: Merck & Co., Inc.
Dec. 21, 2022 4:29 AM ET
By: Ravikash, SA News Editor
12/12/2022
SAN DIEGO, Dec. 12, 2022 /PRNewswire/ -- Mirati Therapeutics, Inc. (NASDAQ: MRTX), a targeted oncology company, today announced that the U.S. Food and Drug Administration (FDA) has granted accelerated approval for KRAZATI™ (adagrasib), a targeted treatment option for adult patients with KRASG12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.
Experience the full interactive Multichannel News Release here: https://www.multivu.com/players/English/8999051-mirati-therapeutics-fda-accelerated-approval-of-krazati-adagrasib/
This indication is approved under accelerated approval based on objective response rate (ORR) and duration of response (DOR). Continued approval for this indication may be contingent upon verification and description of a clinical benefit in a confirmatory trial(s).
To view the multimedia assets associated with this release, please visit: Mirati.com/approval
"The FDA approval of KRAZATI is a positive development for thousands of patients with KRASG12C mutations, including the approximately 14% of patients with NSCLC adenocarcinomas histology that harbor a KRASG12C mutation.1 Mirati is thrilled to make KRAZATI available in a tablet formulation to patients in the U.S. with advanced NSCLC who have progressed beyond a first-line treatment for the historically difficult-to-treat KRAS mutation," David Meek, chief executive officer, Mirati Therapeutics, Inc., continued, "We look forward to continuing to advance our KRAZATI development program including several monotherapy and combination studies in KRASG12C-mutated solid tumors."
KRAZATI has demonstrated a positive benefit-risk profile with accelerated approval based on the Phase 2 registration-enabling cohort of the KRYSTAL-1 study, evaluating KRAZATI 600 mg capsules administered orally twice daily in 116 patients with KRASG12C-mutated advanced NSCLC who previously received treatment with a platinum-based regimen and an immune checkpoint inhibitor. The primary efficacy endpoints were confirmed ORR and DOR as evaluated by blinded independent central review (BICR) according to response evaluation criteria in solid tumors (RECIST v1.1).
The trial demonstrated an ORR of 43% (95% CI: 34-53) with 80% (95% CI: 71-87) of patients achieving disease control. The median DOR was 8.5 months (95% CI: 6.2-13.8).
In a pooled efficacy analysis (n=132) including Phase 1/1b NSCLC and registrational Phase 2 NSCLC cohorts from the KRYSTAL-1 study evaluating adagrasib as a single agent at 600 mg capsules orally twice daily, adagrasib showed an ORR of 44% and a disease control rate of 81% based on BICR, a median DOR of 12.5 months (95% CI, 7.3-NE) and median overall survival of 14.1 months (94% CI, 9.2-19.2).
The safety profile of KRAZATI was evaluated in a pooled patient population with NSCLC and other solid tumors as a single agent at 600 mg orally twice daily in 366 patients enrolled in KRYSTAL-1 and KRYSTAL-12. The most common (≥ 25%) adverse reactions were nausea, diarrhea, vomiting, fatigue, musculoskeletal pain, hepatotoxicity, renal impairment, edema, dyspnea and decreased appetite. Permanent discontinuation of KRAZATI due to an adverse reaction occurred in 13% of patients.
Although KRASG12C is the most common KRAS mutation in NSCLC, patients have had limited options for the treatment of this debilitating and difficult-to-treat condition.2,3
"The approval of KRAZATI offers an effective therapy for patients with advanced NSCLC harboring the KRASG12C mutation. The positive ORR and DOR results, as observed in previously treated patients with NSCLC harboring the KRASG12C mutation, demonstrate the effectiveness of KRAZATI as an option for these difficult-to-treat patients," said Shirish M. Gadgeel, MD, chief of the Division of Hematology and Oncology, Department of Internal Medicine, Henry Ford Cancer Institute/Henry Ford Health System.
"KRASG12C in NSCLC is an area of high unmet need and new treatment options offer patients and our community new hope for survivorship," said Bonnie J. Addario, co-founder and board chair of the GO2 Foundation for Lung Cancer. "I'm pleased that patients have options, there's more awareness of this disease and we are all focused on improving the journeys of people living with KRASG12C-mutated NSCLC."
The Company partnered with Agilent and QIAGEN to develop blood- and tissue-based companion diagnostics (CDx), respectively, for KRAZATI that are now available. With tissue and blood modalities for companion diagnostics, patients have more flexibility, and clinicians have greater options for biomarker testing. These solutions help to personalize a patient's treatment path.
Mirati Therapeutics is launching Mirati & Me, a comprehensive program dedicated to supporting patients, caregivers and the oncology community including coverage and access, financial, educational and emotional support services. Learn more by visiting the Mirati & Me website or 1-844-647-2842.
For more information, visit KRAZATI.com.
About KRAZATI (adagrasib)
Mirati has risen to meet one of the most challenging mutations in cancer research by developing KRAZATI, a highly selective and potent oral small-molecule inhibitor of KRASG12C.
Intentionally designed to meet the challenge of KRASG12C, adagrasib is optimized to sustain target inhibition, an attribute that could be important to treat KRASG12C-mutated cancers, as the KRASG12C protein regenerates every 24−48 hours.4 Adagrasib has shown clinically to be a CNS penetrant, which may be important given that CNS metastases frequently occur in NSCLC and lead to poor prognosis.5,6,7
In the U.S., KRAZATI was reviewed by the FDA for Accelerated Approval (Subpart H), which allows for the approval of drugs that treat serious conditions, and that fill an unmet medical need based on surrogate endpoints. KRAZATI was reviewed under the FDA Real-Time Oncology Review (RTOR) pilot program, which aims to explore a more efficient review process that ensures safe and effective treatments are made available to patients as early as possible. Mirati submitted a Marketing Authorization Application (MAA) in the EU in May 2022. In 2021, adagrasib achieved Breakthrough Therapy Designation in the U.S. as a potential treatment for patients with NSCLC harboring the KRASG12C mutation who have received at least one prior systemic therapy.
Adagrasib continues to be evaluated as monotherapy and in combination with other anti-cancer therapies in patients with advanced KRASG12C-mutated solid tumors, including NSCLC, colorectal cancer, and pancreatic cancer. For more information, visit Mirati.com/science.
Mirati has an Expanded Access Program (EAP) for adagrasib for the treatment of eligible patients with KRASG12C-mutated cancers, regardless of tumor type, including patients with treated or untreated CNS metastases, in the U.S. Learn more about the EAP at Mirati.com/expanded-access-policy.
KRAZATI (adagrasib) U.S. Indication
KRAZATI is indicated for the treatment of adult patients with KRASG12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.
This indication is approved under accelerated approval based on objective response rate (ORR) and duration of response (DOR). Continued approval for this indication may be contingent upon verification and description of a clinical benefit in a confirmatory trial(s).
Please see Full Prescribing Information .
About the KRYSTAL-1 Study
KRYSTAL-1 is an open-label Phase 1/2 multiple-expansion cohort trial evaluating adagrasib as monotherapy and in combination with other anti-cancer therapies in patients with advanced solid tumors harboring the KRASG12C mutation.
About KRASG12C in NSCLC
Lung cancer is one of the most common cancers worldwide, accounting for 2.21 million new cases and 1.8 million deaths worldwide in 2020.8 Lung cancer consists of NSCLC in approximately 85% of cases and small cell lung cancer (SCLC) in approximately 15% of cases.9 KRASG12C is the most common KRAS mutation in NSCLC, present in approximately 14% of patients with lung adenocarcinoma, and is a biomarker mutation of poor prognosis.1,3
About Mirati Therapeutics, Inc.
Mirati Therapeutics, Inc. is a commercial-stage biotechnology company whose mission is to discover, design and deliver breakthrough therapies to transform the lives of patients with cancer and their loved ones. The company is relentlessly focused on bringing forward therapies that address areas of high unmet need, including lung cancer, and advancing a pipeline of novel therapeutics targeting the genetic and immunological drivers of cancer. Unified for patients, Mirati's vision is to unlock the science behind the promise of a life beyond cancer.
For more information about Mirati, visit us at Mirati.com or follow us on Twitter, LinkedIn and Facebook.
SOURCE Mirati Therapeutics, Inc.
Dec. 21, 2022 5:39 PM ET Mirati Therapeutics, Inc. (MRTX)
By: Jonathan Block, SA News Editor
FDA approves Roche’s Actemra for the treatment of COVID-19 in hospitalised adults December 21, 2022
Basel, 21 December 2022 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the U.S. Food and Drug Administration (FDA) has approved Actemra® (tocilizumab) intravenous (IV) for the treatment of COVID-19 in hospitalised adult patients who are receiving systemic corticosteroids and require supplemental oxygen, non-invasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation (ECMO). Actemra is the first FDA-approved monoclonal antibody to treat COVID-19 and is recommended for use as a single 60-minute IV infusion.
“With new variants emerging, FDA-approved treatments including Actemra remain essential to the continued fight against COVID-19,” said Levi Garraway, M.D., Ph.D., Chief Medical Officer and Head of Global Product Development. “Actemra is the first FDA-approved monoclonal antibody for treating patients with severe COVID-19, providing an important option for hospitalised patients and their healthcare providers who continue to be on the frontlines treating COVID-19.”
Four randomised, controlled studies evaluated Actemra for the treatment of COVID-19 in more than 5,500 hospitalised patients. Altogether, the results of these four studies (the University of Oxford-led RECOVERY trial, along with the Roche-sponsored global trials, EMPACTA, COVACTA and REMDACTA) showed that Actemra may improve outcomes in patients receiving corticosteroids and requiring supplemental oxygen or breathing support. The FDA approval is based on the results from the RECOVERY trial, as well as the EMPACTA trial, the first global, Phase III study in COVID-19 to focus on patients from underrepresented racial and ethnic groups. No new warnings and precautions related to Actemra in COVID-19 studies have been identified.
The FDA approval follows the FDA’s Emergency Use Authorization (EUA) for Actemra in hospitalised adults and children (ages 2 and older) with COVID-19, which was granted in June 2021. The use of Actemra to treat hospitalised people ages 2 to less than 18 years old is not FDA approved, however the EUA for this age group currently remains in place after the FDA approval for hospitalised adult patients.
More than one million people hospitalised with COVID-19 have been treated with Actemra worldwide, since the beginning of the pandemic. Around the world, Actemra is approved for use in more than 30 countries for patients hospitalised with severe COVID-19. In the United States, this is the seventh FDA approved indication for Actemra since the medicine was launched in 2010.
Roche stands together with society, governments, healthcare providers and all those working towards the common goal of overcoming the COVID-19 pandemic.
About Actemra/RoActemra in COVID-19
Actemra®/RoActemra® is approved for use in multiple territories including the United States, European Union, Japan, the United Kingdom, New Zealand, Russia and Brazil, provisionally approved in Australia, and authorised for emergency use in Ghana, Mexico and Korea for defined patients hospitalised with severe or critical COVID-19. It has also been recommended and prequalified by the World Health Organization.
About Actemra/Roactemra
Actemra®/RoActemra® was the first humanised interleukin-6 (IL-6) receptor antagonist approved for the treatment of adult patients with moderately to severely active rheumatoid arthritis (RA) who have used one or more disease-modifying antirheumatic drugs (DMARDs), such as methotrexate (MTX), that did not provide enough relief. The extensive Actemra RA IV clinical development program included five Phase III clinical studies and enrolled more than 4,000 people with RA in 41 countries. The Actemra RA subcutaneous clinical development program included two Phase III clinical studies and enrolled more than 1,800 people with RA in 33 countries. Actemra subcutaneous injection is also approved for the treatment of adult patients with giant cell arteritis (GCA), for the treatment of patients two years of age and older with active polyarticular juvenile idiopathic arthritis (PJIA) or active systemic juvenile idiopathic arthritis (SJIA), and for slowing the rate of decline in pulmonary function in adult patients with systemic sclerosis-associated interstitial lung disease (SSc-ILD). In addition, Actemra is also approved in the IV formulation for patients two years of age and older with active PJIA, SJIA, GCA or CAR T cell-induced cytokine release syndrome (CRS). Actemra is not approved for subcutaneous use in people with CRS. Actemra IV is approved for the treatment of COVID-19 in hospitalised adult patients who are receiving systemic corticosteroids and require supplemental oxygen, non-invasive or invasive mechanical ventilation, or extracorporeal membrane oxygenation (ECMO). It is not known if Actemra is safe and effective in children with PJIA, SJIA or CRS under two years of age or in children with conditions other than PJIA, SJIA or CRS.
About Roche’s response to the COVID-19 pandemic
As a leading healthcare company, we are doing all we can to support countries in their fight against COVID-19 and minimising its impact. That is why we are working with governments, policy makers, healthcare professionals and others to help contain the COVID-19 pandemic and make sure patients continue to receive the tests, treatment and care they need.
The pandemic has profoundly raised awareness of the role diagnostics play in COVID-19 diagnosis, treatment development and disease management. Roche has developed and launched more than 20 COVID-19 diagnostics solutions, including polymerase chain reaction (PCR) and rapid antigen and antibody tests. Our solutions serve the entire diagnostic continuum, from high-throughput laboratories to point-of-care and home self-testing, and cover all currently known variants. To help meet global demand, we have supplied more than 1.5 billion tests for COVID-19 since March 2020.
Roche continues to evaluate its existing therapeutic portfolio and is researching future options to help benefit patients with COVID-19. Our IL-6 inhibitor Actemra®/RoActemra® (tocilizumab) has been approved for patients hospitalised with severe COVID-19 in more than 30 countries including the European Union and the United States. The World Health Organization has prequalified Actemra for use in patients with severe COVID-19, facilitating its availability in low- and middle-income countries. In addition, we have been improving access to Actemra by introducing an international differentiated pricing strategy, providing the medicine at cost for use in low- and middle-income countries and non asserting patents in these regions during the pandemic.
We have also been partnering with Regeneron to jointly develop the antibody combination Ronapreve™ (casirivimab and imdevimab, known as REGEN-COV™ in the US). It has been approved in multiple territories including the European Union, Japan, and Switzerland and authorised for emergency or temporary pandemic use in many countries including the US. We are constantly monitoring and assessing Ronapreve’s neutralising activity against emerging variants of concern and entered a dialogue with regulators and industry representatives to discuss the efficacy of monoclonal antibodies in the context of rapidly evolving SARS-CoV-2 variants.
Our utmost goal remains to be a trusted partner who acts with urgency to save and improve the lives of patients with COVID-19 and to reduce its burden on society. For more information please visit our COVID-19 response page.
About Roche
Founded in 1896 in Basel, Switzerland, as one of the first industrial manufacturers of branded medicines, Roche has grown into the world’s largest biotechnology company and the global leader in in-vitro diagnostics. The company pursues scientific excellence to discover and develop medicines and diagnostics for improving and saving the lives of people around the world. We are a pioneer in personalised healthcare and want to further transform how healthcare is delivered to have an even greater impact. To provide the best care for each person we partner with many stakeholders and combine our strengths in Diagnostics and Pharma with data insights from the clinical practice.
In recognising our endeavor to pursue a long-term perspective in all we do, Roche has been named one of the most sustainable companies in the pharmaceuticals industry by the Dow Jones Sustainability Indices for the thirteenth consecutive year. This distinction also reflects our efforts to improve access to healthcare together with local partners in every country we work.
Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan.
For more information, please visit www.roche.com.
Dec. 21, 2022 6:00 PM ET
Roche Holding AG (RHHBF), RHHBY
By: Jonathan Block, SA News Editor
12/20/2022
– This combination has the potential to be the first treatment option combining an antibody-drug conjugate plus an immunotherapy in this treatment setting –
– FDA granted the applications Priority Review with a PDUFA date of April 21, 2023 –
BOTHELL, Wash. & TOKYO & RAHWAY, N.J.--(BUSINESS WIRE)--
Seagen Inc. (Nasdaq: SGEN),Astellas Pharma Inc. (TSE:4503) and Merck (NYSE: MRK), known as MSD outside of the United States and Canada, today announced the U.S. Food and Drug Administration (FDA) has accepted for Priority Review supplemental Biologics License Applications (sBLAs) for PADCEV® (enfortumab vedotin-ejfv) and KEYTRUDA® (pembrolizumab) for use of these two agents in combination for the treatment of patients with locally advanced or metastatic urothelial cancer (la/mUC) who are not eligible to receive cisplatin-containing chemotherapy. The respective applications are intended to expand both labels for PADCEV and KEYTRUDA. The agency set a Prescription Drug User Fee Act (PDUFA) goal date for each application of April 21, 2023.
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20221220005198/en/
“We look forward to working closely with the FDA as we seek potential accelerated approval for this combination in the hopes that it can be another treatment option for patients with locally advanced or metastatic urothelial cancer who are not eligible for cisplatin-containing chemotherapy,” said Ahsan Arozullah, M.D., M.P.H., Senior Vice President and Head of Development Therapeutic Areas, Astellas.
The combination therapy was granted Breakthrough Therapy designation by the FDA in February 2020. The respective sBLAs are supported by efficacy and safety data from the phase 1b/2 EV-103 trial (NCT03288545, also known as KEYNOTE-869) Dose Escalation/Cohort A and Cohort K. Results from Dose Escalation/Cohort A were published in the Journal of Clinical Oncology.1 Results from Cohort K were presented in a late-breaking session at the 2022 European Society for Medical Oncology (ESMO) Congress.2
“Urothelial cancer, the most common type of bladder cancer, is associated with poor survival in the advanced stage,” said Marjorie Green, M.D., Senior Vice President and Head of Late-Stage Development, Seagen. “The investigational results from our clinical development program support the combination of PADCEV and KEYTRUDA as a potential treatment for this patient population.”
Please see Important Safety Information at the end of this press release for both drugs, including a warning and precaution for immune-mediated adverse reactions for pembrolizumab and BOXED WARNING for PADCEV (enfortumab vedotin-ejfv) for serious skin reactions.
“Despite advancements in treatment options, approximately half of advanced bladder cancer patients in the U.S. are ineligible for cisplatin-based chemotherapy, and these patients need new options. We are encouraged by the investigational results of the combination of PADCEV and KEYTRUDA for this patient population and are fully committed to work to bring this new approach forward to patients,” said Dr. Eliav Barr, Senior Vice President, Head of Global Clinical Development and Chief Medical Officer, Merck Research Laboratories.
Seagen, Astellas and Merck are further investigating enfortumab vedotin plus pembrolizumab in the ongoing phase 3 EV-302 study (NCT04223856, also known as KEYNOTE-A39), evaluating the clinical benefit for the investigational treatment combination in patients with previously untreated advanced urothelial cancer. The trial is intended to serve as the confirmatory trial for the potential accelerated approval in the U.S. and serve as the basis for global registration.
The studies are part of an extensive program evaluating this combination in multiple stages of urothelial cancer, including two phase 3 clinical trials in muscle-invasive bladder cancer in EV-304 (NCT04700124, also known as KEYNOTE-B15) and EV-303 (NCT03924895, also known as KEYNOTE-905).
About the EV-103/KEYNOTE-869 Trial
The EV-103 trial (NCT03288545) is an ongoing, multi-cohort, open-label, multicenter phase 1b/2 study of enfortumab vedotin alone or in combination with pembrolizumab and/or chemotherapy in first- or second-line settings in patients with locally advanced or metastatic urothelial cancer (la/mUC) and in patients with muscle-invasive bladder cancer.
About PADCEV
PADCEV (enfortumab vedotin-ejfv) is a first-in-class antibody-drug conjugate (ADC) that is directed against Nectin-4, a protein located on the surface of cells and highly expressed in bladder cancer.7 Nonclinical data suggest the anticancer activity of PADCEV is due to its binding to Nectin-4-expressing cells followed by the internalization and release of the anti-tumor agent monomethyl auristatin E (MMAE) into the cell, which result in the cell not reproducing (cell cycle arrest) and in programmed cell death (apoptosis).8
Indication
PADCEV® is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer (mUC) who:
About KEYTRUDA® (pembrolizumab) injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf.
About Seagen
Seagen Inc. is a global biotechnology company that discovers, develops and commercializes transformative cancer medicines to make a meaningful difference in people’s lives. Seagen is headquartered in the Seattle, Washington area, and has locations in California, Canada, Switzerland and the European Union. For more information on the company’s marketed products and robust pipeline, visit www.seagen.com and follow @SeagenGlobal on Twitter.
About Astellas
Astellas Pharma Inc. is a pharmaceutical company conducting business in more than 70 countries around the world. We are promoting the Focus Area Approach that is designed to identify opportunities for the continuous creation of new drugs to address diseases with high unmet medical needs by focusing on Biology and Modality. Furthermore, we are also looking beyond our foundational Rx focus to create Rx+® healthcare solutions that combine our expertise and knowledge with cutting-edge technology in different fields of external partners. Through these efforts, Astellas stands on the forefront of healthcare change to turn innovative science into VALUE for patients. For more information, please visit our website at https://www.astellas.com/en.
Merck’s focus on cancer
Our goal is to translate breakthrough science into innovative oncology medicines to help people with cancer worldwide. At Merck, the potential to bring new hope to people with cancer drives our purpose and supporting accessibility to our cancer medicines is our commitment. As part of our focus on cancer, Merck is committed to exploring the potential of immuno-oncology with one of the largest development programs in the industry across more than 30 tumor types. We also continue to strengthen our portfolio through strategic acquisitions and are prioritizing the development of several promising oncology candidates with the potential to improve the treatment of advanced cancers. For more information about our oncology clinical trials, visit www.merck.com/clinicaltrials.
About Merck
At Merck, known as MSD outside of the United States and Canada, we are unified around our purpose: We use the power of leading-edge science to save and improve lives around the world. For more than 130 years, we have brought hope to humanity through the development of important medicines and vaccines. We aspire to be the premier research-intensive biopharmaceutical company in the world – and today, we are at the forefront of research to deliver innovative health solutions that advance the prevention and treatment of diseases in people and animals. We foster a diverse and inclusive global workforce and operate responsibly every day to enable a safe, sustainable and healthy future for all people and communities. For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
About the Seagen, Astellas and Merck Collaboration
Seagen and Astellas entered a clinical collaboration agreement with Merck to evaluate the combination of Seagen’s and Astellas’ PADCEV® (enfortumab vedotin-ejfv) and Merck’s KEYTRUDA® (pembrolizumab) in patients with previously untreated metastatic urothelial cancer. KEYTRUDA is a registered trademark of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Rahway, NJ, USA.
View source version on businesswire.com: https://www.businesswire.com/news/home/20221220005198/en/
Source: Seagen Inc.
Dec. 20, 2022 8:41 AM ET
Astellas Pharma Inc. (ALPMF), ALPMY, SGEN, MRK
By: Jonathan Block, SA News Editor
Dec 21, 2022PDF Version
NEW YORK, Dec. 21, 2022 (GLOBE NEWSWIRE) -- Y-mAbs Therapeutics, Inc. (the “Company” or “Y-mAbs”) (Nasdaq: YMAB) a commercial-stage biopharmaceutical company focused on the development and commercialization of novel, antibody-based therapeutic products for the treatment of cancer, today announced that it has entered into a distribution agreement with WEP Clinical Ltd. (“WEP”) in connection with an early access program for DANYELZA (naxitamab-gqgk) 40mg/10mL Injection in Europe.
DANYELZA is a humanized, monoclonal antibody that binds to the glycolipid GD2. GD2 is a disialoganglioside that is overexpressed on neuroblastoma cells and other cells of neuroectodermal origin, including the central nervous system and peripheral nerves. DANYELZA is administered on days 1, 3, and 5 of each treatment cycle as an intravenous infusion after dilution. Treatment cycles are repeated every 4 weeks until complete response or partial response, followed by 5 additional cycles every 4 weeks.
In the U.S., DANYELZA is approved by the FDA and indicated, in combination with granulocyte-macrophage colony-stimulating factor (“GM-CSF”), for the treatment of pediatric patients 1 year of age and older and adult patients with relapsed or refractory high-risk neuroblastoma in the bone or bone marrow who have demonstrated a partial response, minor response, or stable disease to prior therapy.
“We are excited to be able to give European patients access to DANYELZA through this early access program with WEP,” said Thomas Gad, founder, President and Interim CEO. “Initially, DANYELZA will be available through such program in Spain and France, and we hope to add additional territories to the agreement.”
Researchers at Memorial Sloan Kettering Cancer Center (“MSK”) developed DANYELZA, which is exclusively licensed by MSK to Y-mAbs. MSK has institutional financial interests related to the compound and Y-mAbs.
About DANYELZA® (naxitamab-gqgk)
DANYELZA® (naxitamab-gqgk) is indicated, in combination with granulocyte-macrophage colony-stimulating factor (“GM-CSF”), for the treatment of pediatric patients 1 year of age and older and adult patients with relapsed or refractory high-risk neuroblastoma in the bone or bone marrow who have demonstrated a partial response, minor response, or stable disease to prior therapy. This indication was approved in the United States by the FDA under accelerated approval based on overall response rate and duration of response. Continued approval for this indication is contingent upon verification and description of clinical benefits in a confirmatory trial. DANYELZA® includes a Boxed Warning for serious infusion-related reactions, such as cardiac arrest and anaphylaxis, and neurotoxicity, such as severe neuropathic pain and transverse myelitis. See full Prescribing Information (https://labeling.ymabs.com/danyelza) for complete Boxed Warning and other important safety information.
About Y-mAbs
Y-mAbs is a commercial-stage biopharmaceutical company focused on the development and commercialization of novel, antibody-based therapeutic cancer products. In addition to conventional antibodies, the Company’s technologies include bispecific antibodies generated using the Y-BiClone platform and the SADA platform. The Company’s product pipeline includes one FDA-approved product, DANYELZA® (naxitamab-gqgk), which targets tumors that express GD2, and one product candidate at the registration-stage, OMBLASTYS® (omburtamab), which targets tumors that express B7-H3.
DANYELZA®, OMBLASTYS® and Y-mAbs® are registered trademarks of Y-mAbs Therapeutics, Inc.
![]()
Source: Y-mAbs Therapeutics, Inc
Dec. 21, 2022 9:49 AM ET
Y-mAbs Therapeutics, Inc. (YMAB)
By: Ravikash, SA News Editor
12/20/22PDF Version
-- First and only anti-CD19 B-cell-depleting monotherapy for the treatment of adult patients with NMOSD who are anti-aquaporin-4 immunoglobulin G seropositive (AQP4-IgG+) --
-- NMOSD is a devastating rare autoimmune disease that can cause vision loss and paralysis --
DUBLIN--(BUSINESS WIRE)--Dec. 20, 2022-- Horizon Therapeutics plc (Nasdaq: HZNP) today announced the Brazilian Health Regulatory Agency (ANVISA) has approved UPLIZNA as a monotherapy for the treatment of adult patients with NMOSD who are AQP4-IgG+. This rare autoimmune disease is caused by inflammation in the central nervous system, resulting in severe and recurrent attacks that can lead to permanent disability, such as paralysis and vision loss.1 An estimated 10,000 people in Brazil live with the disease.2
“This approval is an important part of Horizon’s commitment to helping people worldwide who are living with challenging rare diseases,” said Vikram Karnani, executive vice president, international and global medical affairs, Horizon. “The NMOSD community in Brazil now has an approved medicine that has been proven in clinical studies to provide sustained protection against NMOSD attacks.”
Unlike other autoimmune diseases, patients with NMOSD typically do not fully recover from attacks.3 It is estimated that 69% of patients have severe vision loss in at least one eye within three years of disease onset and 34% may develop permanent motor impairment.4,5,6 A primary goal of treatment is to help prevent cumulative damage and permanent disability that can be caused by subsequent attacks.
In the N-MOmentum pivotal clinical trial, UPLIZNA demonstrated a significant reduction in the risk of an NMOSD attack with only two infusions per year, following the initial loading doses. Additionally, 89% of patients in the AQP4-IgG+ group remained attack free during the six-month period post-treatment and more than 83% of patients on treatment remained attack free for at least four years.7
“NMOSD is an extremely debilitating and life-changing disease that can impact patients physically, socially and economically,” said Douglas Sato, M.D. Ph.D., neurologist, full professor of medicine, Pontifical Catholic University of Rio Grande do Sul (PUCRS). “UPLIZNA is unique in that it prevents attacks by depleting a broad range of B cells, including plasmablasts and plasma cells, and has a very good safety profile in NMOSD patients. This approval represents a significant advancement in the treatment of NMOSD and can provide hope for those who have been struggling with this disease.”
UPLIZNA was approved by the U.S. Food and Drug Administration (FDA) in June 2020, by the Japanese Ministry of Health, Labor and Welfare in March 2021 and by the European Commission (EC) in April 2022.
About Neuromyelitis Optica Spectrum Disorder (NMOSD)
NMOSD is a unifying term for neuromyelitis optica (NMO) and related syndromes. NMOSD is a rare, severe, relapsing, neuroinflammatory autoimmune disease that attacks the optic nerve, spinal cord, brain and brain stem.1,8 Approximately 80% of all patients with NMOSD test positive for anti-AQP4 antibodies.9 AQP4-IgG binds primarily to astrocytes in the central nervous system and triggers an escalating immune response that results in lesion formation and astrocyte death.10
Anti-AQP4 autoantibodies are produced by plasmablasts and plasma cells. These B-cell populations are central to NMOSD disease pathogenesis, and a large proportion of these cells express CD19.11 Depletion of these CD19+ B cells is thought to remove an important contributor to inflammation, lesion formation and astrocyte damage. Clinically, this damage presents as an NMOSD attack, which can involve the optic nerve, spinal cord and brain.10-12 Loss of vision, paralysis, loss of sensation, bladder and bowel dysfunction, nerve pain and respiratory failure can all be manifestations of the disease.13 Each NMOSD attack can lead to further cumulative damage and disability.14,15 NMOSD occurs more commonly in women and may be more common in individuals of African and Asian descent.16,17
About UPLIZNA (inebilizumab-cdon)
INDICATION
UPLIZNA is indicated for the treatment of neuromyelitis optica spectrum disorder (NMOSD) in adult patients who are anti-aquaporin-4 (AQP4) antibody positive.
About Horizon
Horizon is a global biotechnology company focused on the discovery, development and commercialization of medicines that address critical needs for people impacted by rare, autoimmune and severe inflammatory diseases. Our pipeline is purposeful: We apply scientific expertise and courage to bring clinically meaningful therapies to patients. We believe science and compassion must work together to transform lives. For more information on how we go to incredible lengths to impact lives, visit www.horizontherapeutics.com and follow us on Twitter, LinkedIn, Instagram and Facebook.
View source version on businesswire.com: https://www.businesswire.com/news/home/20221220005190/en/
Source: Horizon Therapeutics plc
Dec. 20, 2022 1:35 PM ET
Horizon Therapeutics Public Limited Company (HZNP)
By: Anuron Mitra, SA News Editor
PUBLISHED19 December 2022 19 December 2022 07:20 GMT
AstraZeneca and Daiichi Sankyo’s Enhertu (trastuzumab deruxtecan) has been recommended for approval in the European Union (EU) as monotherapy for the treatment of adult patients with unresectable or metastatic HER2-low (IHC 1+ or IHC 2+/ISH-) breast cancer who have received prior chemotherapy in the metastatic setting or developed disease recurrence during or within six months of completing adjuvant chemotherapy.
Enhertu is a specifically engineered HER2-directed antibody drug conjugate (ADC) being jointly developed and commercialised by AstraZeneca and Daiichi Sankyo.
The Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency based its positive opinion on results from the DESTINY-Breast04 Phase III trial, which were presented at the American Society of Clinical Oncology 2022 Annual Meeting and simultaneously published in The New England Journal of Medicine.1
In the trial, Enhertu reduced the risk of disease progression or death by 50% versus physician’s choice of chemotherapy (based on a hazard ratio [HR] of 0.50; 95% confidence interval [CI]: 0.40-0.63; p<0.001) in patients with HER2-low metastatic breast cancer with HR-positive or HR-negative disease. A median progression-free survival (PFS) of 9.9 months was seen with Enhertu versus 5.1 months in those treated with chemotherapy, as assessed by blinded independent central review (BICR). A 36% reduction in the risk of death (HR 0.64; 95% CI: 0.49-0.84; p=0.001) was seen with Enhertu compared to chemotherapy with a median overall survival (OS) of 23.4 months versus 16.8 months.
Susan Galbraith, Executive Vice President, Oncology R&D, AstraZeneca, said: “Enhertu is the first-ever HER2-directed medicine to show a survival benefit in patients with HER2-low metastatic breast cancer, confirming the importance of targeting lower levels of HER2 expression in patients previously classified as HER2-negative. The CHMP’s recommendation is encouraging and supports our ambition to evolve the way breast cancer is classified and treated to ultimately improve patient outcomes.”
Ken Takeshita, Global Head, R&D Daiichi Sankyo, said: “This positive CHMP opinion recognises the unmet need in the European Union for patients with HER2-low metastatic breast cancer. Currently, once patients with HR-positive disease progress on hormone therapy there are limited effective treatments, and few targeted options are available for patients with HR-negative disease. We look forward to the European Commission decision and aim to bring Enhertu to eligible patients as soon as possible.”
The safety profile observed in patients treated with Enhertu in the DESTINY-Breast04 trial was consistent with that seen in other trials of Enhertu in breast cancer with no new safety signals identified.
Notes
Breast cancer and HER2 expression
Breast cancer is the most common cancer and is one of the leading causes of cancer-related deaths worldwide.2 More than two million patients with breast cancer were diagnosed in 2020 with nearly 685,000 deaths globally.2 In Europe, approximately 531,000 breast cancer patients are diagnosed annually with nearly 141,000 deaths.3
HER2 is a tyrosine kinase receptor growth-promoting protein expressed on the surface of many types of tumours including breast, gastric, lung and colorectal cancers, and is one of many biomarkers expressed in breast cancer tumours.4
HER2 expression is currently determined by an immunohistochemistry (IHC) test which estimates the amount of HER2 protein on a cancer cell, and/or an in situ hybridisation (ISH) test, which counts the copies of the HER2 gene in cancer cells.4,5 HER2 tests provide IHC and ISH scores across the full HER2 spectrum and are routinely used to determine appropriate treatment options for patients with metastatic breast cancer.
HER2-positive cancers are currently defined as HER2 expression measured as IHC 3+ or IHC 2+/ISH+, and HER2-negative cancers are defined as HER2 expression measured as IHC 0, IHC 1+ or IHC 2+/ISH-.4 However, approximately half of all breast cancers are HER2-low, defined as a HER2 score of IHC1+ or IHC 2+/ISH-.6-8 HER2-low occurs in both HR-positive and HR-negative disease.9
Currently, patients with HR-positive metastatic breast cancer and HER2-low disease have limited effective treatment options following progression on endocrine (hormone) therapy.10 Additionally, few targeted options are available for those with HR-negative disease.11
DESTINY-Breast04
DESTINY-Breast04 is a global, randomised, open-label, Phase III trial evaluating the efficacy and safety of Enhertu (5.4mg/kg) versus physician’s choice of chemotherapy (capecitabine, eribulin, gemcitabine, paclitaxel or nab-paclitaxel) in patients with HR-positive or HR-negative, HER2-low unresectable and/or metastatic breast cancer previously treated with one or two prior lines of chemotherapy. Patients were randomised 2:1 to receive either Enhertu or chemotherapy.
The primary endpoint of DESTINY-Breast04 is PFS in patients with HR-positive disease based on BICR. Key secondary endpoints include PFS based on BICR in all randomised patients (HR-positive and HR-negative disease), OS in patients with HR-positive disease and OS in all randomised patients (HR-positive and HR-negative disease). Other secondary endpoints include PFS based on investigator assessment, objective response rate based on BICR and on investigator assessment, duration of response based on BICR and safety.
DESTINY-Breast04 enrolled 557 patients at multiple sites in Asia, Europe and North America. For more information about the trial, visit ClinicalTrials.gov.
Enhertu
Enhertu is a HER2-directed ADC. Designed using Daiichi Sankyo’s proprietary DXd ADC technology, Enhertu is the lead ADC in the oncology portfolio of Daiichi Sankyo and the most advanced programme in AstraZeneca’s ADC scientific platform. Enhertu consists of a HER2 monoclonal antibody attached to a topoisomerase I inhibitor payload, an exatecan derivative, via a stable tetrapeptide-based cleavable linker.
Enhertu (5.4mg/kg) is approved in more than 40 countries for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received a (or one or more) prior anti-HER2-based regimen, either in the metastatic setting or in the neoadjuvant or adjuvant setting and have developed disease recurrence during or within six months of completing therapy, based on the results from the DESTINY-Breast03 trial. Enhertu also is approved in several countries for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received two or more prior anti-HER2-based regimens based on the results from the DESTINY-Breast01 trial.
Enhertu (5.4mg/kg) is approved in Brazil and the US for the treatment of adult patients with unresectable or metastatic HER2-low (IHC 1+ or IHC 2+/ ISH-) breast cancer who have received a prior systemic therapy in the metastatic setting or developed disease recurrence during or within six months of completing adjuvant chemotherapy, based on the results of the DESTINY-Breast04 trial.
Enhertu (5.4mg/kg) is approved under accelerated approval in the US for the treatment of adult patients with unresectable or metastatic non-small cell lung cancer whose tumours have activating HER2 (ERBB2) mutations, as detected by an FDA-approved test, and who have received a prior systemic therapy based on the results from the DESTINY-Lung02 trial. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
Enhertu (6.4mg/kg) is approved in several countries for the treatment of adult patients with locally advanced or metastatic HER2-positive gastric or gastroesophageal junction adenocarcinoma who have received a prior trastuzumab-based regimen based on the results from the DESTINY-Gastric01 and/or the DESTINY-Gastric02 trial.
Enhertu development programme
A comprehensive global development programme is underway evaluating the efficacy and safety of Enhertu monotherapy across multiple HER2-targetable cancers including breast, gastric, lung and colorectal cancers. Trials in combination with other anticancer treatments, such as immunotherapy, are also underway.
Regulatory applications for Enhertu in breast, non-small cell lung and gastric cancer are currently under review in several countries.
Daiichi Sankyo collaboration
Daiichi Sankyo Company, Limited (TSE: 4568) [referred to as Daiichi Sankyo] and AstraZeneca entered into a global collaboration to jointly develop and commercialise Enhertu (a HER2-directed ADC) in March 2019, and datopotamab deruxtecan (DS-1062; a TROP2-directed ADC) in July 2020, except in Japan where Daiichi Sankyo maintains exclusive rights. Daiichi Sankyo is responsible for the manufacturing and supply of Enhertu and datopotamab deruxtecan.
AstraZeneca in breast cancer
Driven by a growing understanding of breast cancer biology, AstraZeneca is starting to challenge, and redefine, the current clinical paradigm for how breast cancer is classified and treated to deliver even more effective treatments to patients in need – with the bold ambition to one day eliminate breast cancer as a cause of death.
AstraZeneca has a comprehensive portfolio of approved and promising compounds in development that leverage different mechanisms of action to address the biologically diverse breast cancer tumour environment.
With Enhertu (trastuzumab deruxtecan), a HER2-directed ADC, AstraZeneca and Daiichi Sankyo are aiming to improve outcomes in previously treated HER2-positive and HER2-low metastatic breast cancer and are exploring its potential in earlier lines of treatment and in new breast cancer settings.
In HR-positive breast cancer, AstraZeneca continues to improve outcomes with foundational medicines Faslodex (fulvestrant) and Zoladex (goserelin) and aims to reshape the HR-positive space with ngSERD and potential new medicine camizestrant as well as a potential first-in-class AKT kinase inhibitor, capivasertib. AstraZeneca is also collaborating with Daiichi Sankyo to explore the potential of TROP2-directed ADC, datopotamab deruxtecan, in this setting.
PARP inhibitor Lynparza (olaparib) is a targeted treatment option that has been studied in early and metastatic breast cancer with an inherited BRCA mutation. AstraZeneca with MSD (Merck & Co., Inc. in the US and Canada) continue to research Lynparza in these settings and to explore its potential in earlier disease.
To bring much-needed treatment options to patients with triple-negative breast cancer, an aggressive form of breast cancer, AstraZeneca is evaluating the potential of datopotamab deruxtecan alone and in combination with immunotherapy Imfinzi (durvalumab), capivasertib in combination with chemotherapy, and Imfinzi in combination with other oncology medicines, including Lynparza and Enhertu.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Dec. 19, 2022 3:43 PM ET
By: Jonathan Block, SA News Editor1 Comment
tremelimumab
On 15 December 2022, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorisation for the medicinal product Imjudo, intended for the treatment of hepatocellular carcinoma. The applicant for this medicinal product is AstraZeneca AB.
Imjudo will be available as a 20 mg/ml concentrate for solution for infusion. The active substance of Imjudo is tremelimumab, a monoclonal antibody (ATC code: L01FX20). It binds to CTLA-4, which is primarily expressed on the surface of activated T lymphocytes, and thus enhances T-cell activation and proliferation, resulting in increased T-cell diversity and enhanced anti-tumour activity.
The benefit of Imjudo in combination with durvalumab is a significant improvement in overall survival compared with the standard of care, sorafenib, as observed in a randomised, open-label, multicentre Phase III study in patients with unresectable hepatocellular carcinoma. The most common side effects are rash, pruritus, diarrhoea, abdominal pain, AST increased, pyrexia, hypothyroidism, cough/productive cough, oedema peripheral and lipase increased.
The full indication is:
Imjudo in combination with durvalumab is indicated for the first line treatment of adults with advanced or unresectable hepatocellular carcinoma (HCC).
Imjudo should be prescribed by physicians experienced in the treatment of cancer.
Detailed recommendations for the use of this product will be described in the summary of product characteristics (SmPC), which will be published in the European public assessment report (EPAR) and made available in all official European Union languages after the marketing authorisation has been granted by the European Commission.
Dec. 16, 2022 7:01 AM ET
By: Ravikash, SA News Editor
December 16, 2022
- Approval marks fourth indication for VRAYLAR, backed by proven efficacy and well-established tolerability as an adjunctive treatment for major depressive disorder (MDD) with an antidepressant therapy (ADT), showing improvement in symptoms when compared to placebo + ADT
- Designed for specific mood disorders, VRAYLAR is now the first and only dopamine and serotonin partial agonist FDA-approved for the most common forms of depression – as an adjunctive treatment for MDD and the treatment of depressive episodes associated with bipolar I disorder
- About one in five U.S. adults will experience MDD during their lifetime, and many of them may have partial response to the treatment with an ADT
NORTH CHICAGO, Ill., Dec. 16, 2022 /PRNewswire/ -- AbbVie (NYSE: ABBV) today announced that the U.S. Food and Drug Administration (FDA) has approved VRAYLAR® (cariprazine) as an adjunctive therapy to antidepressants for the treatment of major depressive disorder (MDD) in adults. Supported by clinical data demonstrating efficacy and well-established tolerability, this additional indication provides a new option for adults who have a partial response to the treatment of an antidepressant.
Experience the interactive Multimedia News Release here: https://www.multivu.com/players/English/9107351-vraylar-cariprazine-fda-approval-major-depressive-disorder/
"Many living with major depressive disorder find that their ongoing antidepressant therapy doesn't offer meaningful relief from the symptoms they experience every day," said Thomas Hudson, M.D., senior vice president, research and development, chief scientific officer, AbbVie. "Today's approval of VRAYLAR provides an important new treatment option to meet a critical unmet medical need. AbbVie is committed to driving progress and advancing solutions for patients living with complex neuropsychiatric conditions."
MDD is one of the most common mental disorders in the U.S.; approximately one in five adults will experience this disorder during their lifetime.1 In a large U.S. study of adults with MDD, approximately 50 percent still had depressive symptoms with their first antidepressant.2 If some symptoms of depression persist while on an antidepressant, adding a different type of medication, often referred to as an adjunctive treatment, to the existing regimen may help.
"Patients with inadequate response to standard antidepressant medication are often frustrated by the experience of trying multiple medicines and still suffering from unresolved symptoms. Instead of starting over with another standard antidepressant, VRAYLAR works with an existing treatment and can help build on the progress already made," said Gary Sachs, MD, clinical vice president at Signant Health, associate clinical professor of psychiatry at Massachusetts General Hospital, and lead Phase 3 clinical trial investigator. "For adults living with major depressive disorder, because of inadequate improvement in response to standard antidepressants, VRAYLAR is an efficacious adjunctive treatment option with a well-characterized safety profile."
Cariprazine is marketed as VRAYLAR® in the U.S., and in addition to being approved as an adjunctive therapy to antidepressants for the treatment of MDD in adults, it is FDA-approved to treat adults with depressive, acute manic and mixed episodes associated with bipolar I disorder, as well as schizophrenia. Cariprazine is co-developed by AbbVie and Gedeon Richter Plc. More than 8,000 patients worldwide have been treated with cariprazine across more than 20 clinical trials evaluating the efficacy and safety of cariprazine for a broad range of psychiatric disorders.
"When we were in the early stages of development for cariprazine, we focused on designing a compound that covers a range of symptoms for mental health conditions and affects the dopamine D3 receptor," said István Greiner, Ph.D., research and development, director, Gedeon Richter. "While schizophrenia and bipolar manic and mixed episodes were the first indications in the U.S. market, we are thrilled to see the full potential of cariprazine unlocked with approvals in bipolar I depression, and now, as an antidepressant adjunct in major depressive disorder."
Highlights from the clinical program supporting the approval include:
About Study 3111-301-001
Study 3111-301-001 is a randomized, double-blind, placebo-controlled, multicenter trial with 751 participants conducted in the United States, Bulgaria, Estonia, Germany, Hungary, Ukraine and the United Kingdom. Following a screening period of up to 14 days, patients with an inadequate clinical response to their antidepressant monotherapy (ADT) were randomized into three treatment groups (1:1:1). The first group received cariprazine 1.5 mg/day + ADT, the second group received cariprazine 3.0 mg/day + ADT, and the third group received placebo + ADT. For six weeks, the medication was given once daily in addition to the ongoing ADT treatment. Patients treated with cariprazine 3.0 mg/day + ADT demonstrated improvement in MADRS total score at week six over placebo + ADT but did not meet statistical significance.
About Study RGH-MD-75
Study RGH-MD-75 is a randomized, double-blind, placebo-controlled, flexible-dose, outpatient, multicenter trial with 808 participants, conducted in the United States, Estonia, Finland, Slovakia, Ukraine and Sweden. After 7-14 days of screening and washout of prohibited medications, eligible patients entered an 8-week, double-blind treatment period in which they continued antidepressant treatment and were randomized (1:1:1) to adjunctive cariprazine 1-2 mg/day, cariprazine 2-4.5 mg/day, or placebo. Data from Study RGH-MD-75 were published in the Journal of Clinical Psychiatry.7 Patients treated with cariprazine 1-2 mg/day + ADT demonstrated improvement in MADRS total score at week eight over placebo + ADT but did not meet statistical significance.
About VRAYLAR® (cariprazine)
VRAYLAR is an oral, once-daily atypical antipsychotic approved as an adjunctive therapy to antidepressants for the treatment of major depressive disorder (MDD) in adults (1.5 or 3 mg/day), for the treatment of depressive episodes associated with bipolar I disorder (bipolar depression) in adults (1.5 or 3 mg/day), and for the acute treatment of adults with manic or mixed episodes associated with bipolar I disorder (3 to 6 mg/day). VRAYLAR is also approved for the treatment of schizophrenia in adults (1.5 to 6 mg/day).
While the mechanism of action of VRAYLAR is unknown, the efficacy of VRAYLAR is thought to be mediated through a combination of partial agonist activity at central dopamine D₂ and serotonin 5-HT1A receptors and antagonist activity at serotonin 5-HT2A receptors. Pharmacodynamic studies with VRAYLAR have shown that it may act as a partial agonist with high binding affinity at dopamine D3, dopamine D2, and serotonin 5-HT1A receptors. VRAYLAR demonstrated up to ~8-fold greater in vitro affinity for dopamine D3 vs D2 receptors. VRAYLAR also acts as an antagonist at serotonin 5-HT2B and 5-HT2A receptors with high and moderate binding affinity, respectively as well as it binds to the histamine H1 receptors. VRAYLAR shows lower binding affinity to the serotonin 5-HT2C and α1A- adrenergic receptors and has no appreciable affinity for cholinergic muscarinic receptors.8 The clinical significance of these in vitro data is unknown.
VRAYLAR is developed jointly by AbbVie and Gedeon Richter Plc, with AbbVie responsible for commercialization in the U.S., Canada, Japan, Taiwan and certain Latin American countries (including Argentina, Bolivia, Brazil, Chile, Colombia, Ecuador, Mexico, Peru and Venezuela).
Visit www.vraylar.com for more information.
VRAYLAR® (cariprazine) Uses and Important Safety Information
VRAYLAR is a prescription medicine used in adults:
About AbbVie in Mental Health
AbbVie is driving the pursuit of better mental health. Over the last 30 years, the company's scientists and clinicians have worked to tackle the complexity of mental illness and today offer a portfolio of medicines and a pipeline of innovation that spans depression, anxiety, bipolar I disorder, and schizophrenia. To learn more about AbbVie's work to support individuals throughout their mental health journey, please visit www.abbvie.com or follow @abbvie on Twitter, Facebook, Instagram, YouTube and LinkedIn.
About AbbVie
AbbVie's mission is to discover and deliver innovative medicines that solve serious health issues today and address the medical challenges of tomorrow. We strive to have a remarkable impact on people's lives across several key therapeutic areas: immunology, oncology, neuroscience, eye care, virology, women's health and gastroenterology, in addition to products and services across our Allergan Aesthetics portfolio. For more information about AbbVie, please visit us at www.abbvie.com.
Follow @abbvie on Twitter, Facebook, Instagram, YouTube and LinkedIn.
SOURCE AbbVie
Dec. 16, 2022 6:55 PM ET
By: Jonathan Block, SA News Editor2 Comments
December 15, 2022 6:45 am ET
RAHWAY, N.J.--(BUSINESS WIRE)-- AstraZeneca and Merck (NYSE: MRK), known as MSD outside of the United States and Canada, today announced that the U.S. Food and Drug Administration (FDA) has informed AstraZeneca that the agency will extend by three months the Prescription Drug User Fee Act (PDUFA) date for the pending supplemental new drug application (sNDA) for LYNPARZA in combination with abiraterone and prednisone or prednisolone for the treatment of adult patients with metastatic castration-resistant prostate cancer (mCRPC). The purpose of the extension is to provide further time for the full review of the submission. The companies will continue to work with the FDA to facilitate the completion of the agency’s review.
The sNDA for LYNPARZA in combination with abiraterone and prednisone or prednisolone is based on the Phase 3 PROpel trial, results of which were published in NEJM Evidence in June 2022. The application was granted priority review, and AstraZeneca and Merck are committed to working with the FDA to bring this treatment option to patients with mCRPC.
In November, the European Medicines Agency’s Committee for Medicinal Products for Human Use adopted a positive opinion recommending approval of LYNPARZA in combination with abiraterone and prednisone or prednisolone in the European Union (EU) for the treatment of adult patients with mCRPC for whom chemotherapy is not clinically indicated. This combination is also undergoing regulatory reviews in other countries.
Approved indications for LYNPARZA are not impacted by this review period extension. Among these, LYNPARZA is approved in the U.S. for patients with homologous recombination repair (HRR) gene-mutated mCRPC (BRCA-mutated and other HRR gene mutations) who have progressed following prior treatment with enzalutamide or abiraterone and in the EU, Japan and China for patients with BRCA-mutated mCRPC who have progressed following prior therapy that included a new hormonal agent (NHA). These approvals were based on the data from the Phase 3 PROfound trial.
About PROpel
PROpel is a randomized, double-blind Phase 3 trial testing the efficacy, safety and tolerability of LYNPARZA versus placebo when given in addition to abiraterone and prednisone or prednisolone in patients with mCRPC who had not received prior chemotherapy or NHAs in the mCRPC setting. The major efficacy outcome was radiographic progression-free survival as assessed by investigator per RECIST v1.1 and Prostate Cancer Working Group (bone) criteria. Overall survival was an additional efficacy outcome measure.
INDICATIONS for LYNPARZA in the United States
LYNPARZA is a poly (ADP-ribose) polymerase (PARP) inhibitor indicated:
First-Line Maintenance BRCAm Advanced Ovarian Cancer
For the maintenance treatment of adult patients with deleterious or suspected deleterious germline or somatic BRCA-mutated (gBRCAm or sBRCAm) advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in complete or partial response to first-line platinum-based chemotherapy. Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
First-Line Maintenance HRD-Positive Advanced Ovarian Cancer in Combination with Bevacizumab
In combination with bevacizumab for the maintenance treatment of adult patients with advanced epithelial ovarian, fallopian tube or primary peritoneal cancer who are in complete or partial response to first-line platinum-based chemotherapy and whose cancer is associated with homologous recombination deficiency (HRD)-positive status defined by either:
Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
Maintenance Recurrent Ovarian Cancer
For the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in complete or partial response to platinum-based chemotherapy.
Adjuvant Treatment of gBRCAm, HER2-Negative, High-Risk Early Breast Cancer
For the adjuvant treatment of adult patients with deleterious or suspected deleterious gBRCAm, human epidermal growth factor receptor 2 (HER2)-negative high-risk early breast cancer who have been treated with neoadjuvant or adjuvant chemotherapy. Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
gBRCAm, HER2-Negative Metastatic Breast Cancer
For the treatment of adult patients with deleterious or suspected deleterious gBRCAm, human epidermal growth factor receptor 2 (HER2)-negative metastatic breast cancer who have been treated with chemotherapy in the neoadjuvant, adjuvant, or metastatic setting. Patients with hormone receptor (HR)-positive breast cancer should have been treated with a prior endocrine therapy or be considered inappropriate for endocrine therapy. Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
First-Line Maintenance gBRCAm Metastatic Pancreatic Cancer
For the maintenance treatment of adult patients with deleterious or suspected deleterious gBRCAm metastatic pancreatic adenocarcinoma whose disease has not progressed on at least 16 weeks of a first-line platinum-based chemotherapy regimen. Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
HRR Gene-mutated Metastatic Castration-Resistant Prostate Cancer
For the treatment of adult patients with deleterious or suspected deleterious germline or somatic homologous recombination repair (HRR) gene-mutated metastatic castration-resistant prostate cancer (mCRPC) who have progressed following prior treatment with enzalutamide or abiraterone. Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
Please see complete Prescribing Information , including Medication Guide .
About LYNPARZA® (olaparib)
LYNPARZAis a first-in-class PARP inhibitor and the first targeted treatment to potentially exploit DNA damage response (DDR) pathway deficiencies, such as BRCA mutations, to preferentially kill cancer cells. Inhibition of PARP with LYNPARZA leads to the trapping of PARP bound to DNA single-strand breaks, stalling of replication forks, their collapse and the generation of DNA double-strand breaks and cancer cell death. LYNPARZA is being tested in a range of tumor types with defects and dependencies in the DDR.
LYNPARZA, which is being jointly developed and commercialized by AstraZeneca and Merck, has a broad clinical trial development program, and AstraZeneca and Merck are working together to understand how it may affect multiple PARP-dependent tumors as a monotherapy and in combination across multiple cancer types.
About the AstraZeneca and Merck strategic oncology collaboration
In July 2017, AstraZeneca and Merck, known as MSD outside of the United States and Canada, announced a global strategic oncology collaboration to co-develop and co-commercialize certain oncology products including LYNPARZA, the world’s first PARP inhibitor, for multiple cancer types. Working together, the companies will developthese products in combination with other potential new medicines and as monotherapies. Independently, the companies will develop these oncology products in combination with their respective PD-L1 and PD-1 medicines.
Merck’s focus on cancer
Our goal is to translate breakthrough science into innovative oncology medicines to help people with cancer worldwide. At Merck, the potential to bring new hope to people with cancer drives our purpose and supporting accessibility to our cancer medicines is our commitment. As part of our focus on cancer, Merck is committed to exploring the potential of immuno-oncology with one of the largest development programs in the industry across more than 30 tumor types. We also continue to strengthen our portfolio through strategic acquisitions and are prioritizing the development of several promising oncology candidates with the potential to improve the treatment of advanced cancers. For more information about our oncology clinical trials, visit www.merck.com/clinicaltrials.
About Merck
At Merck, known as MSD outside of the United States and Canada, we are unified around our purpose: We use the power of leading-edge science to save and improve lives around the world. For more than 130 years, we have brought hope to humanity through the development of important medicines and vaccines. We aspire to be the premier research-intensive biopharmaceutical company in the world – and today, we are at the forefront of research to deliver innovative health solutions that advance the prevention and treatment of diseases in people and animals. We foster a diverse and inclusive global workforce and operate responsibly every day to enable a safe, sustainable and healthy future for all people and communities. For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Source: Merck & Co., Inc.
Dec. 15, 2022 4:34 AM ET
AstraZeneca PLC (AZN), MRK, JNJ
By: Ravikash, SA News Editor
The U.S. Food and Drug Administration (FDA) will extend the review period by three months of AstraZeneca (NASDAQ:AZN) and Merck's (NYSE:MRK) Lynparza to treat a type of prostate cancer to have more time to review the companies' application.
CHMP recommends expansion of EU label for Hemlibra to include people with moderate haemophilia A
December 16, 2022
Basel, 16 December 2022 - Roche (SIX: RO, ROG; OTCQZ: RHHBY) today announced that the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) has recommended expansion of the Hemlibra® (emicizumab) European Union (EU) marketing authorisation. If approved, Hemlibra would also be indicated for the routine prophylaxis of bleeding episodes in people with haemophilia A (congenital factor VIII deficiency) without factor VIII inhibitors, who have moderate disease (FVIII ≥1% and ≤ 5%) with a severe bleeding phenotype. It is estimated that people with moderate haemophilia A make up 14% of the haemophilia A population.3
“We know that people with moderate haemophilia A can still have bleeds that cause irreversible joint damage and impact quality of life,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “We’re very pleased that the CHMP’s recommendation brings us closer to potentially transforming the day-to-day lives of people in the EU living with moderate haemophilia A.”
While the treatment and management of severe haemophilia A are well-established, there is less information and guidance on prophylaxis for moderate haemophilia A.4 Additionally, the severity of haemophilia A, traditionally measured by factor VIII levels, is not always reflective of bleeding behaviour. Some people with non-severe haemophilia may experience symptoms similar to those with severe haemophilia and would benefit from prophylaxis.5 All severities of haemophilia A may significantly reduce the quality of life for people affected, as well as their family and caregivers. 6 Given that many people with moderate haemophilia A may not receive prophylactic treatments, they may endure a worsened clinical burden, with 85% having bleeds within their lifetime. This can lead to long-term joint problems, requiring interventions and impacting quality of life.2,4,5
The CHMP recommendation is based on the results from the phase III HAVEN 6 study, as well as on real world data. The results from the study showed that Hemlibra demonstrated effective bleed control and a favourable safety profile in people with non-severe haemophilia A without factor VIII inhibitors, where prophylaxis was clinically indicated.1 A final decision regarding the approval of Hemlibra is expected from the European Commission in the near future. If approved, this update will provide an effective and convenient prophylactic treatment option with a favourable safety profile, for people in the EU with moderate haemophilia A with a severe bleeding phenotype.
Hemlibra is approved as a treatment for people with haemophilia A with factor VIII inhibitors in more than 110 countries worldwide and for people without factor VIII inhibitors in more than 100 countries worldwide. It has been studied in one of the largest clinical trial programmes in people with haemophilia A with and without factor VIII inhibitors, including eight phase III studies.
About HAVEN 6
HAVEN 6 is a phase III clinical study designed to evaluate the safety, efficacy, pharmacokinetics and pharmacodynamics of Hemlibra in people with non-severe haemophilia A without factor VIII inhibitors. Data from the primary analysis of HAVEN 6 was presented at the 30th International Society on Thrombosis and Haemostasis (ISTH) Annual Congress, on 11 July 2022.1 The analysis included data from 72 participants, including three women, 51 (70.8%) of whom had moderate haemophilia A without factor VIII inhibitors. The data indicate that Hemlibra has effective bleed control and a favourable safety profile in people with non-severe haemophilia A without factor VIII inhibitors, with no new safety signals identified. Hemlibra achieved clinically meaningful bleed control, with 66.7% of participants experiencing no bleeds that required treatment, 81.9% experiencing no spontaneous bleeds that required treatment, and 88.9% experiencing no joint bleeds that required treatment.1
About Hemlibra® (emicizumab)
Hemlibra is a bispecific factor IXa- and factor X-directed antibody. It is designed to bring together factor IXa and factor X, proteins involved in the natural coagulation cascade, and restore the blood clotting process for people with haemophilia A. Hemlibra is a prophylactic (preventative) treatment that can be administered by an injection of a ready-to-use solution under the skin (subcutaneously) once-weekly, every two weeks, or every four weeks (after an initial once-weekly dose for the first four weeks). Hemlibra was created by Chugai Pharmaceutical Co., Ltd. and is being co-developed globally by Chugai, Roche and Genentech. It is marketed in the United States by Genentech as Hemlibra (emicizumab-kxwh), with kxwh as the suffix designated in accordance with Nonproprietary Naming of Biological Products Guidance for Industry issued by the U.S. Food and Drug Administration.
About Roche in haematology
Roche has been developing medicines for people with malignant and non-malignant blood diseases for more than 20 years; our experience and knowledge in this therapeutic area runs deep. Today, we are investing more than ever in our effort to bring innovative treatment options to patients across a wide range of haematologic diseases. Our approved medicines include MabThera®/Rituxan® (rituximab), Gazyva®/Gazyvaro® (obinutuzumab), Polivy® (polatuzumab vedotin), Venclexta®/Venclyxto® (venetoclax) in collaboration with AbbVie, Hemlibra® (emicizumab) and Lunsumio® (mosunetuzumab). Our pipeline of investigational haematology medicines includes T-cell engaging bispecific antibodies glofitamab, targeting both CD20 and CD3, and cevostamab, targeting both FcRH5 and CD3; Tecentriq® (atezolizumab), a monoclonal antibody designed to bind with PD-L1 and crovalimab, an anti-C5 antibody engineered to optimise complement inhibition. Our scientific expertise, combined with the breadth of our portfolio and pipeline, also provides a unique opportunity to develop combination regimens that aim to improve the lives of patients even further.
About Roche
Founded in 1896 in Basel, Switzerland, as one of the first industrial manufacturers of branded medicines, Roche has grown into the world’s largest biotechnology company and the global leader in in-vitro diagnostics. The company pursues scientific excellence to discover and develop medicines and diagnostics for improving and saving the lives of people around the world. We are a pioneer in personalised healthcare and want to further transform how healthcare is delivered to have an even greater impact. To provide the best care for each person we partner with many stakeholders and combine our strengths in Diagnostics and Pharma with data insights from the clinical practice.
In recognizing our endeavor to pursue a long-term perspective in all we do, Roche has been named one of the most sustainable companies in the pharmaceuticals industry by the Dow Jones Sustainability Indices for the thirteenth consecutive year. This distinction also reflects our efforts to improve access to healthcare together with local partners in every country we work.
Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan.
For more information, please visit www.roche.com.
Dec. 16, 2022 9:51 AM ET
Roche Holding AG (RHHBF), RHHBY
By: Ravikash, SA News Editor

December 15, 2022 at 12:59 AM EST
Approval based on direct-to-Phase 3 program showing more than three times as many Dupixent patients (60% and 58%) experienced clinically meaningful itch reduction at 24 weeks compared to placebo (18% and 20%)
Dupixent also significantly reduced skin lesions and improved health-related quality of life compared to placebo
In Europe, about 70,000 adults living with prurigo nodularis are most in need of new treatment options
Dupixent now approved to treat four chronic inflammatory diseases in the EU
TARRYTOWN, N.Y. and PARIS, Dec. 15, 2022 /PRNewswire/ -- Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) and Sanofi today announced that the European Commission (EC) has expanded the marketing authorization for Dupixent® (dupilumab) in the European Union to treat adults with moderate-to-severe prurigo nodularis who are candidates for systemic therapy. Prurigo nodularis is a chronic, debilitating skin disease with underlying type 2 inflammation and its impact on quality of life is one of the highest among inflammatory skin diseases. With this approval, Dupixent is the first and only targeted medicine specifically indicated to treat prurigo nodularis in Europe and the U.S.
"For the first time, patients with prurigo nodularis in Europe have a medicine that can help relieve the burden of itchy and painful nodules covering their skin, which can have a devastating impact on their day-to-day lives, both physically and mentally," said George D. Yancopoulos, M.D., Ph.D., President and Chief Scientific Officer at Regeneron, and a principal inventor of Dupixent. "Dupixent is now approved for its second dermatological disease and fourth disease overall. We remain committed to further investigating this innovative medicine for diseases – such as chronic urticarias and chronic obstructive pulmonary disease – in which type 2 inflammation may play a role."
"As the first and only targeted medicine approved to treat people living with prurigo nodularis, Dupixent has the potential to transform the standard-of-care for people in Europe living with this debilitating skin disease. In the pivotal trials, patients treated with Dupixent experienced significant improvements in key hallmarks of the disease, such as reduction in itch and achieving clearer skin, as well as broader impacts on their daily lives," Naimish Patel, M.D. Head of Global Development, Immunology and Inflammation at Sanofi. "This approval of Dupixent underscores our continued commitment to bringing Dupixent to patients suffering from chronic skin diseases with underlying type 2 inflammation as quickly as possible."
The EC decision is based on data from two Phase 3 trials, PRIME and PRIME2, evaluating the efficacy and safety of Dupixent (PRIME n=75; PRIME2 n=78) in adults with uncontrolled prurigo nodularis compared to placebo (PRIME n=76; PRIME2 n=82). In these trials, 44% and 37% of Dupixent patients experienced a clinically meaningful reduction in itch at 12 weeks, compared to 16% and 22% for placebo, respectively. The improvement further increased at 24 weeks, with approximately three times as many Dupixent patients (60% and 58%) experiencing a clinically meaningful reduction in itch from baseline, compared to placebo (18% and 20%).
In PRIME and PRIME2, more than twice as many Dupixent patients (48% and 45%) also achieved clear or almost clear skin at 24 weeks, compared to placebo (18% and 16%). Dupixent also significantly improved health-related quality of life, while reducing measures of skin pain and symptoms of anxiety/depression from baseline at 24 weeks compared to placebo.
The safety results of the trial were generally consistent with the known safety profile of Dupixent in its approved indications, with the most common side effects across indications including injection site reactions, conjunctivitis, conjunctivitis allergic, arthralgia, oral herpes and eosinophilia. Adverse events more commonly observed in prurigo nodularis with Dupixent compared to placebo included conjunctivitis (4% Dupixent and 1% placebo).
About Prurigo Nodularis
Prurigo nodularis is a chronic, debilitating skin disease with underlying type 2 inflammation and has one of the highest impacts on a patient's quality of life among inflammatory skin diseases due to the extreme itch it causes. Those with prurigo nodularis experience intense, persistent itch with thick skin lesions (called nodules) that can cover most of the body. The disease is often painful – with burning, stinging and tingling of the skin – and can negatively affect mental health, activities of daily living and social interactions. High-potency topical steroids are commonly prescribed but are associated with safety risks if used long-term. In Europe, about 70,000 adults living with prurigo nodularis are most in need of new treatment options.
About the Dupixent Prurigo Nodularis Trials
The PRIME and PRIME2 Phase 3 double-blind, placebo-controlled trials evaluated the efficacy and safety of Dupixent in 311 adults with uncontrolled prurigo nodularis. In PRIME and PRIME2, the primary endpoint evaluated the proportion of patients with clinically meaningful improvement in itch from baseline (measured by a ≥4-point reduction in Worst-Itch Numeric Rating Scale [WI-NRS] on a 0-10 scale) at 24 and 12 weeks, respectively.
Additional endpoints included the proportion of patients with clear or almost clear skin of nodules at 24 weeks (measured by a score of 0 or 1 on the Investigator's Global Assessment PN-Stage [IGA PN-S] on a 0-4 scale), the proportion of patients who achieved a clinically meaningful response in both WI-NRS and IGA PN-S, improvement from baseline in health-related quality of life (measured by the Dermatology Life Quality Index [DLQI] on a 0-30 scale), improvement from baseline in skin pain (measured by a Numerical Rating Scale from 0-10), and improvement from baseline in symptoms of anxiety and depression (measured by the Hospital Anxiety and Depression Scale [HADS] from 0-42).
About Dupixent
Dupixent is an injection under the skin (subcutaneous injection) at different injection sites. In the EU for adults with prurigo nodularis, Dupixent is administered at 300 mg every two weeks, following a loading dose. It is available as both a pre-filled pen and pre-filled syringe at the 300 mg dose. Dupixent is intended for use under the guidance of a healthcare professional and can be given in a clinic or at home by self-administration after training by a healthcare professional.
Dupixent, which was invented using Regeneron's proprietary VelocImmune® technology, is a fully human monoclonal antibody that inhibits the signaling of the interleukin-4 (IL-4) and interleukin-13 (IL-13) pathways and is not an immunosuppressant. The Dupixent development program has shown significant clinical benefit and a decrease in type 2 inflammation in Phase 3 trials, establishing that IL-4 and IL-13 are key and central drivers of the type 2 inflammation that plays a major role in multiple related and often co-morbid diseases. These diseases include approved indications for Dupixent, such as atopic dermatitis, asthma, chronic rhinosinusitis with nasal polyposis (CRSwNP) and prurigo nodularis, as well as investigational disease eosinophilic esophagitis (EoE) in the EU.
Dupixent has received regulatory approvals in one or more countries around the world for use in certain patients with atopic dermatitis, asthma, CRSwNP, EoE or prurigo nodularis in different age populations. Dupixent is currently approved for one or more of these indications in more than 60 countries, including in Europe, the U.S. and Japan. More than 500,000 patients have been treated with Dupixent globally.
About Regeneron's VelocImmune Technology
Regeneron's VelocImmune technology utilizes a proprietary genetically engineered mouse platform endowed with a genetically humanized immune system to produce optimized fully human antibodies. When Regeneron's co-Founder, President and Chief Scientific Officer George D. Yancopoulos was a graduate student with his mentor Frederick W. Alt in 1985, they were the first to envision making such a genetically humanized mouse, and Regeneron has spent decades inventing and developing VelocImmune and related VelociSuite® technologies. Dr. Yancopoulos and his team have used VelocImmune technology to create approximately one in five of all original, FDA-approved or authorized fully human monoclonal antibodies. This includes REGEN-COV® (casirivimab and imdevimab), Dupixent, Libtayo® (cemiplimab-rwlc), Praluent® (alirocumab), Kevzara® (sarilumab), Evkeeza® (evinacumab-dgnb) and Inmazeb® (atoltivimab, maftivimab and odesivimab-ebgn).
Dupilumab Development Program
Dupilumab is being jointly developed by Regeneron and Sanofi under a global collaboration agreement. To date, dupilumab has been studied across more than 60 clinical trials involving more than 10,000 patients with various chronic diseases driven in part by type 2 inflammation.
In addition to the currently approved indications, Regeneron and Sanofi are studying dupilumab in a broad range of diseases driven by type 2 inflammation or other allergic processes in Phase 3 trials, including pediatric EoE, hand and foot atopic dermatitis, chronic inducible urticaria-cold, chronic spontaneous urticaria, chronic pruritus of unknown origin, chronic obstructive pulmonary disease with evidence of type 2 inflammation, chronic rhinosinusitis without nasal polyposis, allergic fungal rhinosinusitis, allergic bronchopulmonary aspergillosis and bullous pemphigoid. These potential uses of dupilumab are currently under clinical investigation, and the safety and efficacy in these conditions have not been fully evaluated by any regulatory authority.
U.S. INDICATIONS
DUPIXENT is a prescription medicine used:
Please see accompanying full Prescribing Information including Patient Information.
About Regeneron
Regeneron is a leading biotechnology company that invents, develops and commercializes life-transforming medicines for people with serious diseases. Founded and led for nearly 35 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to nine FDA-approved treatments and numerous product candidates in development, almost all of which were homegrown in our laboratories. Our medicines and pipeline are designed to help patients with eye diseases, allergic and inflammatory diseases, cancer, cardiovascular and metabolic diseases, pain, hematologic conditions, infectious diseases and rare diseases.
Regeneron is accelerating and improving the traditional drug development process through our proprietary VelociSuite® technologies, such as VelocImmune®, which uses unique genetically humanized mice to produce optimized fully human antibodies and bispecific antibodies, and through ambitious research initiatives such as the Regeneron Genetics Center®, which is conducting one of the largest genetics sequencing efforts in the world.
For more information, please visit www.Regeneron.com or follow @Regeneron on Twitter.
About Sanofi
We are an innovative global healthcare company, driven by one purpose: we chase the miracles of science to improve people's lives. Our team, across some 100 countries, is dedicated to transforming the practice of medicine by working to turn the impossible into the possible. We provide potentially life-changing treatment options and life-saving vaccine protection to millions of people globally, while putting sustainability and social responsibility at the center of our ambitions.
Sanofi is listed on EURONEXT: SAN and NASDAQ: SNY.
View original content:https://www.prnewswire.com/news-releases/dupixent-dupilumab-approved-by-european-commission-as-the-first-and-only-targeted-medicine-indicated-for-prurigo-nodularis-301703698.html
SOURCE Regeneron Pharmaceuticals, Inc.
Dec. 15, 2022 5:02 AM ET
Regeneron Pharmaceuticals, Inc. (REGN), SNY
By: Ravikash, SA News Editor
12/12/2022
– ADCETRIS in Combination with Nivolumab, Doxorubicin and Dacarbazine Delivers 88% Complete Response (CR) Rate in Advanced-Stage cHL and 92% CR Rate in Early-Stage cHL –
– 95% of Advanced-Stage Patients Remain Progression Free at 12 Months with 17 Months Median Follow-Up –
– Regimen Was Well-Tolerated in Both Cohorts with No Treatment-Related Deaths and Less Than Half of Patients in Either Group Developed Any Grade Peripheral Sensory Neuropathy –
BOTHELL, Wash.--(BUSINESS WIRE)-- Seagen Inc. (Nasdaq: SGEN) today announced results from two parts of a phase 2 trial (SGN35-027) evaluating ADCETRIS® (brentuximab vedotin) in combination with the PD-1 inhibitor nivolumab and standard chemotherapy agents doxorubicin and dacarbazine (AN+AD) for the frontline treatment of patients with classical Hodgkin lymphoma (cHL). Part B of the trial evaluated patients with advanced-stage disease and was presented as an oral presentation, and Part C evaluated patients with early-stage disease and was presented as a poster at the 64th American Society of Hematology (ASH) Annual Meeting and Exposition in New Orleans.
Results demonstrated a complete response (CR) rate of 88% and an overall response rate (ORR) of 93% in patients with advanced-stage disease (Part B), and a CR rate of 92% and an ORR of 95% in patients with early-stage disease (Part C). The data showed that the combination was well-tolerated, with no new safety signals observed.
“We are encouraged that ADCETRIS in combination with the immunotherapy nivolumab and standard chemotherapy agents showed promising efficacy and safety as a first-line treatment in patients with early- and advanced-stage classical Hodgkin lymphoma,” said Marjorie Green, M.D., Senior Vice President and Head of Late-Stage Development at Seagen. “These agents with complementary mechanisms of action warrant further investigation as a potential frontline treatment option for patients with Hodgkin lymphoma.”
Part B Phase 2 Study of Brentuximab Vedotin, Nivolumab, Doxorubicin, and Dacarbazine (AN+AD) for Advanced Stage Classic Hodgkin Lymphoma (Abstract #314)
Part B of SGN35-027 is evaluating the novel ADCETRIS combination in 57 patients with advanced-stage cHL (Stage II with bulky disease, Stage III or IV). Results showed:
Part C Phase 2 Study of Brentuximab Vedotin, Nivolumab, Doxorubicin, and Dacarbazine (AN+AD) for Early-Stage Classic Hodgkin Lymphoma (Abstract #4230)
Part C of SGN35-027 is evaluating the novel ADCETRIS combination in 125 patients with early-stage (non-bulky Stage I or II) cHL. Of 125 patients in the study, 76 were included at the time of efficacy assessment. Results showed:
About the SGN35-027 Clinical Study
SGN35-027 is an ongoing open-label, multiple part, multicenter, phase 2 clinical trial evaluating two brentuximab vedotin treatment combinations in patients with advanced and early-stage cHL. The trial includes three parts (Parts A, B, and C). Part A includes a combination of brentuximab vedotin and doxorubicin, vinblastine and dacarbazine (A+AVD), while Parts B and C include brentuximab vedotin in combination with nivolumab, doxorubicin, and dacarbazine (AN+AD). The primary endpoint for Part A is the proportion of patients with treatment-emergent febrile neutropenia. The primary endpoint for Parts B and C is the proportion of participants with complete response at end of treatment according to the Lymphoma Response to Immunomodulatory Therapy Criteria (LYRIC). Incidence of adverse events is a secondary endpoint for Parts B and C.
About ADCETRIS
ADCETRIS is an antibody-drug conjugate (ADC) directed to CD30, a defining marker of cHL and expressed on the surface of several types of lymphomas. It is approved in seven indications in the U.S.:
Seagen and Takeda jointly develop ADCETRIS. Under the terms of the collaboration agreement, Seagen has U.S. and Canadian commercialization rights, and Takeda has rights to commercialize ADCETRIS in the rest of the world. Seagen recognizes royalty revenues from Takeda based on a percentage of Takeda's net sales of ADCETRIS in its licensed territories based on annual net sales tiers. Seagen and Takeda are funding joint development costs for ADCETRIS on a 50:50 basis, except in Japan where Takeda is solely responsible for development costs.
ADCETRIS® (brentuximab vedotin) for injection U.S. Important Safety Information
BOXED WARNING
PROGRESSIVE MULTIFOCAL LEUKOENCEPHALOPATHY (PML): JC virus infection resulting in PML and death can occur in ADCETRIS-treated patients.
CONTRAINDICATION
Contraindicated with concomitant bleomycin due to pulmonary toxicity (e.g., interstitial infiltration and/or inflammation)
Please see full Prescribing information, including BOXED WARNING, for ADCETRIS here.
View source version on businesswire.com: https://www.businesswire.com/news/home/20221212005222/en/
Source: Seagen Inc.
Dec. 12, 2022 1:50 PM ET
By: Jonathan Block, SA News Editor
DECEMBER, 13, 2022 DOWNLOAD(OPENS IN NEW WINDOW)
mRNA-4157/V940, in combination with KEYTRUDA, demonstrated a statistically significant and clinically meaningful reduction in the risk of disease recurrence or death compared to KEYTRUDA monotherapy in stage III/IV melanoma patients with high risk of recurrence following complete resection
Results are the first demonstration of efficacy for an investigational mRNA cancer treatment in a randomized clinical trial
Companies plan to discuss results with regulatory authorities and initiate a Phase 3 study in melanoma in 2023 and rapidly expand to additional tumor types
CAMBRIDGE, MA and RAHWAY, NJ / ACCESSWIRE / December 13, 2022 / Moderna, Inc. (NASDAQ:MRNA), a biotechnology company pioneering messenger RNA (mRNA) therapeutics and vaccines, and Merck (NYSE:MRK), known as MSD outside of the United States and Canada, today announced that the Phase 2b KEYNOTE-942/mRNA-4157-P201 trial of mRNA-4157/V940, an investigational personalized mRNA cancer vaccine, in combination with KEYTRUDA®, Merck's anti-PD-1 therapy, demonstrated a statistically significant and clinically meaningful improvement in the primary endpoint of recurrence-free survival (RFS) versus KEYTRUDA alone for the adjuvant treatment of patients with stage III/IV melanoma following complete resection. Adjuvant treatment with mRNA-4157/V940 in combination with KEYTRUDA reduced the risk of recurrence or death by 44% (HR=0.56 [95% CI, 0.31-1.08]; one-sided p-value=0.0266) compared with KEYTRUDA alone.
"Today's results are highly encouraging for the field of cancer treatment. mRNA has been transformative for COVID-19, and now, for the first time ever, we have demonstrated the potential for mRNA to have an impact on outcomes in a randomized clinical trial in melanoma," said Stéphane Bancel, Moderna's Chief Executive Officer. "We will begin additional studies in melanoma and other forms of cancer with the goal of bringing truly individualized cancer treatments to patients. We look forward to publishing the full data set and sharing the results at an upcoming oncology medical conference, as well as with health authorities."
"These positive findings represent an important milestone in our collaboration with Moderna," said Dr. Dean Y. Li, president, Merck Research Laboratories. "Over the last six years, our teams have worked closely together combining our respective expertise in mRNA and immuno-oncology with a focus on improving outcomes for patients with cancer. We look forward to advancing this program into the next phase of development."
"The results of this randomized Phase 2b trial are exciting for the field. These data provide the first evidence that we can improve on the rates of recurrence-free survival achieved by PD-1 blockade in resected high-risk melanoma. These findings also provide the first randomized evidence that a personalized neoantigen approach may be beneficial in melanoma," said Jeffrey S. Weber, MD, PhD, principal investigator of the study and Deputy Director of the Perlmutter Cancer Center at NYU Langone. Dr. Weber is a paid consultant for Merck and Moderna.
Adverse events observed with mRNA-4157/V940 in KEYNOTE-942 were consistent with those previously reported in a Phase 1 clinical trial. The safety profile of KEYTRUDA was consistent with that observed in previously reported studies. Serious treatment-related adverse events occurred in 14.4% of patients who received the combination arm of mRNA-4157/V940 and KEYTRUDA versus 10% with KEYTRUDA alone.
The companies plan to discuss the results with regulatory authorities and initiate a Phase 3 study in melanoma patients in 2023.
In October of this year, the companies announced that Merck had exercised its option to jointly develop and commercialize mRNA-4157/V940. Merck and Moderna will share costs and any profits equally under this worldwide collaboration.
Personalized cancer vaccines are designed to prime the immune system so that a patient can generate a tailored antitumor response specific to their tumor mutation signature. mRNA-4157/V940 is designed to stimulate an immune response by generating specific T cell responses based on the unique mutational signature of a patient's tumor. KEYTRUDA is an immunotherapy that works by increasing the ability of the body's immune system to help detect and fight tumor cells. Based on early clinical studies, combining mRNA-4157/V940 with KEYTRUDA may potentially provide an additive benefit and enhance T cell-mediated destruction of tumor cells.
About mRNA-4157/V940
mRNA-4157/V940 is a novel investigational messenger ribonucleic acid (mRNA)-based personalized cancer vaccine consisting of a single synthetic mRNA coding for up to 34 neoantigens that is designed and produced based on the unique mutational signature of the DNA sequence of the patient's tumor. Upon administration into the body, the algorithmically derived and RNA-encoded neoantigen sequences are endogenously translated and undergo natural cellular antigen processing and presentation, a key step in adaptive immunity.
About KEYNOTE-942/mRNA-4157-P201 ( NCT03897881 )
KEYNOTE-942 is an ongoing randomized, open-label Phase 2b trial that enrolled 157 patients with stage III/IV melanoma. Following complete surgical resection, patients were randomized to receive mRNA-4157/V940 (nine total doses of mRNA-4157) and KEYTRUDA (200 mg every three weeks up to 18 cycles [for approximately one year]) versus KEYTRUDA alone for approximately one year until disease recurrence or unacceptable toxicity. The primary endpoint is recurrence-free survival, and secondary endpoints include distant metastasis-free survival and safety.
Key eligibility criteria for the trial included: patients with resectable cutaneous melanoma metastatic to a lymph node and at high risk of recurrence, patients with complete resection within 13 weeks prior to the first dose of KEYTRUDA, patients were disease free at study entry (after surgery) with no loco-regional relapse or distant metastasis and no clinical evidence of brain metastases, patients had a formalin fixed paraffin embedded (FFPE) tumor sample available suitable for sequencing, Eastern Cooperative Oncology Group (ECOG) Performance Status 0 or 1 and patients with normal organ and marrow function reported at screening.
About KEYTRUDA® (pembrolizumab) injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body's immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry's largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA®(pembrolizumab) Indications in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of adult and pediatric (12 years and older) patients with stage IIB, IIC, or III melanoma following complete resection.
See additional selected indications for KEYTRUDA in the U.S. after the Selected Important Safety Information
Selected Important Safety Information for KEYTRUDA
Severe and Fatal Immune-Mediated Adverse Reactions
KEYTRUDA is a monoclonal antibody that belongs to a class of drugs that bind to either the PD-1 or the PD-L1, blocking the PD-1/PD-L1 pathway, thereby removing inhibition of the immune response, potentially breaking peripheral tolerance and inducing immune-mediated adverse reactions. Immune-mediated adverse reactions, which may be severe or fatal, can occur in any organ system or tissue, can affect more than one body system simultaneously, and can occur at any time after starting treatment or after discontinuation of treatment. Important immune-mediated adverse reactions listed here may not include all possible severe and fatal immune-mediated adverse reactions.
Monitor patients closely for symptoms and signs that may be clinical manifestations of underlying immune-mediated adverse reactions. Early identification and management are essential to ensure safe use of anti-PD-1/PD-L1 treatments. Evaluate liver enzymes, creatinine, and thyroid function at baseline and periodically during treatment. For patients with TNBC treated with KEYTRUDA in the neoadjuvant setting, monitor blood cortisol at baseline, prior to surgery, and as clinically indicated. In cases of suspected immune-mediated adverse reactions, initiate appropriate workup to exclude alternative etiologies, including infection. Institute medical management promptly, including specialty consultation as appropriate.
Withhold or permanently discontinue KEYTRUDA depending on severity of the immune-mediated adverse reaction. In general, if KEYTRUDA requires interruption or discontinuation, administer systemic corticosteroid therapy (1 to 2 mg/kg/day prednisone or equivalent) until improvement to Grade 1 or less. Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Consider administration of other systemic immunosuppressants in patients whose adverse reactions are not controlled with corticosteroid therapy.
Immune-Mediated Pneumonitis
KEYTRUDA can cause immune-mediated pneumonitis. The incidence is higher in patients who have received prior thoracic radiation. Immune-mediated pneumonitis occurred in 3.4% (94/2799) of patients receiving KEYTRUDA, including fatal (0.1%), Grade 4 (0.3%), Grade 3 (0.9%), and Grade 2 (1.3%) reactions. Systemic corticosteroids were required in 67% (63/94) of patients. Pneumonitis led to permanent discontinuation of KEYTRUDA in 1.3% (36) and withholding in 0.9% (26) of patients. All patients who were withheld reinitiated KEYTRUDA after symptom improvement; of these, 23% had recurrence. Pneumonitis resolved in 59% of the 94 patients.
Pneumonitis occurred in 8% (31/389) of adult patients with cHL receiving KEYTRUDA as a single agent, including Grades 3-4 in 2.3% of patients. Patients received high-dose corticosteroids for a median duration of 10 days (range: 2 days to 53 months). Pneumonitis rates were similar in patients with and without prior thoracic radiation. Pneumonitis led to discontinuation of KEYTRUDA in 5.4% (21) of patients. Of the patients who developed pneumonitis, 42% interrupted KEYTRUDA, 68% discontinued KEYTRUDA, and 77% had resolution.
About Merck
At Merck, known as MSD outside of the United States and Canada, we are unified around our purpose: We use the power of leading-edge science to save and improve lives around the world. For more than 130 years, we have brought hope to humanity through the development of important medicines and vaccines. We aspire to be the premier research-intensive biopharmaceutical company in the world - and today, we are at the forefront of research to deliver innovative health solutions that advance the prevention and treatment of diseases in people and animals. We foster a diverse and inclusive global workforce and operate responsibly every day to enable a safe, sustainable and healthy future for all people and communities. For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
About Moderna
In over 10 years since its inception, Moderna has transformed from a research-stage company advancing programs in the field of messenger RNA (mRNA), to an enterprise with a diverse clinical portfolio of vaccines and therapeutics across seven modalities, a broad intellectual property portfolio in areas including mRNA and lipid nanoparticle formulation, and an integrated manufacturing plant that allows for rapid clinical and commercial production at scale. Moderna maintains alliances with a broad range of domestic and overseas government and commercial collaborators, which has allowed for the pursuit of both groundbreaking science and rapid scaling of manufacturing. Most recently, Moderna's capabilities have come together to allow the authorized use and approval of one of the earliest and most effective vaccines against the COVID-19 pandemic.
Moderna's mRNA platform builds on continuous advances in basic and applied mRNA science, delivery technology and manufacturing, and has allowed the development of therapeutics and vaccines for infectious diseases, immuno-oncology, rare diseases, cardiovascular diseases and auto-immune diseases. Moderna has been named a top biopharmaceutical employer by Science for the past eight years. To learn more, visit www.modernatx.com.
Moderna's Focus on Cancer
At Moderna, we are delivering on the promise of mRNA science to create a new generation of transformative medicines for patients. We are relentlessly working to grow our cancer therapeutic modality by discovering mRNA medicines that harness the body's immune system to identify and kill cancer cells in the same way the immune system identifies and targets infections. One example of a promising oncology candidate is the creation of individualized, mRNA-based personalized cancer vaccines that deliver individualized medicine to one patient at a time. We also continue to strengthen our portfolio through strategic collaborations that increase our potential to improve treatment options for patients with cancer.
SOURCE: Moderna,Inc.
View source version on accesswire.com:
https://www.accesswire.com/731571/Moderna-and-Merck-Announce-mRNA-4157V940-an-Investigational-Personalized-mRNA-Cancer-Vaccine-in-Combination-with-KEYTRUDAR-pembrolizumab-Met-Primary-Efficacy-Endpoint-in-Phase-2b-KEYNOTE-942-Trial
Dec. 13, 2022 8:17 AM ETMerck & Co
By: Ravikash, SA News Editor4 Comments
Merck (NYSE:MRK) and Moderna (NASDAQ:MRNA) said their mRNA cancer vaccine mRNA-4157/V940, in combination with Keytruda, met the main goal of a phase 2b study in certain patients with melanoma, a type of skin cancer.
The personalized cancer vaccine mRNA-4157/V940, in combination with Keytruda, showed a statistically significant and clinically meaningful improvement in the primary goal of recurrence-free survival (RFS), compared to Keytruda alone as an adjuvant treatment for patients with stage 3/4 melanoma following complete resection.
Dec 13, 2022
Basel, December 13, 2022 — Novartis today announced the European Commission (EC) approved Pluvicto® (INN: lutetium (177Lu) vipivotide tetraxetan), a targeted radioligand therapy. Pluvicto® is approved in combination with androgen deprivation therapy (ADT) with or without androgen receptor (AR) pathway inhibition, for the treatment of adult patients with prostate-specific membrane antigen (PSMA)–positive metastatic castration-resistant prostate cancer (mCRPC)3. These patients have been treated with AR pathway inhibition and taxane-based chemotherapy3.
The approval follows a positive opinion issued in October by the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) and is applicable to all 27 European Union member states plus Iceland, Norway, Northern Ireland and Liechtenstein.
The EC approval was granted based on results from the pivotal Phase III VISION trial, where participants, previously treated with AR pathway inhibition and taxane based chemotherapy, receiving Pluvicto® plus best standard of care (BSoC) had a 38% reduction in the risk of death and a statistically significant reduction (60%) in the risk of radiographic disease progression or death (rPFS) compared to BSoC alone1. About a third (30%) of patients with evaluable disease at baseline demonstrated an objective response (per RECIST 1.1) with Pluvicto® plus BSoC, compared to the 2% in the BSoC-alone arm1.
“Today’s approval of Pluvicto® by the European Commission marks a major milestone for patients with advanced prostate cancer who have few alternative treatments at this stage of their disease,” said Haseeb Ahmad, President Europe, Novartis. “We are excited by the potential of Pluvicto® to bring groundbreaking clinical benefits to these patients, transforming cancer care for the third-most diagnosed cancer globally4.”
Approximately 473,300 prostate cancer cases and 108,000 prostate cancer-related deaths occurred across Europe in 20205. Patients with metastatic prostate cancer have an approximate 3 in 10 chances of surviving 5 years6, indicating a high unmet need for new targeted treatment options for these patients2. Novartis is committed to addressing this need by reimagining cancer care with radioligand therapy and precision medicine, with a goal to reduce the global disease burden, extend the lives of patients with prostate cancer and elevate current standards of care.
About Pluvicto® (lutetium (177Lu) vipivotide tetraxetan)
Pluvicto® is an intravenous radioligand therapy combining a targeting compound (a ligand) with a therapeutic radionuclide (a radioactive particle, in this case lutetium-177)1. After administration into the bloodstream, Pluvicto® binds to target cells, including prostate cancer cells that express PSMA, a transmembrane protein1. Once bound, energy emissions from the radioisotope damage the target cells and nearby cells disrupting their ability to replicate and/or triggering cell death3.
Pluvicto® is approved in the US and other countries including Great Britain and Canada to treat adults with a type of advanced cancer called prostate-specific membrane antigen–positive metastatic castration-resistant prostate cancer (PSMA–positive mCRPC) and who have already been treated with other anticancer treatments (androgen receptor pathway inhibition and taxane-based chemotherapy)3,7,8.
Novartis is also evaluating opportunities to investigate Pluvicto® radioligand therapy in earlier stages of prostate cancer9,10.
About VISION
VISION is an international, prospective, randomized, open-label, multicenter, phase III study that assessed the efficacy and safety of Pluvicto® (lutetium (177Lu) vipivotide tetraxetan) (7.4 GBq administered by IV infusion every 6 weeks for a maximum of 6 cycles) plus investigator-chosen standard of care (BSoC) in the investigational arm, versus BSoC in the control arm1. Patients with PSMA PET-scan positive mCRPC who have received androgen receptor (AR) pathway inhibition and taxane-based chemotherapy, were randomized in a 2:1 ratio in favor of the investigational arm1. The alternate primary endpoints were rPFS and OS1. The study enrolled 831 patients1.
Novartis and Prostate Cancer
With more 1.4 million new cases and 375,000 deaths in 2020 alone, prostate cancer is the most frequently diagnosed cancer in men in 112 countries—more than half the world5.
At Novartis, we are harnessing the innovation of our world-class scientists, strategic partnerships, and one of the industry’s most competitive pipelines to explore the potential of new, targeted therapies and precision medicine platforms to address the greatest unmet needs in prostate cancer.
Through the bold science of targeted therapies, our goal is to reduce the global disease burden, extend the lives of patients with prostate cancer, and elevate current standards of care.
Novartis and Radioligand Therapy (RLT)
Novartis is reimagining cancer care with radioligand therapy for patients with advanced cancers. By harnessing the power of radioactive atoms and applying it to advanced cancers, RLT is theoretically able to deliver radiation to target cells anywhere in the body11,12. Novartis has established global expertise, specialized supply chain and manufacturing capabilities across its network of radioligand therapy production sites. In order to support growing demand for our RLT platform, we are investing in the expansion of our RLT production capabilities in Millburn, New Jersey (US), Zaragosa (Spain) and Ivrea (Italy), as well as building a new radioligand manufacturing facility in Indianapolis, Indiana (US), that is planned to be operational in 2023. We are continually evaluating additional opportunities to expand capacity.
About Novartis
Novartis is reimagining medicine to improve and extend people’s lives. We deliver high-value medicines that alleviate society’s greatest disease burdens through technology leadership in R&D and novel access approaches. In our quest to find new medicines, we consistently rank among the world’s top companies investing in research and development. About 108,000 people of more than 140 nationalities work together to bring Novartis products to nearly 800 million people around the world. Find out more at https://www.novartis.com
Novartis is on Twitter. Sign up to follow @Novartis at https://twitter.com/novartisnews
For Novartis multimedia content, please visit https://www.novartis.com/news/media-library
For questions about the site or required registration, please contact media.relations@novartis.com
Dec. 13, 2022 4:37 AM ET
By: Ravikash, SA News Editor
The European Commission (EC) approved Novartis (NYSE:NVS) Pluvicto to treat certain patients with advanced prostate cancer.
December 7, 2022 Download PDF
INGELHEIM, Germany and RIDGEFIELD, Conn. and INDIANAPOLIS, Dec. 7, 2022 /PRNewswire/ -- The DINAMO phase III clinical trial met its primary endpoint by demonstrating a statistically significant reduction in HbA1c (a marker of average blood sugar) with Jardiance® (empagliflozin) compared with placebo for children and adolescents aged 10-17 years living with type 2 diabetes. When Jardiance was added to other baseline treatments (diet, exercise, metformin and/or insulin) HbA1c was reduced by 0.84% compared with placebo at week 26 (95% CI –1.50 to –0.19; P=0.012). The results were presented today at the International Diabetes Federation (IDF) World Diabetes congress 2022, Boehringer Ingelheim and Eli Lilly and Company (NYSE: LLY) announced.
The DINAMO (DIabetes study of liNAgliptin and eMpagliflozin in children and adOlescents) trial included youth aged 10-17 years with type 2 diabetes and HbA1c ≥6.5% and ≤10.5%. Participants were randomly assigned treatment with Jardiance (10 or 25 mg) (n=52), Tradjenta® (linagliptin) (5 mg) (n=53) or placebo (n=53) once daily. All participants were treated with diet and exercise plus, when appropriate, metformin and/or insulin.
The overall safety data was generally consistent with previous findings in adults with type 2 diabetes, confirming the well-established safety profile of both Jardiance and Tradjenta.
"Across the lifespan, we know that people living with type 2 diabetes have a high risk for many diabetes complications, so it's important to recognize and treat diabetes early in its course," said Lori Laffel, M.D., principal investigator of the DINAMO study and chief of the Pediatric, Adolescent, and Young Adult Section at the Joslin Diabetes Center and professor of pediatrics at Harvard Medical School. "Today's results from the DINAMO global clinical trial demonstrated that the SGLT2 inhibitor Jardiance compared with placebo significantly improved overall blood sugar control in children and adolescents with type 2 diabetes. These findings are particularly important given the need for more therapeutic options, especially oral agents, to manage type 2 diabetes in young people as, to date, metformin is the only globally available oral treatment for youth."
A secondary endpoint from the trial showed that at week 26, Jardiance reduced fasting plasma glucose (–35.2 mg/dL; P=0.0035).
"With more than 41,000 new cases worldwide annually, type 2 diabetes in today's young people is a global public health issue, especially in light of the rise of risk factors such as obesity," said Lykke Hinsch Gylvin, M.D., chief medical officer at Boehringer Ingelheim. "The clinically meaningful benefit and consistent safety profile demonstrated with Jardiance in the DINAMO trial is an encouraging outcome for the vulnerable population of children and adolescents."
"The rise in the prevalence of type 2 diabetes in the pediatric population highlights a clear unmet need," said Jeff Emmick, M.D., Ph.D., vice president, Product Development, Eli Lilly & Company. "Having overcome the challenges in recruitment and design that pediatric trials tend to face, the outcomes of DINAMO represent another positive step forward in Boehringer Ingelheim and Lilly's commitment to improve the lives of people living with cardio-renal and metabolic conditions such as type 2 diabetes."
Reduction in HbA1c in participants treated with Tradjenta was not statistically significant when compared with placebo. A numerical reduction of 0.34% (P=0.2935) was observed.
The findings have been submitted for publication in a peer-reviewed journal.
About DINAMO: DIabetes study of liNAgliptin and eMpagliflozin in children and adOlescents
DINAMO (NCT03429543) enrolled children and adolescents aged 10-17 years with type 2 diabetes (HbA1c ≥6.5% and ≤10.5%). Of the 262 participants screened, 158 were randomly assigned to treatment with Jardiance (10 or 25 mg) (n=52), Tradjenta (5 mg) (n=53) or placebo (n=53) once daily. All participants were treated with diet and exercise plus metformin and/or insulin (or no background if metformin intolerant). The trial compared efficacy and safety of a Jardiance dosing regimen compared with placebo and Tradjenta compared with placebo on durable glycemic control in youth with type 2 diabetes.
What is JARDIANCE?
JARDIANCE is a prescription medicine used to:
JARDIANCE is not for people with type 1 diabetes. It may increase their risk of diabetic ketoacidosis (increased ketones in the blood or urine).
JARDIANCE is not for use to lower blood sugar in adults with type 2 diabetes who have severe kidney problems, because it may not work.
https://www.jardiance.com/type-2-diabetes/
For more information, please see Prescribing Information and Medication Guide.
CL-JAR-100131 10.17.2022
Boehringer Ingelheim and Eli Lilly and Company
In January 2011, Boehringer Ingelheim and Eli Lilly and Company announced an Alliance that centers on compounds representing several of the largest diabetes treatment classes. Depending on geographies, the companies either co-promote or separately promote the respective molecules each contributing to the Alliance. The Alliance leverages the strengths of two of the world's leading pharmaceutical companies to focus on patient needs. By joining forces, the companies demonstrate their commitment, not only to the care of people with diabetes, but also to investigating the potential to address areas of unmet medical need.
About Boehringer Ingelheim
Boehringer Ingelheim is working on breakthrough therapies that improve the lives of humans and animals. As a leading research-driven biopharmaceutical company, the company creates value through innovation in areas of high unmet medical need. Founded in 1885 and family-owned ever since, Boehringer Ingelheim takes a long-term perspective. Around 52,000 employees serve more than 130 markets in the three business areas, Human Pharma, Animal Health, and Biopharmaceutical Contract Manufacturing. Learn more at www.boehringer-ingelheim.us.
About Eli Lilly and Company
Lilly unites caring with discovery to create medicines that make life better for people around the world. We've been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world's most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer's disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we're motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram and LinkedIn.
This press release contains forward-looking statements (as that term is defined in the Private Securities Litigation Reform Act of 1995) about Jardiance® as a treatment for adults with type 2 diabetes, to reduce the risk of cardiovascular death in adults with type 2 diabetes and known cardiovascular disease, and to reduce the risk of cardiovascular death and hospitalization for heart failure in adults with heart failure, and as a potential treatment for adults with cardio-kidney-metabolic conditions and reflects Lilly's current beliefs and expectations. However, as with any pharmaceutical product, there are substantial risks and uncertainties in the process of drug research, development and commercialization. Among other things, there can be no guarantee that planned or ongoing studies will be completed as planned, that future study results will be consistent with the results to date or that Jardiance® will receive additional regulatory approvals. For a further discussion of these and other risks and uncertainties that could cause actual results to differ from Lilly's expectations, please see Lilly's most recent Forms 10-K and 10-Q filed with the U.S. Securities and Exchange Commission. Lilly undertakes no duty to update forward-looking statements.
Jardiance®, EMPEROR-Reduced®, EMPEROR-Preserved®, EMPA-REG OUTCOME® and EMPACT-MI® are registered trademarks of Boehringer Ingelheim.
![]()
![]() View original content to download multimedia:https://www.prnewswire.com/news-releases/phase-iii-trial-demonstrated-jardiance-is-the-first-sglt2-inhibitor-to-show-statistically-significant-reduction-in-blood-sugar-levels-in-children-and-adolescents-with-type-2-diabetes-301697025.html
View original content to download multimedia:https://www.prnewswire.com/news-releases/phase-iii-trial-demonstrated-jardiance-is-the-first-sglt2-inhibitor-to-show-statistically-significant-reduction-in-blood-sugar-levels-in-children-and-adolescents-with-type-2-diabetes-301697025.html
SOURCE Eli Lilly and Company
Dec. 07, 2022 8:48 AM ET
By: Dulan Lokuwithana, SA News Editor
Dec 08, 2022 PDF Version
NEW YORK, Dec. 08, 2022 (GLOBE NEWSWIRE) -- Y-mAbs Therapeutics, Inc. (the “Company” or “Y-mAbs”) (Nasdaq: YMAB) a commercial-stage biopharmaceutical company focused on the development and commercialization of novel, antibody-based therapeutic products for the treatment of cancer, today announced that the National Medical Products Administration (“NMPA”) in China has granted DANYELZA (naxitamab-gqgk) 40mg/10ml conditional approval. DANYELZA will be marketed in China by Y-mAbs’ partner SciClone Pharmaceuticals (Holdings) Limited (“SciClone Pharmaceuticals”).
DANYELZA is a humanized, monoclonal antibody that targets the ganglioside GD2, which is highly expressed in various neuroectoderm-derived tumors and sarcomas. DANYELZA is administered to patients three times in a week in an outpatient setting and the treatment is repeated every four weeks.
DANYELZA is indicated, in combination with granulocyte-macrophage colony-stimulating factor (“GM-CSF”), for the treatment of pediatric patients 1 year of age and older and adult patients with relapsed or refractory high-risk neuroblastoma in the bone or bone marrow who have demonstrated a partial response, minor response, or stable disease to prior therapy. Continued approval for this indication may be contingent upon verification and description of clinical benefits in a confirmatory trial, Study 201, where no less than 10 patients shall be enrolled in China.
“Today is an important day for children living with refractory/relapsed high-risk neuroblastoma in China. It’s very exciting to see this treatment conditionally approved in such a substantial territory after a solid joint effort by our Chinese partner SciClone Pharmaceuticals and the development team at Y-mAbs,” said Thomas Gad, founder, President and Interim CEO.
Researchers at Memorial Sloan Kettering Cancer Center (“MSK”) developed DANYELZA, which is exclusively licensed by MSK to Y-mAbs. MSK has institutional financial interests related to the compound and Y-mAbs.
About DANYELZA® (naxitamab-gqgk)
DANYELZA® (naxitamab-gqgk) is indicated, in combination with granulocyte-macrophage colony-stimulating factor (“GM-CSF”), for the treatment of pediatric patients 1 year of age and older and adult patients with relapsed or refractory high-risk neuroblastoma in the bone or bone marrow who have demonstrated a partial response, minor response, or stable disease to prior therapy. This indication was approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefits in a confirmatory trial. DANYELZA® includes a Boxed Warning for serious infusion-related reactions, such as cardiac arrest and anaphylaxis, and neurotoxicity, such as severe neuropathic pain and transverse myelitis. See full Prescribing Information for complete Boxed Warning and other important safety information.
About Y-mAbs
Y-mAbs is a commercial-stage biopharmaceutical company focused on the development and commercialization of novel, antibody-based therapeutic cancer products. In addition to conventional antibodies, the Company’s technologies include bispecific antibodies generated using the Y-BiClone platform and the SADA platform. The Company’s broad and advanced product pipeline includes one FDA-approved product, DANYELZA® (naxitamab-gqgk), which targets tumors that express GD2, and one product candidate at the registration-stage, OMBLASTYS® (omburtamab), which targets tumors that express B7-H3.
DANYELZA®, OMBLASTYS® and Y-mAbs® are registered trademarks of Y-mAbs Therapeutics, Inc.
![]()
Source: Y-mAbs Therapeutics, Inc
Dec. 08, 2022 9:45 AM ET
Y-mAbs Therapeutics, Inc. (YMAB)
By: Ravikash, SA News Editor
Dec 06, 2022
Basel, December 6, 2022 — Novartis today announced results from the RIGHT Choice Phase II trial evaluating Kisqali® (ribociclib) plus endocrine therapy (ET) against combination chemotherapy (CT) in the first-line setting for pre- and perimenopausal patients with aggressive forms of hormone receptor-positive, human epidermal growth factor receptor-2 negative (HR+/HER2−) metastatic breast cancer (MBC), including patients with visceral crisis. CT has remained the preferred option for patients with rapidly progressing disease and visceral crisis, despite the widespread adoption of CDK4/6 inhibitors (CDK4/6i) plus ET as first-line treatment for HR+/HER2- MBC. Kisqali demonstrated a nearly one-year progression-free survival (PFS) benefit in the study, supporting the superiority of Kisqali plus ET for this hard-to-treat patient population. RIGHT Choice is the first randomized study comparing a CDK4/6i plus ET vs. combination CT in aggressive HR+/HER2− MBC; data from this open-label, multi-national trial will be presented as a late-breaker oral presentation at the 2022 San Antonio Breast Cancer Symposium (SABCS) and included in the SABCS press program.
“Younger patients with aggressive disease often show resistance to treatment, resulting in worse prognoses – so it is encouraging to see RIGHT Choice data demonstrating a significant one-year benefit for this patient population when using ribociclib plus endocrine therapy compared to combination chemotherapy. Patients on the ribociclib arm had also lower rates of adverse events, such as diarrhea and fatigue, compared to chemotherapy, which could potentially impact quality of life,” said Dr. Yen-Shen Lu, Division Chief of Medical Oncology at Department of Oncology, National Taiwan University Hospital. “With these improvements in outcomes and tolerability, oncologists should consider ribociclib plus ET as a treatment option for patients with aggressive forms of HR+/HER2− MBC, including patients with visceral crisis.”
The study enrolled 222 patients with aggressive forms of HR+/HER2− MBC (i.e., with symptomatic visceral metastases, rapid disease progression or markedly symptomatic non-visceral metastases), including more than 50% of patients with visceral crisis as determined by investigators; Kisqali plus ET doubled the median PFS vs. combination CT at 24.0 months compared to 12.3 months (HR=0.54; 95% CI: 0.36-0.79; p=.0007) in the first-line setting. Median time to treatment failure with Kisqali plus ET was 18.6 months compared to 8.5 months with combination CT (HR=0.45; 95% CI: 0.32-0.63). Furthermore, patients in the Kisqali plus ET arm of the trial reported lower rates of treatment-related serious adverse events (AEs) and lower rates of discontinuation due to treatment-related AEs, compared to patients in the combination CT trial arm. Overall, the Kisqali safety profile was consistent with previously reported data1.
“Kisqali is a unique CDK4/6 inhibitor with the most robust evidence demonstrating overall survival and quality of life benefits for a wide spectrum of patients, including those with aggressive disease,” said Jeff Legos, Executive Vice President, Global Head of Oncology and Hematology at Novartis. “RIGHT Choice adds to the breadth of data that supports Kisqali as the first-line treatment of choice for patients with MBC, including those with visceral crisis.”
About Kisqali® (ribociclib)
Kisqali is the only CDK4/6 inhibitor with proven overall survival benefit across all three pivotal Phase III advanced breast cancer trials2-13 and is recognized by the National Comprehensive Cancer Network (NCCN) guidelines as the only CDK4/6i with overall survival benefit in first-line HR+/HER2- advanced breast cancer14. Additionally, Kisqali has the highest rating of any CDK4/6i on the ESMO Magnitude of Clinical Benefit Scale, achieving a score of five out of five for first-line premenopausal patients with HR+/HER2- advanced breast cancer15. Further, Kisqali in combination with either letrozole or fulvestrant has uniquely, among other CDK4/6i, received a score of four out of five for postmenopausal patients with HR+/HER2- advanced breast cancer treated in the first line16.
Kisqali has been approved in more than 95 countries worldwide, including by the United States Food and Drug Administration (FDA) and the European Commission, for the treatment of women with HR+/HER2- advanced or metastatic breast cancer in combination either with an aromatase inhibitor or with fulvestrant as initial endocrine-based therapy or following disease progression on endocrine therapy. Kisqali in combination with fulvestrant is approved as initial endocrine-based therapy or following disease progression on endocrine therapy in men by the FDA17.
Novartis is committed to continuing to study Kisqali in breast cancer. NATALEE is a large Phase III clinical trial of Kisqali plus endocrine therapy in the adjuvant treatment of HR+/HER2- early breast cancer being conducted in collaboration with Translational Research In Oncology (TRIO)18. Additionally, Novartis is collaborating with SOLTI, who is leading HARMONIA, to test whether Kisqali changes tumor biology to enable a better response to endocrine-based therapy compared to Ibrance®* for patients with advanced HR+/HER2-, HER2-enriched subtype19, and with the Akershus University Hospital in Norway on the NEOLETRIB trial, a neoadjuvant Phase II trial studying the effects of Kisqali in HR+/HER2- early breast cancer to discover the potentially unique underlying mechanism of action20.
Kisqali was developed by the Novartis Institutes for BioMedical Research (NIBR) under a research collaboration with Astex Pharmaceuticals.
Please see full Prescribing Information for Kisqali, available at www.Kisqali.com.
About Novartis in Advanced Breast Cancer
Novartis tackles breast cancer with superior science, collaboration and a passion for transforming patient care. We've taken a bold approach to our research by including patient populations often neglected in clinical trials, identifying new pathways or mutations that may play a role in disease progression and developing therapies that not only maintain, but also improve, quality of life for patients. Our priority over the past 30 years and today is to deliver treatments proven to improve and extend lives for those diagnosed with metastatic breast cancer.
About Novartis
Novartis is reimagining medicine to improve and extend people’s lives. As a leading global medicines company, we use innovative science and digital technologies to create transformative treatments in areas of great medical need. In our quest to find new medicines, we consistently rank among the world’s top companies investing in research and development. Novartis products reach nearly 800 million people globally and we are finding innovative ways to expand access to our latest treatments. About 108,000 people of more than 140 nationalities work at Novartis around the world. Find out more at https://www.novartis.com.
Novartis is on Twitter. Sign up to follow @Novartis at https://twitter.com/novartisnews
For Novartis multimedia content, please visit https://www.novartis.com/news/media-library
For questions about the site or required registration, please contact media.relations@novartis.com.
Dec. 07, 2022 5:16 AM ET
By: Ravikash, SA News Editor
Novartis (NYSE:NVS) on Tuesday said its drug Kisqali plus endocrine therapy (ET) showed a statistically significant progression-free survival (PFS) benefit of one year compared to combination chemotherapy (CT) in patients with a type of aggressive breast cancer in a phase 2 trial.
https://www.us.kisqali.com/metastatic-breast-cancer/
NOVEMBER 29, 2022 • INVESTOR RELATIONS
TOKYO and CAMBRIDGE, Eisai Co., Ltd. (Headquarters: Tokyo, CEO: Haruo Naito, "Eisai") and Biogen Inc. (Nasdaq: BIIB, Corporate headquarters: Cambridge, Massachusetts, CEO: Christopher A. Viehbacher, "Biogen") announced today that the results from Eisai’s large global Phase 3 confirmatory Clarity AD clinical study of lecanemab (development code: BAN2401), an investigational anti-amyloid beta (Aβ) protofibril antibody for the treatment of mild cognitive impairment (MCI) due to Alzheimer’s disease (AD) and mild AD (collectively known as early AD) with confirmed presence of amyloid pathology in the brain, were presented at the 2022 Clinical Trials on Alzheimer’s Disease (CTAD) conference, in San Francisco, California and virtually.
Summary of Presentations in the Scientific Session featuring Lecanemab at CTAD
Design of Clarity AD Study
Eisai’s Clarity AD was a global confirmatory Phase 3 placebo-controlled, double-blind, parallel-group, randomized study in 1,795 people with early AD (lecanemab group: 898 placebo group: 897) at 235 sites in North America, Europe, and Asia. The participants were randomized 1:1 to receive either placebo or lecanemab 10-mg/kg IV biweekly, and the randomization was stratified according to clinical subgroup (MCI due to AD or mild AD), presence or absence of concomitant approved AD symptomatic medication at baseline (e.g., acetylcholinesterase inhibitors, memantine, or both), ApoE4 status and geographical region. Eligibility criteria allowed patients with a broad range of comorbidities/comedications, including but not limited to hypertension, diabetes, heart disease, obesity, renal disease and anti-coagulants. As a result of Eisai’s recruitment strategy of diversity in the Clarity AD study, 4.5% and 22.5% of the randomized participants in the U.S. were Black and Hispanic, respectively.
The primary endpoint was change from baseline at 18 months in the CDR-SB1 (Clinical Dementia Rating Sum of Boxes), the global cognitive and functional scale, and key secondary endpoints were the change from baseline at 18 months in amyloid Positron Emission Tomography (PET) using Centiloids, AD Assessment Scale - Cognitive Subscale 14 (ADAS-Cog142), AD Composite Score (ADCOMS3) and AD Cooperative Study-Activities of Daily Living Scale for Mild Cognitive Impairment (ADCS MCI-ADL4). In addition, longitudinal changes in brain tau pathology as measured by tau PET (n=257) and cerebrospinal fluid (CSF) biomarkers of AD pathology (n=281) were evaluated in optional sub-studies.
Efficacy Results of Clarity AD
Mean change of CDR-SB from baseline at 18 months as the primary endpoint was 1.21 and 1.66 for lecanemab and placebo groups, respectively. Lecanemab treatment resulted in highly statistically significant results, reducing clinical decline on the global cognitive and functional scale, compared with placebo at 18 months by -0.45 (95% Confidence Interval (CI): -0.67, -0.23; P=0.00005), representing a 27% slowing of decline. Starting as early as six months (difference: -0.17 [95% CI: -0.29, -0.05]; P<0.01), and increasing in absolute difference over time across all time points every 3 months, the treatment showed highly statistically significant changes in CDR-SB from baseline compared to placebo (all p-values are less than 0.01) (Figure 1).
All key secondary endpoints also showed highly statistically significant results compared with placebo (P<0.001). In the amyloid PET sub-study, treatment with lecanemab showed statistically significant reduction in amyloid plaque burden at all timepoints starting at 3 months. Mean change in Centiloids at 18 months was -55.5 and 3.6 for lecanemab and placebo groups, respectively (mean difference: -59.1 [95%CI: -62.6, -55.6]; P<0.00001). Lecanemab slowed decline of cognitive function by 26% on ADAS-Cog14 at 18 months (mean difference: -1.44 [95%CI: -2.27, -0.61]; P=0.00065). In the ADCOMS assessment, lecanemab slowed disease progression by 24% at 18 months (mean difference: -0.050 [95% CI: -0.074, -0.027; P=0.00002]). Lecanemab slowed decline of activities of daily living by 37% on ADCS MCI-ADL at 18 months (mean difference: 2.016 [95%CI: 1.208, 2.823]; P<0.00001). In addition, the primary stratified analysis showed consistent results in CDR-SB, ADAS-Cog14 and ADCS MCI-ADL at 18 months of treatment with lecanemab in all subgroups of disease stage (MCI due to AD or mild AD), ApoE4 status (non-carriers, carriers), presence or absence of concomitant approved AD symptomatic medication, and region (North America, Asia, Europe).

Figure1. CDR-SB as Primary endpoint change (18 months)
Safety Results of Clarity AD
The most common adverse events (>10%) in the lecanemab group were infusion reactions (lecanemab: 26.4%; placebo: 7.4%), ARIA-H (combined cerebral microhemorrhages, cerebral macrohemorrhages, and superficial siderosis; lecanemab: 17.3%; placebo: 9.0%), ARIA-E (edema/effusion; lecanemab: 12.6%; placebo: 1.7%), headache (lecanemab: 11.1%; placebo: 8.1%), and fall (lecanemab: 10.4%; placebo: 9.6%). Infusion reactions were largely mild-to-moderate (grade 1-2: 96%) and occurred on the first dose (75%).
During the study period, deaths occurred in 0.7% and 0.8% of participants in the lecanemab and placebo groups, respectively and no deaths were related to lecanemab or occurred with amyloid-related imaging abnormalities (ARIA) in 18-month double-blind study period. Serious adverse events were experienced by 14.0% of participants in the lecanemab group and 11.3% of participants in the placebo group. Treatment-emergent adverse events occurred in 88.9% and 81.9% of participants in the lecanemab and placebo groups, respectively. Treatment-emergent adverse events leading to drug withdrawal occurred in 6.9% and 2.9% of participants in the lecanemab and placebo groups, respectively.
Overall, lecanemab’s ARIA incidence profile was within expectations based on the Phase 2 trial results. ARIA-E events were largely mild-to-moderate radiographically (91% of those who had ARIA-E), asymptomatic (78% of those who had ARIA-E), occurred within the first 3 months of treatment (71% of those who had ARIA-E) and resolved within 4 months of detection (81% of those who had ARIA-E). Among the 2.8% of lecanemab-treated subjects with symptomatic ARIA-E, the most commonly reported symptoms were headache, visual disturbance, and confusion. The incidence of symptomatic ARIA-H was 0.7% in the lecanemab group and 0.2% in the placebo group. No imbalance was observed in isolated ARIA-H (i.e., ARIA-H in participants who did not also experience ARIA-E) between lecanemab (8.9%) and placebo (7.8%). ARIA-E and ARIA-H were less common in ApoE4 non-carriers versus carriers, with higher frequency in ApoE4 homozygous carriers vs ApoE4 heterozygous carriers. In the core study and subsequent open-label extension study, rates of deaths with concurrent cerebral macrohemorrhage were 0.1% in both the placebo group (1/897) and the lecanemab group (2/1608). The two cases on lecanemab occurred in the open-label extension study. Both cases had significant comorbidities and risk factors including anticoagulation contributing to macrohemorrhage or death. Therefore, it is Eisai’s assessment that the deaths cannot be attributed to lecanemab.
Imaging, Plasma, and CSF Biomarkers Assessments of Clarity AD
Biomarkers assessments on amyloid, tau and neurodegeneration with lecanemab administration were conducted using imaging, plasma and CSF. Amyloid biomarkers showed early and sustained amyloid reversal effects in CSF and plasma Aβ 42/40 ratio with lecanemab treatment. Mean amyloid PET was 22.99 Centiloids at 18 months of lecanemab treatment which was below threshold for amyloid positivity of 30 Centiloids. Tau biomarkers showed that removing amyloid improved CSF and plasma p-tau (p-tau181), downstream of amyloid in the AD pathology pathway. Tau PET analysis showed that lecanemab treatment slowed tau accumulation in the temporal lobe as well as improved total tau (t-tau) in compared to the placebo. As for biomarkers of neurodegeneration, lecanemab improved glial fibrillary acidic protein (GFAP) in plasma, a marker of astrocyte activation, and neurogranin in CSF, a marker of synaptic dysfunction, improved to normal levels by treatment, while there was no significant difference in neurofilament light chains in CSF or plasma between lecanemab and placebo.
Clarity AD Results in Context
AD is a progressive neurological disorder that severely impacts people living with the condition and their loved ones. With the increased global aging population, AD has become a critical issue for society and healthcare systems. New therapeutic agents that act on the disease pathology are needed. The treatment goals for early AD are to have sustained effects on cognitive function, activities of daily living and psychiatric symptoms, to maintain independence longer by slowing progression of the disease and to improve or maintain quality of life.
In Eisai’s confirmatory Clarity AD study, lecanemab demonstrated consistency of results across scales of cognition and function and subgroups (race, ethnicity, comorbidities). Lecanemab treatment showed 31% lower risk of converting to next stage of disease by Global CDR assessment (Hazard Ratio: 0.69). A slope analysis using CDR-SB based on observed data and extrapolation to 30 months showed that lecanemab takes 25.5 months to reach same level as placebo at 18 months, indicating a 7.5 month slowing of progression. Modeling simulations based on the phase 2 trial data suggest that lecanemab may slow the rate of disease progression by 2.5-3.1 years and has the potential to help people remain in the earlier stages of AD for a longer period of time. In addition, it was shown to maintain the health-related quality of life and reduce the burden on caregivers (23-56% reduction in score worsening). The convergence of evidence across cognition and function, disease progression, health related quality of life, and caregiver burden demonstrate that lecanemab treatment may provide meaningful benefits to patients, their care partners, physicians and society.
Eisai is hosting a live webcast of the scientific session featuring the lecanemab presentations, which can be viewed live on the investors section of the Eisai Co., Ltd. website. The content will be available on demand afterward.
Eisai serves as the lead of lecanemab development and regulatory submissions globally with both Eisai and Biogen co-commercializing and co-promoting the product and Eisai having final decision-making authority.
This release discusses investigational uses of an agent in development and is not intended to convey conclusions about efficacy or safety. There is no guarantee that such an investigational agent will successfully gain health authority approval.
1 CDR-SB is a numeric scale used to quantify the various severity of symptoms of dementia. Based on interviews of people living with AD and family/caregivers, qualified healthcare professionals assess cognitive and functional performance in six areas: memory, orientation, judgment and problem solving, community affairs, home and hobbies, and personal care. The total score of the six areas is the score of CDR-SB, and CDR-SB is also used as an appropriate item for evaluating the effectiveness of therapeutic drugs targeting the early stages of AD.
2 ADAS-Cog is the most common cognitive assessment instrument used in AD clinical trials all over the world. ADAS-Cog14 consists of 14 competencies: word recall, commands, constructional praxis, object and finger naming, ideational praxis, orientation, word recognition, remembering word recognition instructions, comprehension of spoken language, word finding difficulty, spoken language ability, delayed word recall, number cancellation, and maze task. ADAS-Cog has been used in clinical trials for earlier stages of AD including MCI.
3 Developed by Eisai, ADCOMS combines items from the ADAS-Cog scale for assessing cognitive functions, MMSE and the CDR scale for evaluating the severity of dementia to enable highly sensitive detection of changes in clinical functions of early AD symptoms and changes in memory
4 ADCS MCI-ADL assesses the competence of patients with MCI in activities of daily living (ADLs), based on 24 questions to the patient’s partner about actual recent activities of daily living.
2. About Lecanemab
Lecanemab is an investigational humanized monoclonal antibody for AD that is the result of a strategic research alliance between Eisai and BioArctic. Lecanemab selectively binds to neutralize and eliminate soluble, toxic amyloid-beta (Aβ) aggregates (protofibrils) that are thought to contribute to the neurodegenerative process in AD. As such, lecanemab may have the potential to have an effect on disease pathology and to slow down the progression of the disease. Currently, lecanemab is being developed as the only anti- Aβ antibody that can be used for the treatment of early AD without the need for titration. The Phase 2 clinical study (Study 201) in early AD subjects demonstrated reduction of brain Aβ accumulation (P<0.0001) and slowing of disease progression measured by ADCOMS (P<0.05) based on the pre-specified analysis at 18 months of treatment. The study did not achieve its primary outcome measure* at 12 months of treatment. The Study 201 open-label extension was initiated after completion of the Core period and a Gap period off treatment of 9-59 months (average of 24 months, n=180 from core study enrolled) to evaluate safety and efficacy and is underway.
Since July 2020 the Phase 3 clinical study (AHEAD 3-45) for individuals with preclinical AD, meaning they are clinically normal and have intermediate or elevated levels of amyloid in their brains, is ongoing. AHEAD 3-45 is conducted as a public-private partnership between the Alzheimer’s Clinical Trial Consortium that provides the infrastructure for academic clinical trials in AD and related dementias in the U.S, funded by the National Institute on Aging, part of the National Institutes of Health, Eisai and Biogen.
Since January 2022, the Tau NexGen clinical study for Dominantly Inherited AD (DIAD), that is conducted by Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU), led by Washington University School of Medicine in St. Louis, is ongoing.
Furthermore, Eisai has initiated a lecanemab subcutaneous dosing Phase 1 study.
In July 2022, the U.S. Food and Drug Administration (FDA) accepted Eisai’s Biologics License Application (BLA) for lecanemab under the accelerated approval pathway and granted Priority Review. The Prescription Drug User Fee Act action date (PDUFA) is set for January 6, 2023. The FDA has agreed that the results of Clarity AD can serve as the confirmatory study to verify the clinical benefit of lecanemab. In an effort to secure traditional FDA approval for lecanemab as soon as possible, Eisai submitted the BLA through the FDA’s Accelerated Approval Pathway so that the agency could complete its review of all lecanemab data with the exception of the data from the confirmatory Clarity AD study. In March 2022, Eisai began submitting application data, with the exception of Clarity AD data, to Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) under the prior assessment consultation system. Eisai will discuss the results of Clarity AD study with regulatory authorities in the U.S., Japan and Europe with the aim to file for traditional approval in the U.S. and for marketing authorization applications in Japan and Europe by the end of Eisai’s FY2022, which ends March 31, 2023.
* An 80% or higher estimated probability of demonstrating 25% or greater slowing in clinical decline at 12 months treatment measured by ADCOMS from baseline compared to placebo.
3. About the Collaboration between Eisai and Biogen for AD
Eisai and Biogen have been collaborating on the joint development and commercialization of AD treatments since 2014. Eisai serves as the lead of lecanemab development and regulatory submissions globally with both companies co-commercializing and co-promoting the product and Eisai having final decision-making authority.
4. About the Collaboration between Eisai and BioArctic for AD
Since 2005, Eisai and BioArctic have had a long-term collaboration regarding the development and commercialization of AD treatments. Eisai obtained the global rights to study, develop, manufacture and market lecanemab for the treatment of AD pursuant to an agreement with BioArctic in December 2007. The development and commercialization agreement on the antibody lecanemab back-up was signed in May 2015.
5. About Eisai Co., Ltd.
Eisai’s Corporate Concept is “to give first thought to patients and people in the daily living domain, and to increase the benefits that health care provides.” Under this Concept (also known as human health care (hhc) Concept), we aim to effectively achieve social good in the form of relieving anxiety over health and reducing health disparities. With a global network of R&D facilities, manufacturing sites and marketing subsidiaries, we strive to create and deliver innovative products to target diseases with high unmet medical needs, with a particular focus in our strategic areas of Neurology and Oncology.
In addition, we demonstrate our commitment to the elimination of neglected tropical diseases (NTDs), which is a target (3.3) of the United Nations Sustainable Development Goals (SDGs), with working on various activities together with global partners.
For more information about Eisai, please visit www.eisai.com (for global headquarters: Eisai Co., Ltd.), and connect with us on Twitter @Eisai_SDGs.
6. About Biogen
As pioneers in neuroscience, Biogen discovers, develops, and delivers worldwide innovative therapies for people living with serious neurological diseases as well as related therapeutic adjacencies. One of the world’s first global biotechnology companies, Biogen was founded in 1978 by Charles Weissmann, Heinz Schaller, Sir Kenneth Murray, and Nobel Prize winners Walter Gilbert and Phillip Sharp. Today, Biogen has a leading portfolio of medicines to treat multiple sclerosis, has introduced the first approved treatment for spinal muscular atrophy, and developed the first and only approved treatment to address a defining pathology of Alzheimer’s disease. Biogen is also commercializing biosimilars and focusing on advancing one of the industry’s most diversified pipelines in neuroscience that will transform the standard of care for patients in several areas of high unmet need.
We routinely post information that may be important to investors on our website at www.biogen.com. Follow us on social media - Twitter, LinkedIn, Facebook, YouTube.
Nov. 29, 2022 8:58 PM ET
Eisai Co., Ltd. (ESALF), ESALY, BIIB
By: Jonathan Block, SA News Editor5 Comments
Fresh data on Biogen (NASDAQ:BIIB) and Eisai's (OTCPK:ESALY) (OTCPK:ESALF) experimental Alzheimer's treatment lecanemab indicated that while the biologic led to moderately less decline in cognition and function compared to placebo, it was also associated with higher rates of brain swelling and bleeding.
Eisai Co., Ltd. (ESALF), ESALY, BIIB
02 December 2022
Issued: London, UK
For media and investors only
GSK plc (LSE/NYSE: GSK) today announced positive headline results from the planned interim analysis of Part 1 of the RUBY/ENGOT-EN6/GOG3031/NSGO phase III trial investigating Jemperli (dostarlimab-gxly) plus standard-of-care chemotherapy (carboplatin-paclitaxel) followed by Jemperli compared to chemotherapy plus placebo followed by placebo in adult patients with primary advanced or recurrent endometrial cancer. The trial met its primary endpoint of investigator-assessed progression-free survival (PFS). It showed a statistically significant and clinically meaningful benefit in the prespecified mismatch repair deficient (dMMR)/microsatellite instability-high (MSI-H) patient subgroup and in the overall population. A clinically relevant benefit in PFS was also observed in the mismatch repair proficient (MMRp)/microsatellite stable (MSS) patient subgroup.
While the overall survival (OS) data were immature at the time of this analysis, a favorable trend was observed in the overall population, including both the dMMR/MSI-H and MMRp/MSS subgroups.
The safety and tolerability profile of dostarlimab-gxly in the RUBY phase III trial was consistent with clinical trials of similar regimens. The most common treatment-emergent adverse events in patients receiving dostarlimab-gxly plus chemotherapy were nausea, alopecia, fatigue, peripheral neuropathy, anemia, arthralgia, constipation and diarrhea.
Hesham Abdullah, Senior Vice President, Global Head of Oncology Development, GSK said: “Patients with primary advanced or recurrent endometrial cancer have limited treatment options. Long-term outcomes remain poor, and new treatment options are urgently needed to evolve the current standard of care, which is platinum-based chemotherapy. Based on these positive headline results from the RUBY phase III trial, GSK intends to seek regulatory approvals for a potential new indication for dostarlimab-gxly in the treatment of primary advanced or recurrent endometrial cancer.”
Regulatory submissions based on the trial results are anticipated in the first half of 2023. Full results from the trial will be published in a medical journal and presented at an upcoming scientific meeting.
RUBY is part of an international collaboration between the European Network of Gynaecological Oncological Trial groups (ENGOT), a research network of the European Society of Gynaecological Oncology (ESGO) that consists of 22 trial groups from 31 European countries that perform cooperative clinical trials, and the GOG Foundation, a non-profit organization dedicated to transforming the standard of care in gynecologic oncology.
About endometrial cancer
Endometrial cancer is found in the inner lining of the uterus, known as the endometrium. It is the most common gynecologic cancer in the US and the second most common gynecologic cancer globally.1 Approximately 15-20% of women with endometrial cancer will be diagnosed with advanced disease at the time of diagnosis.2
About RUBY
RUBY is a two-part global, randomized, double-blind, multicenter phase III trial of patients with primary advanced or recurrent endometrial cancer. Part 1 is evaluating dostarlimab-gxly plus carboplatin-paclitaxel followed by dostarlimab-gxly versus carboplatin-paclitaxel plus placebo followed by placebo. Part 2 is evaluating dostarlimab-gxly plus carboplatin-paclitaxel followed by dostarlimab-gxly plus niraparib versus placebo plus carboplatin-paclitaxel followed by placebo. The primary endpoints in Part 1 are investigator-assessed PFS based on the Response Evaluation Criteria in Solid Tumors v1.1 and OS. In Part 2, the primary endpoint is investigator-assessed PFS. Secondary endpoints in Part 1 and Part 2 include PFS per blinded independent central review, overall response rate, duration of response, disease control rate, patient-reported outcomes, and safety and tolerability.
About Jemperli (dostarlimab-gxly)
Jemperli is a programed death receptor-1 (PD-1)-blocking antibody that binds to the PD-1 receptor and blocks its interaction with the PD-1 ligands PD-L1 and PD-L2.3 Dostarlimab-gxly is being investigated in registrational enabling studies, as monotherapy and as part of combination regimens, including in women with recurrent or primary advanced endometrial cancer, women with Stage III or IV non-mucinous epithelial ovarian cancer, and patients with other advanced solid tumors or metastatic cancers.
Dostarlimab-glxy was discovered by AnaptysBio and licensed to TESARO, Inc., under a collaboration and exclusive license agreement signed in March 2014. The collaboration has resulted in three monospecific antibody therapies that have progressed into the clinic. These are: dostarlimab-gxly (GSK4057190), a PD-1 antagonist; cobolimab, (GSK4069889), a TIM-3 antagonist; and GSK4074386, a LAG-3 antagonist. GSK is responsible for the ongoing research, development, commercialization, and manufacturing of each of these medicines under the agreement.
Indications and Important Safety Information for JEMPERLI (dostarlimab-gxly)
JEMPERLI is indicated for the treatment of adult patients with mismatch repair deficient (dMMR) recurrent or advanced:
These indications are approved under accelerated approval based on tumor response rate and durability of response. Continued approval for these indications may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
Please see the full US Prescribing Information.
About GSK
GSK is a global biopharma company with a purpose to unite science, technology, and talent to get ahead of disease together. Find out more at us.gsk.com/en-us/company
References
Dec. 02, 2022 5:40 AM ET
By: Ravikash, SA News Editor
GSK (NYSE:GSK) said its medicine Jemperli (dostarlimab) met the main goal of progression-free survival (PFS) in patients with primary advanced or recurrent endometrial cancer in a phase 3 trial.
NOVEMBER 30, 2022
MHRA Approval follows European Commission marketing authorization of LUPKYNIS to treat adults with active lupus nephritis in 27 European Union Member States
LUPKYNIS is the first oral medicine approved in the U.S. and Europe for the treatment of adults living with active lupus nephritis
VICTORIA, British Columbia--(BUSINESS WIRE)--
Aurinia Pharmaceuticals Inc. (NASDAQ:AUPH) (Aurinia or the Company), a biopharmaceutical company committed to delivering therapeutics that change the trajectory of autoimmune disease, announced that the U.K. Medicines and Healthcare products Regulatory Agency (MHRA) has granted the Great Britain marketing authorization of LUPKYNIS® (voclosporin) to treat adults with active lupus nephritis (LN), a serious complication of systemic lupus erythematosus (SLE).
“The MHRA authorization of LUPKYNIS for individuals with lupus nephritis in Great Britain, on the heels of the marketing authorization in the EU, further expands the availability of LUPKYNIS as a treatment option for people with lupus nephritis,” said Peter Greenleaf, President and Chief Executive Officer, Aurinia. “Otsuka has been a valued partner in these efforts, and we are pleased to work with them to reach new patients.”
The marketing authorization by the MHRA, which follows the European Commission (EC) authorization on September 19, 2022, is based on the Phase 3 AURORA 1 study and the AURORA 2 continuation study, which demonstrated voclosporin, in combination with mycophenolate mofetil (MMF) and low-dose corticosteroids, led to statistically superior complete renal response rates at 52 weeks compared to MMF and low-dose corticosteroids alone. The safety profile of voclosporin and MMF and low-dose corticosteroids was generally comparable to MMF and low-dose corticosteroids alone.
In addition, a marketing authorization application for LUPKYNIS was submitted to the Swiss Agency for Therapeutic Products (Swissmedic) and is currently under review. Swissmedic previously granted orphan drug status to voclosporin in LN in February 2022.
Aurinia and Otsuka Pharmaceutical Co., Ltd., (Otsuka) entered a collaboration and licensing agreement in December 2020 for the development and commercialization of voclosporin for the treatment of LN in the United Kingdom, EU, Japan, Russia, Switzerland, Norway, Belarus, Iceland, Liechtenstein, and Ukraine.
About Lupus Nephritis
LN is a serious manifestation of SLE, a chronic and complex autoimmune disease. About 200,000-300,000 people live with SLE in the U.S. and about one-third of these people are diagnosed with lupus nephritis at the time of their SLE diagnosis. About 50 percent of all people with SLE may develop lupus nephritis. If poorly controlled, LN can lead to permanent and irreversible tissue damage within the kidney. Black and Asian individuals with SLE are four times more likely to develop LN and individuals of Hispanic ancestry are approximately twice as likely to develop the disease when compared with Caucasian individuals. Black and Hispanic individuals with SLE also tend to develop LN earlier and have poorer outcomes when compared to Caucasian individuals.
About LUPKYNIS
LUPKYNIS® is the first U.S. FDA- and EC-approved oral medicine for the treatment of adult patients with active LN. LUPKYNIS is a novel, structurally modified calcineurin inhibitor (CNI) with a dual mechanism of action, acting as an immunosuppressant through inhibition of T-cell activation and cytokine production and promoting podocyte stability in the kidney. The recommended starting dose of LUPKYNIS is three capsules twice daily with no requirement for serum drug monitoring. Dose modifications can be made based on Aurinia’s proprietary personalized eGFR-based dosing protocol. Boxed Warning, warnings, and precautions for LUPKYNIS are consistent with those of other CNI-immunosuppressive treatments.
About Aurinia
Aurinia Pharmaceuticals is a fully integrated biopharmaceutical company focused on delivering therapies to treat targeted patient populations that are impacted by serious diseases with a high unmet medical need. In January 2021, the Company introduced LUPKYNIS® (voclosporin), the first FDA-approved oral therapy dedicated for the treatment of adult patients with active lupus nephritis. The Company’s head office is in Victoria, British Columbia, its U.S. commercial office is in Rockville, Maryland. The Company focuses its development efforts globally.
INDICATION AND IMPORTANT SAFETY INFORMATION
INDICATIONS
LUPKYNIS is indicated in combination with a background immunosuppressive therapy regimen for the treatment of adult patients with active LN. Limitations of Use: Safety and efficacy of LUPKYNIS have not been established in combination with cyclophosphamide. Use of LUPKYNIS is not recommended in this situation.
Source: Aurinia Pharmaceuticals Inc.
Released November 30, 2022
Dec. 01, 2022 5:33 AM ET
Aurinia Pharmaceuticals Inc. (AUPH)OTSKF, OTSKY
By: Ravikash, SA News Editor1 Comment

November 30, 2022 at 8:05 AM EST
If approved, Evkeeza would be the first and only treatment of its kind to help children as young as 5 years old control dangerously high levels of LDL cholesterol caused by homozygous familial hypercholesterolemia
TARRYTOWN, N.Y., Nov. 30, 2022 /PRNewswire/ -- Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) today announced the U.S. Food and Drug Administration (FDA) has accepted for Priority Review the supplemental Biologics License Application (sBLA) for Evkeeza® (evinacumab-dgnb) as an adjunct to other lipid-lowering therapies to treat children aged 5 to 11 years with homozygous familial hypercholesterolemia (HoFH). The FDA target action date is March 30, 2023.
HoFH is an ultra-rare inherited condition that affects approximately 1,300 patients in the U.S. and is the most severe form of familial hypercholesterolemia (FH). The disease occurs when two copies of the FH-causing genes are inherited, one from each parent, resulting in dangerously high levels (usually >400 mg/dL) of low-density lipoprotein-cholesterol (LDL-C or bad cholesterol). Those living with HoFH are at risk for premature atherosclerotic disease and cardiac events even in their teenage years.
The sBLA is supported by data from a three-part trial evaluating Evkeeza in children aged 5 to 11 years with HoFH. Efficacy was assessed in 14 children enrolled in the Part B portion of the trial. Despite treatment with other lipid-lowering therapies, these children entered the trial with an average LDL-C level of 264 mg/dL, more than twice the target (<110 mg/dL) for pediatric patients with HoFH. The trial met its primary endpoint, showing children who added Evkeeza to other lipid-lowering therapies reduced their LDL-C by 48% at week 24 on average. Furthermore, 79% (n=11) saw their LDL-C reduced by at least half at 24 weeks following Evkeeza treatment, with an average absolute reduction in LDL-C from baseline of 132 mg/dL.
Among 20 children evaluated for long-term safety across Parts A, B and C of the trial (mean exposure: 52 weeks, range: 42-64 weeks), the most common adverse events (AEs) occurring in ≥15% of patients included COVID-19 (n=15), pyrexia (n=5), headache (n=4), throat pain (oropharyngeal pain, n=4) as well as upper abdominal pain, diarrhea, vomiting, fatigue, nasopharyngitis, rhinitis and cough (all n=3). Most reported AEs were mild or moderate, and none led to study discontinuation. The safety profile of Evkeeza observed in these patients was generally consistent to those seen in adults and pediatric patients aged 12 years and older.
Evkeeza is the first angiopoietin-like 3 (ANGPTL3) targeted therapy approved by the FDA, European Commission and the United Kingdom's Medicines and Healthcare products Regulatory Agency as an adjunct therapy for patients aged 12 years and older with HoFH. The potential use of Evkeeza in HoFH patients aged 5 to 11 years has not been fully evaluated by any regulatory authority.
About the Trial
The three-part, single-arm, open-label trial evaluated Evkeeza in pediatric patients with HoFH aged 5 to 11 years. Part A (n=6) was a Phase 1b trial designed to assess the pharmacokinetics (PK), safety and tolerability of Evkeeza.
Efficacy was evaluated in the 24-week treatment period in the Phase 3 Part B portion, which enrolled 14 patients with an average age of 9 years. Among them, 86% were on statins, 93% were on ezetimibe, 50% were on LDL apheresis and 14% were on lomitapide. Patients received Evkeeza 15 mg/kg every four weeks delivered intravenously alongside their lipid-lowering treatment regimen. The primary endpoint was change in LDL-C at week 24. Secondary endpoints included the effect of Evkeeza on other lipid parameters (i.e., apolipoprotein B, non-high-density lipoprotein cholesterol, lipoprotein[a] and total cholesterol), efficacy by mutation status, safety and tolerability, immunogenicity and PK.
Patients who completed Part A or B were allowed to continue treatment in Part C (n=20), an ongoing Phase 3 extension trial. Parts A, B and C were not designed to evaluate the effect of Evkeeza on cardiovascular events.
About Evkeeza® (evinacumab)
Evkeeza was invented using Regeneron's VelocImmune® technology and is a fully human monoclonal antibody that binds to and blocks the function of ANGPTL3, a protein that inhibits lipoprotein lipase (LPL) and endothelial lipase (EL) and regulates circulating lipids, including LDL-C.
Regeneron scientists discovered the angiopoietin gene family more than two decades ago. Human genetics research published in New England Journal of Medicine in 2017 by scientists from the Regeneron Genetics Center® found that patients whose ANGPTL3 gene did not function properly (called a "loss-of function mutation") have significantly lower levels of key blood lipids, including LDL-C, and that this is associated with a significantly lower risk of coronary artery disease.
The generic name for Evkeeza in its approved U.S. indications is evinacumab-dgnb, with dgnb the suffix designated in accordance with Nonproprietary Naming of Biological Products Guidance for Industry issued by the FDA. The safety and effectiveness of Evkeeza have not been established in patients with other causes of hypercholesterolemia, including those with heterozygous familial hypercholesterolemia (HeFH). The effect of Evkeeza on cardiovascular morbidity and mortality has not been determined.
Regeneron is responsible for the development and distribution of Evkeeza in the U.S. and is collaborating with Ultragenyx to clinically develop, commercialize and distribute Evkeeza outside of the U.S.
Please see full Prescribing Information, including Patient Information.
About Regeneron
Regeneron (NASDAQ: REGN) is a leading biotechnology company that invents, develops and commercializes life-transforming medicines for people with serious diseases. Founded and led for nearly 35 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to nine FDA-approved treatments and numerous product candidates in development, almost all of which were homegrown in our laboratories. Our medicines and pipeline are designed to help patients with eye diseases, allergic and inflammatory diseases, cancer, cardiovascular and metabolic diseases, pain, hematologic conditions, infectious diseases and rare diseases.
Regeneron is accelerating and improving the traditional drug development process through our proprietary VelociSuite® technologies, such as VelocImmune®, which uses unique genetically humanized mice to produce optimized fully human antibodies and bispecific antibodies, and through ambitious research initiatives such as the Regeneron Genetics Center, which is conducting one of the largest genetics sequencing efforts in the world.
For more information, please visit www.Regeneron.com or follow @Regeneron on Twitter.
View original content:https://www.prnewswire.com/news-releases/evkeeza-evinacumab-dgnb-sbla-for-children-with-ultra-rare-inherited-form-of-high-cholesterol-accepted-for-fda-priority-review-301690183.html
SOURCE Regeneron Pharmaceuticals, Inc.
Nov. 30, 2022 9:06 AM ET
Regeneron Pharmaceuticals, Inc. (REGN)RARE
By: Dulan Lokuwithana, SA News Editor
November 29, 2022
– EC Authorizes a Low-Dose Tablet for HIV Treatment in Virologically Suppressed Children at Least Two Years of Age and Weighing at Least 14 kg, Helping to Address a Critical Unmet Need –
– Biktarvy Provides an Effective Therapy Choice for a Diverse Range of People Living with HIV, including Children with Limited Treatment Options –
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the European Commission (EC) has authorized a new low-dose tablet dosage form of Biktarvy® (bictegravir 30 mg/emtricitabine 120 mg/tenofovir alafenamide 15 mg tablets) and an extension of the indication for Biktarvy to treat HIV infection in virologically suppressed children who are at least two years of age and weigh at least 14 kg. The European Marketing Authorization is the first pediatric approval for Biktarvy in the European Union (EU) and applies to all 27 member states of the EU, as well as Norway, Iceland and Liechtenstein.
“The European Commission’s approval is a significant milestone to address what is sadly an important unmet need, namely children with HIV requiring new treatment options,” said Jared Baeten, MD, PhD, Vice President, HIV Clinical Development, Gilead Sciences. “Additional therapy choices help to ensure children can access care and expand their HIV treatment options, which helps advance the collective efforts to overcome the HIV epidemic. Through the Gilead Global Pediatric Center of Excellence, we are committed to applying our decades of antiviral expertise to drive innovation in pediatric HIV research.”
While there have been many advances in the treatment of HIV in children and adolescents, there remains a need to prioritize, evaluate and develop options for the millions of the children worldwide. In 2021, an estimated 800,000 children under the age of 19 living with HIV were still not receiving HIV treatment. Children comprised 4% of people with HIV in 2021 but 15% of AIDS-related deaths, and the gap in HIV treatment coverage between children and adults is increasing rather than narrowing.
The authorization of an extended indication and line extension for Biktarvy for the treatment of HIV in children at least two years of age and weighing at least 14 kg is based on an open-label study (NCT02881320), which found Biktarvy to be effective and generally well-tolerated through 24 weeks in virologically suppressed adolescents and children with HIV. In Study 1474, treatment outcomes with Biktarvy were evaluated in virologically suppressed adolescents between the ages of 12 to less than 18 years weighing at least 35 kg (treatment cohort 1, N=50) and in virologically suppressed children between the ages of 6 to less than 12 years weighing at least 25 kg (treatment cohort 2, N=50). Treatment outcomes with Biktarvy were also evaluated in virologically suppressed children at least 2 years of age and weighing at least 14 kg to less than 25 kg (treatment cohort 3, N=22). At Week 2 or Week 4, select study participants from the three treatment cohorts underwent an intensive pharmacokinetic (PK) evaluation to confirm the dose to be administered in each treatment cohort. All participants from the three treatment cohorts went on to receive Biktarvy for 48 weeks. Beyond Week 48, participants can receive Biktarvy in an active open-label extension phase for up to 96 weeks.
After switching to Biktarvy, 98% (49/50) of participants in treatment cohort 1 remained suppressed (HIV-1 RNA < 50 copies/mL) at Week 48. After switching to Biktarvy, 98% (49/50) of participants in treatment cohort 2 remained suppressed (HIV-1 RNA < 50 copies/mL) at Week 48. After switching to Biktarvy, 91% (20/22) of participants in treatment cohort 3 remained virologically suppressed at Week 24. In this study, no new adverse reactions have been observed in pediatric subjects aged 2 years and older living with HIV-1 as compared to those observed in adults.
Biktarvy is indicated in the EU for the treatment of HIV infection in adults and paediatric patients at least 2 years of age and weighing at least 14 kg without present or past evidence of viral resistance to the integrase inhibitor class, emtricitabine or tenofovir. For important safety information for Biktarvy, including dosing and method of administration, special warnings, drug interactions and adverse drug reactions, please see the Summary of Product Characteristics (SmPC) for Biktarvy, available from the European Medicines Agency website at www.ema.europa.eu.
Biktarvy does not cure HIV or AIDS.
About Biktarvy
Biktarvy is a complete HIV treatment that combines three powerful medicines to form the smallest 3-drug, integrase strand transfer inhibitor (INSTI)-based single-tablet regimen (STR) available, offering simple once-daily dosing with or without food, with a limited drug interaction potential and a high barrier to resistance. Biktarvy combines the novel, unboosted INSTI bictegravir, with the Descovy® (emtricitabine 200 mg/tenofovir alafenamide 25 mg tablets, F/TAF) backbone. Biktarvy is a complete STR and should not be taken with other HIV medicines.
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer.
For more than 30 years, Gilead has been a leading innovator in the field of HIV, driving advances in treatment, prevention and cure research. Gilead researchers have developed 12 HIV medications, including the first single-tablet regimen to treat HIV, and the first antiretroviral for pre-exposure prophylaxis (PrEP) to reduce the risk of acquiring HIV infection, and the first, long-acting injectable HIV treatment medication administered twice-yearly. Our advances in medical research have helped to transform HIV into a preventable, chronic condition for millions of people.
Gilead is committed to continued scientific innovation to provide solutions for the evolving needs of people affected by HIV around the world. Through partnerships and collaborations, the company also aims to improve education, expand access and address barriers to care, with the goal of ending the HIV epidemic for everyone, everywhere. Gilead was recognized as the number one philanthropic funder of HIV-related programs in a report released by Funders Concerned About AIDS.
Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California.
View source version on businesswire.com: https://www.businesswire.com/news/home/20221128005858/en/
Source: Gilead Sciences, Inc.
Nov. 29, 2022 10:10 AM ET
By: Anuron Mitra, SA News Editor1 Comment
November 23, 2022
- Third approved indication for SKYRIZI® (risankizumab) and the first specific IL-23 inhibitor for the treatment of Crohn's disease in the European Union (EU)
- A significantly higher proportion of patients on SKYRIZI achieved clinical remission, endoscopic response, mucosal healing and endoscopic remission at week 12 in induction studies compared to placebo1,2,3
- A significantly higher proportion of patients achieved clinical remission and endoscopic response at week 52 with SKYRIZI maintenance1,2,3
- Crohn's disease is a chronic, systemic inflammatory disease that manifests as inflammation within the gastrointestinal tract, causing persistent diarrhea, abdominal pain and can require urgent medical care4,5,6
NORTH CHICAGO, Ill., Nov. 23, 2022 /PRNewswire/ -- AbbVie (NYSE: ABBV) announced the European Commission (EC) approved SKYRIZI® (risankizumab, 600 mg intravenous [IV] induction and 360 mg subcutaneous [SC] maintenance therapy) as the first specific interleukin-23 (IL-23) inhibitor for the treatment of adults with moderately to severely active Crohn's disease who have had inadequate response, lost response or were intolerant to conventional or biologic therapy.1,2,3
"There are still many patients suffering from debilitating symptoms associated with Crohn's disease, such as abdominal pain and stool frequency, which is why we've embraced the challenge of serving these patients in need," said Thomas Hudson, M.D., senior vice president, research and development, chief scientific officer, AbbVie. "The approval of SKYRIZI in the European Union is a significant milestone in our pursuit to expand our IBD portfolio."
The EC approval for SKYRIZI in Crohn's disease is supported by results from the global Phase 3 program, which included three studies: ADVANCE induction, MOTIVATE induction and FORTIFY maintenance.1 The three Phase 3 studies are multicenter, randomized, double-blind, placebo-controlled studies and include assessments of efficacy, safety and tolerability of SKYRIZI.1,2,3
Clinical Remission & Endoscopic Response*,†
Mucosal Healing & Endoscopic Remission‡,§
"Beyond managing daily symptoms, clinical remission and endoscopic goals are key treatment targets in Crohn's disease," said Marc Ferrante, M.D., Ph.D., Department of Gastroenterology and Hepatology, University Hospitals Leuven, Belgium. "Research advancements have made it possible for patients to aim for higher treatment goals, including mucosal healing. The approval of SKYRIZI as the first IL-23 inhibitor for moderate to severe Crohn's disease is a critical step forward towards a treatment option that can support a patient's health goals."
Additionally, across all three studies, safety results of SKYRIZI in Crohn's disease were consistent with the known safety profile of SKYRIZI, with no new safety risks observed.1,2,3 In ADVANCE, the most common adverse events (AEs) observed in the SKYRIZI treatment groups were headache, nasopharyngitis and fatigue.2 In MOTIVATE, the most common AEs observed were headache, arthralgia and nasopharyngitis.2 In FORTIFY, the most common AEs were exacerbation of Crohn's disease, nasopharyngitis and arthralgia.3
SKYRIZI is also approved in the EU for the treatment of adults with psoriasis and psoriatic arthritis.
SKYRIZI is part of a collaboration between Boehringer Ingelheim and AbbVie, with AbbVie leading development and commercialization globally.
About the ADVANCE Induction, MOTIVATE Induction & FORTIFY Maintenance Studies2,3
The three Phase 3 studies are multicenter, randomized, double-blind, placebo-controlled studies to evaluate the efficacy and safety of SKYRIZI 600 mg and 1200 mg as induction therapy, and SKYRIZI 180 mg and 360 mg as maintenance therapy in subjects with moderately to severely active Crohn's disease. More information can be found on www.clinicaltrials.gov (ADVANCE: NCT03105128; MOTIVATE: NCT03104413, FORTIFY: NCT03105102).
About SKYRIZI® (risankizumab)
SKYRIZI is an interleukin-23 (IL-23) inhibitor that selectively blocks IL-23 by binding to its p19 subunit.7 IL-23, a cytokine involved in inflammatory processes, is thought to be linked to a number of chronic immune-mediated diseases.7 Phase 3 trials of SKYRIZI in psoriasis, psoriatic arthritis, Crohn's disease and ulcerative colitis are ongoing.8,9,10
EU Indications and Important Safety Information about SKYRIZI® (risankizumab)1
Indications
Skyrizi (risankizumab) is indicated for the treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy.
Skyrizi, alone or in combination with methotrexate (MTX), is indicated for the treatment of active psoriatic arthritis in adults who have had an inadequate response or who have been intolerant to one or more disease-modifying antirheumatic drugs (DMARDs).
Skyrizi is indicated for the treatment of adult patients with moderately to severely active Crohn's disease who have had an inadequate response to, lost response to, or were intolerant to conventional therapy or a biologic therapy.
About AbbVie
AbbVie's mission is to discover and deliver innovative medicines that solve serious health issues today and address the medical challenges of tomorrow. We strive to have a remarkable impact on people's lives across several key therapeutic areas: immunology, oncology, neuroscience, eye care, virology, women's health and gastroenterology, in addition to products and services across our Allergan Aesthetics portfolio. For more information about AbbVie, please visit us at www.abbvie.com. Follow @abbvie on Twitter, Facebook, LinkedIn or Instagram.
![]()
SOURCE AbbVie

Nov. 23, 2022 5:43 AM ET
By: Ravikash, SA News Editor2 Comments
November 22, 2022 6:45 am ET
In KEYNOTE-859, KEYTRUDA® (pembrolizumab) combined with chemotherapy demonstrated statistically significant overall survival, progression-free survival, and overall response rate for patients regardless of PD-L1 expression
RAHWAY, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside of the United States and Canada, today announced positive topline results from the pivotal Phase 3 KEYNOTE-859 trial investigating KEYTRUDA, Merck’s anti-PD-1 therapy, in combination with chemotherapy for the first-line treatment of patients with human epidermal growth factor receptor 2 (HER2)-negative locally advanced unresectable or metastatic gastric or GEJ adenocarcinoma. KEYTRUDA in combination with chemotherapy showed a statistically significant and clinically meaningful improvement in the trial’s primary endpoint of overall survival (OS) versus chemotherapy alone in the all-randomized patient population at a pre-specified interim analysis conducted by an independent Data Monitoring Committee. Statistically significant and clinically meaningful improvements in progression-free survival (PFS) and overall response rate (ORR) were also observed in the all-randomized patient population.
The safety profile of KEYTRUDA in this trial was consistent with that observed in previously reported studies; no new safety signals were identified. Results will be presented at an upcoming medical meeting and will be submitted to regulatory authorities.
“Despite improvements in cancer care, advanced gastric cancer continues to have one of the lowest five-year survival rates, and new interventions are urgently needed. The results from KEYNOTE-859 show the potential of KEYTRUDA plus chemotherapy to improve survival beyond chemotherapy alone for patients with HER2-negative locally advanced unresectable or metastatic gastric or gastroesophageal junction adenocarcinoma, regardless of PD-L1 expression,” said Dr. Eliav Barr, senior vice president and head of global clinical development, chief medical officer, Merck Research Laboratories. “We are excited by these new results that demonstrate our commitment to exploring new treatment options for patients fighting gastrointestinal cancers with KEYTRUDA and thank all investigators and patients who participated in this trial.”
Merck has an extensive clinical development program evaluating KEYTRUDA in gastrointestinal cancers, which includes KEYNOTE-811 in first-line advanced HER2-positive gastric cancer, KEYNOTE-585 in early-stage gastric cancer, and further exploration in advanced/metastatic gastric cancer in LEAP-015. Merck is continuing to study KEYTRUDA for multiple uses in hepatobiliary, esophageal, pancreatic and colorectal cancers.
About KEYNOTE-859
KEYNOTE-859 is a randomized, double-blind Phase 3 trial (ClinicalTrials.gov, NCT03675737) evaluating KEYTRUDA in combination with chemotherapy compared to placebo in combination with chemotherapy for the first-line treatment of patients with HER2-negative locally advanced unresectable or metastatic gastric or GEJ adenocarcinoma. The primary endpoint is OS, and secondary endpoints include PFS, ORR, duration of response and safety. The trial enrolled 1,579 patients who were randomized to receive KEYTRUDA (200 mg every three weeks for up to approximately two years) in combination with fluoropyrimidine- and platinum-containing chemotherapy, or placebo in combination with chemotherapy.
About KEYTRUDA® (pembrolizumab) injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of adult and pediatric (12 years and older) patients with stage IIB, IIC, or III melanoma following complete resection.
Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [tumor proportion score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [Combined Positive Score (CPS) ≥1] as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy.
KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Carcinoma
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
Non-muscle Invasive Bladder Cancer
KEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC) as determined by an FDA-approved test.
Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or gastroesophageal junction (GEJ) (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
KEYTRUDA, in combination with chemotherapy, with or without bevacizumab, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
KEYTRUDA is indicated for the adjuvant treatment of patients with RCC at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions.
Endometrial Carcinoma
KEYTRUDA, as a single agent, is indicated for the treatment of patients with advanced endometrial carcinoma that is MSI-H or dMMR, as determined by an FDA-approved test, who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation.
Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
About Merck
At Merck, known as MSD outside of the United States and Canada, we are unified around our purpose: We use the power of leading-edge science to save and improve lives around the world. For more than 130 years, we have brought hope to humanity through the development of important medicines and vaccines. We aspire to be the premier research-intensive biopharmaceutical company in the world – and today, we are at the forefront of research to deliver innovative health solutions that advance the prevention and treatment of diseases in people and animals. We foster a diverse and inclusive global workforce and operate responsibly every day to enable a safe, sustainable and healthy future for all people and communities. For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf .
Source: Merck & Co., Inc.
Nov. 22, 2022 7:18 AM ET
By: Ravikash, SA News Editor2 Comments
Merck's (NYSE:MRK) blockbuster drug Keytruda, in combination with chemotherapy, met the main goal of overall survival (OS) in patients with a type of gastric cancer in a phase 3 trial.
Aug. 16, 2022 4:41 AM ET
Roche Holding AG (RHHBY), RHHBF
By: Ravikash, SA News Editor1 Comment
Sharebaloxavir marboxil
On 10 November 2022, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion recommending a change to the terms of the marketing authorisation for the medicinal product Xofluza. The marketing authorisation holder for this medicinal product is Roche Registration GmbH.
The CHMP adopted a change to the existing indications to include the treatment of paediatric patients from 1 year of age.
For information, the full indications for Xofluza will be as follows:1
Treatment of influenza
Xofluza is indicated for the treatment of uncomplicated influenza in patients aged 1 12 years and above.
Post exposure prophylaxis of influenza
Xofluza is indicated for post-exposure prophylaxis of influenza in individuals aged 1 12 years and above.
Xofluza should be used in accordance with official recommendations.
Detailed recommendations for the use of this product will be described in the updated summary of product characteristics (SmPC), which will be published in the revised European public assessment report (EPAR), and will be available in all official European Union languages after a decision on this change to the marketing authorisation has been granted by the European Commission.
1 New text in bold, removed text as strikethrough
Nov. 11, 2022 8:58 AM ET
Roche Holding AG (RHHBY), RHHBF
By: Ravikash, SA News Editor
TZIELD™ (teplizumab-mzwv) approved by FDA as the first and only treatment indicated to delay the onset of Stage 3 type 1 diabetes (T1D) in adult and pediatric patients aged 8 years and older with Stage 2 T1DNovember 17, 2022
RED BANK, N.J., Nov. 17, 2022 /PRNewswire/ -- Provention Bio, Inc. (Nasdaq: PRVB) (the "Company"), a biopharmaceutical company dedicated to intercepting and preventing immune-mediated diseases, today announced that the United States Food and Drug Administration (FDA) approved the Biologics License Application (BLA) for TZIELD (teplizumab-mzwv), an anti-CD3-directed antibody, for intravenous use, as the first and only immunomodulatory treatment to delay the onset of Stage 3 T1D in adult and pediatric patients aged 8 years and older with stage 2 T1D. The most common adverse reactions (>10%) that occurred during treatment and through 28 days after the last study drug administration from the TN-10 study were lymphopenia (73% TZIELD, 6% Placebo), rash (TZIELD 36%, Placebo 0%), leukopenia (TZIELD 21%, Placebo 0%) and headache (TZIELD 11%, Placebo 6%).
"This is a historic occasion for the T1D community and a paradigm shifting breakthrough for individuals aged 8 years and older with Stage 2 T1D who now have a therapy approved by the FDA to delay the onset of Stage 3 disease. It cannot be emphasized enough how precious a delay in the onset of Stage 3 T1D can be from a patient and family perspective; more time to live without and, when necessary, prepare for the burdens, complications and risks associated with Stage 3 disease" noted Ashleigh Palmer, Co-Founder and CEO of Provention Bio. "We would especially like to acknowledge and thank all the researchers, scientists, developers, and investigators, especially TrialNet and NIDDK; all the clinical trial participants and their families; the FDA and its reviewers; and JDRF and other patient advocacy groups and champions who have, over more than two decades, tirelessly and selflessly contributed to the development and approval of this first and only therapy to address the underlying autoimmunity of T1D, thereby fundamentally changing the course of the disease rather than just treating its symptoms."
"The progression of T1D can be particularly onerous; patients who progress from Stage 2 to Stage 3 T1D can develop diabetic ketoacidosis, which can be life threatening and is experienced by up to 50% of Stage 3 patients at the time of presentation. The onset of Stage 3 T1D is a life-changing moment - once insulin-producing cells are no longer capable of maintaining normal glycemic control, this irreversible condition can lead to the need, in just one year, for a patient, 1,460 finger sticks to check blood glucose levels, around 1,100 insulin injections, and experiencing an average of 127 episodes of hypoglycemia. These complications can cause stress, fear, and anxiety in patients as they work to manage their T1D diagnosis and provide perspective on the meaning of a delay in the onset of Stage 3 T1D." notes Dr. Eleanor Ramos, Chief Medical Officer at Provention Bio. "As described in the approved label, TZIELD binds to CD3 (a cell surface antigen present on T lymphocytes) and its mechanism is believed to involve partial agonistic signaling and deactivation of pancreatic beta cell autoreactive T lymphocytes. TZIELD leads to an increase in the proportion of regulatory T cells and exhausted CD8+ T cells in peripheral blood."
In October 2022 the company announced a co-promotion agreement for the U.S. launch of TZIELD for delay in onset of clinical T1D in at-risk individuals with Sanofi. Olivier Bogillot, Head of U.S. General Medicines, Sanofi, stated, "This approval is a profound and long-awaited victory for the diabetes community. We applaud Provention Bio for its unwavering determination to bring the first ever disease-modifying therapy for T1D to patients. We look forward to leveraging Sanofi's established infrastructure and expertise in endocrinology to deliver for individuals in need across the U.S."
Provention Bio has launched COMPASS, a patient support program with a staff of dedicated personnel available to answer questions and help navigate coverage, reimbursement and access for patients that are prescribed TZIELD. Provention Bio offers financial assistance options (e.g. copay assistance) to eligible patients for out-of-pocket costs. Patients are encouraged to speak to their healthcare providers to find out whether TZIELD is appropriate for them, and patients who have been prescribed TZIELD and their healthcare providers can call 1-844-778-2246, Monday through Friday from 8AM-8PM EST or email a COMPASS Navigator at COMPASS@proventionbio.com.
The Unmet Need in T1D
Over 1.8 million Americans have T1D, an autoimmune disease caused by the destruction of beta cells. Diagnosis of T1D usually occurs in children and young adults, but it can happen at any age after symptoms appear when a person cannot make enough insulin. However, T1D starts in the body long before any symptoms appear and can be detected at this pre-symptomatic stage through a blood test. The psychological impact of T1D is hard to quantify, but a diagnosis is life-altering, and regular monitoring and maintenance can be extremely stressful. T1D typically takes more than a decade off a person's life, and life expectancy is reduced by 16 years on average for people diagnosed before the age of 10. Insulin therapy and glucose monitoring are currently the standard of care for treating clinical-stage, or Stage 3 T1D, and are necessary to keep T1D patients alive. The constant monitoring and administration of insulin represents a significant life-long burden for patients.
About TZIELD®
TZIELD (teplizumab-mzwv) is a CD3-directed antibody indicated to delay the onset of Stage 3 T1D in adults and pediatric patients aged 8 years and older with Stage 2 T1D. TZIELD injection is supplied as a sterile, preservative-free, clear and colorless solution in a 2 mg/2 mL (1 mg/mL) single-dose vial for intravenous use. TZIELD should be administered by intravenous infusion (over a minimum of 30 minutes) once daily for 14 days. Please see full prescribing information for the dosing schedule.
If a patient needs help paying for TZIELD, Provention Bio's Patient Assistance Program may be able to help. While co-pay amounts vary based on individual coverage, with the Provention Bio Copay Program, commercially or privately insured individuals enrolled in COMPASS may pay as little as $0 for TZIELD. If a patient qualifies, their COMPASS Navigator can help enroll them into the program so they may be able to lower their out-of-pocket costs. Interested patients and healthcare providers can contact COMPASS for questions about our available assistance programs at COMPASS@proventionbio.com and 1-844-778-2246, Monday through Friday from 8AM-8PM EST.
Please see accompanying Prescribing Information.
TN-10 Study
TZIELD was investigated in the TN-10 Study, a pivotal randomized, double-blind, event driven, placebo controlled clinical trial which evaluated TZIELD for the delay of T1D (Stage 3, or clinical T1D) in Stage 2 T1D patients, defined by the presence of two or more T1D-related autoantibodies and dysglycemia. Seventy-six patients (TZIELD N=44, placebo N=32) were enrolled ages 8 to 49, with 72% under the age of 18, and randomized to receive a single 14-day course of either teplizumab or placebo by IV infusion. The primary efficacy endpoint in this study was the time from randomization to development of Stage 3 T1D diagnosis.
In Study TN-10, Stage 3 T1D was diagnosed in 20 (45%) of the TZIELD-treated patients and in 23 (72%) of the placebo-treated patients. A Cox proportional hazards model was used to analyze the time to Stage 3 T1D diagnosis, stratified by age and oral glucose tolerance test status at randomization. The median time from randomization to Stage 3 T1D diagnosis was 50 months in the TZIELD group and 25 months in the placebo group, for a difference of 25 months. With a median follow-up time of 51 months, therapy with TZIELD resulted in a statistically significant delay in the development of Stage 3 T1D, hazard ratio 0.41 (95% CI: 0.22 to 0.78; p=0.0066).
The most common adverse reactions (>10%) that occurred during treatment and through 28 days after the last study drug administration from the TN-10 study were lymphopenia (73% TZIELD, 6% Placebo), rash (TZIELD 36%, Placebo 0%), leukopenia (TZIELD 21%, Placebo 0%) and headache (TZIELD 11%, Placebo 6%).
About Sanofi US Co-Promotion
In October 2022, Provention entered into a co-promotion agreement with Sanofi U.S. for the launch of TZIELD. Under the terms of the agreement, Sanofi will commit commercial resources in the United States, including diabetes field specialists, account directors, field-based reimbursement, and medical science liaisons to expand the number of key healthcare professionals reached in the United States. In exchange, Provention will reimburse field force-related expenses that Sanofi will incur in connection with commercializing teplizumab under the agreement.
Provention retains all rights to TZIELD and maintains responsibility for the commercialization strategy.
About Provention Bio, Inc.
Provention Bio, Inc. (Nasdaq: PRVB) is a biopharmaceutical company focused on advancing the development of investigational therapies that may intercept and prevent debilitating and life-threatening immune-mediated diseases. The Company's pipeline includes clinical-stage product candidates that have demonstrated in pre-clinical or clinical studies proof-of-mechanism and/or proof-of-concept in autoimmune diseases, including T1D, celiac disease and lupus. Visit www.ProventionBio.com for more information and follow us on Twitter: @ProventionBio.
Provention Bio and the Provention Bio logo are registered Trademarks of Provention Bio, Inc. TZIELD and Provention Bio COMPASS are trademarks of Provention Bio, Inc.
SOURCE Provention Bio, Inc.
Nov. 17, 2022 6:01 PM ET
By: Jonathan Block, SA News Editor9 Comments
Nov. 16, 2022 9:00 AM ET AbbVie Inc. (ABBV)
- UBRELVY® is the first and only oral CGRP receptor antagonist (gepant) approved by Health Canada for the acute treatment of migraine
-- UBRELVY® was demonstrated to reduce or eliminate migraine pain with a single oral tablet, with the flexibility to take an optional second dose for persistent pain
MONTREAL, Nov. 16, 2022 /CNW/ - AbbVie (NYSE: ABBV) announced today that Health Canada has approved UBRELVY® (ubrogepant tablet) for the acute treatment of migraine, with or without aura, in adults.1 UBRELVY® is the first orally-administered calcitonin gene-related peptide (CGRP) receptor antagonist (gepant) for the treatment of migraine attacks once they start.

An estimated 2.7 million Canadians have been diagnosed with migraine, which is estimated to underrepresent today's migraine prevalence.2 Migraine is a complex neurological disease with recurrent attacks that lasts 4-72 hours. It can be defined by symptoms such as moderate to severe pain intensity, nausea, vomiting, photophobia and phonophobia.3 It is one of the leading causes of disability in the world.4
"I see first-hand the incredible impact migraines can have on my patients. When you suffer an attack, some patients are forced to seek solitude and remove themselves from everything in their lives. It can be unbearable and impact one's interaction with family, friends, school and work," said Dr. Jonathan Gladstone, MD, FRCPC, neurologist and Director of the Gladstone Headache Clinic in Toronto. "Health Canada's approval of UBRELVY is an important step forward allowing physicians to provide a new migraine-specific option for migraine sufferers. The gepant class is one of the most evaluated targeted therapies for migraine treatment. I'm excited to have another treatment option available to help patients successfully manage their migraines."
Calcitonin gene-related peptide (CGRP) is a neuropeptide present in the peripheral and central nervous system. CGRP is released from sensory nerve endings during a migraine attack, particularly the nerve endings of sensory trigeminal ganglion neurons. Ubrogepant is a small molecule, high affinity (Ki = 0.07nM) CGRP receptor antagonist (gepant) that blocks the binding of CGRP to its receptor and antagonizes CGRP receptor function. UBRELVY® was demonstrated to reduce or eliminate migraine pain with a single oral tablet, with the flexibility to take an optional second dose for persistent pain.1
"Migraine can often be a debilitating disease that can impact a person's ability to function and perform their daily routines. Being able to successfully address migraine attacks as they occur is essential," says Wendy Gerhart, Executive Director, Migraine Canada. "Migraine Canada welcomes the approval of UBRELVY® in Canada as a new and innovative treatment option. This will have a positive impact for the migraine community across the country."
The efficacy of UBRELVY® for the acute treatment of migraine was demonstrated in two multicenter, randomized, double-blind, placebo-controlled, single migraine attack studies. These studies enrolled patients with a history of migraine with and without aura, according to the ICHD-3 beta diagnostic criteria, and who experienced 2 to 8 migraine attacks per month with moderate to severe headache pain. In Study 1 (ACHIEVE I), patients were randomized to receive UBRELVY 50 mg or 100 mg or placebo; and in Study 2 (ACHIEVE II), patients were randomized to receive UBRELVY 50 mg or placebo. Patients were permitted to use standard migraine preventive medications during the study. At baseline, 23% percent of patients were taking preventive medications for migraine. The most commonly used preventive medications were topiramate, onabotulinumtoxinA, propranolol, and amitriptyline.1
"UBRELVY® as the first oral gepant approved in Canada for the acute treatment of migraine is an important milestone in our commitment to bring innovative new medicines to Canadians with the goal make a meaningful difference for patients," says Tracey Ramsay, Vice President and General Manager, AbbVie Canada.
About UBRELVY® (ubrogepant tablet) 1
UBRELVY® (ubrogepant tablet) is indicated for the acute treatment of migraine, with or without aura, in adults.
Calcitonin gene-related peptide (CGRP) is a neuropeptide present in the peripheral and central nervous system. CGRP is released from sensory nerve endings during a migraine attack, particularly the nerve endings of sensory trigeminal ganglion neurons. Ubrogepant is a small molecule, high affinity (Ki = 0.07nM) CGRP receptor antagonist (gepant) that blocks the binding of CGRP to its receptor and antagonizes CGRP receptor function. UBRELVY® was demonstrated to reduce or eliminate migraine pain with a single oral tablet, with the flexibility to take an optional second dose for persistent pain.
UBRELVY® is contraindicated:
Please consult the UBRELVY® Product Monograph here.
About AbbVie
AbbVie's mission is to discover and deliver innovative medicines that solve serious health issues today and address the medical challenges of tomorrow. We strive to have a remarkable impact on people's lives across several key therapeutic areas: immunology, oncology, neuroscience, eye care, virology, women's health and gastroenterology, in addition to products and services across its Allergan Aesthetics portfolio. For more information about AbbVie, please visit us at www.abbvie.ca. Follow AbbVie Canada on Twitter, on Instagram or find us on LinkedIn.
SOURCE AbbVie Canada
Nov 17, 2022 7:00 AM
BRUKINSA is the only Bruton’s Tyrosine Kinase (BTK) inhibitor to achieve superiority over IMBRUVICA® (ibrutinib) in relapsed/refractory CLL.
BRUKINSA also showed superiority to chemoimmunotherapy in treatment naive CLL.
BRUKINSA had a favorable safety profile, including lower rates of atrial fibrillation/flutter compared with IMBRUVICA.
CAMBRIDGE, Mass. & BASEL, Switzerland & BEIJING--(BUSINESS WIRE)-- BeiGene (NASDAQ: BGNE; HKEX: 06160; SSE: 688235), a global biotechnology company, today announced that the European Commission (EC) has approved BRUKINSA® (zanubrutinib) for the treatment of adult patients with treatment-naïve (TN) or relapsed/refractory (R/R) CLL.
“This approval represents an important milestone for CLL patients and their physicians who now have a new chemotherapy-free treatment option, and an alternative to current BTKi treatment options,” said Mehrdad Mobasher, M.D., M.P.H., Chief Medical Officer, Hematology at BeiGene. “Given that BRUKINSA has demonstrated consistent benefit across patient subgroups, regardless of risk status, we believe BRUKINSA could now be the preferred treatment option for newly diagnosed and relapsed/refractory CLL patients.”
The EC approval is based on positive results from two Phase 3 clinical trials: SEQUOIA (NCT03336333), in patients with previously untreated CLL, and ALPINE (NCT03734016), in patients with R/R CLL. In these two trials, BRUKINSA demonstrated superior efficacy versus either bendamustine plus rituximab (B+R) or ibrutinib in first-line or R/R CLL, respectively. BRUKINSA is the only BTKi to achieve superiority versus ibrutinib in R/R CLL, as assessed by independent review committee, with an overall response rate (ORR) of 80.4% vs 72.9% (p=0.0264).i Additionally, more BRUKINSA patients than ibrutinib patients had a sustained response at 1 year with rates of 90% vs 78%.i The adverse events within the two trials were consistent with the overall safety profile of BRUKINSA. Subsequent to the regulatory submission, BeiGene announced topline results of the final PFS analysis of the head-to-head ALPINE trial, in which BRUKINSA demonstrated superior PFS compared with ibrutinib in patients with R/R CLL.
Prof. Clemens Wendtner, Head of Hematology and Oncology at Munich Clinic, an academic teaching hospital of the University of Munich, Germany, commented, “BRUKINSA has demonstrated clinically meaningful improvements as a next-generation BTKi over the first generation BTKi, and is proven to be significantly more effective and tolerable. Ensuring medicines are safe and tolerable for this patient population is critical, given the long-term treatment needed for CLL. Combined with the flexible dosing options, this approval offers a practice-changing option for patients with CLL, one of the most common types of leukemia in adults.”
“We’re pleased with the significant progress we’ve made to date bringing BRUKINSA to patients with hematological malignancies globally,” noted Gerwin Winter, Senior Vice President, Head of Europe at BeiGene. “With this notable approval, we welcome the opportunity to expand BeiGene’s presence in Europe and provide this innovative treatment option to CLL patients across the region.”
BRUKINSA is currently approved in the EU for the treatment of adult patients with WM who have received at least one prior therapy or as the first-line treatment for patients unsuitable for chemoimmunotherapy and adult patients with MZL who have received at least one prior anti-CD20-based therapy.
In Europe, BeiGene has now obtained reimbursement for BRUKINSA for the treatment of WM in Austria, Belgium, Denmark, England and Wales, Germany, Iceland, Ireland, Italy, Luxembourg, Scotland, Spain, Switzerland, and The Netherlands, while additional EU countries are currently going through the reimbursement process.
About Chronic Lymphocytic Leukemia (CLL)
A slow-growing, life-threatening and incurable cancer of adults, CLL is a type of mature B-cell malignancy in which abnormal leukemic B lymphocytes (a type of white blood cells) arise from the bone marrow and flood peripheral blood, bone marrow, and lymphoid tissues.ii-iv CLL is one of the most common types of leukemia, accounting for about one-quarter of new cases of leukemia.v In Europe, the estimated incidence is 4.92/100,000 persons per year.vi,vii
About BRUKINSA
BRUKINSA is a small-molecule inhibitor of Bruton’s tyrosine kinase (BTK) discovered by BeiGene scientists that is currently being evaluated globally in a broad clinical program as a monotherapy and in combination with other therapies to treat various B-cell malignancies. BRUKINSA was specifically designed to deliver targeted and sustained inhibition of the BTK protein by optimizing bioavailability, half-life, and selectivity. With differentiated pharmacokinetics compared to other approved BTK inhibitors, BRUKINSA has been demonstrated to inhibit the proliferation of malignant B cells within a number of disease-relevant tissues.
BRUKINSA is supported by a broad clinical program which includes more than 4,700 subjects in 35 trials in more than 25 countries and regions. To date, BRUKINSA is approved in 58 markets, including the United States, China, the European Union Great Britain, Canada, Australia, South Korea, Switzerland, and additional international markets.
BeiGene Oncology
BeiGene is committed to advancing best- and first-in-class clinical candidates internally or with like-minded partners to develop impactful and affordable medicines for patients across the globe. We have a growing R&D and medical affairs team of approximately 3,500 colleagues dedicated to advancing more than 100 clinical trials that have involved more than 20,000 subjects. Our expansive portfolio is directed predominantly by our internal colleagues supporting clinical trials in more than 45 countries and regions. Hematology-oncology, and solid tumor targeted therapies, and immuno-oncology are key focus areas for the Company, with both monotherapies and combination therapies prioritized in our research and development. BeiGene currently has three approved medicines discovered and developed in our own labs: BTK inhibitor BRUKINSA® in the U.S., China, the European Union, Great Britain, Canada, Australia, South Korea, Switzerland, and additional international markets; and the non-FC-gamma receptor binding anti-PD-1 antibody tislelizumab as well as the poly adenosine diphosphate-ribose polymerase (PARP) inhibitor pamiparib in China.
BeiGene also partners with innovative companies who share our goal of developing therapies to address global health needs. We commercialize a range of oncology medicines in China licensed from Amgen, Bristol Myers Squibb, EUSA Pharma, and Bio-Thera. We also plan to address greater areas of unmet need globally through our other collaborations including Mirati Therapeutics, Seagen, and Zymeworks.
In January 2021 BeiGene and Novartis announced a collaboration granting Novartis rights to co-develop, manufacture, and commercialize BeiGene’s anti-PD-1 antibody tislelizumab in North America, Europe, and Japan. Building upon this productive collaboration, BeiGene and Novartis announced an option, collaboration, and license agreement in December 2021 for BeiGene’s TIGIT inhibitor ociperlimab that is in Phase 3 development. Novartis and BeiGene also entered into a strategic commercial agreement through which BeiGene will promote five approved Novartis oncology products across designated regions of China.
About BeiGene
BeiGene is a global biotechnology company that is developing and commercializing innovative and affordable oncology medicines to improve treatment outcomes and access for far more patients worldwide. With a broad portfolio, we are expediting development of our diverse pipeline of novel therapeutics through our internal capabilities and collaborations. We are committed to radically improving access to medicines for far more patients who need them. Our growing global team of more than 9,000 colleagues spans five continents, with administrative offices in Beijing, China; Cambridge, U.S.; and Basel, S
View source version on businesswire.com: https://www.businesswire.com/news/home/20221117005394/en/
Source: BeiGene
Nov. 17, 2022 7:59 AM ET BeiGene, Ltd. (BGNE)
By: Ravikash, SA News Editor
olaparib
On 10 November 2022, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion recommending a change to the terms of the marketing authorisation for the medicinal product Lynparza. The marketing authorisation holder for this medicinal product is AstraZeneca AB.
The CHMP adopted a new indication as follows:
Lynparza is indicated in combination with abiraterone and prednisone or prednisolone for the treatment of adult patients with mCRPC in whom chemotherapy is not clinically indicated (see section 5.1).
For information, the full indications for Lynparza tablets will therefore be as follows:1
Ovarian cancer
Lynparza is indicated as monotherapy for the:
Lynparza in combination with bevacizumab is indicated for the:
Breast cancer
Lynparza is indicated as:
Adenocarcinoma of the pancreas
Lynparza is indicated as monotherapy for the maintenance treatment of adult patients with germline BRCA1/2-mutations who have metastatic adenocarcinoma of the pancreas and have not progressed after a minimum of 16 weeks of platinum treatment within a first-line chemotherapy regimen.
Prostate cancer
Lynparza is indicated:
Detailed recommendations for the use of this product will be described in the updated summary of product characteristics (SmPC), which will be published in the revised European public assessment report (EPAR), and will be available in all official European Union languages after a decision on this change to the marketing authorisation has been granted by the European Commission.
1New text in bold
Nov. 11, 2022 8:35 AM ET
By: Ravikash, SA News Editor
trastuzumab deruxtecan
On 10 November 2022, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion recommending a change to the terms of the marketing authorisation for the medicinal product Enhertu. The marketing authorisation holder for this medicinal product is Daiichi Sankyo Europe GmbH.
The CHMP adopted a new indication for the treatment of gastric cancer. For information, the full indications for Enhertu will therefore be as follows:1
Breast cancer
Enhertu as monotherapy is indicated for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received one or more prior anti-HER2-based regimens.
Gastric cancer
Enhertu as monotherapy is indicated for the treatment of adult patients with advanced HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma who have received a prior trastuzumab-based regimen.
Detailed recommendations for the use of this product will be described in the updated summary of product characteristics (SmPC), which will be published in the revised European public assessment report (EPAR), and will be available in all official European Union languages after a decision on this change to the marketing authorisation has been granted by the European Commission.
1 New text in bold.
Nov. 11, 2022 8:50 AM ET
Daiichi Sankyo Company, Limited (DSKYF), DSNKY, AZN
By: Dulan Lokuwithana, SA News Editor
November 11, 2022
OSAKA, Japan and CAMBRIDGE, Massachusetts, November 11, 2022 – Takeda (TSE:4502/NYSE:TAK) today announced that the European Commission (EC) has granted Marketing Authorization for LIVTENCITYTM (maribavir) for the treatment of cytomegalovirus (CMV) infection and/or disease that are refractory (with or without resistance) to one or more prior therapies, including ganciclovir, valganciclovir, cidofovir or foscarnet, in adult patients who have undergone a haematopoietic stem cell transplant (HSCT) or solid organ transplant (SOT).4 LIVTENCITY is the first and only oral treatment that inhibits CMV-specific UL97 protein kinase and its natural substrates.1
CMV is one of the most common infections experienced by transplant patients with a global estimated incidence rate of 16-56% in SOT and 30-80% in HSCT recipients.5,6 More than 34,000 SOTs,7 including liver, kidney, and heart transplant, and more than 48,000 HSCTs8 were performed in Europe and neighboring countries in 2019. Although prevention and management of CMV infection in SOT and HSCT patients with available therapies may help improve outcomes,5 breakthrough infections can still occur with prophylaxis,9 and some CMV infections may not respond to treatment.10
“The European Society for Organ Transplantation (ESOT) understands that the transplant patient journey extends well beyond the transplant itself. When not successfully treated, CMV poses a challenge to transplant recipients and their physicians and often leads to increased organ rejection, higher hospitalization rates, and greater burden on healthcare resources, contributing to inequities for patients across the system,” said Dr. Luciano Potena, ESOT President. “The approval of LIVTENCITY by the EC recognizes the need for a new antiviral approach for managing CMV infection that is refractory (with or without resistance) to one or more prior CMV therapies.”
The centralized marketing authorization is valid in all EU member states as well as in Iceland, Liechtenstein, Norway, and Northern Ireland, and was based on the Phase 3 SOLSTICE trial, which evaluated the safety and efficacy of LIVTENCITY versus conventional antiviral therapies—ganciclovir, valganciclovir, cidofovir or foscarnet—for the treatment of adult HSCT and SOT recipients with CMV infection refractory (with or without resistance) to prior therapies.
The EC approval marks the fourth approval of LIVTENCITY for post-transplant refractory (with or without resistance) CMV infection, following the U.S., Canada, and Australia.13-15
“Patients who receive a transplant can face a difficult journey on the road to recovery that involves medicines to suppress their immune system. The additional burden of a CMV infection that has become refractory to treatment, and which could threaten their transplant, poses a challenge to patients being offered a second chance at life,” said Ramona Sequeira, President, Global Portfolio Division, Takeda. “With the EC approval of LIVTENCITY, we are privileged to offer healthcare providers in the EU and EEA* with an additional oral antiviral treatment for post-transplant refractory CMV.”
LIVTENCITYTM (maribavir), an orally bioavailable anti-CMV compound, is the first and only antiviral agent that targets and inhibits the UL97 protein kinase and its natural substrates.16 It is approved by the U.S. Food and Drug Administration for the treatment of adults and pediatric patients (12 years of age or older and weighing at least 35 kg) with post-transplant CMV infection/disease that is refractory to treatment (with or without genotypic resistance) with ganciclovir, valganciclovir, cidofovir, or foscarnet.12 It is approved by the EC for the treatment of CMV infection and/or disease that are refractory (with or without resistance) to one or more prior therapies, including ganciclovir, valganciclovir, cidofovir or foscarnet in adult patients who have undergone a HSCT or SOT.4 It is also approved by Health Canada for the treatment of adults with post-transplant cytomegalovirus (CMV) infection/disease who are refractory (with or without genotypic resistance) to one or more prior antiviral therapies.11 LIVTENCITY is also approved in Australia for the treatment of adults with post-transplant CMV infection and disease resistant, refractory, or intolerant to one or more prior therapies.13
The TAK-620-303 (SOLSTICE) trial (NCT02931539, EudraCT 2015-004725-13) was a global, multicenter, randomized, open-label, active-controlled superiority trial to assess the efficacy and safety of treatment with either maribavir or conventional antiviral therapy in 352 haematopoietic stem cell transplant and solid organ transplant recipients with CMV infection refractory, with or without resistance, to one or a combination of the conventional antiviral therapies: ganciclovir, valganciclovir, foscarnet, or cidofovir. Adult patients underwent a 2-week screening period, followed by randomization 2:1 to maribavir (n=235) (400 mg, twice daily) or conventional antiviral therapies (n=117) (as dosed by the investigator) for up to 8 weeks. After completion of the treatment period, subjects entered a 12-week follow-up phase.16
The trial’s primary efficacy endpoint was confirmed CMV DNA level <LLOQ (lower limit of quantification, [i.e., <137 IU/mL] in 2 consecutive samples separated by at least 5 days as assessed by COBAS® AmpliPrep/COBAS® TaqMan® CMV test) at the end of Week 8. The key secondary endpoint was CMV DNA level <LLOQ and CMV infection symptom control† at the end of Study Week 8 with maintenance of this treatment effect through Study Week 16.16
Takeda is a global, values-based, R&D-driven biopharmaceutical leader headquartered in Japan, committed to discover and deliver life-transforming treatments, guided by our commitment to patients, our people and the planet. Takeda focuses its R&D efforts on four therapeutic areas: Oncology, Rare Genetics and Hematology, Neuroscience, and Gastroenterology (GI). We also make targeted R&D investments in Plasma-Derived Therapies and Vaccines. We are focusing on developing highly innovative medicines that contribute to making a difference in people’s lives by advancing the frontier of new treatment options and leveraging our enhanced collaborative R&D engine and capabilities to create a robust, modality-diverse pipeline. Our employees are committed to improving quality of life for patients and to working with our partners in health care in approximately 80 countries and regions.
For more information, visit https://www.takeda.com.
Nov. 11, 2022 5:00 AM ET
Takeda Pharmaceutical Company Limited (TAK)
By: Ravikash, SA News Editor
November 8, 2022 at 4:41 PM EST
Approval based on superior survival outcomes of Libtayo plus chemotherapy, compared to chemotherapy alone, in a patient population with a wide range of disease characteristics
Second advanced NSCLC indication expands patient population eligible for a Libtayo-based regimen to include combination treatment with chemotherapy irrespective of PD-L1 expression levels
TARRYTOWN, N.Y., Nov. 8, 2022 /PRNewswire/ -- Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) today announced that the U.S. Food and Drug Administration (FDA) has approved the PD-1 inhibitor Libtayo® (cemiplimab-rwlc) in combination with platinum-based chemotherapy for the first-line treatment of adult patients with advanced non-small cell lung cancer (NSCLC) with no EGFR, ALK or ROS1 aberrations. Patients must either have metastatic or locally advanced tumors that are not candidates for surgical resection or definitive chemoradiation. Patients may be treated with this combination irrespective of PD-L1 expression or histology.
"This second FDA approval for cemiplimab-rwlc in advanced non-small cell lung cancer greatly broadens the scope in which a cemiplimab-rwlc-based regimen can be prescribed to encompass a wide range of patients, either as single agent in those with PD-L1 ≥50% or now in combination with chemotherapy irrespective of PD-L1 expression or tumor histology," said David R. Gandara, M.D., Professor Emeritus and Senior Advisor of the Thoracic Oncology Program at the University of California Davis Comprehensive Cancer Center. "The approval is based on a Phase 3 trial designed to closely resemble a patient population with varied disease presentations that physicians manage in everyday clinical practice. Even with these diverse disease presentations, cemiplimab-rwlc combined with chemotherapy demonstrated a marked increase in overall survival, at a median of 22 months versus 13 months with chemotherapy alone. Clearly, this is an advance which is clinically meaningful for our patients with advanced stage non-small cell lung cancer."
The FDA approval is based on data from the global Phase 3 trial, EMPOWER-Lung 3, that investigated Libtayo in combination with a physician's choice of platinum-doublet chemotherapy (Libtayo combination), compared to platinum-doublet chemotherapy alone. The trial enrolled 466 patients with locally advanced or metastatic NSCLC, irrespective of PD-L1 expression or tumor histology, and with no ALK, EGFR or ROS1 aberrations. Among those enrolled, 43% had tumors with squamous histology, 67% had tumors with <50% PD-L1 expression, 15% had inoperable locally advanced disease not eligible for definitive chemoradiation, and 7% had pretreated and clinically stable brain metastases.
"Libtayo is now approved for extending the survival of patients with advanced non-small cell lung cancer as both a monotherapy in high PD-L1 expressors and in combination with chemotherapy irrespective of PD-L1 expression levels, achieving a high bar that has only been met by one other PD-1 targeting agent," said Israel Lowy, M.D., Ph.D., Senior Vice President, Translational and Clinical Sciences, Oncology at Regeneron. "With this FDA approval, Libtayo can expand its role as a key treatment option for advanced non-small lung cancer, in addition to serving as a standard-of-care for two advanced non-melanoma skin cancers. We are committed to investigating Libtayo through ongoing trials as a monotherapy and as a backbone of combination treatments in multiple cancers. We thank the many investigators, patients and their families who have participated in our pivotal trials."
Efficacy in EMPOWER-Lung 3 was assessed in 466 patients who were randomized 2:1 to receive either Libtayo 350 mg (n=312) or placebo (n=154) intravenously every 3 weeks, plus histology-specific platinum-doublet chemotherapy. The trial was stopped early based on a recommendation by the Independent Data Monitoring Committee after the Libtayo combination demonstrated a significant improvement in overall survival (OS), the primary endpoint. Efficacy results comparing the Libtayo combination to chemotherapy alone showed a:
Safety was assessed in 312 patients in the Libtayo combination group (median duration of exposure: 38 weeks) and 153 patients in the chemotherapy group (median duration of exposure: 21 weeks). The most common adverse reactions occurring in >15% of patients were alopecia (37% Libtayo combination, 43% placebo), musculoskeletal pain (30% Libtayo combination, 36% placebo), nausea (25% Libtayo combination, 16% placebo), fatigue (23% Libtayo combination, 18% placebo), peripheral neuropathy (23% Libtayo combination, 19% placebo) and decreased appetite (17% Libtayo combination, 12% placebo). Serious adverse reactions occurred in 25% of patients, with treatment discontinuations due to adverse reactions in 5% and fatal adverse reactions in 6%. No new Libtayo safety signals were observed.
"The approval of Libtayo in the combination setting builds on the monotherapy indication in advanced non-small cell lung cancer and furthers our excitement for what's to come as we continue our potentially transformative oncology research," said George D. Yancopoulos, M.D., Ph.D., President and Chief Scientific Officer at Regeneron. "Libtayo is the backbone of our oncology strategy, designed to synergistically combine multiple modalities to provide more options for more patients. We look forward to delivering on the promise of our research in other meaningful combinations that leverage Libtayo and our homegrown pipeline of investigational bispecific antibodies."
"We welcome this latest approval for Libtayo as a first-line combination treatment for appropriate patients with advanced lung cancer," said Andrea Ferris, President and CEO at the LUNGevity Foundation. "Lung cancer remains one of the most common cancers worldwide, and every new treatment option is an important step forward against this deadly cancer."
About Libtayo
Libtayo is a fully human monoclonal antibody targeting the immune checkpoint receptor PD-1 on T-cells and was invented using Regeneron's proprietary VelocImmune® technology. By binding to PD-1, Libtayo has been shown to block cancer cells from using the PD-1 pathway to suppress T-cell activation. In the U.S. and other countries Libtayo is indicated in certain patients with advanced basal cell carcinoma (BCC), advanced cutaneous squamous cell carcinoma (CSCC) and advanced NSCLC, as well as in advanced cervical cancer in Canada and Brazil. As of July 1, 2022, Libayo is developed and marketed globally by Regeneron.
In the U.S., the generic name for Libtayo in its approved indications is cemiplimab-rwlc, with rwlc as the suffix designated in accordance with Nonproprietary Naming of Biological Products Guidance for Industry issued by the FDA. Outside of the U.S., the generic name of Libtayo in its approved indication is cemiplimab.
The extensive clinical program for Libtayo is focused on difficult-to-treat cancers. Libtayo is currently being investigated in trials as a monotherapy, as well as in combination with either conventional or novel therapeutic approaches for other solid tumors and blood cancers. These potential uses are investigational, and their safety and efficacy have not been evaluated by any regulatory authority.
U.S. FDA-approved Indications
Libtayo is a prescription medicine used to treat:
It is not known if Libtayo is safe and effective in children.
Please see full Prescribing Information, including Medication Guide.
About Regeneron's VelocImmune Technology
Regeneron's VelocImmune technology utilizes a proprietary genetically engineered mouse platform endowed with a genetically humanized immune system to produce optimized fully human antibodies. When Regeneron's co-Founder, President and Chief Scientific Officer George D. Yancopoulos was a graduate student with his mentor Frederick W. Alt in 1985, they were the first to envision making such a genetically humanized mouse, and Regeneron has spent decades inventing and developing VelocImmune and related VelociSuite® technologies. Dr. Yancopoulos and his team have used VelocImmune technology to create approximately one in five of all original, FDA-approved or authorized fully human monoclonal antibodies. This includes REGEN-COV® (casirivimab and imdevimab), Dupixent® (dupilumab), Libtayo, Praluent® (alirocumab), Kevzara® (sarilumab), Evkeeza® (evinacumab-dgnb) and Inmazeb™ (atoltivimab, maftivimab and odesivimab-ebgn).
About Regeneron
Regeneron is a leading biotechnology company that invents, develops and commercializes life-transforming medicines for people with serious diseases. Founded and led for nearly 35 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to nine FDA-approved treatments and numerous product candidates in development, almost all of which were homegrown in our laboratories. Our medicines and pipeline are designed to help patients with eye diseases, allergic and inflammatory diseases, cancer, cardiovascular and metabolic diseases, pain, hematologic conditions, infectious diseases and rare diseases.
Regeneron is accelerating and improving the traditional drug development process through our proprietary VelociSuite technologies, such as VelocImmune, which uses unique genetically humanized mice to produce optimized fully human antibodies and bispecific antibodies, and through ambitious research initiatives such as the Regeneron Genetics Center, which is conducting one of the largest genetics sequencing efforts in the world.
For more information, please visit www.Regeneron.com or follow @Regeneron on Twitter.
Cision View original content:https://www.prnewswire.com/news-releases/libtayo-cemiplimab-rwlc-in-combination-with-chemotherapy-approved-by-the-fda-as-first-line-treatment-for-advanced-non-small-cell-lung-cancer-nsclc-301672231.html
SOURCE Regeneron Pharmaceuticals, Inc.
Nov. 08, 2022 5:52 PM ET
Regeneron Pharmaceuticals, Inc. (REGN)
By: Jonathan Block, SA News Editor
PUBLISHED11 November 2022
AstraZeneca today announced the availability of the first rare disease therapy from Alexion, AstraZeneca’s Rare Disease group, in China. The availability of Soliris (eculizumab) for the treatment of paroxysmal nocturnal haemoglobinuria (PNH) and atypical haemolytic uraemic syndrome (aHUS) in adults and children in China represents a significant expansion of the company’s global footprint in rare disease, following the acquisition of Alexion Pharmaceuticals, Inc. in 2021.
In addition to announcing the availability of Soliris, AstraZeneca announced that the National Medicine Products Administration of China (NMPA) has accepted its supplementary application for Soliris for the treatment of adults with refractory generalised myasthenia gravis (gMG) in patients who are anti-acetylcholine receptor (AchR) antibody positive. The NMPA has also granted AstraZeneca approval to start ongoing, global clinical trials in China for investigational therapies being evaluated for the treatment of lupus nephritis (LN), immunoglobulin A nephropathy (IgAN) and light chain (AL) amyloidosis.
Marc Dunoyer, Chief Executive Officer, Alexion, said: “These milestones represent significant progress against our commitment to expand access to our rare disease medicines globally, including the introduction of innovative rare disease therapies in China. Expanding access to our medicines is possible because of the combined strength of Alexion’s rare disease expertise and AstraZeneca’s vast global footprint, and we look forward to continue bringing our therapies to people living with rare diseases around the world in the future.”
Leon Wang, Executive Vice President, International and China President of AstraZeneca, said: “This milestone reflects our ambition to bring transformative, rare disease medicines to the significant number of patients and families living with rare diseases in China who currently have limited treatment options or no available treatment at all. We look forward to advancing our commitment in China and aim to bring additional, innovative therapies to even more rare disease patients across the country.”
As the first C5 complement inhibitor, Soliris works by inhibiting the C5 protein in the terminal complement cascade, a part of the body’s immune system. Soliris is approved for multiple indications in many countries around the world. The availability of Soliris in China provides people living with PNH or aHUS with a new treatment option that can reduce disease symptoms and prevent the dysregulated complement system from causing further damage.
AstraZeneca established a rare disease business unit in China in September 2021. In the future, the company aims to introduce more innovative medicines in China, targeting the complement system and beyond, for the treatment of rare diseases including PNH, aHUS, gMG, neuromyelitis optica spectrum disorder (NMOSD), hypophosphatasia, IgAN, LN and amyloidosis.
Soliris
Soliris (eculizumab) is a first-in-class C5 complement inhibitor. The medication works by inhibiting the C5 protein in the terminal complement cascade, a part of the body’s immune system. When activated in an uncontrolled manner, the terminal complement cascade over-responds, leading the body to attack its own healthy cells. Soliris is administered intravenously every two weeks, following an introductory dosing period.
Soliris is approved in the US, EU and Japan for the treatment of PNH, aHUS, certain adults with gMG and certain adults with NMOSD.
Soliris is not indicated for the treatment of patients with Shiga-toxin E. coli-related haemolytic uraemic syndrome.
Alexion
Alexion, AstraZeneca Rare Disease, is the group within AstraZeneca focused on rare diseases, created following the 2021 acquisition of Alexion Pharmaceuticals, Inc. As a leader in rare diseases for 30 years, Alexion is focused on serving patients and families affected by rare diseases and devastating conditions through the discovery, development and commercialisation of life-changing medicines. Alexion focuses its research efforts on novel molecules and targets in the complement cascade and its development efforts on haematology, nephrology, neurology, metabolic disorders, cardiology and ophthalmology. Headquartered in Boston, Massachusetts, Alexion has offices around the globe and serves patients in more than 50 countries.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Nov. 11, 2022 4:33 AM ET
By: Ravikash, SA News Editor
Early skin and enthesitis responses in active psoriatic arthritis patients treated with TREMFYA predicted long-term clinical response, measured at two years, including disease remission
Bio-naïve, TREMFYA-treated patients maintained several disease control endpoints through two years, regardless of baseline characteristics or dosing regimen
SPRING HOUSE, PENNSYLVANIA, November 11, 2022 – The Janssen Pharmaceutical Companies of Johnson & Johnson today announced a new post-hoc analysis of the Phase 3 DISCOVER program (DISCOVER-1 and DISCOVER-2) evaluating TREMFYA® (guselkumab) in adult patients with active psoriatic arthritis (PsA), which showed that early skin and enthesitis responsesa,b predicted longer-term clinical response,c including disease remission, at week 52.1 TREMFYA is the first fully human selective interleukin (IL)-23 inhibitor therapy approved in the U.S. for adults with moderate to severe plaque psoriasis (PsO) and adults with active PsA.2
In another post-hoc analysis of DISCOVER-2, a diverse population of bio-naïve, TREMFYA-treated patients sustained several PsA disease control endpoints through two years, regardless of their baseline characteristicsd or dosing regimen,e including minimal disease activity (MDA) response,f skin clearance (IGA 0)g and resolution of dactylitis.3,h
“Active psoriatic arthritis is a chronic, lifelong condition that can be progressive for many patients, leading to extensive and permanent joint damage,” said Philip J. Mease, M.D.,i Swedish Medical Center/Providence St. Joseph Health and University of Washington in Seattle, Washington. “The data derived from TREMFYA-treated patients in the DISCOVER program give us more tools to evaluate treatment plan options that may help facilitate long-term relief as we work toward the ultimate goal of disease remission."
Presented at the 2022 American College of Rheumatology (ACR) Convergence meeting, taking place in Philadelphia, PA and virtually, November 10-14, 2022, these new analyses of DISCOVER-1 and DISCOVER-2 dataj show:
Early Skin and Entheseal Responses in TREMFYA-Treated Active PsA Patients May Predict Future Achievement of Rigorous Measures of Disease Control:1
Treatment with TREMFYA Sustained Stringent PsA Disease Control Endpoints Through Two Years Among a Diverse Population of Adults with Active PsA in Post-hoc Analysis of DISCOVER-23:
“These analyses from the DISCOVER program give us greater insight into the potential for TREMFYA to provide long-term efficacy across multiple stringent disease control endpoints in diverse patient groups with active psoriatic arthritis,” said Terence Rooney, M.D., Vice President, Rheumatology and Maternal-Fetal Immunology Disease Area Leader, Janssen Research & Development, LLC. “The data underscore Janssen’s ongoing commitment to extensive research in active psoriatic arthritis, so that we may provide effective therapeutic options for patients living with the disease.”
About DISCOVER-1 (NCT03162796)10
DISCOVER-1 was a randomized, double-blind, multicenter Phase 3 study evaluating the efficacy and safety of TREMFYA administered by SC injection in participants with active PsA, including those previously treated with one or two TNF inhibitors. DISCOVER-1 evaluated 381 participants who were treated and followed through approximately one year. The study consisted of a screening phase of up to six weeks, a blinded treatment phase of 52 weeks that included a placebo-controlled period from week 0 to week 24 and a blinded active treatment period from week 24 to week 52. It also included a safety follow-up phase through week 60 (i.e., approximately 12 weeks from the last administration of study agent at week 48). Efficacy, safety, pharmacokinetic, immunogenicity and biomarker evaluations were performed in the study on a defined schedule.
About DISCOVER-2 (NCT03158285)12
DISCOVER-2 is a randomized, double-blind, multicenter Phase 3 study evaluating the efficacy and safety of TREMFYA administered by SC injection in biologic-naïve patients with active PsA. DISCOVER-2 evaluated 739 participants who were treated and followed through approximately two years. The study consisted of a screening phase of up to six weeks, a blinded treatment phase of approximately 100 weeks that included a placebo-controlled period from week 0 to week 24 and a blinded active treatment period from week 24 to week 100. It also included a safety follow-up phase through week 112 (i.e., approximately 12 weeks after the last administration of study agent at week 100). Clinical efficacy, radiographic efficacy, health economics, safety, pharmacokinetics, immunogenicity, biomarker, and pharmacogenomics evaluations were performed in the study on a defined schedule.
About TREMFYA® (guselkumab)3
Developed by Janssen, TREMFYA is the first approved fully human monoclonal antibody that selectively binds to the p19 subunit of IL-23 and inhibits its interaction with the IL-23 receptor. IL-23 is an important driver of the pathogenesis of inflammatory diseases such as moderate to severe plaque PsO and active PsA. TREMFYA is approved in the U.S., Canada, Japan, and a number of other countries worldwide for the treatment of adults with moderate to severe plaque PsO who are candidates for injections or pills (systemic therapy) or phototherapy (treatment using ultraviolet light), and for the treatment of adult patients with active PsA. It is also approved in the EU for the treatment of moderate to severe plaque PsO in adults who are candidates for systemic therapy and for the treatment of active PsA in adult patients who have had an inadequate response or who have been intolerant to a prior csDMARD therapy.
The Janssen Pharmaceutical Companies of Johnson & Johnson maintain exclusive worldwide marketing rights to TREMFYA®.
https://www.tremfya.com/plaque-psoriasis
Please read the full Prescribing Information, including Medication Guide for TREMFYA®, and discuss any questions that you have with your doctor.
About the Janssen Pharmaceutical Companies of Johnson & Johnson
At Janssen, we’re creating a future where disease is a thing of the past. We’re the Pharmaceutical Companies of Johnson & Johnson, working tirelessly to make that future a reality for patients everywhere by fighting sickness with science, improving access with ingenuity, and healing hopelessness with heart. We focus on areas of medicine where we can make the biggest difference: Cardiovascular, Metabolism, & Retina; Immunology; Infectious Diseases & Vaccines; Neuroscience; Oncology; and Pulmonary Hypertension.
Learn more at www.janssen.com. Follow us at www.twitter.com/JanssenGlobal and www.twitter.com/JanssenUS.
Janssen Research & Development, LLC; Janssen Biotech, Inc.; and Janssen Scientific Affairs, LLC are part of the Janssen Pharmaceutical Companies of Johnson & Johnson.
Nov. 11, 2022 12:12 PM ET
By: Jonathan Block, SA News Editor
PUBLISHED11 November 2022 11 November 2022 07:00 GMT
AstraZeneca’s Imfinzi (durvalumab) in combination with Imjudo (tremelimumab) plus platinum-based chemotherapy has been approved in the US for the treatment of adult patients with Stage IV (metastatic) non-small cell lung cancer (NSCLC).
The approval by the Food and Drug Administration (FDA) was based on the results from the POSEIDON Phase III trial. Patients treated with a limited course of five cycles of the anti-CTLA-4 antibody Imjudo added to Imfinzi plus four cycles of platinum-based chemotherapy experienced a 23% reduction in the risk of death versus a range of chemotherapy options (based on a hazard ratio [HR] of 0.77; 95% CI 0.65-0.92; p=0.00304). An estimated 33% of patients were alive at two years versus 22% for chemotherapy. This treatment combination also reduced the risk of disease progression or death by 28% compared to chemotherapy alone (HR 0.72; 95% CI 0.60-0.86; p=0.00031).
Updated results from the POSEIDON Phase III trial after approximately four years of follow-up presented at the European Society of Medical Oncology (ESMO) Congress 2022 and published in the Journal of Clinical Oncology demonstrated sustained survival benefit, improving overall survival (OS) by 25% compared to chemotherapy alone (HR 0.75; 95% CI 0.63-0.88). An estimated 25% of patients treated with the combination were alive at three years versus 13.6% for those treated with chemotherapy alone. The safety profile for Imjudo plus Imfinzi and chemotherapy was consistent with the known profiles of each medicine, and no new safety signals were identified.
In the US, lung cancer is the second most commonly diagnosed cancer, with more than 236,000 patients expected to be diagnosed in 2022.1 For patients with metastatic NSCLC, prognosis is particularly poor, as only approximately 8% will live beyond five years after diagnosis.2
Melissa Johnson, MD, Director of Lung Cancer Research, Sarah Cannon Research Institute at Tennessee Oncology in Nashville, Tennessee, and a lead investigator in the POSEIDON Phase III trial, said: “Metastatic non-small cell lung cancer remains a significant treatment challenge because many patients’ tumours do not respond well to standard therapies, including checkpoint inhibitors. The approval of this dual immunotherapy regimen with chemotherapy introduces a new, generally well-tolerated treatment option for patients with this devastating disease and gives them the chance to benefit from the long-term survival advantage seen with CTLA-4 inhibition.”
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “This approval underscores the importance of delivering novel treatment combinations that extend survival in metastatic non-small cell lung cancer, a complex setting where many patients still face a dismal prognosis. This marks the second indication for Imjudo added to Imfinzi in just a few weeks following its approval in unresectable liver cancer, reinforcing the benefits of this new medicine and our commitment to improving patient outcomes in cancer settings with continued unmet need.”
Regulatory applications are also currently under review in Europe, Japan and several other countries for this indication based on the POSEIDON results.
Imfinzi is the only approved immunotherapy and the global standard of care in the curative-intent setting of unresectable, Stage III NSCLC in patients whose disease has not progressed after chemoradiation therapy based on the PACIFIC Phase III trial. Imfinzi is also approved in the US, the EU, Japan, China and many other countries around the world for the treatment of extensive-stage small cell lung cancer (ES-SCLC) based on the CASPIAN Phase III trial. Imfinzi is approved in the US and several other countries in combination with chemotherapy for the treatment of locally advanced or metastatic biliary tract cancer based on the TOPAZ-1 Phase III trial, and it is approved with Imjudo in the US for the treatment of unresectable hepatocellular carcinoma based on the HIMALAYA Phase III trial.
Notes
Stage IV NSCLC
Lung cancer is the second most common form of cancer globally, with more than two million patients diagnosed in 2020.3 Lung cancer is broadly split into NSCLC and SCLC, with 80-85% classified as NSCLC. Within NSCLC, patients are classified as squamous, representing 25-30% of patients, or non-squamous, in approximately 70-75% of patients.4-6
POSEIDON
The POSEIDON trial was a randomised, open-label, multi-centre, global, Phase III trial of Imfinzi plus platinum-based chemotherapy, or Imfinzi, Imjudo and chemotherapy, versus chemotherapy alone in the 1st-line treatment of 1,013 patients with metastatic NSCLC. The trial population included patients with either non-squamous or squamous disease, and the full range of PD-L1 expression levels. POSEIDON excluded patients with certain epidermal growth factor receptor (EGFR) mutations or anaplastic lymphoma kinase (ALK) fusions.
In the experimental arms, patients were treated with a flat dose of 1,500mg of Imfinzi, or Imfinzi and 75mg of Imjudo with up to four cycles of chemotherapy every three weeks, followed by maintenance treatment with Imfinzi once every four weeks, or Imfinzi and a fifth dose of 75mg of Imjudo given at week 16. In comparison, the control arm allowed up to six cycles of chemotherapy. Pemetrexed maintenance treatment was allowed in all arms in patients with non-squamous disease if given during the induction phase. Nearly all patients with non-squamous disease (95.5%) had pemetrexed and platinum, while the majority of patients with squamous disease receiving chemotherapy (88.3%) received gemcitabine and platinum.
Primary endpoints included progression-free survival (PFS) and OS for the Imfinzi plus chemotherapy arm. Key secondary endpoints included PFS and OS in the Imfinzi plus Imjudo and chemotherapy arm. As both PFS endpoints were met for Imfinzi plus chemotherapy and Imfinzi, Imjudo and chemotherapy, the prespecified statistical analysis plan allowed for testing OS in the Imfinzi plus Imjudo and chemotherapy arm. The trial was conducted in more than 150 centres across 18 countries, including the US, Europe, South America, Asia and South Africa.
Imfinzi
Imfinzi (durvalumab) is a human monoclonal antibody that binds to the PD-L1 protein and blocks the interaction of PD-L1 with the PD-1 and CD80 proteins, countering the tumour's immune-evading tactics and releasing the inhibition of immune responses.
In addition to its approved indications in lung cancer, Imfinzi is also the only approved immunotherapy in unresectable or metastatic biliary tract cancer and hepatocellular carcinoma (in combination with Imjudo), and is also approved for previously treated patients with advanced bladder cancer in several countries.
As part of a broad development programme, Imfinzi is being tested as a single treatment and in combinations with other anti-cancer treatments for patients with SCLC, NSCLC, bladder cancer, several gastrointestinal (GI) cancers, ovarian cancer, endometrial cancer and other solid tumours.
Imjudo
Imjudo (tremelimumab) is a human monoclonal antibody that targets the activity of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). Imjudo blocks the activity of CTLA-4, contributing to T-cell activation, priming the immune response to cancer and fostering cancer cell death.
Imjudo is also approved in combination with Imfinzi for the treatment of unresectable hepatocellular carcinoma (HCC) and is being tested in combination with Imfinzi across multiple tumour types including locoregional HCC (EMERALD-3), SCLC (ADRIATIC) and bladder cancer (VOLGA and NILE).
AstraZeneca in lung cancer
AstraZeneca is working to bring patients with lung cancer closer to cure through the detection and treatment of early-stage disease, while also pushing the boundaries of science to improve outcomes in the resistant and advanced settings. By defining new therapeutic targets and investigating innovative approaches, the Company aims to match medicines to the patients who can benefit most.
The Company's comprehensive portfolio includes leading lung cancer medicines and the next wave of innovations, including Tagrisso (osimertinib) and Iressa (gefitinib); Imfinzi (durvalumab) and Imjudo (tremelimumab); Enhertu (trastuzumab deruxtecan) and datopotamab deruxtecan in collaboration with Daiichi Sankyo; Orpathys (savolitinib) in collaboration with HUTCHMED; as well as a pipeline of potential new medicines and combinations across diverse mechanisms of action.
AstraZeneca is a founding member of the Lung Ambition Alliance, a global coalition working to accelerate innovation and deliver meaningful improvements for people with lung cancer, including and beyond treatment.
AstraZeneca in immuno-oncology (IO)
AstraZeneca has a comprehensive and diverse IO portfolio and pipeline anchored in immunotherapies designed to overcome evasion of the anti-tumour immune response and stimulate the body’s immune system to attack tumours.
AstraZeneca aims to reimagine cancer care and help transform outcomes for patients with Imfinzi as a single treatment and in combination with Imjudo as well as other novel immunotherapies and modalities. The Company is also exploring next-generation immunotherapies like bispecific antibodies and therapeutics that harness different aspects of immunity to target cancer.
AstraZeneca is boldly pursuing an innovative clinical strategy to bring IO-based therapies that deliver long-term survival to new settings across a wide range of cancer types. With an extensive clinical programme, the Company also champions the use of IO treatment in earlier disease stages, where there is the greatest potential for cure.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Nov. 10, 2022 6:17 PM ET
By: Jonathan Block, SA News Editor1 Comment
Nov. 10, 2022 9:00 AM ET
Bristol-Myers Squibb Company (BMY)
CAMZYOSTM is the only Health Canada approved reversible cardiac myosin inhibitor indicated for the treatment of symptomatic obstructive hypertrophic cardiomyopathy of New York Heart Association (NYHA) Class II-III in adults.
MONTREAL, Nov. 10, 2022 /CNW/ - Today, Bristol Myers Squibb Canada (BMS) announced Health Canada's approval of CAMZYOSTM (mavacamten capsules) for the treatment of symptomatic obstructive hypertrophic cardiomyopathy (oHCM) of New York Heart Association (NYHA) Class II-III in adult patients.i Hypertrophic Cardiomyopathy (HCM) is a chronic disease where the heart's walls become thickened, making it harder for the heart to pump blood. CAMZYOSTM is the first Canadian-approved allosteric and selective cardiac myosin inhibitor that targets the underlying pathophysiology of oHCM. i

"Until now, the standard of care for patients has been to control the symptoms of obstructive HCM using drugs that were not designed for HCM. Patients often require invasive therapies," said Dr Rafik Tadros, Cardiologist, Montreal Heart Institute. "CAMZYOSTM has now brought an option that is specifically developed to treat the underlying pathophysiological mechanism of HCM and represents an important advance to improve functional capacity and quality of life in many patients."
Clinical Data
The Canadian authorization was based on data from the Phase 3 EXPLORER-HCM trial which was a double-blind, randomized, placebo-controlled parallel group trial that enrolled a total of 251 adult patients with symptomatic (NYHA class II or III), oHCM.viii In EXPLORER HCM trial, 37% of patients achieved the primary endpoint a composite of functional capacity and symptoms compared to 17% of patients in the placebo group and all had greater improvement across the secondary endpoints at Week 30 in the CAMZYOSTM group compared to the placebo group.vii In the Phase 3 EXPLORER-HCM trial, serious adverse events were observed in 8.1% of mavacamten-treated patients and 8.6% of placebo-treated patients to Week 30. Overall safety and tolerability similar to placebo.ix
CAMZYOSTM reduces left ventricular ejection fraction (LVEF) and can cause heart failure due to systolic dysfunction. Echocardiogram assessments of LVEF and Left Ventricular Outflow tract (LVOT) gradient are required prior to, and regularly during, treatment with CAMZYOSTM. Initiation of CAMZYOSTM in patients with LVEF <55% is not recommended. Interrupt CAMZYOSTM treatment if LVEF is <50% at any visit or if the patient experiences heart failure symptoms or worsening clinical status. Concomitant use of CAMZYOSTM with certain cytochrome P450 inhibitors and inducers may increase the risk of heart failure due to systolic dysfunction or may lead to a loss of therapeutic response.i
About Bristol Myers Squibb Canada Co.
Bristol Myers Squibb Canada Co. is an indirect wholly-owned subsidiary of Bristol Myers Squibb Company, a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. Bristol Myers Squibb Canada Co. employs close to 300 people across the country. For more information, please visit https://www.bms.com/ca/en.
About Bristol Myers Squibb
Bristol Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information about Bristol Myers Squibb, visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
SOURCE Bristol Myers Squibb
Bristol-Myers Squibb Company (BMY)
11/10/22
Download this Press Release PDF Format (opens in new window) (PDF)
- Median Overall Survival for GPS Combination (GPS in Combination with Keytruda) was 18.4 Months Compared to 13.8 months in a Checkpoint Inhibitor Single Agent Study in a Similar Patient Population Treated with Checkpoint Inhibitor Alone -
- Median Progression Free Survival for GPS Combination (GPS in Combination with Keytruda) was 12 Weeks Compared to Eight Weeks in a Checkpoint Inhibitor Single Agent Study in a Similar Patient Population Treated with Checkpoint Inhibitor Alone -
- Clinical Benefit (Increased Disease Control Rate, Progression Free Survival and Overall Survival) Observed in Patients Harboring Tumors with Detectable PD-L1 Biomarker –
- Full Clinical Trial Data to be Presented at a Major Medical Conference in 1H 2023 –
NEW YORK, Nov. 10, 2022 (GLOBE NEWSWIRE) -- SELLAS Life Sciences Group, Inc. (NASDAQ: SLS) (“SELLAS” or the “Company”), a late-stage clinical biopharmaceutical company focused on the development of novel therapies for a broad range of cancer indications, today announced confirmatory top-line clinical data from the final analysis of results from its Phase 1/2 clinical trial of galinpepimut-S (GPS), the Company’s Wilms Tumor-1 (WT1)-targeting peptide immunotherapeutic, in combination with Merck’s anti-PD-1 therapy, pembrolizumab (KEYTRUDA®), in patients diagnosed with WT1(+) relapsed or refractory platinum-resistant advanced metastatic ovarian cancer.
Data from 17 patients enrolled in the study, conducted under a Clinical Trial Collaboration and Supply Agreement with Merck & Co., Inc., Rahway, N.J., USA (known as MSD outside the United States and Canada), have undergone final analysis for safety and efficacy. All enrolled patients were resistant to standard of care platinum-based therapy and 76.5 percent (13/17) of the patients were refractory to or had failed their first- or second-line therapies, with 23.5 percent (4/17) having failed three or more lines of therapy, including one patient (5.9 percent) who failed five previous lines of therapy. Of the 17 patients, 16 received at least three doses of GPS and had follow-up cross-sectional imaging (CT/MRI) to determine tumor status.
Summary of Top-Line Overall Survival and Progression-Free Survival Data:
Importantly, survival and disease control benefits were observed in patients harboring tumors with any level of detectable PD-L1 expression, i.e., those with Combined Positive Score (CPS) of one or higher. The DCR is 63.6 percent in patients with a CPS of one or higher in this study.
In the Phase 1/2 study, patients with a CPS score of less than one showed a median OS of 3.2 months vs. patients with a CPS greater than or equal to one who had a median OS of 18.4 months and, as it relates to time to progression, patients with a CPS score of less than one had a median PFS of 1.9 months and patients with a CPS score of greater than or equal than one showed a median PFS of 3.8 months.
In 16 evaluable patients in whom serial peripheral blood samples were available, a correlation was observed between PFS and OS and WT1-specific immune response after GPS vaccination across more than one channel with intracellular cytokine flow-cytometry assays in peripheral blood lymphocytes assaying reactivity against the four pooled WT1 antigens comprising GPS. The data were consistent with those seen in previous studies of GPS.
The safety profile of GPS in combination with pembrolizumab was similar to pembrolizumab alone, with the only addition of low-grade, rapidly resolving local reactions at the GPS injection site, consistent with observations from other GPS clinical studies.
“GPS has been primarily studied as maintenance therapy to provide an overall survival benefit after patients reach a state of minimal residual disease or complete remission. In contrast, in this very difficult to treat patient population with relapsed or refractory measurable advanced platinum-resistant ovarian cancer, who underwent intensive chemotherapy with no apparent enduring clinical benefit, the data suggests that the combination of GPS plus pembrolizumab may be effective in stabilizing active disease. We are excited that both the disease control rate and overall survival results support our belief that GPS has the potential to become an important therapeutic addition to a backbone of checkpoint blockade for WT1(+) ovarian cancer patients, as well as other WT1-expressing tumor types,” said Angelos Stergiou, MD, ScD h.c., President and Chief Executive Officer of SELLAS. “SELLAS plans to submit the full dataset at a major medical conference by mid-next year. I would like to extend my sincere gratitude to all patients who have participated in this clinical trial, as well as the collective study teams at SELLAS and Merck,” concluded Dr. Stergiou.
“What I find particularly important for patients with advanced platinum-resistant ovarian cancer is that GPS appears to enhance efficacy of checkpoint inhibitors in patients with tumors with detectable PD-L1 expression, whilst demonstrating a low adverse event burden. Until now, in immunotherapy-recalcitrant tumor types, like ovarian cancer, the impression by many experts has been that patients with CPS scores equal to or higher than 10 may be the most probable candidates to derive clinically meaningful benefit from checkpoint inhibitor therapy, either alone or in combination,“ stated Bruno Bastos, MD, Medical Oncologist and Study Investigator at the Miami Cancer Institute, Physician Lead of the Multiple Tumors/Phase 1 Clinic, and Assistant Professor at the Herbert Wertheim College of Medicine. “Based on these results, it appears that any ovarian cancer patient whose tumor expresses any level of PD-L1, as long as this biomarker is detected, may benefit from the addition of GPS to a checkpoint inhibitor regimen. If these data are confirmed in larger studies in PD-L1+, WT1+ advanced ovarian cancer, it could enable us to bring the benefits of immunotherapy drugs to a broader patient population in a very difficult setting. This is indeed exciting for the ovarian cancer field, as well as patients suffering from other types of WT1+ malignancies,” concluded Dr. Bastos.
About SELLAS Life Sciences Group, Inc.
SELLAS Life Sciences Group, Inc. (NASDAQ: SLS) is a late-stage clinical biopharmaceutical company focused on the development of novel therapeutics for a broad range of cancer indications. SELLAS’ lead product candidate, GPS, is licensed from Memorial Sloan Kettering Cancer Center and targets the WT1 protein, which is present in an array of tumor types. GPS has potential as a monotherapy or in combination with other therapies to address a broad spectrum of hematologic malignancies and solid tumor indications. The Company is also developing GFH009, a small molecule, highly selective CDK9 inhibitor, which is licensed from GenFleet Therapeutics (Shanghai), Inc., for all therapeutic and diagnostic uses in the world outside of Greater China.
For more information on SELLAS, please visit www.sellaslifesciences.com.
Keytruda® is a registered trademark of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA and is not a trademark of SELLAS. The manufacturer of this brand is not affiliated with and does not endorse SELLAS or its products.
Source: SELLAS Life Sciences Group, Inc.
Nov. 10, 2022 9:31 AM ET
SELLAS Life Sciences Group, Inc. (SLS), MRK
By: Ravikash, SA News Editor
Sellas Life Sciences (NASDAQ:SLS) stock rose ~6% on Thursday after the company reported data from the final analysis of a phase 1/2 trial of its drug galinpepimut-S (GPS) in combination with Merck's (NYSE:MRK) Keytruda (pembrolizumab) to treat patients with WT1(+) relapsed or refractory platinum-resistant advanced metastatic ovarian cancer.
SELLAS Life Sciences Group, Inc. (SLS), MRK
https://www.sellaslifesciences.com/galinpepimut-s-gps-therapy/default.aspx#section=therapy
https://www.sellaslifesciences.com/Home/default.aspx
November 4, 2022Download PDF
- EMPA-KIDNEY, the largest and broadest dedicated SGLT2 inhibitor trial in chronic kidney disease, provides new evidence for patients commonly seen in clinical practice
- Phase III trial also demonstrated a statistically significant reduction (14%) in hospitalization for any cause, bringing potential relief for patients and reducing burden on healthcare systems
OXFORD, United Kingdom and INGELHEIM, Germany and RIDGEFIELD, Conn. and INDIANAPOLIS, Nov. 4, 2022 /PRNewswire/ -- EMPA-KIDNEY phase III clinical trial met its primary endpoint by demonstrating a significant kidney and cardiovascular benefit for adults living with chronic kidney disease (CKD). When treated with Jardiance® (empagliflozin), the risk of kidney disease progression or cardiovascular death was significantly reduced by 28% vs. placebo (HR; 0.72; 95% CI 0.64 to 0.82; P<0.0001). The results were announced today during the American Society of Nephrology (ASN)'s Kidney Week 2022 by the Medical Research Council Population Health Research Unit (MRC PHRU) at the University of Oxford, which designed, conducted and analyzed EMPA-KIDNEY in a scientific collaboration with Boehringer Ingelheim and Eli Lilly and Company (NYSE: LLY). The results were also published simultaneously in The New England Journal of Medicine.
EMPA-KIDNEY is the first SGLT2 inhibitor CKD trial to demonstrate a significant reduction in all-cause hospitalizations (14%) (HR; 0.86; 95% CI 0.78 to 0.95; p=0.0025) vs. placebo, one of the pre-specified key secondary confirmatory endpoints. CKD doubles a person's risk for hospitalization and is a leading cause of death globally. Hospitalizations account for 35%-55% of total healthcare costs for people with CKD in the U.S.
The overall safety data was generally consistent with previous findings, confirming the well-established safety profile of Jardiance.
"We know that there is an urgent need for new therapies proven to delay CKD progression which can lead to the need for dialysis or transplantation. Today's results demonstrate that Jardiance may benefit adults at risk of progression, including those with or without diabetes, and across a wide range of kidney function," said William Herrington, associate professor at MRC PHRU (part of Oxford Population Health), and honorary consultant nephrologist, and EMPA-KIDNEY co-principal investigator. "By reducing the risk of kidney disease progression or cardiovascular death, Jardiance has the potential to positively impact healthcare systems worldwide."
"The design of the EMPA-KIDNEY trial included a wider range of patients than ever before," said Professor Richard Haynes, co-principal investigator. "Previous SGLT2 inhibitor trials focused on certain groups of people living with CKD, such as those with diabetes or high levels of protein in their urine. Today's positive trial results across a broad CKD population reflect an opportunity to improve the treatment of this disease and prevent people from needing dialysis."
EMPA-KIDNEY is the largest and broadest dedicated SGLT2 inhibitor trial to date. It included 6,609 participants across a wide range of underlying causes, many with co-morbidities across the spectrum of cardiovascular, kidney or metabolic conditions. The trial assessed both kidney and cardiovascular outcomes in people across the spectrum of CKD severity.
"The Boehringer Ingelheim and Lilly Alliance is incredibly proud that EMPA-KIDNEY has provided another pivotal moment for Jardiance," said Carinne Brouillon, head of Human Pharma and member of the Board of Managing Directors, Boehringer Ingelheim. "Today's data adds to the body of evidence from our clinical program which includes more than 700,000 adults with cardiovascular, kidney and metabolic conditions. EMPA-KIDNEY reinforces the potential role of Jardiance in changing the way these interconnected conditions may be managed."
Reductions in other key secondary endpoints of hospitalization for heart failure or cardiovascular death or all-cause death were not statistically significant, however the power to detect this was limited by the number of events observed. Reduction in the risk of these endpoints is consistent with the totality of the evidence from other trials which have shown statistical significance of these outcomes.
"Today's EMPA-KIDNEY trial results will be welcomed by people living with CKD and the medical community. We are also encouraged by the risk reduction for hospitalization after just two years, as this finding is in line with the significant reductions seen in prior Jardiance cardiovascular outcomes trials," said Jeff Emmick, M.D., Ph.D., vice president, Product Development, Lilly. "The Alliance looks forward to discussing plans for marketing authorization for CKD with regulators worldwide in due course."
Notes to editors
Kidney disease progression: Defined as end-stage kidney disease (the initiation of maintenance dialysis or receipt of a kidney transplant), a sustained decline in estimated glomerular filtration rate (eGFR) to below 10 mL/min/1.73 m2, kidney death or a sustained decline of at least 40% in eGFR from randomization).
End stage kidney disease: Includes initiation of maintenance dialysis or receipt of a kidney transplant
About EMPA-KIDNEY: The study of heart and kidney protection with Jardiance
EMPA-KIDNEY (NCT03594110) is a multinational, randomized, double-blind, placebo-controlled clinical trial, designed to evaluate the effect of Jardiance on kidney disease progression and cardiovascular mortality risk. The primary outcome is defined as time to a first event of either cardiovascular death or kidney disease progression, defined as end-stage kidney disease (the need for kidney replacement therapy such as dialysis or kidney transplantation), a sustained decline in eGFR to <10 mL/min/1.73 m2, kidney death, or a sustained decline of ≥40 percent in eGFR from randomization. Key secondary outcomes include cardiovascular death or hospitalization for heart failure, all-cause hospitalization and all-cause mortality. EMPA-KIDNEY includes 6,609 adults randomized from eight countries with established CKD both with and without diabetes, as well as with and without albuminuria, receiving either Jardiance 10 mg or placebo, on top of current standard of care.
About the EMPOWER program
The Boehringer Ingelheim and Lilly Alliance has developed the EMPOWER program to explore the impact of Jardiance on major clinical cardiovascular and kidney outcomes in a spectrum of cardio-kidney-metabolic conditions. Cardio-renal-metabolic conditions are the leading cause of mortality worldwide and account for up to 20 million deaths annually. Through the EMPOWER program, Boehringer Ingelheim and Lilly are working to advance knowledge of these interconnected systems and create care which offers integrated, multi-organ benefits. Comprised of nine clinical trials and two real-world evidence studies, EMPOWER reinforces the long-term commitment of the Alliance to improve outcomes for people living with cardio-kidney-metabolic conditions. With more than 700,000 adults enrolled worldwide in clinical trials, it is the broadest and most comprehensive clinical program for an SGLT2 inhibitor to date.
What is JARDIANCE?
JARDIANCE is a prescription medicine used to:
JARDIANCE is not for people with type 1 diabetes. It may increase their risk of diabetic ketoacidosis (increased ketones in the blood or urine).
JARDIANCE is not for use to lower blood sugar in adults with type 2 diabetes who have severe kidney problems, because it may not work.
IMPORTANT SAFETY INFORMATION
Do not take JARDIANCE if you are allergic to empagliflozin or any of the ingredients in JARDIANCE.
Do not take JARDIANCE if you are on dialysis.
For more information, please see Prescribing Information and Medication Guide.
Boehringer Ingelheim and Eli Lilly and Company
In January 2011, Boehringer Ingelheim and Eli Lilly and Company announced an Alliance that centers on compounds representing several of the largest diabetes treatment classes. Depending on geographies, the companies either co-promote or separately promote the respective molecules each contributing to the Alliance. The Alliance leverages the strengths of two of the world's leading pharmaceutical companies to focus on patient needs. By joining forces, the companies demonstrate their commitment, not only to the care of people with diabetes, but also to investigating the potential to address areas of unmet medical need.
About Boehringer Ingelheim
Boehringer Ingelheim is working on breakthrough therapies that improve the lives of humans and animals. As a leading research-driven biopharmaceutical company, the company creates value through innovation in areas of high unmet medical need. Founded in 1885 and family-owned ever since, Boehringer Ingelheim takes a long-term perspective. Around 52,000 employees serve more than 130 markets in the three business areas, Human Pharma, Animal Health, and Biopharmaceutical Contract Manufacturing. Learn more at www.boehringer-ingelheim.us
About Eli Lilly and Company
Lilly unites caring with discovery to create medicines that make life better for people around the world. We've been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world's most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer's disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we're motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more about Lilly, please visit us at lilly.com and lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn.
![]()

![]()
View original content to download multimedia:https://www.prnewswire.com/news-releases/landmark-empa-kidney-trial-showed-significant-benefit-of-jardiance-in-reducing-kidney-disease-progression-or-cardiovascular-death-by-28-vs-placebo-in-people-with-chronic-kidney-disease-301669038.html
SOURCE Eli Lilly and Company
Nov. 04, 2022 1:26 PM ET
By: Dulan Lokuwithana, SA News Editor1 Comment
Jardiance, a cardiovascular therapy developed by Eli Lilly (NYSE:LLY) in partnership with German pharma company Boehringer Ingelheim has met the main goal in a late-stage trial for adults with chronic kidney disease (CKD), according to data presented on Friday.
https://seekingalpha.com/symbol/LLY
NOVEMBER 9, 2022
PARIS, FRANCE, 9 November 2022 – Ipsen (Euronext: IPN; ADR: IPSEY) announced the Phase III NAPOLI 3 trial of Onivyde® (irinotecan liposome injection) plus 5 fluorouracil/leucovorin and oxaliplatin (NALIRIFOX regimen) met its primary endpoint demonstrating clinically meaningful and statistically significant improvement in overall survival compared to nab-paclitaxel plus gemcitabine in 770 previously untreated patients with metastatic pancreatic ductal adenocarcinoma (mPDAC) and key secondary efficacy outcome of progression-free survival (PFS) also showed significant improvement over the comparator arm. The safety profile of Onivyde in the NAPOLI 3 trial was consistent with those observed in the previous phase I/II mPDAC study.
Ipsen intends to file a supplemental New Drug Application with the U.S. Food and Drug Administration for Onivyde in combination with oxaliplatin plus 5- fluorouracil/leucovorin for the treatment of patients with previously untreated mPDAC following the Fast Track Designation granted in 2020. The clinical trial results will be presented at an upcoming medical conference.
“The positive results from the NAPOLI 3 trial demonstrate that compared with the standard-of-care, the investigational Onivyde treatment regimen extended the lives of people living with metastatic pancreatic ductal adenocarcinoma who were previously untreated,” said Howard Mayer, Executive Vice President and Head of Research and Development for Ipsen. “The prognosis for people diagnosed with pancreatic cancer is extremely poor and we plan to submit these new findings to the regulatory authority as, if approved, we believe this regimen could offer up an important new treatment option for people living with an aggressive and hard-to-treat cancer. We thank the patients who participated in the study, their families and their healthcare teams.”
PDAC is the most common type of cancer that forms in the pancreas with approximately 60,000 people diagnosed in the U.S. each year and nearly 500,000 people globally.1,2 Since there are no specific symptoms in the early stages, PDAC is often detected late and after the disease has spread to other parts of the body (metastatic or stage IV).3 Even in later stages, weight loss, abdominal pain and jaundice are the most common symptoms making PDAC difficult to detect.4 Currently, fewer than 20 percent of people diagnosed with PDAC survive longer than one year and overall, pancreatic cancer has the lowest five-year survival rate of all cancer types globally and in the U.S.1,2
Phase III NAPOLI 3 Study
NAPOLI 3 is a randomized, open-label Phase III trial of Onivyde treatment regimen (NALIRIFOX) in patients who have not previously received chemotherapy for metastatic pancreatic ductal adenocarcinoma. Onivyde plus oxaliplatin and 5 FU/LV was administered twice in a month (days 1 and 15 of 28 day cycle) compared to an injection of nab-paclitaxel and gemcitabine administered three times a month (days 1, 8, 15 of a 28 day cycle). Approximately 770 patients were enrolled in the trial with the primary outcome measure of overall survival. Secondary outcome measures included progression-free survival, objective response rate, quality of life assessment, incidence of treatment-emergent adverse events, serious adverse events and laboratory abnormalities. Adverse events and laboratory analyses were also analyzed.
Onviyde® (irinotecan liposome injection)
Onivyde is a long-circulating, liposomal topoisomerase inhibitor designed to interrupt DNA replication in cancer cells. Onivyde enters cancer cells using a naturally occurring process (enhanced permeability and retention or EPR effect) and as macrophages unpack the liposomes, Onivyde is activated facilitating the release of the cytotoxic payload into the tumor, including irinotecan and its conversion into SN-38, its active metabolite. Ipsen has exclusive commercialization rights for the current and potential future indications for Onivyde in the U.S. Servier, an independent international pharmaceutical company with a strong international presence in 150 countries, is responsible for the commercialization of Onivyde outside of the U.S. and Taiwan. PharmaEngine is a commercial stage oncology company headquartered in Taipei and is responsible for the commercialization of Onivyde in Taiwan.
Onivyde is currently approved in most major markets including the U.S., Europe and Asia in combination with fluorouracil (5-FU) and leucovorin (LV) for the treatment of patients with metastatic adenocarcinoma of the pancreas after disease progression following gemcitabine-based therapy. Onivyde is not indicated as a single agent for the treatment of patients with metastatic adenocarcinoma of the pancreas.
https://www.onivyde.com/en-us/for-patients
Please see full U.S. Prescribing Information including Boxed WARNING for Onivyde.
About Ipsen
Ipsen is a global, mid-sized biopharmaceutical company focused on transformative medicines in Oncology, Rare Disease and Neuroscience. With Specialty Care sales of €2.6bn in FY 2021, Ipsen sells medicines in over 100 countries. Alongside its external-innovation strategy, the Company’s research and development efforts are focused on its innovative and differentiated technological platforms located in the heart of leading biotechnological and life-science hubs: Paris-Saclay, France; Oxford, U.K.; Cambridge, U.S.; Shanghai, China. Ipsen, excluding its Consumer HealthCare business, has around 4,500 colleagues worldwide and is listed in Paris (Euronext: IPN) and in the U.S. through a Sponsored Level I American Depositary Receipt program (ADR: IPSEY). For more information, visit ipsen.com
Nov. 09, 2022 5:16 AM ET
By: Ravikash, SA News Editor3 Comments
Ipsen (OTCPK:IPSEY) (OTCPK:IPSEF) said its drug Onivyde met the main goal of overall survival in patients with pancreatic cancer in a phase 3 trial.
https://www.onivyde.com/en-us/for-patients
Nov 07, 2022
Basel, November 7, 2022 — Novartis today announced results from the Phase II open-label extension ORION-3 trial, which showed that Leqvio provides effective low-density lipoprotein cholesterol (LDL-C) reduction over a four-year period in patients with either atherosclerotic cardiovascular disease (ASCVD) or ASCVD risk equivalent, and elevated LDL-C despite maximally tolerated statin therapy1,2. Leqvio is the first and only small interfering RNA (siRNA) therapy to lower LDL-C and is administered with two doses a year*. Results were presented at the American Heart Association (AHA) Scientific Sessions 20221,2.
“The results we’ve seen in patients after four years of treatment demonstrate that inclisiran is well-tolerated and can help patients achieve LDL-C reduction while also maintaining and sustaining their levels,” said Kausik Ray, M.D., Professor of Public Health in the Department of Public Health and Primary Care at Imperial College London and Honorary Consultant Cardiologist at the Imperial College NHS Trust. “Still too many patients are struggling to reach their LDL-C target levels. A therapy that provides sustained LDL-C reduction with a twice-yearly maintenance dosing schedule may be a turning point in the treatment of ASCVD.”
In ORION-3, an open-label extension of the Phase II ORION-1 trial, LDL-C level reduction was sustained over the four-year study period: patients treated with Leqvio achieved an average 47.5% reduction in LDL-C from baseline (Day 1 of ORION-1) to Day 210 (95% Cl:-50.69,-44.27) and a time-averaged reduction in LDL-C of 44.2% over the four years through twice-yearly dosing1,2.
“Patients with ASCVD are a large, high-risk population in the US, and a majority are not achieving their LDL-C target levels,” said Norman Lepor, M.D., a Los Angeles based cardiologist and Director of the National Heart Institute. “The sustained benefit of a twice-yearly treatment is a major advance in meeting the needs of these patients.”
ORION-3 provides the longest safety follow-up in a Leqvio study to date. After four years of therapy, Leqvio was well-tolerated, with a safety profile consistent with previous 18-month Phase III LDL-C lowering studies1,4. The most common drug-related treatment-emergent adverse events were general disorders and injection site reactions that were mostly mild-tomoderate, consistent with previous studies1-4. In addition to this ORION-3 data presented at the AHA Scientific Sessions, there was a pooled exploratory safety analysis of the Phase III ORION trials entitled, “Inclisiran and cardiovascular events: a patient-level analysis of Phase III trials” published in the European Heart Journal on November 4, adding to the growing body of safety evidence for Leqvio11.
“The ORION-3 results show that Leqvio consistently helped patients lower their LDL-C, and with a well-tolerated safety profile,” said David Soergel, M.D., Global Head of Cardiovascular, Renal and Metabolic Drug Development, Novartis. “With two maintenance doses a year, Leqvio is an important option for ASCVD patients who are not reaching recommended LDL-C target levels despite taking other cholesterol-lowering medications.”
Current evidence suggests that elevated LDL-C is the most readily modifiable risk factor of ASCVD5-8.
*After an initial dose and one at three months.
About Leqvio
Leqvio is approved for the treatment of primary hyperlipidemia (including heterozygous familial hypercholesterolemia) as an adjunct to diet and maximally tolerated statin therapy to reduce LDL-C. Of note, the indication may vary depending on countries. Leqvio is a subcutaneous injection given by a healthcare provider with an initial dose, another at three months, and then every six months. This approach may help those who have trouble sticking to medicines that are self-administered and have greater dosing frequency3,4. Leqvio is approved in more than 60 countries worldwide, including the US, and the EU.
Novartis has obtained global rights to develop, manufacture and commercialize Leqvio under a license and collaboration agreement with Alnylam Pharmaceuticals, a leader in RNAi therapeutics.
About ORION-3
ORION-3 (NCT03060577) is an open-label, non-randomized, extension of the Phase II ORION-1 trial which evaluated Leqvio the long-term safety and efficacy of Leqvio in 233 patients with ASCVD (~1210 patient years), ASCVD risk equivalents, and elevated LDL-C despite maximally tolerated lipid-lowering therapy1,2,12. The primary endpoint of the study was percentage LDL-C change from baseline (Day 1 of ORION-1) to day 210 of ORION-3. LDL-C levels were assessed up to Day 1440 (4 years). Patients received twice-yearly doses of 300 mg inclisiran sodium in this study1,2.
About VictORION
Novartis envisions a world where ASCVD is eliminated so patients can live longer and healthier lives. ORION-3 is part of VictORION, an innovative and robust clinical program for Leqvio, comprising 27 trials and enrolling 47,000 patients in more than 50 countries worldwide. The program is designed to expand on the foundational evidence of LDL-C reduction with Leqvio in diverse patient populations to include implementation research, real-world evidence, and trials that establish its benefits on cardiovascular outcomes. A growing number of studies are planned to generate a vast array of data with major trials such as ORION-4, V (VictORION)-2-PREVENT, V-INITIATE, V-INCEPTION, V-REAL, V-DIFFERENCE, and V-PLAQUE. The VictORION program reinforces our commitment to stopping premature death from cardiovascular disease and to leading a generational decline in cardiovascular morbidity and mortality.
About Novartis in Cardiovascular
Cardiovascular (CV) disease is a global health crisis13,17.
CV disease is the number one killer in the world13,17. Taking more lives than all cancers combined, it contributes to one in every three deaths globally13,17. Of all CV events, 80% can be prevented18. Patients and their families deserve better, and our society deserves more.
Thanks to a combination of our legacy, global footprint and leading science, Novartis is uniquely positioned to help change this landscape. We are transforming the way we think about how CV disease is managed throughout life. Our efforts include the use of early interventions and the development of pioneering treatments that address the spectrum of CV disease, from prevention to management, as well as the creation of innovative access models. By re-writing the way we work with society, we will lead a worldwide effort to improve health outcomes and roll back the crisis of CV death.
Our goal is to bend the curve of life by reducing and stopping premature death from CV disease.
About Novartis
Novartis is reimagining medicine to improve and extend people’s lives. We deliver high-value medicines that alleviate society’s greatest disease burdens through technology leadership in R&D and novel access approaches. In our quest to find new medicines, we consistently rank among the world’s top companies investing in research and development. About 108,000 people of more than 140 nationalities work together to bring Novartis products to nearly 800 million people around the world. Find out more at https://www.novartis.com
November 02, 2022
– Approval Expands on Previous FDA Approval of Vemlidy in Adults Living With This Chronic Liver Disease –
–Efficacy and Safety of Once-Daily Vemlidy Demonstrated in Individuals 12 Years of Age and Older –
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the U.S. Food and Drug Administration (FDA) has approved the supplemental new drug application (sNDA) for Vemlidy® (tenofovir alafenamide) 25 mg tablets as a once-daily treatment for chronic hepatitis B virus (HBV) infection in pediatric patients 12 years of age and older with compensated liver disease.
Vemlidy is a novel, targeted prodrug of tenofovir that was previously approved by the FDA in 2016 as a once-daily treatment for adults with chronic HBV infection with compensated liver disease. It is recommended as a preferred or first-line treatment for adults with chronic HBV with compensated liver disease in guidelines from the American Association for the Study of Liver Diseases (AASLD) and European Association for the Study of the Liver (EASL).1,2
Vemlidy’s approval in this pediatric patient population is supported by 24-week data from a Phase 2 clinical trial (Trial 1092) comparing treatment with Vemlidy 25 mg to placebo among 70 treatment-naïve and treatment-experienced patients aged 12 to less than 18 years weighing at least 35 kg. The study met its primary endpoint of percentage of patients with HBV DNA levels below 20 IU/mL at 24 weeks of therapy; overall, 21% (10/47) of subjects treated with Vemlidy 25 mg achieved HBV DNA <20 IU/mL at 24 weeks compared to 0% (0/23) of subjects treated with placebo.
“Chronic hepatitis B can have a significant long-term health impact on children, including the development of liver cancer later in life if the disease is left untreated, which is compounded by treatment challenges in this population,” said Kathleen Schwarz, MD, Pediatric Gastroenterologist, Rady Children’s Hospital-San Diego, an investigator in the Vemlidy clinical trial. “As a clinician, I recognize the critical importance of treating this disease as quickly as possible to help avoid complications and potential damage to the liver. In the clinical trial, we saw that tenofovir alafenamide may represent an effective treatment option for people as young as 12 years of age living with this chronic disease.”
“While pediatric hepatitis B prevalence has dropped significantly in the U.S., children who develop chronic hepatitis B following an acute infection can experience lifelong health impact,” said Merdad Parsey, MD, PhD, Chief Medical Officer, Gilead Sciences. “Gilead is focused on meeting the biggest challenges in liver disease and impacting the course of disease. With an established safety profile and once-daily dosing, Vemlidy provides physicians a new option to address the treatment needs of pediatric patients living with hepatitis B.”
Vemlidy has a boxed warning in its product label regarding post-treatment severe acute exacerbation of hepatitis B. See below for important safety information.
Indication
VEMLIDY is indicated for the treatment of chronic hepatitis B virus infection in adults and pediatric patients 12 years of age and older with compensated liver disease.
About the 1092 Study
Trial 1092 was a Phase 2 clinical trial that randomized 70 treatment-naïve and treatment-experienced patients between the ages of 12 to less than 18 years weighing at least 35 kg to receive either Vemlidy 25 mg (N=47) or placebo (N=23) once daily. The study met its primary endpoint of percentage of patients with HBV DNA levels below 20 IU/mL at 24 weeks of therapy; overall, 21% (10/47) of subjects treated with Vemlidy 25 mg achieved HBV DNA <20 IU/mL at 24 weeks compared to 0% (0/23) of subjects treated with placebo. Investigators also observed higher rates of serum alanine aminotransferase (ALT) normalization based on AASLD criteria at 24 weeks; 44% of Vemlidy-treated patients and 0% of placebo-treated patients achieved ALT normalization, with median (Q1, Q3) changes from baseline in ALT of -32.0 U/L (-63.0, -13.0 U/L) and 1.0 U/L (-10.0, 25.0 U/mL). Serum ALT is an enzyme used to monitor liver damage. Importantly, the mean percent changes in bone mineral density from baseline to Week 24 were numerically similar for Vemlidy- treated patients and placebo-treated patients (2.4% (N=37) and 1.9% (N=18) for lumbar spine, and 1.5% (N=39) and 1.9% (N=18) for whole body, respectively). At Week 24, mean changes from baseline BMD Z-scores were -0.03 and -0.09 for lumbar spine, and -0.05 and -0.01 for whole body, for the Cohort 1 VEMLIDY and placebo groups, respectively.
About Gilead Sciences in Liver Disease
For more than 20 years, Gilead has sought to address some of the biggest challenges in liver disease. The company has transformed the trajectory of multiple liver diseases through a relentless pursuit of innovation and pioneering access programs to bring meaningful therapies to people around the world. More work is required, and Gilead is committed to advancing innovative therapeutics to address the most pressing unmet needs in liver disease and overcoming barriers to better care.
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer. Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California.
U.S. Prescribing Information for Vemlidy, including BOXED WARNINGS, is available at www.gilead.com
Vemlidy, Gilead and the Gilead logo are registered trademarks of Gilead Sciences, Inc., or its related companies.
For more information about Gilead, please visit the company’s website at www.gilead.com
View source version on businesswire.com: https://www.businesswire.com/news/home/20221102005034/en/
Source: Gilead Sciences, Inc.
Nov. 02, 2022 7:40 AM ET
By: Ravikash, SA News Editor
PUBLISHED4 November 2022
AstraZeneca and Sanofi’s Beyfortus (nirsevimab) has been approved in the European Union (EU) for the prevention of respiratory syncytial virus (RSV) lower respiratory tract disease in newborns and infants during their first RSV season.1 Beyfortus is the first and only single-dose RSV passive immunisation for the broad infant population, including those born healthy, at term or preterm, or with specific health conditions.
RSV is a common and highly contagious seasonal virus, infecting nearly all children by the age of two.2,3
The European Commission is the first regulatory body to grant approval to Beyfortus.1 The approval was based on results from the Beyfortus clinical development programme, including the MELODY Phase III, MEDLEY Phase II/III and Phase IIb trials,1,4-11and follows the recommendation by The Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency in September 2022.12
In the pivotal MELODY efficacy trial, Beyfortus met its primary endpoint of reducing the incidence of medically attended lower respiratory tract infections (LRTI) caused by RSV by 74.5% (95% CI 49.6, 87.1; p<0.001) vs. placebo through day 151 (a typical RSV season) with a single dose.1,4-9 Beyfortus also demonstrated a comparable safety and tolerability profile to Synagis (palivizumab) in the MEDLEY Phase II/III trial, with occurrence of treatment emergent adverse events (TEAEs) or treatment emergent serious adverse events (TESAEs) similar between groups.1,10-13
Silke Mader, Chairwoman of the Executive Board and Co-founder of the European Foundation for the Care of Newborn Infants (EFCNI), said: “Respiratory syncytial virus represents a health threat among infants, and each year we see the impact it can have on families, healthcare providers and the healthcare system. At EFCNI, we are excited about the opportunity to expand prevention efforts to all infants, as we believe this can help ease the current emotional, physical and financial burdens of RSV.”
Iskra Reic, Executive Vice President, Vaccines and Immune Therapies, AstraZeneca, said: “Beyfortus is the first single-dose preventative option against respiratory syncytial virus to gain approval in Europe and is also the first and only preventative option approved for a broad infant population. Today’s marketing authorisation of Beyfortus marks a significant achievement for the scientific community and addresses a persistent, global unmet need in RSV prevention.”
Thomas Triomphe, Executive Vice President, Vaccines, Sanofi, said: “Today is a landmark day for RSV prevention, as decades of research and development come together in the world’s first approval of a broadly protective option against respiratory syncytial virus disease. Once launched, Beyfortus will offer parents the ability to help protect their babies during their first RSV season.”
RSV is the most common cause of LRTI, including bronchiolitis and pneumonia in infants.14 It is also a leading cause of hospitalisation in all infants.15-18 Globally, in 2019, there were approximately 33 million cases of acute lower respiratory infections leading to more than three million hospitalisations, and it was estimated that there were 26,300 in-hospital deaths of children younger than five years.19 RSV-related direct medical costs, globally – including hospital, outpatient and follow-up care – were estimated at €4.82 billion in 2017.21
Notes
Beyfortus
Beyfortus (nirsevimab), a long-acting antibody designed for all infants for protection against RSV disease from birth through their first RSV season with a single dose, is being developed jointly by AstraZeneca and Sanofi using AstraZeneca’s YTE technology.
Beyfortus has been developed to offer newborns and infants direct RSV protection via an antibody to help prevent LRTI caused by RSV. Monoclonal antibodies do not require the activation of the immune system to help offer timely, rapid and direct protection against disease.20
Beyfortus has been granted marketing authorisation in the European Union for the prevention of RSV LRTI disease in newborns and infants from birth during their first RSV season. The recommended dose of Beyfortus is a single intramuscular injection of 50 mg for infants with body weight <5 kg and a single intramuscular injection of 100 mg for infants with body weight ≥5 kg.12
Beyfortus has also been granted regulatory designations to facilitate expedited development by several major regulatory agencies around the world. These include Breakthrough Therapy Designation by the China Center for Drug Evaluation under the National Medical Products Administration; Breakthrough Therapy Designation from the US Food and Drug Administration; access granted to the European Medicines Agency (EMA PRIority Medicines (PRIME) scheme; and named “a medicine for prioritized development” under the Project for Drug Selection to Promote New Drug Development in Pediatrics by the Japan Agency for Medical Research and Development (AMED). The safety and efficacy of Beyfortus was evaluated under an accelerated assessment procedure by the EMA.
Pivotal clinical trials
The Phase IIb study was a randomised, placebo-controlled trial designed to measure the efficacy of Beyfortus (nirsevimab) against medically attended LRTI through 150 days postdose. Healthy preterm infants of 29–35 weeks’ gestation were randomised (2:1) to receive a single 50mg intramuscular injection of Beyfortus or placebo.1,4,5
The dosing regimen was recommended based on further exploration of the Phase IIb data. The subsequent Phase III study, MELODY applied the recommended dosing regimen.1,3,6
The MELODY Phase III study was a randomised, placebo-controlled trial conducted across 21 countries designed to determine efficacy of Beyfortus against medically attended LRTI due to RSV confirmed by reverse transcriptase polymerase chain reaction testing through 150 days after dosing, versus placebo, in healthy late preterm and term infants (35 weeks gestational age or greater) entering their first RSV season.1-3
MEDLEY was a Phase II/III, randomised, double-blind, Synagis-controlled trial with the primary objective of assessing safety and tolerability for Beyfortus in preterm infants and infants with congenital heart disease (CHD) and/or chronic lung disease of prematurity (CLD) eligible to receive Synagis.1,8,9 Between July 2019 and May 2021 approximately 918 infants entering their first RSV season were randomised to receive a single 50mg (in infants weighing <5kg) or 100mg (in infants weighing ≥5kg) intramuscular injection of Beyfortus or Synagis. Safety was assessed by monitoring the occurrence of TEAEs and TESAEs through 360 days post-dose.1,8,9 Serum levels of Beyfortus following dosing (on day 151) in this trial were comparable with those observed in the MELODY Phase III trial, indicating similar protection in this population to that in the healthy term and late preterm infants is likely. Data was published in the New England Journal of Medicine (NEJM) in March 2022.
The results of MELODY, MEDLEY Phase II/III and the Phase IIb trials demonstrate that Beyfortus helps protect infants during their first RSV season against RSV disease with a single dose.1-9 This all-infant population includes preterm, healthy late preterm and term infants, as well as infants with specific conditions.
These trials form the basis of regulatory submissions which began in 2022.
Results from the Phase IIb trial
The primary endpoint of the Phase IIb study was met, reducing the incidence of medically attended LRTI, caused by RSV by 70.1% (95% CI: 52.3, 81.2) compared to placebo. Between November 2016 and December 2017, 1,453 infants were randomised (Beyfortus, n=969; placebo, n=484) at the RSV season start. Research was conducted by AstraZeneca in both hemispheres, at 164 sites in 23 countries.1,4,5 Data was published in NEJM in July 2020.
Medically Attended LRTI and Hospitalisation for RSV LRTI Through 150 Days Postdose (ITT population)
Results from the MELODY Phase III trial
The primary endpoint of the MELODY Phase III trial was met, reducing the incidence of medically attended LRTI, such as bronchiolitis or pneumonia, caused by RSV by 74.5% (95% CI 49.6, 87.1; P<0.001) compared to placebo. Infants were randomised (2:1) to receive a single 50mg (in infants weighing <5kg) or 100mg (in infants weighing ≥5kg) intramuscular injection of Beyfortus or placebo. Between July 2019 and March 2020, 1,490 infants were randomised to either Beyfortus or placebo at the RSV season start.1-3 Data was published in NEJM in March 2022.
Medically Attended LRTI and Hospitalisation for RSV LRTI Through 150 Days Postdose (ITT population)
Endpoints and analyses, n (%) Nirsevimab (N = 994)
Participants requiring imputation of data*
*Data were imputed for participants who had no events and were not followed through 150 days postdose. Analyses were conducted using Poisson regression with robust variance. CI, confidence interval; ITT, intent-to-treat; LRTI, lower respiratory tract infection; RRR, relative risk reduction; RSV, respiratory syncytial virus.
Results from the pre-specified pooled analysis of the Phase IIb and MELODY trials
A prespecified pooled analysis of the MELODY Phase III trial and the recommended dose from the Phase IIb trial, in which an efficacy (relative risk reduction versus placebo) of 79.5% (95% CI 65.9, 87.7; P<0.0001) was seen against medically attended LRTI, such as bronchiolitis or pneumonia, caused by RSV in infants born at term or preterm entering their first RSV season.1,8 The pooled analysis studied healthy preterm and term infants who received the recommended dose of Beyfortus based on weight compared to placebo through Day 151 and showed an efficacy of 77.3% (95% CI 50.3, 89.7; P<0.001) against RSV LRTI hospitalisations.1,4,8
Medically Attended LRTI and Hospitalisation for RSV LRTI Through 150 Days Postdose (ITT population)
Endpoints and analyses, n (%) Nirsevimab (N = 1564) )
*Data were imputed for participants who had no events and were not followed through 150 days postdose. Analyses were conducted using Poisson regression with robust variance. CI, confidence interval; ITT, intent-to-treat; LRTI, lower respiratory tract infection; RRR, relative risk reduction; RSV, respiratory syncytial virus.
Sanofi Alliance
In March 2017, AstraZeneca and Sanofi announced an agreement to develop and commercialise nirsevimab. Under the terms of the agreement, AstraZeneca leads all development and manufacturing activities, and Sanofi leads commercialisation activities and records revenue. Under the terms of the global agreement, Sanofi made an upfront payment of €120m, has paid a development milestone of €30m and will pay up to a further €465m upon achievement of certain development and sales-related milestones. The two companies share costs and profits. Revenue from the agreement is reported as Collaboration Revenue in the Company’s financial statements.
Sobi agreement
Related, in November 2018, AstraZeneca agreed to sell US commercial rights for Synagis (palivizumab) to Swedish Orphan Biovitrum AB (publ) (Sobi) in addition to the right to participate in payments that may be received by AstraZeneca from the US profits or losses for nirsevimab. Under the agreement AstraZeneca received upfront consideration and also received non-contingent payments for nirsevimab during 2019-2021. AstraZeneca is also entitled to receive certain milestone payments for nirsevimab, including a $175m milestone following the date on which the Biologics License Application (BLA) for nirsevimab is accepted for filing by the FDA and a $90m milestone payment following the date on which BLA approval in the US occurs. AstraZeneca will continue to manufacture and supply nirsevimab globally and is entitled to an additional royalty from Sobi if profits from nirsevimab in the US exceed a pre-specified level.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Nov. 04, 2022 4:37 AM ET
By: Ravikash, SA News Editor
The European Commission (EC) approved AstraZeneca (NASDAQ:AZN) and Sanofi's (NASDAQ:SNY) antibody therapy Beyfortus to prevent respiratory syncytial virus (RSV) lower respiratory tract disease in newborns and infants during their first RSV season.
https://seekingalpha.com/symbol/AZN
https://seekingalpha.com/symbol/SNY
Nov 02, 2022 7:00 AM
BRUKINSA is the first and only Bruton’s Tyrosine Kinase (BTK) inhibitor for marginal zone lymphoma approved in the European Union
CAMBRIDGE, Mass. & BASEL, Switzerland & BEIJING--(BUSINESS WIRE)-- BeiGene (NASDAQ: BGNE; HKEX: 06160; SSE: 688235), a global biotechnology company, today announced that the European Commission (EC) has granted marketing authorization of BRUKINSA® (zanubrutinib) for the treatment of adult patients with relapsed/refractory (R/R) marginal zone lymphoma (MZL) who have received at least one prior anti-CD20-based therapy. The approval is applicable to all 27 member states of the European Union (EU), plus Iceland and Norway. BeiGene is focused on developing innovative and affordable oncology medicines to improve treatment outcomes and access for patients worldwide.
Notably, the EC granted an additional year of marketing protection because the data submitted for the therapeutic indication demonstrated a significant clinical benefit for BRUKINSA in comparison with existing therapies.
“We are proud of what this approval means for European MZL patients, who previously did not have an approved BTK inhibitor as a treatment option for this rare hematological malignancy,” said Mehrdad Mobasher, M.D., M.P.H., Chief Medical Officer, Hematology at BeiGene. “This milestone builds on the track record we’ve built with BRUKINSA to date, with approvals in more than 55 countries and regions, as we continue to fulfill our commitment to build a transformational global R&D model that enables broader, faster access to novel medicines.”
The EC approval follows a positive opinion granted in September by the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) based on results from the multicenter, global, single-arm, open-label, Phase 2 MAGNOLIA trial in patients with R/R MZL who received at least one anti-CD-20 based regimen. In the trial, BRUKINSA achieved a high overall response rate of 68% with 26% of patients achieving complete remission, as assessed by an Independent Review Committee (IRC). Responses were observed in all patients regardless of MZL subtypes. BRUKINSA also delivered rapid and durable disease control with a median time to response of 2.8 months.i
BRUKINSA was generally well-tolerated and safety in MZL was consistent with its established profile. The most common grade ≥3 adverse events (>5%) included neutropenia (23%), pneumonia (11%), thrombocytopenia (8%) and anemia (8%). There were low rates of discontinuation due to adverse events at 3.5%, highlighting that BRUKINSA continued to be well-tolerated.ii
“This milestone marks the first and only approved BTK inhibitor for marginal zone lymphoma in Europe,” comments Pier Luigi Zinzani, MD., PhD., Full Professor of Haematology at the Institute of Haematology “Seràgnoli”, University of Bologna, Italy. “As there is no current standard of care in Europe, the approval of BRUKINSA provides a chemotherapy-free treatment option for people with MZL that has shown meaningful efficacy with durable and high response rates across MZL subtypes.”
Gerwin Winter, Senior Vice President, Head of Europe at BeiGene, notes, “We are excited to bring the first and only BTKi approved for MZL to patients in Europe and look forward to continuing to work with our growing and dedicated teams to make our medicine accessible to patients who need them across Europe.”
BRUKINSA is also approved in the EU for the treatment of adult patients with Waldenström’s macroglobulinemia (WM) who have received at least one prior therapy or for the first-line treatment of patients unsuitable for chemo-immunotherapy, and last month, CHMP issued a positive opinion recommending approval of BRUKINSA for the treatment of adult patients with chronic lymphocytic leukemia (CLL).
BeiGene has obtained reimbursement for BRUKINSA for the treatment of WM in Austria, Belgium, Denmark, England and Wales, Germany, Italy, Iceland, Ireland, The Netherlands, Spain, and Switzerland, while additional countries across Europe are currently going through the reimbursement process.
About BRUKINSA
BRUKINSA is a small molecule inhibitor of Bruton’s tyrosine kinase (BTK) discovered by BeiGene scientists that is currently being evaluated globally in a broad clinical program as a monotherapy and in combination with other therapies to treat various B-cell malignancies. BRUKINSA was specifically designed to deliver targeted and sustained inhibition of the BTK protein by optimizing bioavailability, half-life, and selectivity. With differentiated pharmacokinetics compared to other approved BTK inhibitors, BRUKINSA has been demonstrated to inhibit the proliferation of malignant B cells within a number of disease relevant tissues.
BRUKINSA is supported by a broad clinical program which includes more than 4,500 subjects in 35 trials across 28 markets. To date, BRUKINSA has received approvals covering more 55 countries and regions, including the United States, China, the EU, Great Britain, Switzerland, Canada, Australia, and additional international markets.
BeiGene Oncology
BeiGene is committed to advancing best- and first-in-class clinical candidates internally or with like-minded partners to develop impactful and affordable medicines for patients across the globe. We have a growing R & D and medical affairs team of approximately 3,300 colleagues dedicated to advancing more than 100 clinical trials that have involved more than 16,000 subjects. Our expansive portfolio is directed predominantly by our internal colleagues supporting clinical trials in more than 45 countries and regions. Hematology-oncology and solid tumor targeted therapies and immuno-oncology are key focus areas for the Company, with both mono- and combination therapies prioritized in our research and development. BeiGene currently has three approved medicines discovered and developed in our own labs: BTK inhibitor BRUKINSA in the U.S., China, the European Union, Great Britain, Switzerland, Canada, Australia, and additional international markets; and the non-FC-gamma receptor binding anti-PD-1 antibody, tislelizumab, as well as the PARP inhibitor, pamiparib, in China.
BeiGene also partners with innovative companies who share our goal of developing therapies to address global health needs. We commercialize a range of oncology medicines in China licensed from Amgen, Bristol Myers Squibb, EUSA Pharma and Bio-Thera. We also plan to address greater areas of unmet need globally through our other collaborations including with Mirati Therapeutics, Seagen, and Zymeworks.
In January 2021, BeiGene and Novartis announced a collaboration granting Novartis rights to co-develop, manufacture, and commercialize BeiGene’s anti-PD1 antibody, tislelizumab, in North America, Europe, and Japan. Building upon this productive collaboration, BeiGene and Novartis announced an option, collaboration, and license agreement in December 2021 for BeiGene’s TIGIT inhibitor, ociperlimab, that is in Phase 3 development. Novartis and BeiGene also entered into a strategic commercial agreement through which BeiGene will promote five approved Novartis Oncology products across designated regions of China.
About BeiGene
BeiGene is a global biotechnology company that is developing and commercializing innovative and affordable oncology medicines to improve treatment outcomes and access for far more patients worldwide. With a broad portfolio, we are expediting development of our diverse pipeline of novel therapeutics through our internal capabilities and collaborations. We are committed to radically improving access to medicines for far more patients who need them. Our growing global team of more than 9,000 colleagues spans five continents, with administrative offices in Beijing, China; Cambridge, U.S.; and Basel, Switzerland. To learn more about BeiGene, please visit www.beigene.com and follow us on Twitter at @BeiGeneGlobal.
View source version on businesswire.com: https://www.businesswire.com/news/home/20221102005244/en/
Source: BeiGene
Nov. 02, 2022 7:59 AM ET
By: Ravikash, SA News Editor
Karyopharm and Menarini Group Announce Orphan Medicinal Product Designation from the European Commission for Selinexor for the Treatment of Myelofibrosis
NEWTON, Mass. and FLORENCE, Italy, Oct. 31, 2022 /PRNewswire/ -- Karyopharm Therapeutics Inc. (Nasdaq:KPTI), a commercial-stage pharmaceutical company pioneering novel cancer therapies, and the Menarini Group ("Menarini"), a privately-held, leading international pharmaceutical company, today announced that the European Commission (EC) has granted orphan medicinal product designation for selinexor for the treatment of myelofibrosis (MF). Selinexor was granted orphan drug designation in MF by the U. S. Food and Drug Administration (FDA) in May 2022. Karyopharm is currently evaluating selinexor, a first-in-class XP01 inhibitor, as monotherapy in patients with previously treated MF, and in combination with ruxolitinib in treatment-naïve patients. In December 2021, Karyopharm and Menarini entered into an exclusive licensing agreement whereby Menarini is responsible for commercializing all current and future indications of NEXPOVIO® in the European Economic Area, United Kingdom and Switzerland, CIS countries, Turkey and Latin America. Stemline Therapeutics B.V., a wholly owned subsidiary of Menarini, is leading all commercialization activities in Europe.
"We are very pleased to receive orphan medicinal product designation from the EC for selinexor for the treatment of myelofibrosis," said Reshma Rangwala, MD, PhD, Chief Medical Officer of Karyopharm. "Building on our recent orphan drug designation from the FDA, this recognition continues to reinforce the significant unmet need for a drug with a novel mechanism of action like selinexor for this devastating disease. Our clinical plans remain on track, and we look forward to the continued development of selinexor in MF."
"Myelofibrosis is a difficult-to-treat and complex disorder of the bone marrow with limited therapeutic options and we are committed to bringing novel treatments to patients through our collaboration with Karyopharm. We are excited about the potential to bring selinexor to myelofibrosis patients in Europe, pending positive study read-outs and regulatory approval," said Olivia del Puerto, MD LMS, Head of Medical Affairs Oncology - EMEA of Menarini.
About the EMA Orphan Designation
Orphan medicinal product designation in the European Union (EU) is granted by the European Commission which adopts the positive opinion issued by the European Medicines Agency (EMA) Committee for Orphan Medicinal Products. The EMA's orphan designation is available to companies developing treatments for life-threatening or chronically debilitating conditions that affect fewer than five in 10,000 persons in the EU. Medicines that meet the EMA's orphan designation criteria qualify for financial and regulatory incentives that include a 10-year period of marketing exclusivity in the EU after product approval, reduced fees and access to centralized marketing authorization.
About NEXPOVIO® (selinexor)
NEXPOVIO®, which is marketed as XPOVIO® in the U.S., has been approved in the following oncology indications by the European Commission: (i) in combination with dexamethasone for the treatment of multiple myeloma in adult patients who have received at least four prior therapies and whose disease is refractory to at least two proteasome inhibitors, two immunomodulatory agents and an anti-CD38 monoclonal antibody, and who have demonstrated disease progression on the last therapy; and (ii) in combination with bortezomib and dexamethasone for the treatment of adults with multiple myeloma who have received at least one prior therapy. The marketing authorization of NEXPOVIO is valid in the EU Member States as well as Iceland, Liechtenstein, Norway, and Northern Ireland. NEXPOVIO has been commercially available in Germany since October 1, 2022.
NEXPOVIO is a first-in-class, oral exportin 1 (XPO1) inhibitor. NEXPOVIO functions by selectively binding to and inhibiting the nuclear export protein exportin 1 (XPO1, also called CRM1). NEXPOVIO blocks the nuclear export of tumor suppressor, growth regulatory and anti-inflammatory proteins, leading to accumulation of these proteins in the nucleus and enhancing their anti-cancer activity in the cell. The forced nuclear retention of these proteins can counteract a multitude of the oncogenic pathways that, unchecked, allow cancer cells with severe DNA damage to continue to grow and divide in an unrestrained fashion.
Please see NEXPOVIO® Summary of Product Characteristics and European Public Assessment Report at https://ec.europa.eu/health/documents/community-register/html/h1537.htm
Please refer to local prescribing information where XPOVIO/NEXPOVIO is approved for full information.
About Karyopharm Therapeutics
Karyopharm Therapeutics Inc. (Nasdaq: KPTI) is a commercial-stage pharmaceutical company pioneering novel cancer therapies. Since its founding, Karyopharm has been the industry leader in oral Selective Inhibitor of Nuclear Export (SINE) compound technology, which was developed to address a fundamental mechanism of oncogenesis: nuclear export dysregulation. Karyopharm's lead SINE compound and first-in-class, oral exportin 1 (XPO1) inhibitor, XPOVIO® (selinexor), is approved in the U.S. and marketed by the Company in three oncology indications and has received regulatory approvals in various indications in a growing number of ex-U.S. territories and countries, including Europe and the United Kingdom (as NEXPOVIO®), China, Singapore, Canada, Israel, South Korea, and Australia. Karyopharm has a focused pipeline targeting multiple high unmet need cancer indications, including in multiple myeloma, endometrial cancer, myelodysplastic syndromes and myelofibrosis. For more information about our people, science and pipeline, please visit www.karyopharm.com, and follow us on Twitter at @Karyopharm and LinkedIn.
About Menarini Group
The Menarini Group is a leading international pharmaceutical and diagnostics company, with a turnover of over $4 billion and over 17,000 employees. Menarini is focused on therapeutic areas with high unmet needs with products for oncology, cardiology, pneumology, gastroenterology, infectious diseases, diabetology, inflammation, and analgesia. With 18 production sites and 9 Research and Development centers, Menarini's products are available in 140 countries worldwide. For further information, please visit www.menarini.com and LinkedIn.
XPOVIO® and NEXPOVIO® are registered trademarks of Karyopharm Therapeutics Inc. Any other trademarks referred to in this release are the property of their respective owners.
1 NORD, Rare Disease Database, Primary Myelofibrosis, Accessed on 10/4/22, https://rarediseases.org/rare-diseases/primary-myelofibrosis/
SOURCE Karyopharm Therapeutics Inc.
Nov. 01, 2022 10:18 AM ET
Karyopharm Therapeutics Inc. (KPTI)
Karyopharm Therapeutics Inc. (KPTI)
10/31/2022 CATEGORY:
Reblozyl, the first erythroid maturation agent, met primary and key secondary endpoints in the first-line treatment of patients with very low/low/intermediate-risk myelodysplastic syndromes
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) today announced the COMMANDS study, a Phase 3, open-label, randomized trial evaluating Reblozyl®(luspatercept-aamt), met its primary endpoint, demonstrating a highly statistically significant and clinically meaningful improvement in red blood cell transfusion independence (RBC-TI) with concurrent hemoglobin (Hb) increase in the first-line treatment of adult patients with very low-, low- or intermediate-risk myelodysplastic syndromes (MDS) who require RBC transfusions. This result was based on a pre-specified interim analysis conducted through an independent review committee. Safety results in the trial were consistent with the safety profile of Reblozyl previously demonstrated in the MEDALIST study (NCT02631070), and no new safety signals were reported.
“While advancements have been made in the treatment of anemia for patients with myelodysplastic syndromes, there remains a significant need for new and better first-line treatment options for patients with transfusion-dependent MDS,” said Noah Berkowitz, M.D., Ph.D., senior vice president, Hematology Development, Bristol Myers Squibb. “We are pleased with the positive results of the COMMANDS study and look forward to presenting these important data.”
Bristol Myers Squibb will complete a full evaluation of the COMMANDS data and work with investigators to present detailed results at an upcoming medical meeting, as well as discuss these results with health authorities. Bristol Myers Squibb thanks the patients and investigators who are participating in the COMMANDS clinical trial.
Reblozyl is being developed and commercialized through a global collaboration with Merck following Merck’s acquisition of Acceleron Pharma, Inc. in November 2021.
About COMMANDS
COMMANDS (NCT03682536) is a Phase 3, open-label, randomized study evaluating the efficacy and safety of Reblozyl versus epoetin alfa, for the treatment of anemia due to very low-, low- or intermediate-risk (IPSS-R) myelodysplastic syndrome (MDS) in patients who are red blood cell (RBC) transfusion dependent and were erythropoiesis stimulating agent (ESA) naïve.
The primary endpoint evaluated in this study is RBC transfusion independence (RBC-TI) for 12 weeks with a mean hemoglobin increase ≥1.5 g/dL. Key secondary endpoints include RBC-TI for 24 weeks, RBC-TI ≥12 weeks and erythroid response of at least 8 weeks during weeks 1-24 of the study.
About Reblozyl® (luspatercept-aamt)
Reblozyl, a first-in-class therapeutic option, promotes late-stage red blood cell maturation in animal models.5 Reblozyl is being developed and commercialized through a global collaboration with Merck following Merck’s acquisition of Acceleron Pharma, Inc. in November 2021. Reblozyl is currently approved in the U.S. for the treatment of:
Reblozyl is not indicated for use as a substitute for red blood cell transfusions in patients who require immediate correction of anemia.
Please see full Prescribing Information for REBLOZYL.
About Bristol Myers Squibb
Bristol Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over seriousdiseases. For more information about Bristol Myers Squibb,visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
Source: Bristol Myers Squibb
Oct. 31, 2022 7:42 AM ET
Bristol-Myers Squibb Company (BMY), MRKGERN
By: Dulan Lokuwithana, SA News Editor1 Comment
Bristol Myers Squibb (NYSE:BMY) announced Monday that Reblozyl, an FDA-approved anemia therapy the company develops in partnership with Merck (NYSE:MRK), reached the primary and key secondary endpoints in a Phase 3 trial involving patients with myelodysplastic syndromes.
https://seekingalpha.com/symbol/BMY
https://seekingalpha.com/symbol/MRK
PUBLISHED27 October 2022 27 October 2022 09:05 BST
Detailed positive results from the Phase III CHAMPION-NMOSD trial showed that Ultomiris (ravulizumab) significantly reduced relapse risk in adults with anti-aquaporin-4 (AQP4) antibody-positive (Ab+) neuromyelitis optica spectrum disorder (NMOSD), compared to the external placebo arm from the pivotal Soliris PREVENT clinical trial. Data were presented today at the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) Congress.
NMOSD is a rare and debilitating autoimmune disease that affects the central nervous system (CNS), including the spine and optic nerves.1-3 Most people living with NMOSD experience unpredictable relapses, characterised by a new onset of neurologic symptoms or worsening of existing neurologic symptoms, which tend to be severe and recurrent and may result in permanent disability.4-6
Sean J. Pittock, MD, Director of Mayo Clinic's Center for Multiple Sclerosis and Autoimmune Neurology and of Mayo's Neuroimmunology Laboratory and lead primary investigator in the CHAMPION-NMOSD trial, said: “The CHAMPION-NMOSD trial showed zero relapses with a median treatment duration of 73 weeks, providing evidence that ravulizumab may offer patients sustained reduction in the risk of relapse with dosing every eight weeks and underscoring the efficacy of C5 inhibition in managing NMOSD.”
Michael Yeaman, PhD, Professor of Medicine at the UCLA School of Medicine, Director of the Institute for Infection and Immunity, Lundquist Institute at Harbor–UCLA and Chair Medical Advisor to the Guthy-Jackson Charitable Foundation for NMOSD, said: “In recent years, we have seen meaningful progress in bringing safe and effective treatments to patients with AQP4 Ab+ NMOSD, a rare disease that can disrupt many aspects of daily life, including mobility, vision, strength and balance. Based on insights from working with patients and their families every day, continued innovative research and clinical trials help empower people living with NMOSD by advancing new treatment options that may be more compatible with their individual needs and lifestyles.”
Gianluca Pirozzi, MD, PhD, Senior Vice President, Head of Development and Safety, Alexion, said: “CHAMPION-NMOSD is a remarkable example of the innovation required to design and execute rare disease clinical trials that are both scientifically rigorous and clinically meaningful. By seeking input from patients and coordinating with health authorities, this Phase III trial put the needs of patients first and evaluated measures that mattered most to them. We are excited by the significance of the results and the potential of Ultomiris to advance care for the NMOSD community.”
CHAMPION-NMOSD is a global Phase III, open-label, multicentre trial evaluating the safety and efficacy of Ultomiris in adults (n=58). Due to the potential long-term functional impact of NMOSD relapses and available effective treatment options, a direct placebo comparator arm was precluded for ethical reasons. Ultomiris, the active treatment, was compared to the external placebo arm from the pivotal Soliris PREVENT clinical trial.7
Data showed zero adjudicated relapses were observed among Ultomiris patients with a median treatment duration of 73 weeks (relapse risk reduction: 98.6%, hazard ratio (95% CI): 0.014 (0.000, 0.103), p<0.0001). Additionally, 100% of patients receiving Ultomiris remained relapse-free at 48 weeks, compared to 63% of patients in the external placebo arm.7
The CHAMPION-NMOSD trial also met key secondary efficacy endpoints, including adjudicated on-trial annualised relapse rate (total number of relapses in the study divided by total number of patient years) and clinically important change from baseline in mobility (ability to walk) as measured by Hauser Ambulation Index (a scale to assess mobility).7
Overall, the safety and tolerability of Ultomiris was consistent with previous clinical studies and real-world use and no new safety signals were observed. The most common adverse events (AEs) (greater than or equal to 10% of patients) were COVID-19 (24%), headache (24%), back pain (12%), arthralgia (10%) and urinary tract infection (10%). All cases of COVID-19 were non-serious and considered to be unrelated to Ultomiris. There were two meningococcal infections reported; both patients recovered fully with no sequelae and one continued in the trial. Fifty-six patients are continuing to receive treatment in an ongoing long-term extension period.7
CHAMPION-NMOSD subgroup and sensitivity analyses
Additional results from the CHAMPION-NMOSD trial were also presented at ECTRIMS in two posters, detailing subgroup and sensitivity analyses.
In the subgroup analysis, based on time to first adjudicated on-trial relapse, Ultomiris was superior to the external placebo arm in patients receiving monotherapy (n=30; hazard ratio [HR]: 0.021; 95% confidence interval [CI]: 0.000–0.176; relapse risk reduction [RRR]: 97.9%; p<0.0001) and in patients receiving concomitant therapy (n=28; HR: 0.031; 95% CI: 0.000–0.234; RRR: 96.9%; p<0.0001). Significant differences were observed in patients who had received rituximab in the previous year (n=20; HR: 0.063; 95% CI: 0.000–0.562; RRR: 93.7%; p=0.0078) or had not (n=38; HR: 0.019; 95% CI: 0.000-0.142; RRR 98.1%; p<0.0001) compared to placebo.
The robust treatment effect of Ultomiris was also observed across pre-specified efficacy subgroups, including age (<45 years or ≥45 years: RRR: 95.7–97.9%; p≤0.0012), sex (RRR: 94.3–98.2%; p≤0.0068), Asian and white races (RRR: 95.1–97.8%; p≤0.0027) and geographic region (RRR: 91.5–96.1%; p≤0.025).8
Further, pre-specified sensitivity analyses were conducted to account for potential differences in baseline patient characteristics that could impact treatment efficacy. Time to first adjudicated relapse and relapse risk reduction were analysed using a stabilised inverse probability of treatment weighting approach. The results were consistent with the primary analysis, suggesting that any differences in baseline characteristics between the Ultomiris and external placebo groups did not impact the treatment effect.9
Regulatory submissions for Ultomiris for the treatment of NMOSD are currently under review with multiple health authorities, including in the United States (US), European Union (EU) and Japan.
CHAMPION-NMOSD
CHAMPION-NMOSD is a global Phase III, open-label, multicentre trial evaluating the safety and efficacy of Ultomiris in adults with NMOSD. The trial enrolled 58 patients across North America, Europe, Asia-Pacific and Japan. Participants were required to have a confirmed NMOSD diagnosis with a positive anti-AQP4 antibody test, at least one attack or relapse in the twelve months prior to the screening visit, an Expanded Disability Status Scale Score of 7 or less and body weight of at least 40 kilograms at trial entry. Participants could stay on stable supportive immunosuppressive therapy for the duration of the trial.20
Due to the potential long-term functional impact of NMOSD relapses and available effective treatment options, a direct placebo comparator arm was precluded for ethical reasons. The active treatment was compared to an external placebo arm from the pivotal Soliris PREVENT clinical trial.
Over a median treatment duration of 73 weeks, all enrolled patients received a single weight-based loading dose of Ultomiris on Day 1, followed by regular weight-based maintenance dosing beginning on Day 15, every eight weeks. The primary endpoint was time to first on-trial relapse, as confirmed by an independent adjudication committee. The end of the primary treatment period could have occurred either when all patients completed or discontinued prior to the Week 26 visit and two or more adjudicated relapses were observed, or when all patients completed or discontinued prior to the Week 50 visit if fewer than two adjudicated relapses were observed. In the trial, there were zero adjudicated relapses so the end of the primary treatment period occurred when the last enrolled participant completed the 50 week visit.
Patients who completed the primary treatment period were eligible to continue into a long-term extension period, which is ongoing.
Ultomiris
Ultomiris (ravulizumab), the first and only long-acting C5 complement inhibitor, provides immediate, complete and sustained complement inhibition. The medication works by inhibiting the C5 protein in the terminal complement cascade, a part of the body’s immune system. When activated in an uncontrolled manner, the complement cascade over-responds, leading the body to attack its own healthy cells. Ultomiris is administered intravenously every eight weeks in adult patients, following a loading dose.
Ultomiris is approved in the US, EU and Japan for the treatment of certain adults with generalised myasthenia gravis.
Ultomiris is also approved in the US, EU and Japan for the treatment of certain adults with paroxysmal nocturnal haemoglobinuria (PNH) and for certain children with PNH in the US and EU.
Additionally, Ultomiris is approved in the US, EU and Japan for certain adults and children with atypical haemolytic uraemic syndrome to inhibit complement-mediated thrombotic microangiopathy.
As part of a broad development programme, Ultomiris is being assessed for the treatment of additional haematology and neurology indications.
Alexion
Alexion, AstraZeneca Rare Disease, is the group within AstraZeneca focused on rare diseases, created following the 2021 acquisition of Alexion Pharmaceuticals, Inc. As a leader in rare diseases for 30 years, Alexion is focused on serving patients and families affected by rare diseases and devastating conditions through the discovery, development and commercialisation of life-changing medicines. Alexion focuses its research efforts on novel molecules and targets in the complement cascade and its development efforts on haematology, nephrology, neurology, metabolic disorders, cardiology and ophthalmology. Headquartered in Boston, Massachusetts, Alexion has offices around the globe and serves patients in more than 50 countries.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Oct. 27, 2022 6:38 AM ET
By: Ravikash, SA News Editor
AstraZeneca (NASDAQ:AZN) said its medicine Ultomiris reduced the risk of relapse by 98.6%, compared to placebo, in certain patients with a type of central nervous system (CNS) disorder in a phase 3 trial.
https://seekingalpha.com/symbol/AZN
https://alexion.com/Documents/Ultomiris_USPI.pdf

Oct 26, 2022 7:00 AM
BRUKINSA® received Marketing Authorization as a treatment for rare blood cancers in six countries in Central and South America
CAMBRIDGE, Mass., & BASEL, Switzerland & BEIJING--(BUSINESS WIRE)-- BeiGene, (NASDAQ: BGNE; HKEX: 06160; SSE: 688235), a global biotechnology company focused on developing innovative and affordable oncology medicines to improve treatment outcomes and access for patients worldwide, today announced significant progress in efforts to unlock global opportunities for BRUKINSA® (zanubrutinib) with recent regulatory approvals in six Latin American countries:
“It has only been one year since the first approval for BRUKINSA in Latin America and these approvals, including the most recent in Argentina, demonstrate our drive to accomplish our mission and broaden access to innovative medicines,” said Eduardo Molinari, Senior Director of New Market Development in Latin America at BeiGene. “I look forward to collaborating with our partner, Adium, on commercialization activities to provide this important treatment option to people living with MCL, MZL, and WM in Latin America.”
Dr. Maria Silvana Cugliari, Head of Hematology, Angel Roffo Institute of Oncology University of Buenos Aires, Argentina commented, “BTK inhibition has proven to be a highly effective treatment strategy for a number of indolent B-cell malignancies and BeiGene’s expansive clinical development program for BRUKINSA in multiple indications has provided evidence of strong efficacy and durable response rates, along with a consistent safety profile.”
The announcement of regulatory approvals for BRUKINSA in Latin America follows a recent positive CHMP Opinion for BRUKINSA as a treatment for chronic lymphocytic leukemia (CLL) in the European Union.
About BRUKINSA
BRUKINSA is a small-molecule inhibitor of Bruton’s tyrosine kinase (BTK) discovered by BeiGene scientists that is currently being evaluated globally in a broad clinical program as a monotherapy and in combination with other therapies to treat various B-cell malignancies. BRUKINSA was specifically designed to deliver targeted and sustained inhibition of the BTK protein by optimizing bioavailability, half-life, and selectivity. With differentiated pharmacokinetics compared to other approved BTK inhibitors, BRUKINSA has been demonstrated to inhibit the proliferation of malignant B cells within a number of disease-relevant tissues.
BRUKINSA is supported by a broad clinical program which includes more than 4,500 subjects in 35 trials across 28 markets. To date, BRUKINSA has received approvals covering more than 55 countries and regions, including the United States, China, the EU, Switzerland, Great Britain, Canada, Australia, and additional international markets.
BeiGene Oncology
BeiGene is committed to advancing best- and first-in-class clinical candidates internally or with like-minded partners to develop impactful and affordable medicines for patients across the globe. We have a growing R&D and medical affairs team of approximately 3,300 colleagues dedicated to advancing more than 100 clinical trials that have involved more than 16,000 subjects. Our expansive portfolio is directed predominantly by our internal colleagues supporting clinical trials in more than 45 countries and regions. Hematology-oncology, and solid tumor targeted therapies, and immuno-oncology are key focus areas for the Company, with both monotherapies and combination therapies prioritized in our research and development. BeiGene currently has three licensed medicines discovered and developed in our own labs: BTK inhibitor BRUKINSA® in the U.S., China, the European Union, Switzerland, Great Britain, Canada, Australia, and additional international markets; and the non-FC-gamma receptor binding anti-PD-1 antibody tislelizumab as well as the poly adenosine diphosphate-ribose polymerase (PARP) inhibitor pamiparib in China.
BeiGene also partners with innovative companies who share our goal of developing therapies to address global health needs. We commercialize a range of oncology medicines in China licensed from Amgen, Bristol Myers Squibb, EUSA Pharma, and Bio-Thera. We also plan to address greater areas of unmet need globally through our other collaborations including Mirati Therapeutics, Seagen, and Zymeworks.
In January 2021 BeiGene and Novartis announced a collaboration granting Novartis rights to co-develop, manufacture, and commercialize BeiGene’s anti-PD-1 antibody tislelizumab in North America, Europe, and Japan. Building upon this productive collaboration, BeiGene and Novartis announced an option, collaboration, and license agreement in December 2021 for BeiGene’s TIGIT inhibitor ociperlimab that is in Phase 3 development. Novartis and BeiGene also entered into a strategic commercial agreement through which BeiGene will promote five approved Novartis oncology products across designated regions of China.
About BeiGene
BeiGene is a global biotechnology company that is developing and commercializing innovative and affordable oncology medicines to improve treatment outcomes and access for far more patients worldwide. With a broad portfolio, we are expediting development of our diverse pipeline of novel therapeutics through our internal capabilities and collaborations. We are committed to radically improving access to medicines for far more patients who need them. Our growing global team of more than 9,000 colleagues spans five continents, with administrative offices in Beijing, China; Cambridge, U.S.; and Basel, Switzerland. To learn more about BeiGene, please visit www.beigene.com and follow us on Twitter at @BeiGeneGlobal.
About Adium
Adium is a private pharmaceutical company based in Montevideo, Uruguay. Adium distributes its products in 18 Latin American & Caribbean countries including Brazil, Mexico and Colombia. Adium has been distributing products from leading international companies in the field of Oncology, Urology, Hematology and Rare Diseases, for more than 20 years. Adium provides its partners a full set of local capabilities including commercial, market access, regulatory and pharmacovigilance.
View source version on businesswire.com: https://www.businesswire.com/news/home/20221026005423/en/
Source: BeiGene
Oct. 26, 2022 7:40 AM ET
By: Ravikash, SA News Editor
Oct. 26, 2022 7:27 AM ETAstraZeneca PLC (AZN)
First and only biologic to consistently and significantly reduce exacerbations in a broad population of severe asthma patients
MISSISSAUGA, ON, Oct. 26, 2022 /CNW/ - Today, AstraZeneca (AZN) and Amgen announced the Canadian availability of Tezspire™ (tezepelumab injection), indicated for the add-on maintenance treatment of adults and adolescents 12 years and older with severe asthma.1
Tezspire was approved based on results from the PATHWAY Phase llb trial and NAVIGATOR Phase III trial in which Tezspire demonstrated significant improvements across every primary and key secondary endpoint in patients with severe asthma, compared to placebo, when added to standard therapy.2
Tezspire is a first-in-class biologic for severe asthma that acts at the top of the inflammatory cascade by targeting thymic stromal lymphopoietin (TSLP), an epithelial cytokine.2-5 It is the first and only biologic to consistently and significantly reduce asthma exacerbations across Phase II and III clinical trials which included a broad population of severe asthma patients irrespective of key biomarkers, including blood eosinophil counts, allergic status and fractional exhaled nitric oxide (FeNO).2,3,6-13 Tezspire is the only biologic approved for severe asthma with no phenotype (e.g., eosinophilic or allergic) or biomarker limitation within its approved label.1
"Patients with severe asthma often have difficulty achieving control of their disease because of its complex and heterogeneous nature. They face a significantly reduced quality of life, frequent exacerbations, and an increased risk of hospitalization," said Dr. Anne Ellis, Professor of Medicine at Queen's University, Chair of the Allergy/Immunology Department, and President of the Canadian Society of Allergy and Clinical Immunology. "Tezspire represents an important new treatment for many Canadian patients who remain underserved and continue to struggle with severe asthma."
"With the approval of Tezspire, physicians are able to offer a new treatment that has the potential to transform care for a broad population of severe asthma patients in Canada," said Kiersten Combs, President of AstraZeneca Canada. "Tezspire marks the first biologic treatment for severe asthma without phenotypic limitations and irrespective of biomarker levels."
"Tezspire introduces a new era in severe asthma management, in which a broad patient population will have a treatment option that controls inflammation at the source, regardless of the cause," said Dr. Daniel Martinez, Executive Medical Director, Amgen Canada. "This is a true win for Canadian patients, providing a much-needed treatment option for a complex and debilitating disease."
In clinical studies, the most frequently reported adverse reactions in patients who received Tezspire were pharyngitis, arthralgia, and injection site reaction.1
Results from the NAVIGATOR Phase III trial were published in The New England Journal of Medicine in May 2021. There were no clinically meaningful differences in safety results between the Tezspire and placebo groups in the NAVIGATOR trial.2
Tezspire
Tezspire (tezepelumab injection) is being developed by AstraZeneca in collaboration with Amgen and is a first-in-class human monoclonal antibody that inhibits the action of TSLP, a key epithelial cytokine that sits at the top of the inflammatory cascade and is critical in the initiation and persistence of allergic, eosinophilic and other types of airway inflammation associated with severe asthma, including airway hyperresponsiveness.3,4 TSLP is released in response to multiple triggers associated with asthma exacerbations, including allergens, viruses and other airborne particles.3,4 Expression of TSLP is increased in the airways of patients with asthma and has been correlated with disease severity.3,5 Blocking TSLP may prevent the release of pro-inflammatory cytokines by immune cells, resulting in the prevention of asthma exacerbations and improved asthma control.2,3,5 Tezspire acts at the top of the inflammation cascade and has the potential to help address a broad population of severe asthma patients irrespective of biomarker levels.2,3
Tezspire was approved by Health Canada in July 2022. Tezspire has also been approved in the US, EU, and Japan for the treatment of severe asthma, and regulatory reviews are ongoing in additional countries around the world.27
Tezspire is also in development for other potential indications including chronic obstructive pulmonary disease (COPD), chronic rhinosinusitis with nasal polyps, chronic spontaneous urticaria and eosinophilic esophagitis (EoE).
AstraZeneca
AstraZeneca is a global, innovation-driven biopharmaceutical business with a focus on the discovery, development, and commercialization of medicines that transform lives. Our core scientific focus is in the areas of Cardiovascular, Renal and Metabolic (CVRM) disease; Oncology; Rare Disease; Respiratory & Immunology; and Vaccine & Immune Therapies. AstraZeneca operates in more than 100 countries and its innovative medicines are used by millions of patients worldwide. In Canada, the company employs more than 1,200 people across Canada, including roughly 700 employees at our head office and clinical research hub in Mississauga, Ontario. For more information, please visit the company's website at www.astrazeneca.ca.
Amgen
As a leader in innovation, Amgen Canada understands the value of science. With main operations located in Mississauga, Ontario's vibrant biomedical cluster, and its research facility in Burnaby, B.C., Amgen Canada has been an important contributor to advancements in science and innovation in Canada since 1991. The company contributes to the development of new therapies and new ways of using existing medicines in partnership with many of Canada's leading healthcare, academic, research, government and patient organizations. To learn more about Amgen Canada, visit www.amgen.ca and follow us on www.twitter.com/amgencanadagm.
https://seekingalpha.com/symbol/AZN
https://seekingalpha.com/symbol/AMGN
PUBLISHED24 October 2022 24 October 2022 07:00 BST
AstraZeneca’s Imjudo (tremelimumab) in combination with Imfinzi (durvalumab) has been approved in the US for the treatment of adult patients with unresectable hepatocellular carcinoma (HCC), the most common type of liver cancer. The novel dose and schedule of the combination, which includes a single dose of the anti-CTLA-4 antibody Imjudo 300mg added to the anti-PD-L1 antibody Imfinzi 1500mg followed by Imfinzi every four weeks, is called the STRIDE regimen (Single Tremelimumab Regular Interval Durvalumab).
The approval by the US Food and Drug Administration (FDA) was based on positive results from the HIMALAYA Phase III trial. In this trial, patients treated with the combination of Imjudo and Imfinzi experienced a 22% reduction in the risk of death versus sorafenib (based on a hazard ratio [HR] of 0.78, 95% confidence interval [CI] 0.66-0.92 p=0.0035).1 Results were also published in the New England Journal of Medicine Evidence showing that an estimated 31% of patients treated with the combination were still alive after three years, with 20% of patients treated with sorafenib still alive at the same duration of follow-up.2
Liver cancer is the third-leading cause of cancer death and the sixth most commonly diagnosed cancer worldwide.3,4 It is the fastest rising cause of cancer-related deaths in the US, with approximately 36,000 new diagnoses each year.5,6
Ghassan Abou-Alfa, MD, MBA, Attending Physician at Memorial Sloan Kettering Cancer Center (MSK), and principal investigator in the HIMALAYA Phase III trial, said: “Patients with unresectable liver cancer are in need of well-tolerated treatments that can meaningfully extend overall survival. In addition to this regimen demonstrating a favourable three-year survival rate in the HIMALAYA trial, safety data showed no increase in severe liver toxicity or bleeding risk for the combination, important factors for patients with liver cancer who also have advanced liver disease.”
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “With this first regulatory approval for Imjudo, patients with unresectable liver cancer in the US now have an approved dual immunotherapy treatment regimen that harnesses the potential of CTLA-4 inhibition in a unique combination with a PD-L1 inhibitor to enhance the immune response against their cancer.”
Andrea Wilson Woods, President & Founder, Blue Faery: The Adrienne Wilson Liver Cancer Foundation, said: “In the past, patients living with liver cancer had few treatment options and faced poor prognoses. With today's approval, we are grateful and optimistic for new, innovative, therapeutic options. These new treatments can improve long-term survival for those living with unresectable hepatocellular carcinoma, the most common form of liver cancer. We appreciate the patients, their families, and the broader liver cancer community who continue to fight for new treatments and advocate for others.”
The safety profiles of the combination of Imjudo added to Imfinzi and for Imfinzi alone were consistent with the known profiles of each medicine, and no new safety signals were identified.
Regulatory applications for Imjudo in combination with Imfinzi are currently under review in Europe, Japan and several other countries for the treatment of patients with advanced liver cancer based on the HIMALAYA results.
HIMALAYA
HIMALAYA was a randomised, open-label, multicentre, global Phase III trial of Imfinzi monotherapy and a regimen comprising a single priming dose of Imjudo 300mg added to Imfinzi 1500mg followed by Imfinzi every four weeks versus sorafenib, a standard-of-care multi-kinase inhibitor.
The trial included a total of 1,324 patients with unresectable, advanced HCC who had not been treated with prior systemic therapy and were not eligible for locoregional therapy (treatment localised to the liver and surrounding tissue).
The trial was conducted in 181 centres across 16 countries, including in the US, Canada, Europe, South America and Asia. The primary endpoint was overall survival (OS) for the combination versus sorafenib and key secondary endpoints included OS for Imfinzi versus sorafenib, objective response rate and progression-free survival (PFS) for the combination and for Imfinzi alone.
Imfinzi
Imfinzi (durvalumab) is a human monoclonal antibody that binds to the PD-L1 protein and blocks the interaction of PD-L1 with the PD-1 and CD80 proteins, countering the tumour’s immune-evading tactics and releasing the inhibition of immune responses.
Imfinzi was recently approved to treat patients with advanced biliary tract cancer in the US based on results from the TOPAZ-1 Phase III trial. It is the only approved immunotherapy in the curative-intent setting of unresectable, Stage III non-small cell lung cancer (NSCLC) in patients whose disease has not progressed after chemoradiotherapy and is the global standard of care in this setting based on the PACIFIC Phase III trial.
Imfinzi is also approved in the US, EU, Japan, China and many other countries around the world for the treatment of extensive-stage small cell lung cancer (ES-SCLC) based on the CASPIAN Phase III trial. In 2021, updated results from the CASPIAN trial showed Imfinzi plus chemotherapy tripled patient survival at three years versus chemotherapy alone.
Imfinzi is also approved for previously treated patients with advanced bladder cancer in several countries.
Since the first approval in May 2017, more than 100,000 patients have been treated with Imfinzi.
As part of a broad development programme, Imfinzi is being tested as a single treatment and in combinations with other anti-cancer treatments for patients with SCLC, NSCLC, bladder cancer, several gastrointestinal (GI) cancers, ovarian cancer, endometrial cancer, and other solid tumours.
Imfinzi combinations have also demonstrated clinical benefit in metastatic NSCLC in the POSEIDON Phase III trial.
Imjudo
Imjudo (tremelimumab) is a human monoclonal antibody that targets the activity of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). Imjudo blocks the activity of CTLA-4, contributing to T-cell activation, priming the immune response to cancer and fostering cancer cell death.
Beyond HIMALAYA, Imjudo is being tested in combination with Imfinzi across multiple tumour types including locoregional HCC (EMERALD-3), SCLC (ADRIATIC) and bladder cancer (VOLGA and NILE).
Imjudo is also under review by global regulatory authorities in combination with Imfinzi and chemotherapy in 1st-line metastatic NSCLC based on the results of the POSEIDON Phase III trial, which showed the addition of a short course of Imjudo to Imfinzi plus chemotherapy improved both overall and progression-free survival compared to chemotherapy alone.
AstraZeneca in immuno-oncology (IO)
Immunotherapy is a therapeutic approach designed to stimulate the body’s immune system to attack tumours. The Company’s immuno-oncology (IO) portfolio is anchored in immunotherapies that have been designed to overcome evasion of the anti-tumour immune response. AstraZeneca is invested in using IO approaches that deliver long-term survival for new groups of patients across tumour types.
The Company is pursuing a comprehensive clinical trial programme that includes Imfinzi as a single treatment and in combination with Imjudo (tremelimumab) and other novel antibodies in multiple tumour types, stages of disease, and lines of treatment, and where relevant using the PD-L1 biomarker as a decision-making tool to define the best potential treatment path for a patient.
In addition, the ability to combine the IO portfolio with radiation, chemotherapy, and targeted small molecules from across AstraZeneca’s oncology pipeline, and from research partners, may provide new treatment options across a broad range of tumours.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Oct. 24, 2022 4:26 AM ET
By: Ravikash, SA News Editor
10/26/2022
CATEGORY:
Retrospective analyses from the Phase 3 DAYBREAK open-label extension study showed more than 90% of participants who received Zeposia mounted a serologic response to COVID-19 vaccination
All adverse events related to COVID-19 in vaccinated study participants were nonserious
Data are among 13 abstracts being presented at ECTRIMS 2022 that further reinforce the safety and efficacy profile of Zeposia and Bristol Myers Squibb’s commitment to the multiple sclerosis community
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) today announced new retrospective analyses on serologic responses and clinical outcomes with COVID-19 vaccination in participants treated with Zeposia (ozanimod) from the ongoing Phase 3 DAYBREAK open-label extension (OLE) study in relapsing multiple sclerosis (MS). More than 92% (137/148) of all study participants in these analyses mounted a serological response following vaccination. In addition, among participants with prior COVID-19 exposure, seroconversion was observed in 100% (39/39) of individuals following full COVID-19 mRNA or non-mRNA vaccination. COVID-19-related adverse events were reported in 10% (15/148) of vaccinated participants (12/148 confirmed; 3/148 suspected), all of which were nonserious.
The new analyses (Presentation #P1199) will be featured in the late-breaking research session on October 27, 2022, at the 38th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS), taking place in Amsterdam, the Netherlands.
“These data are of clinical importance for physicians treating multiple sclerosis, because they provide a greater understanding of how COVID-19 infections and vaccinations interplay with Zeposia treatment,” said Bruce Cree, MD, PhD, MAS, study investigator and professor of Clinical Neurology, University of California San Francisco (UCSF) Weill Institute for Neurosciences and Clinical Research Director, UCSF MS Center. “Regardless of prior COVID-19 exposure, most participants mounted an immune response following COVID-19 vaccination without experiencing adverse events, in contrast to some reports of other S1P modulators that described no response, adding insight to the treatment decision making process.”
Additional key Bristol Myers Squibb data presentations at ECTRIMS 2022 include:
“We remain dedicated to pursuing pathbreaking science and collaborating with the scientific and patient communities to deepen our understanding on the use of our medicines and to help individuals with multiple sclerosis live healthier, more fulfilling lives,” said Jonathan Sadeh, MD, MSc, senior vice president of Immunology and Fibrosis Development, Bristol Myers Squibb. “Our COVID-19 and disability progression findings, as well as our presentations demonstrating an association between Zeposia use and reduced brain volume loss along with improved cognitive processing speed, reinforce the importance of early intervention in the treatment of multiple sclerosis and Zeposia’s profile in the treatment armamentarium.”
About DAYBREAK
DAYBREAK is a Phase 3, multi-center, long-term open-label extension (OLE), randomized, double-blind, double-dummy, active-controlled, parallel group study to evaluate the safety and efficacy of Zeposia (ozanimod) administered orally to patients with relapsing forms of multiple sclerosis (MS).
Eligible patients from the RADIANCE, SUNBEAM and RPC01-1001 trials diagnosed with relapsing forms of MS are enrolled to receive treatment until the end of the DAYBREAK trial or until the development program is discontinued. Patients in the trial are receiving Zeposia 0.92 mg (equivalent to ozanimod HCl 1 mg).
About RADIANCE
RADIANCE Part B was a pivotal, Phase 3, multicenter, randomized, double-blind, double-dummy, active-controlled trial evaluating the efficacy, safety and tolerability of oral Zeposia (0.92 mg, equivalent to 1 mg) against weekly intramuscular AVONEX® (interferon beta-1a) over a 24-month treatment period. The study included 1,320 people living with relapsing forms of MS across 150 sites in 21 countries.
The primary endpoint of the trial was ARR over 24 months. The secondary MRI endpoints included the number of new or enlarging hyperintense T2-weighted brain MRI lesions over 24 months.
About SUNBEAM
SUNBEAM was a pivotal, Phase 3, multicenter, randomized, double-blind, double-dummy, active-controlled trial evaluating the efficacy, safety and tolerability of two doses of oral Zeposia (0.92 mg and 0.46 mg, equivalent to 1 mg and 0.5 mg ozanimod HCl, respectively) against weekly intramuscular AVONEX® (interferon beta-1a) for at least a 12-month treatment period. The study included 1,346 people living with relapsing forms of MS across 152 sites in 20 countries.
The primary endpoint of the trial was annualized relapse rates during the treatment period. The secondary MRI endpoints included the number of new or enlarging hyperintense T2-weighted brain MRI lesions over 12 months, number of gadolinium-enhanced brain MRI lesions at Month 12 and percent change from baseline in whole brain volume at Month 12. Cortical grey and thalamic volume changes were also prospectively assessed versus active comparator.
About Zeposia (ozanimod)
Zeposia (ozanimod) is an oral, sphingosine 1-phosphate (S1P) receptor modulator that binds with high affinity to S1P receptors 1 and 5. Zeposia blocks the capacity of lymphocytes to egress from lymph nodes, reducing the number of lymphocytes in peripheral blood. The mechanism by which Zeposia exerts therapeutic effects in multiple sclerosis (MS) is unknown but may involve the reduction of lymphocyte migration into the central nervous system.
The U.S. Food and Drug Administration approved Zeposia for the treatment of adults with relapsing forms of multiple sclerosis in March 2020 and adults with moderately to severely active UC in May 2021. The European Commission approved Zeposia for the treatment of adult patients with relapsing remitting multiple sclerosis with active disease as defined by clinical or imaging features in May 2020 and for the treatment of adults with moderately to severely active UC who have had an inadequate response, lost response, or were intolerant to either conventional therapy or a biologic agent in November 2021.
U.S. FDA APPROVED INDICATIONS
ZEPOSIA® (ozanimod) is indicated for the treatment of:
About Bristol Myers Squibb
Bristol Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information about Bristol Myers Squibb, visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
Oct. 26, 2022 6:18 AM ET
Bristol-Myers Squibb Company (BMY)
By: Ravikash, SA News Editor
Bristol Myers Squibb (NYSE:BMY) said retrospective analyses from a phase 3 open-label extension (OLE) study showed that more than 92% of people who received its multiple sclerosis (MS) drug Zeposia mounted a serologic response to COVID-19 vaccination.
https://seekingalpha.com/symbol/BMY
Oct. 24, 2022 7:37 AM ETAstraZeneca PLC (AZN)
MISSISSAUGA, ON, Oct. 24, 2022 /CNW/ - Today, AstraZeneca (AZN) announced the Canadian availability of Breztri™ Aerosphere® (budesonide/glycopyrronium/formoterol fumarate dihydrate), indicated for the long-term maintenance treatment to reduce exacerbations of chronic obstructive pulmonary disease (COPD) and treat airflow obstruction in patients with COPD, including chronic bronchitis and/or emphysema who (FREEW) are not adequately treated by a combination of an ICS/LABA or a combination of a LAMA/LABA.1

It is estimated that two million Canadians are living with COPD2, a lung condition which includes chronic bronchitis and emphysema, that causes debilitating bouts of breathlessness3,4,5 largely due to the narrowing of the airways.6 An exacerbation occurs when the symptoms of a person living with COPD intensify suddenly, requiring medical treatment.7 In Canada, COPD is the eighth leading cause of hospitalization; and in people over 65 years old, it is the fourth leading cause of hospitalization.8
"Preventing exacerbations is a key clinical priority, as even a single exacerbation can cause further deterioration of a patient's lung function, increase the risks of hospitalization and premature death," said Dr. Jean Bourbeau, Respirologist and Professor in the Departments of Medicine and Epidemiology & Biostatistics at McGill University. "Triple fixed-dose combination therapies, like Breztri Aerosphere, have demonstrated benefits in reducing exacerbations and mortality for eligible patients and represent an important advance in the field."
About 70 per cent of exacerbations are not reported by patients to their physicians.9 During a severe exacerbation, symptoms like shortness of breath and increased coughing suddenly intensify, airway muscles tighten significantly and cut off air supply. This can lead to blue lips or fingers, confusion, drowsiness, extreme shortness of breath and, when not treated, death.10 One in four patients die within a year of a severe exacerbation.11
"Quality of life is everything for people living with COPD in Canada and their families," said Peter Glazier, Executive Vice President of the Lung Health Foundation. "We're pleased to know there is a new treatment option to help people live life to the fullest extent possible with COPD."
"The availability of Breztri Aerosphere in Canada marks a significant advance in care for COPD patients, and reinforces our commitment to this community," said Dr. Alex Romanovschi, Vice President, Scientific Affairs, AstraZeneca Canada. "We are proud of the potential Breztri Aerosphere has to help improve the clinical outcomes and the quality of life for people living with COPD."
Health Canada's approval was based on positive results from ETHOS and KRONOS Phase III trials in moderate to very severe COPD patients. In the ETHOS trial, Breztri Aerosphere showed a statistically significant reduction in the rate of moderate or severe exacerbations compared with dual-combination therapies (glycopyrronium/formoterol fumarate) and PT009 (budesonide/formoterol fumarate) over 52 weeks.12 In the KRONOS trial, which included patients with no history of exacerbations in the prior 12 months, Breztri Aerosphere demonstrated a statistically significant reduction in the rate of moderate or severe exacerbation over 24 weeks vs. a dual-combination therapy (glycopyrronium/formoterol fumarate).13
Results from the ETHOS Phase III trial were published in The New England Journal of Medicine in June 202012 and results from the KRONOS Phase III trial were published in The Lancet Respiratory Medicine in September 2018.13 In both trials, the safety profile and tolerability of Breztri Aerosphere were consistent with the profiles of the dual comparators.12,13 The most frequently reported adverse reactions were pneumonia, oral candidiasis, muscle spasms, and dysphonia.1
Breztri Aerosphere1
Breztri Aerosphere is a triple therapy in a single inhaler that demonstrates a statistically significant reduction in the rate of moderate or severe exacerbations. The triple therapy combines inhaled corticosteroid (ICS), long-acting muscarinic antagonist (LAMA), and a long-acting beta2-adrenergic agonist (LABA), to help reduce inflammation in the airways making it easier to breathe. Breztri Aerosphere was approved by Health Canada in July 2021.
Breztri Aerosphere is also approved for patients with COPD in Japan, China, and the US, and as Trixeo Aerosphere in Europe.
About AstraZeneca
AstraZeneca is a global, innovation-driven biopharmaceutical business with a focus on the discovery, development, and commercialization of medicines that transform lives. Our core scientific focus is in the areas of Cardiovascular, Renal and Metabolic (CVRM) disease; Oncology; Rare Disease; Respiratory & Immunology; and Vaccine & Immune Therapies. AstraZeneca operates in more than 100 countries and its innovative medicines are used by millions of patients worldwide. In Canada, the company employs more than 1,200 people across Canada, including roughly 700 employees at our head office and clinical research hub in Mississauga, Ontario. For more information, please visit the company's website at www.astrazeneca.ca.
https://seekingalpha.com/symbol/AZN
https://www.nasdaq.com/market-activity/stocks/azn/dividend-history
October 24, 2022
– New Clinical Outcomes from BICSTaR Study Show Sustained Impact of Biktarvy for People with HIV –
– Five-Year Data from Studies 1489 and 1490 Solidify the Robust and Durable Efficacy and Safety Profile of Biktarvy –
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced the presentation of real-world results from the BICSTaR study, highlighting Biktarvy® (bictegravir 50 mg/emtricitabine 200 mg/tenofovir alafenamide 25 mg tablets, B/F/TAF) as a generally well tolerated and efficacious regimen regardless of prior treatment and comorbidity status in people with HIV. The latest five-year data from two Phase 3 studies (Study 1489 and Study 1490) provide evidence of the long-term safety and efficacy profile of Biktarvy in those who switch from a dolutegravir-containing regimen. The data were presented at the 30th International Congress on Drug Therapy in HIV Infection (HIV Glasgow 2022).
New real-world data was presented from the 24-month BICSTaR follow-up analysis, evaluating the effectiveness and safety of Biktarvy in clinical practice across nine countries. The analysis included follow-up during the COVID-19 pandemic and considered age, race, sex, adherence, and late diagnosis in the population group. Trial participants who initiated treatment with Biktarvy experienced high viral suppression (HIV-1 RNA <50 copies/mL). Overall, 97% (104/107) of treatment-naïve and 95% (497/521) of treatment-experienced participants achieved viral suppression (missing=excluded analysis) at 24 months. There were no reports of treatment-emergent resistance. Treatment discontinuations (14% overall) were low, and few people (7%) discontinued Biktarvy as a result of drug-related AEs (DRAEs). The most commonly reported drug-related adverse events were weight change (3%) and depression (1%). These data reinforce the safety and durability of Biktarvy for people with HIV with a high level of comorbidities.
“These latest data demonstrate how innovation and improvement in HIV treatment options can help people living with HIV and their clinicians identify an HIV treatment regimen that supports their treatment over the long-term,” said Benoit Trottier, MD, Physician and Director of Research at Clinique de Médecine Urbaine du Quartier Latin, Montreal, Canada. “Factors such as aging and comorbidities are vital components of long-term health discussions. The BICSTaR study reinforces the real-world effectiveness of Biktarvy across populations with a range of comorbidities and the findings are consistent with evidence from randomized clinical trials of Biktarvy treatment.”
Additional data from Study 1489 and Study 1490 presented at the conference show Biktarvy to have high efficacy and sustained safety for people switching to the treatment, with a continued high barrier to resistance. These outcomes were reported in participants 96 weeks after switching to open-label Biktarvy following 144 weeks of blinded dolutegravir + 2 NRTIs. At Week 240, more than 99% of participants in both Study 1489 (217/218; missing=excluded) and Study 1490 (232/234; missing=excluded) achieved viral suppression. Additionally, at every visit through 240 weeks, the study showed that following the switch to Biktarvy, efficacy was >96% (missing=excluded), demonstrating that Biktarvy may provide sustained viral suppression for people with HIV, even after switching treatments. Biktarvy was generally well tolerated, with 0.4% (2/519) of switch participants in both studies experiencing an AE that led to drug discontinuation in the open-label extension period. There were no renal discontinuations. The most commonly reported adverse events during the open-label extension phase were diarrhea (0.6%) and weight change (0.6%).
On October 14, 2022, the U.S. Food and Drug Administration (FDA) approved a label change for Biktarvy, updating the prescribing information to include efficacy data from 144 weeks and safety data from 240 weeks of clinical trial data in adults with HIV who were initiating therapy in Study 1489 and Study 1490.
“As we strive to advance scientific innovation with the goal of helping to end the HIV epidemic, we’re committed to a treatment research program that addresses the individual needs of all people with HIV,” said Jared Baeten, MD, PhD, Vice President, HIV Clinical Development, Gilead Sciences. “Gilead’s ongoing, person-centered research is focused on the evolving needs and preferences of people living with HIV. These latest data presented at HIV Glasgow 2022 demonstrate the clinical use of innovative medicines like Biktarvy to help a broad range of people with HIV, regardless of their burden of comorbidities.”
Please see below for the U.S. Indication and Important Safety Information, including Boxed Warning, for Biktarvy.
There is currently no cure for HIV or AIDS.
About the BICSTaR Study
The Bictegravir Single Tablet Regimen (BICSTaR) study is an ongoing, multinational, observational single-arm, non-comparative real-world cohort study, which aims to evaluate the effectiveness, safety, tolerability, and patient-reported outcomes of treatment with Biktarvy in treatment‐naïve and treatment‐experienced people with HIV. Among the people with HIV enrolled in the BICSTaR study, there is a high baseline prevalence of comorbidities.
About Studies 1489 and 1490
Study 1489 and Study 1490 are Phase 3, randomized, double-blind, active-controlled studies. For 144 weeks, treatment-naïve participants were blinded to receive either Biktarvy (n=634) or a dolutegravir-containing triple therapy (n=640). The primary endpoint was the proportion of adults with HIV-1 RNA <50 copies/mL at Week 48 using the FDA snapshot algorithm. Secondary endpoints included efficacy, safety, and tolerability assessed through Weeks 96 and 144. Beyond week 144, participants were given the option to receive Biktarvy in an active open-label extension phase for up to 96 weeks.
About Biktarvy
Biktarvy is a complete HIV treatment that combines three powerful medicines to form the smallest 3-drug, integrase strand transfer inhibitor (INSTI)-based single-tablet regimen (STR) available, offering simple once-daily dosing with or without food, with a limited drug interaction potential and a high barrier to resistance. Biktarvy combines the novel, unboosted INSTI bictegravir, with the Descovy® (emtricitabine 200 mg/tenofovir alafenamide 25 mg tablets, F/TAF) backbone. Biktarvy is a complete STR and should not be taken with other HIV medicines.
U.S. Indication for Biktarvy
Biktarvy is indicated as a complete regimen for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults and pediatric patients weighing at least 14 kg who have no antiretroviral treatment history or to replace the current antiretroviral regimen in those who are virologically-suppressed (HIV-1 RNA less than 50 copies per mL) on a stable antiretroviral regimen with no history of treatment failure and no known substitutions associated with resistance to the individual components of Biktarvy.
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer.
For 35 years, Gilead has been a leading innovator in the field of HIV, driving advances in treatment, prevention and cure research. Gilead researchers have developed 12 HIV medications, including the first single-tablet regimen to treat HIV and the first antiretroviral for pre-exposure prophylaxis (PrEP) to reduce the risk of acquiring HIV infection, and the first, long-acting injectable HIV treatment medication administered twice-yearly. These advances in medical research have helped to transform HIV into a preventable, chronic condition for millions of people.
Gilead is committed to continued scientific innovation to provide solutions for the evolving needs of people affected by HIV around the world. Through partnerships and collaborations, the company also aims to improve education, expand access and address barriers to care, with the goal of ending the HIV epidemic for everyone, everywhere. Gilead was recognized as the number one philanthropic funder of HIV-related programs in a report released by Funders Concerned About AIDS.
Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California.
U.S. Prescribing Information for Biktarvy, including BOXED WARNING, is available at www.gilead.com .
View source version on businesswire.com: https://www.businesswire.com/news/home/20221023005062/en/
Source: Gilead Sciences, Inc.
Oct. 24, 2022 7:20 AM ET
Gilead Sciences, Inc. (GILD)GSK, PFE, SGIOF, SGIOY
By: Ravikash, SA News Editor
Gilead Sciences (NASDAQ:GILD) reported real-world data from a study called BICSTaR and new five-year data from two phase 3 trials of HIV therapy Biktarvy.
https://seekingalpha.com/symbol/GILD
See All Press Releases Sign up for Email Alerts
10/19/2022CATEGORY:
Results from CheckMate -76K Demonstrate Statistically Significant and Clinically Meaningful Improvement in Recurrence-Free Survival
Data reinforce the benefits of Opdivo in earlier stages of melanoma
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) today announced results from the Phase 3 CheckMate -76K trial, in which Opdivo (nivolumab) as an adjuvant therapy demonstrated a statistically significant and clinically meaningful benefit in recurrence-free survival (RFS) versus placebo in patients with completely resected stage IIB or IIC melanoma. At a pre-specified interim analysis, the trial met its primary endpoint of recurrence-free survival (RFS); Opdivo reduced the risk of recurrence or death by 58% versus placebo (hazard ratio [HR] 0.42; 95% CI 0.30-0.59; p < 0.0001). Twelve-month RFS rates for Opdivo were 89% (95% CI: 86-92) versus 79% (95% CI: 74-84) for placebo. The RFS benefit was observed across predefined subgroups in the trial, including T category and disease stage. These results are being presented as late-breaking data at the 2022 Society for Melanoma Research (SMR) Annual Meeting taking place October 17-20, 2022, during a plenary session on October 19 in Edinburgh, Scotland.
Twelve-month RFS rates by stage for patients who received Opdivo were 93% in stage IIB (versus 84% in placebo) and 84% in stage IIC (versus 72% with placebo).
The safety profile of Opdivo was consistent with that seen in previously reported studies and no new safety signals were observed at the time of the analysis. Grade 3/4 treatment-related adverse events (TRAEs) were 10% in the Opdivo arm and 2% in the placebo arm. TRAE-related discontinuation was 15% for those in the Opdivo arm and 3% for those in the placebo arm.
“Within five years after surgery, one third of stage IIB and one half of IIC patients see their cancer return. Helping reduce that risk remains a need to be addressed when it comes to treating melanoma,” said Professor Georgina Long, AO, MD, PhD, Co-Medical Director of Melanoma Institute Australia (MIA) and chair of melanoma medical oncology and translational research at MIA, The University of Sydney, and Royal North Shore and Mater Hospitals. “The data from CheckMate -76K show that treating with nivolumab in the adjuvant setting for stage IIB and IIC melanoma patients has yielded significant recurrence-free survival benefits and could be an important treatment option for this patient population.”
CheckMate -76K is part of Bristol Myers Squibb’s development program studying Opdivo and Opdivo-based combinations in earlier stages of cancer. The program currently spans seven different tumor types.
“These data add to our growing body of evidence supporting the clinical benefit of Opdivo for the treatment of melanoma, from the metastatic setting to earlier stages of cancer,” said Gina Fusaro, PhD, development program lead, melanoma, Bristol Myers Squibb. “We are continually seeking to advance our science to develop medicines that can help improve outcomes for people living with cancer.”
About CheckMate -76K
CheckMate -76K is a randomized Phase 3, double-blind study evaluating adjuvant Opdivo (nivolumab) 480 mg Q4W for up to 12 months versus placebo in 790 patients with completely resected stage IIB or IIC melanoma.
The primary endpoint of the trial is recurrence-free survival (RFS). Secondary endpoints of the trial include overall survival (OS), distant metastases-free survival (DMFS), progression-free survival on next-line therapy (PFS2), and safety endpoints.
Bristol Myers Squibb: Creating a Better Future for People with Cancer
Bristol Myers Squibb is inspired by a single vision — transforming patients’ lives through science. The goal of the company’s cancer research is to deliver medicines that offer each patient a better, healthier life and to make cure a possibility. Building on a legacy across a broad range of cancers that have changed survival expectations for many, Bristol Myers Squibb researchers are exploring new frontiers in personalized medicine, and through innovative digital platforms, are turning data into insights that sharpen their focus. Deep scientific expertise, cutting-edge capabilities and discovery platforms enable the company to look at cancer from every angle. Cancer can have a relentless grasp on many parts of a patient’s life, and Bristol Myers Squibb is committed to taking actions to address all aspects of care, from diagnosis to survivorship. Because as a leader in cancer care, Bristol Myers Squibb is working to empower all people with cancer to have a better future.
About Opdivo
Opdivo is a programmed death-1 (PD-1) immune checkpoint inhibitor that is designed to uniquely harness the body’s own immune system to help restore anti-tumor immune response. By harnessing the body’s own immune system to fight cancer, Opdivo has become an important treatment option across multiple cancers.
Opdivo’s leading global development program is based on Bristol Myers Squibb’s scientific expertise in the field of Immuno-Oncology and includes a broad range of clinical trials across all phases, including Phase 3, in a variety of tumor types. To date, the Opdivo clinical development program has treated more than 35,000 patients. The Opdivo trials have contributed to gaining a deeper understanding of the potential role of biomarkers in patient care, particularly regarding how patients may benefit from Opdivo across the continuum of PD-L1 expression.
In July 2014, Opdivo was the first PD-1 immune checkpoint inhibitor to receive regulatory approval anywhere in the world. Opdivo is currently approved in more than 65 countries, including the United States, the European Union, Japan and China. In October 2015, the Company’s Opdivo and Yervoy combination regimen was the first Immuno-Oncology combination to receive regulatory approval for the treatment of metastatic melanoma and is currently approved in more than 50 countries, including the United States and the European Union.
U.S. INDICATIONS
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of adult patients with unresectable or metastatic melanoma.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adult patients with unresectable or metastatic melanoma.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of adult patients with melanoma with involvement of lymph nodes or metastatic disease who have undergone complete resection.
OPDIVO® (nivolumab), in combination with platinum-doublet chemotherapy, is indicated as neoadjuvant treatment of adult patients with resectable (tumors ≥4 cm or node positive) non-small cell lung cancer (NSCLC).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) whose tumors express PD-L1 (≥1%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab) and 2 cycles of platinum-doublet chemotherapy, is indicated for the first-line treatment of adult patients with metastatic or recurrent non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) with progression on or after platinum-based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving OPDIVO.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with unresectable malignant pleural mesothelioma (MPM).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with intermediate or poor risk advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab), in combination with cabozantinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with advanced renal cell carcinoma (RCC) who have received prior anti-angiogenic therapy.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with classical Hodgkin lymphoma (cHL) that has relapsed or progressed after autologous hematopoietic stem cell transplantation (HSCT) and brentuximab vedotin or after 3 or more lines of systemic therapy that includes autologous HSCT. This indication is approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN) with disease progression on or after platinum-based therapy.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
OPDIVO® (nivolumab), as a single agent, is indicated for the adjuvant treatment of adult patients with urothelial carcinoma (UC) who are at high risk of recurrence after undergoing radical resection of UC.
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of adult and pediatric (12 years and older) patients with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adults and pediatric patients 12 years and older with microsatellite instability-high (MSI-H) or mismatch repair deficient
(dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adult patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with unresectable advanced, recurrent or metastatic esophageal squamous cell carcinoma (ESCC) after prior fluoropyrimidine- and platinum-based chemotherapy.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of completely resected esophageal or gastroesophageal junction cancer with residual pathologic disease in adult patients who have received neoadjuvant chemoradiotherapy (CRT).
OPDIVO® (nivolumab), in combination with fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of adult patients with unresectable advanced or metastatic esophageal squamous cell carcinoma (ESCC).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with unresectable advanced or metastatic esophageal squamous cell carcinoma (ESCC).
OPDIVO® (nivolumab), in combination with fluoropyrimidine- and platinum- containing chemotherapy, is indicated for the treatment of adult patients with advanced or metastatic gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma.
Please see US Full Prescribing Information for OPDIVO and YERVOY.
About the Bristol Myers Squibb and Ono Pharmaceutical Collaboration
In 2011, through a collaboration agreement with Ono Pharmaceutical Co., Bristol Myers Squibb expanded its territorial rights to develop and commercialize Opdivo globally, except in Japan, South Korea and Taiwan, where Ono had retained all rights to the compound at the time. On July 23, 2014, Ono and Bristol Myers Squibb further expanded the companies’ strategic collaboration agreement to jointly develop and commercialize multiple immunotherapies – as single agents and combination regimens – for patients with cancer in Japan, South Korea and Taiwan.
About Bristol Myers Squibb
Bristol Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information about Bristol Myers Squibb, visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
Source: Bristol Myers Squibb
Oct. 19, 2022 5:36 PM ET
Bristol-Myers Squibb Company (BMY)
By: Anuron Mitra, SA News Editor4 Comments
October 24, 2022 6:45 am ET
RAHWAY, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside of the United States and Canada, announced today that the European Commission (EC) has approved an expanded indication for VAXNEUVANCE™ (Pneumococcal 15-valent Conjugate Vaccine) to include active immunization for the prevention of invasive disease, pneumonia and acute otitis media caused by Streptococcus pneumoniae (S. pneumoniae) in infants, children and adolescents from 6 weeks to less than 18 years of age. The approval facilitates availability of VAXNEUVANCE for this population in all 27 European Union (EU) Member States plus Iceland, Norway and Lichtenstein. VAXNEUVANCE is also indicated in the EU for active immunization for the prevention of invasive disease and pneumonia caused by S. pneumoniae in individuals 18 years of age and older. The use of VAXNEUVANCE in the EU should be in accordance with official recommendations.
“VAXNEUVANCE was developed to maintain a strong immune response to serotypes included in currently available pneumococcal conjugate vaccines, or PCVs, while expanding coverage to disease-causing serotypes that can pose substantial risk to infants and children,” said Dr. Eliav Barr, senior vice president, head of global clinical development and chief medical officer, Merck Research Laboratories. “With this approval, we are pleased to bring an important new PCV option to a vulnerable population in Europe, including infants less than one year of age, who typically experience the highest rates of disease.”
The EC’s decision follows a positive opinion from the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP), who reviewed data from eight randomized, double-blind clinical studies that enrolled approximately 8,400 individuals, including 5,400 who received VAXNEUVANCE. The studies evaluated the use of VAXNEUVANCE in various pediatric populations at risk for pneumococcal disease, including healthy infants, children and adolescents, pre-term infants and children living with HIV infection or sickle cell disease. The use of VAXNEUVANCE was also evaluated across a variety of clinical circumstances, such as interchangeable use following initiation of an infant vaccination schedule with the currently licensed 13-valent pneumococcal conjugate vaccine (PCV13) or in a catch-up setting for older children who are either pneumococcal vaccine-naïve or who previously received an incomplete series of another PCV.
The data supporting the approval included findings from the pivotal PNEU-PED-EU-1 study which evaluated the safety, tolerability and immunogenicity of a two-dose infant series followed by a toddler dose of VAXNEUVANCE in healthy infants (n=1,184). Results showed that immune responses for VAXNEUVANCE were noninferior to PCV13 for the 13 serotypes shared between the two vaccines and superior for the two additional serotypes in VAXNEUVANCE, 22F and 33F, as assessed by serotype-specific anti-pneumococcal polysaccharide immunoglobulin G (IgG) response rates and geometric mean concentrations (GMCs) at 30 days post-toddler dose.
Pneumococcal disease is an infection caused by the bacterium S. pneumoniae, or pneumococcus. While there are more than 100 different types of S. pneumoniae, called serotypes, a selected number of serotypes are responsible for the majority of pneumococcal infections. Invasive pneumococcal disease (IPD) can cause serious and potentially life-threatening infections such as bacteremia (infection in the bloodstream); bacteremic pneumonia (pneumonia with bacteremia); and meningitis (infection of the linings of the brain and spinal cord).
In July 2021, VAXNEUVANCE received approval from the U.S. Food and Drug Administration (FDA) for active immunization for the prevention of invasive disease caused by S. pneumoniae serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, 22F, 23F and 33F in adults 18 years of age and older, and in June 2022, the FDA approved an expanded indication for VAXNEUVANCE to include individuals 6 weeks through 17 years of age.
About VAXNEUVANCE (Pneumococcal 15-valent Conjugate Vaccine)
VAXNEUVANCE, Merck’s 15-valent pneumococcal conjugate vaccine, consists of purified capsular polysaccharides from S.pneumoniae serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, 22F, 23F and 33F individually conjugated to CRM197 carrier protein.
VAXNEUVANCE is indicated in the EU for active immunization for the prevention of invasive disease, pneumonia and acute otitis media caused by S. pneumoniae in infants, children and adolescents from 6 weeks to less than 18 years of age, and for active immunization for the prevention of invasive disease and pneumonia caused by S. pneumoniae in individuals 18 years of age and older.
VAXNEUVANCE is indicated in the U.S. for active immunization of individuals 6 weeks of age and older for the prevention of invasive disease caused by the S. pneumoniae serotypes contained in the vaccine.
https://www.vaxneuvance.com/pediatrics/
About Merck
At Merck, known as MSD outside of the United States and Canada, we are unified around our purpose: We use the power of leading-edge science to save and improve lives around the world. For more than 130 years, we have brought hope to humanity through the development of important medicines and vaccines. We aspire to be the premier research-intensive biopharmaceutical company in the world – and today, we are at the forefront of research to deliver innovative health solutions that advance the prevention and treatment of diseases in people and animals. We foster a diverse and inclusive global workforce and operate responsibly every day to enable a safe, sustainable and healthy future for all people and communities. For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Please see U.S. Prescribing Information for VAXNEUVANCE (Pneumococcal 15-valent Conjugate Vaccine) at https://www.merck.com/product/usa/pi_circulars/v/vaxneuvance/vaxneuvance_pi.pdf and Patient Information/Medication Guide for VAXNEUVANCE at https://www.merck.com/product/usa/pi_circulars/v/vaxneuvance/vaxneuvance_ppi.pdf .
Source: Merck & Co., Inc.
Oct. 24, 2022 7:41 AM ET
By: Ravikash, SA News Editor
The European Commission (EC) approved the expanded use of Merck's (NYSE:MRK) pneumococcal 15-valent conjugate vaccine Vaxneuvance for use in children aged 6 weeks to less than 18 years of age.
October 21, 2022
NORTH CHICAGO, Ill., Oct. 21, 2022 /PRNewswire/ -- AbbVie (NYSE: ABBV) today announces that the U.S. Food and Drug Administration (FDA) has approved RINVOQ® (upadacitinib 15 mg, once daily), an oral therapy, for the treatment of adults with active non-radiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation who have had an inadequate response or intolerance to tumor necrosis factor (TNF) blocker therapy. This additional indication follows the FDA approval of RINVOQ in April of this year for adults with active ankylosing spondylitis (AS) who have had an inadequate response or intolerance to one or more TNF blockers, making it the first and only JAK inhibitor that is approved for both conditions.1
Experience the full interactive Multichannel News Release here:
https://www.multivu.com/players/English/9092951-abbvie-fda-active-non-radiographic-axial-spondyloarthritis/
Nr-axSpA is a chronic, progressive inflammatory rheumatic disease that causes joint inflammation, leading to back pain and stiffness, and cannot be detected by x-ray.5,6 Nr-axSpA and AS are two sub-types of a broader condition called axial spondyloarthritis. Approximately five percent of patients with nr-axSpA will progress to AS after five years, and 19 percent will progress after ten years.7
"This latest FDA approval of RINVOQ in active nr-axSpA provides a new oral, once-daily treatment option for patients who historically have had limited treatment options for this painful, chronic disease," said Thomas Hudson, M.D., senior vice president, research and development, chief scientific officer, AbbVie. "RINVOQ is now approved to treat patients across the spectrum of axial spondyloarthritis. This further underscores AbbVie's commitment to advancing the standards of care for patients living with these diseases."
The FDA approval decision is supported by data from the Phase 3 SELECT-AXIS 2 clinical trial, which assessed the efficacy, safety, and tolerability of RINVOQ in adults with active nr-axSpA. Among patients who received RINVOQ 15 mg, nearly half achieved an ASAS40* response, the primary endpoint, at week 14 compared to placebo (44.9 percent vs. 22.3 percent respectively).1,2 ASAS40 responses were observed as early as two weeks in nr-axSpA patients treated with RINVOQ. The safety profile in patients with nr-axSpA treated with RINVOQ 15 mg was consistent with the safety profile in patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis.1
"Many patients living with nr-axSpA continue to experience symptoms and are unable to control disease with current treatments. In the SELECT-AXIS 2 trials, RINVOQ demonstrated efficacy in both nr-axSpA and AS with safety that was consistent across indications," said Atul Deodhar, M.D., professor of medicine and medical director of the Rheumatology Clinics for the Division of Arthritis and Rheumatic Diseases at Oregon Health & Science University, and lead investigator of the SELECT-AXIS 2 nr-axSpA trial. "Today's FDA approval offers an important new therapeutic option for patients and their caregivers to help take control of their symptoms and disease."
"There are limited treatment options for people living with nr-axSpA, a condition that can pose many challenges for patients and significantly impact their quality of life as their symptoms persist despite treatment," said Cassie Shafer, chief executive officer, Spondylitis Association of America (SAA). "This approval of RINVOQ is a tremendous step forward in providing our patient community with another option to help them reach their treatment goals, and to find relief."
This FDA approval marks the sixth indication for RINVOQ in the United States across chronic immune-mediated diseases, including four in rheumatology.1
Additional study results include the following:
About RINVOQ® (upadacitinib)
Discovered and developed by AbbVie scientists, RINVOQ is a selective JAK inhibitor that is being studied in several immune-mediated inflammatory diseases. Based on enzymatic and cellular assays, RINVOQ demonstrated greater inhibitory potency for JAK-1 vs JAK-2, JAK-3, and TYK-2.1 The relevance of inhibition of specific JAK enzymes to therapeutic effectiveness and safety is not currently known.
In the U.S., RINVOQ 15 mg is approved for adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more TNF blockers; adults with active psoriatic arthritis who have had an inadequate response or intolerance to one or more TNF blockers; adults with active ankylosing spondylitis (AS) who have had an inadequate response or intolerance to one or more tumor necrosis factor (TNF) blockers and adults with active non-radiographic axial spondyloarthritis with objective signs of inflammation who have had an inadequate response or intolerance to TNF blocker therapy.1 RINVOQ 45 mg is approved for use in adult patients with moderately to severely active ulcerative colitis who have had an inadequate response or intolerance to one or more TNF blockers as an induction therapy once daily for 8 weeks. The recommended dose of RINVOQ for maintenance treatment is 15 mg once daily. A dosage of 30 mg once daily may be considered for patients with refractory, severe or extensive disease. Discontinue RINVOQ if an adequate response is not achieved with the 30 mg dose. Use the lowest effective dosage needed to maintain response. RINVOQ 15 mg once daily can also be initiated in adults and children 12 years of age and older weighing at least 40 kg with refractory, moderate to severe atopic dermatitis whose disease is not adequately controlled with other system drug products, including biologics or when use of those therapies is inadvisable. In these children and adults less than 65 years of age who do not achieve an adequate response, the dose may be increased to 30 mg once daily.
Phase 3 trials of RINVOQ in rheumatoid arthritis, atopic dermatitis, psoriatic arthritis, axial spondyloarthritis, Crohn's disease, ulcerative colitis, giant cell arteritis and Takayasu arteritis are ongoing.4,8-14
U.S. Uses and Important Safety Information About RINVOQ® (upadacitinib) 1
RINVOQ is a prescription medicine used to treat:
It is not known if RINVOQ is safe and effective in children with juvenile idiopathic arthritis, psoriatic arthritis, ulcerative colitis, ankylosing spondylitis, or non-radiographic axial spondyloarthritis.
About AbbVie
AbbVie's mission is to discover and deliver innovative medicines that solve serious health issues today and address the medical challenges of tomorrow. We strive to have a remarkable impact on people's lives across several key therapeutic areas: immunology, oncology, neuroscience, eye care, virology, and women's health, in addition to products and services across our Allergan Aesthetics portfolio. For more information about AbbVie, please visit us at www.abbvie.com. Follow @abbvie on Twitter, Facebook, LinkedIn or Instagram.
Oct. 21, 2022 4:59 PM ET
By: Jonathan Block, SA News Editor1 Comment
10/21/2022CATEGORY:
U.S. FDA has assigned a target action date of June 16, 2023
Application based on results from the Phase 3 VALOR-HCM study
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) today announced that the U.S. Food and Drug Administration (FDA) has accepted its supplemental new drug application (sNDA) for CAMZYOS® (mavacamten) for an expanded indication to reduce the need for septal reduction therapy (SRT). CAMYZOS is currently FDA approved for the treatment of adults with symptomatic New York Heart Association (NYHA) class II-III obstructive hypertrophic cardiomyopathy (HCM) to improve functional capacity and symptoms. The FDA assigned a Prescription Drug User Fee Act (PDUFA) goal date of June 16, 2023.
“Currently, it is recommended that many patients with severe symptomatic obstructive hypertrophic cardiomyopathy undergo SRT. This often requires either an open-heart surgical procedure or septal ablation procedure – both specialized care options,” said Roland Chen, MD, senior vice president and head of cardiovascular development, Global Drug Development at Bristol Myers Squibb. “The approval of CAMZYOS earlier this year marked a significant milestone for patients. FDA acceptance of the filing for this expanded indication has the potential to strengthen the profile of CAMZYOS, while further reinforcing our commitment to delivering transformative cardiovascular therapies to patients.”
The sNDA submission was based on the results of the Phase 3 VALOR-HCM study, a randomized, double-blind, placebo-controlled study, which evaluated CAMZYOS in patients with symptomatic, obstructive HCM (NYHA class III-IV) who met the 2011 ACC/AHA guideline criteria for SRT and who have been referred for an invasive procedure. VALOR-HCM met its primary and all secondary endpoints with a high degree of statistical significance, with no new safety signals observed.
About the Phase 3 VALOR-HCM Trial
VALOR-HCM (NCT04349072) is a randomized, double-blind, placebo-controlled, multicenter Phase 3 study of patients with symptomatic, obstructive HCM (NYHA class III-IV) who meet guideline criteria for septal reduction therapy (SRT) and have been referred for an invasive procedure. The study enrolled 112 patients randomized on a 1:1 basis to receive mavacamten or placebo. VALOR-HCM includes three treatment periods over 128 weeks: a 16-week placebo-controlled period, a 16-week active treatment period where all patients received mavacamten and a 96-week long-term extension period where all patients received mavacamten.
The primary endpoint of VALOR-HCM is a composite of the number of patients who decide to proceed with SRT prior to or at Week 16 and the number of patients who remain SRT-guideline eligible (LVOT gradient of ≥50mmHg and NYHA Class III-IV) at Week 16 in the mavacamten group compared with the placebo group. Key secondary endpoints include impact on exercise gradient LVOT, NYHA Class and Kansas City Cardiomyopathy Questionnaire (KCCQ) and biomarkers at Week 16.
About CAMZYOS (mavacamten)
CAMZYOS(mavacamten) is the first and only cardiac myosin inhibitor approved by the U.S. Food and Drug Administration (FDA) indicated for the treatment of adults with symptomatic New York Heart Association (NYHA) class II-III obstructive hypertrophic cardiomyopathy (HCM) to improve functional capacity and symptoms. CAMZYOS is an allosteric and reversible inhibitor selective for cardiac myosin. CAMZYOS modulates the number of myosin heads that can enter “on actin” (power-generating) states, thus reducing the probability of force-producing (systolic) and residual (diastolic) cross-bridge formation. Excess myosin actin cross-bridge formation and dysregulation of the super-relaxed state are mechanistic hallmarks of HCM. CAMZYOS shifts the overall myosin population towards an energy-sparing, recruitable, super-relaxed state. In HCM patients, myosin inhibition with CAMZYOS reduces dynamic LVOT obstruction and improves cardiac filling pressures.
About CAMZYOS REMS Program
CAMZYOSis only available through a restricted program called the CAMZYOS REMS Program because of the risk of heart failure due to systolic dysfunction. Notable requirements of the CAMZYOS REMS Program include the following:
Further information is available by telephone at 1-833-628-7367.
INDICATION
CAMZYOS (mavacamten) is indicated for the treatment of adults with symptomatic New York Heart Association (NYHA) class II-III obstructive hypertrophic cardiomyopathy (HCM) to improve functional capacity and symptoms.
Further information is available at www.CAMZYOSREMS.com or by telephone at 1-833-628-7367.
Please see US Full Prescribing Information, including Boxed WARNING and Medication Guide.
About Bristol Myers Squibb
Bristol Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information about Bristol Myers Squibb, visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
Source: Bristol Myers Squibb
Oct. 21, 2022 7:24 AM ET
Bristol-Myers Squibb Company (BMY)
By: Ravikash, SA News Editor1 Comment

19 October 2022
Issued: London UK
For media and investors only
GSK plc (LSE/NYSE: GSK) today announced positive interim results from the ZOSTER-049 extension study showing that overall Shingrix (Zoster Vaccine Recombinant, Adjuvanted) can provide at least a decade of protection against shingles (herpes zoster) after initial vaccination.[i] The interim analysis data will be presented on 20 October 2022 at the IDWeek congress in Washington, DC, USA.
These results come from ZOSTER-049 (ZOE-LTFU), an extension from two phase III clinical trials[1] ZOE-50 and ZOE-70. From those trials, vaccine efficacy was 97%[ii] in adults 50 years and above and 91%[iii] in adults 70 years and above over a follow-up period of approximately four years.[1] The ZOE-LTFU study, which follows participants from the ZOE-50 and ZOE-70 clinical trials for an additional six years, is ongoing and will continue to evaluate the longer-term efficacy, immunogenicity and safety of the vaccine.
Dr. Javier Díez-Domingo, Principal Investigator, FISABIO (Foundation for the Promotion of Health and Biomedical Research of the Valencian Community, Spain), said: “Shingles is a painful disease that one in three adults will develop in their lifetime.[iv] We can now – for the first time – confirm that the clinical benefit of the Recombinant Zoster Vaccine overall, continues for at least 10 years after vaccination, giving patients and their healthcare providers peace of mind about the duration of protection against shingles.”
Sabine Luik, Chief Medical Officer & SVP Global Medical Regulatory & Quality, GSK, said: “We are delighted to see the continuing longevity of protection from our shingles vaccine. The findings from ZOE-LTFU demonstrate that it can provide a decade of protection against the pain, debilitating impact and potentially severe complications that shingles can cause in people aged 50 and over. These data significantly add to, and complement, the existing body of evidence demonstrating the long-term benefit of the vaccine, and we look forward to seeing additional results from this ongoing study.”
Shingles is caused by the reactivation of the varicella zoster virus (VZV), the same virus that causes chicken pox.[ii],[iv],[vi] As people age, the immune system loses the ability to mount a strong and effective immune response, increasing the risk of developing shingles.[iv],[v],[vi] The disease can cause unbearable pain and, in some cases, intense pain continues after the shingles rash fades, that nerve pain (called post-herpetic neuralgia [PHN]) can last for months or even years.[iv]
The Recombinant Zoster Vaccine (RZV) is the first approved shingles vaccine to combine a non-live antigen with GSK’s adjuvant and may help overcome the natural age-related decline in immunity that contributes to the challenge of protecting adults aged 50 years and above from this disease.[vii],[viii]
About ZOSTER-049i
ZOSTER-049 is an open-label, long-term follow-up (LTFU) study from two pivotal phase III randomised clinical trials (ZOE-50, ZOE-70). The study is evaluating the efficacy, safety and immunogenicity for six additional years after completion of the ZOE-50 and ZOE-70 studies. In the interim analysis conducted over the >4 years of long-term follow-up, representing up to 10 years since immunisation (mean: from 5.6 (±0.3) to 9.6 (±0.3) years post-vaccination), vaccine efficacy was 81.6%. From 1 month post-second dose in those initial studies up to year 10 post-vaccination (mean: 9.6 (±0.3) years post-vaccination), vaccine efficacy was 89.0%. The safety profile observed in this extension study is consistent with the established safety profile of the vaccine. No new safety concerns were identified. The incidence of serious adverse events was consistent with the age of the study population. No deaths or other Safety Adverse Events (SAE) considered related to vaccination were reported. Five cases of HZ-related complications (PHN - 3 cases and HZ disseminated disease - 2 cases) were reported.
A total of 7,413 participants were enrolled in the study’s Safety cohort. The participants were 60.7% female and 39.3% male. Participants were 76.0% White-Caucasian/European heritage, 18.7% Asian and 5.3% Other. ZOSTER-049 is being conducted in 18 countries/regions including Australia, Brazil, Canada, Czech Republic, Estonia, Finland, France, Germany, Hong Kong, Italy, Japan, Republic of Korea, Mexico, Spain, Sweden, Taiwan, the United Kingdom and the United States.
About Shingles
Shingles typically presents as a rash, with painful blisters across the chest, abdomen or face.[vi] The pain is often described as aching, burning, stabbing or shock-like.[iv] Following the rash, a person can also experience post-herpetic neuralgia (PHN) pain lasting from at least three months up to several years from the onset of rash. PHN is the most common complication of shingles, occurring in 5-25% of all shingles cases, depending on the patients’ age.[iv]
About Shingrix
Shingrix [Herpes Zoster vaccine (recombinant, AS01B adjuvanted)] is a non-live, recombinant subunit vaccine which combines a recombinant antigen, glycoprotein E and the adjuvant system, AS01B.[vii],[viii]
In the EU, RZV is indicated for the prevention of shingles (herpes zoster) and, in some markets, for post-herpetic neuralgia (PHN), in adults 50 years of age and above. Full EU prescribing information is available here.
In several countries, including the European Economic Area (EEA), the vaccine is also approved for adults aged 18 or above who are at increased risk of shingles. It is the only shingles vaccine approved for this at-risk patient population.
In the US, RZV is licensed for prevention of HZ (shingles) in adults aged 50 years and above, and in adults aged 18 years and above who are or will be at increased risk of HZ due to immunodeficiency or immunosuppression caused by known disease or therapy. It is not indicated for prevention of primary varicella infection (chickenpox). Full US prescribing information is available at us.gsk.com or US Prescribing Information for RZV.
About GSK
GSK is a global biopharma company with a purpose to unite science, technology and talent to get ahead of disease together. Find out more at gsk.com/company
Cautionary statement regarding forward-looking statements
GSK cautions investors that any forward-looking statements or projections made by GSK, including those made in this announcement, are subject to risks and uncertainties that may cause actual results to differ materially from those projected. Such factors include, but are not limited to, those described in the Company's Annual Report on Form 20-F for 2021, GSK’s Q2 Results for 2022 and any impacts of the COVID-19 pandemic.
Oct. 20, 2022 5:00 AM ET
By: Ravikash, SA News Editor
GSK (NYSE:GSK) on Wednesday said its vaccine Shingrix was able to provide at least a decade of protection against shingles (herpes zoster) after initial vaccination in adults aged 50 years and above.
https://seekingalpha.com/symbol/GSK
10/17/2022 Download(opens in new window)
Smaller Sheath Simplifies Access and Improves Ease-of-Use
DANVERS, Mass.--(BUSINESS WIRE)-- The United States Food and Drug Administration (FDA) has granted 510(k) clearance to Abiomed (Nasdaq: ABMD) for its Impella Low Profile Sheath. Compared to the existing 14 French (Fr) sheath used for placement of Impella CP, the new sheath maintains the same inner diameter, but reduces the outer diameter by nearly 2 Fr. As a result of its smaller size and other technological advancements, the Low Profile Sheath will facilitate easier Impella insertion and removal, reduce procedural steps and help improve outcomes.

Abiomed’s 14 Fr Impella Low Profile Sheath, in its assembled state (Photo: Business Wire)
The Low Profile Sheath is the first sheath specifically engineered to be compatible with the Impella single-access technique, which removes the need for an additional access site. Additionally, it is designed to:
“Abiomed’s Low Profile Sheath is a game changing technological achievement that will further improve patient outcomes by making it even easier for physicians to insert, manage and remove Impella heart pumps,” said Chuck Simonton, MD, Abiomed’s chief medical officer.
Abiomed will begin a phased roll-out of the Impella Low Profile Sheath this quarter.
ABOUT IMPELLA HEART PUMPS
Impella CP with SmartAssist® is U.S. FDA approved to treat certain advanced heart failure patients undergoing elective and urgent percutaneous coronary interventions (PCI), such as stenting or balloon angioplasty, to reopen blocked coronary arteries.
Impella CP with SmartAssist® and Impella 5.5® with SmartAssist® are U.S. FDA approved to treat heart attack or cardiomyopathy patients in cardiogenic shock, and have the unique ability to enable native heart recovery, allowing patients to return home with their own heart.
ABOUT ABIOMED
Based in Danvers, Massachusetts, USA, Abiomed (Nasdaq: ABMD) is a leading provider of medical technology that provides circulatory support and oxygenation. Our products are designed to enable the heart to rest and recover by improving blood flow and/or provide sufficient oxygenation to those in respiratory failure. For additional information, please visit abiomed.com.
Source: Abiomed, Inc.
Oct. 17, 2022 12:21 PM ET
By: Ravikash, SA News Editor
October 17, 2022
-- First Treatment in 30 Years to Improve Upon Standard of Care (SOC) for Second-Line Treatment of DLBCL –
- - Based on Landmark ZUMA-7 Study, Patients with DLBCL Treated Second-Line with Yescarta Had Event-Free Survival of 8.3 Months versus Two Months for SOC [4-fold greater improvement] --
-- In ZUMA-7, Yescarta Patients with DLBCL were 2.5 Times More Likely than SOC to be Alive at Two Years Without Cancer Progression or Need for Additional Treatments --
SANTA MONICA, Calif.--(BUSINESS WIRE)-- Kite, a Gilead Company (Nasdaq: GILD), today announces that the European Commission (EC) has granted approval for the use of Yescarta® (axicabtagene ciloleucel) for the treatment of adult patients with diffuse large B-cell lymphoma (DLBCL) and high-grade B-cell lymphoma (HGBL) who relapse within 12 months from completion of, or are refractory to, first-line chemoimmunotherapy. The approval is based on results from the pivotal Phase 3 ZUMA-7 study, the largest and longest trial of a CAR T-cell therapy versus SOC in this patient population. Yescarta is now the first Chimeric Antigen Receptor (CAR) T-cell therapy approved for patients in Europe who do not respond to first-line treatment. This provides an important additional treatment option for the most common form of non-Hodgkin lymphoma. Although 60% of newly diagnosed LBCL patients, including those with DLBCL, will respond to their initial treatment, 40% will relapse or will not respond and need second-line treatment.
“We are very proud to announce Kite’s fifth approved indication in Europe in our continued commitment to the research and delivery of cell therapies with curative potential to patients who might benefit around the world,” said Christi Shaw, CEO, Kite. “Today’s approval marks an important step by providing patients in Europe this option of CAR T-cell therapy earlier in their treatment journey.”
SOC therapy for this patient population has historically been a multi-step process expected to end with a stem cell transplant. The process starts with chemoimmunotherapy, and if a patient responds to and can tolerate further treatment, they move on to high-dose chemotherapy (HDT) followed by a stem cell transplant (ASCT).
“This approval marks a major shift in the treatment of LBCL when initial treatment has failed. In ZUMA-7, treatment with axicabtagene ciloleucel resulted in an overall better outcome for patients than standard of care, especially in terms of event-free survival, marking a new era for treatment earlier in the disease pathway for more patients,” said Professor John Gribben, Professor of Medical Oncology at the Cancer Research UK Barts Centre, London. “The ZUMA-7 data has also broadened our understanding of this CAR T-cell therapy, allowing us to better manage or prevent side-effects, which is important as it moves earlier in the treatment pathway and for older patients and those with medical conditions for whom the standard of care might have been difficult.”
The ZUMA-7 study demonstrated that at a median follow-up of two years, Yescarta-treated patients had a four-fold greater improvement in the primary endpoint of event-free survival (EFS; hazard ratio 0.40; 95% CI: 0.31-0.51, P<0.001) over the current SOC (8.3 months vs 2.0 months). Additionally, Yescarta demonstrated a 2.5 fold increase in patients who were alive at two years without disease progression or need for additional cancer treatment vs SOC (41% v 16%). Improvements in EFS with Yescarta were consistent across key patient subgroups, including elderly patients (HR: 0.28 [95% CI: 0.16-0.46]), primary refractory patients (HR: 0.43 [95% CI: 0.32- 0.57]), high-grade B-cell lymphoma (HR: 0.28 [95% CI: 0.14-0.59]), and double-expressor lymphoma patients (HR: 0.42 [95% CI: 0.27-0.67]).
In the ZUMA-7 trial, Yescarta had a safety profile that was consistent with previous studies. Among the 170 Yescarta-treated patients evaluable for safety, Grade ≥3 cytokine release syndrome (CRS) and neurologic events were observed in 6% and 21% of patients, respectively. No Grade 5 CRS or neurologic events occurred. In the SOC arm, 83% of patients had Grade ≥3 events, mostly cytopenias (low blood counts).
About ZUMA-7
ZUMA-7 is an ongoing, randomized, open-label, global, multicenter (US, Australia, Canada, Europe, Israel) Phase 3 study of 359 patients at 77 centers, evaluating the safety and efficacy of a single-infusion of Yescarta versus current SOC for second-line therapy (platinum-based salvage combination chemotherapy regimen followed by high-dose chemotherapy and autologous stem cell transplant in those who respond to salvage chemotherapy) in adult patients with relapsed or refractory LBCL within 12 months of first-line therapy. The primary endpoint is event free survival (EFS). Key secondary endpoints include objective response rate (ORR) and overall survival (OS). Additional secondary endpoints include patient reported outcomes and safety.
In the analysis of patient reported outcomes (PROs), patients receiving Yescarta and eligible for the PROs portion of the study (n=165), showed statistically significant improvements in Quality of Life (QoL) at Day 100 compared with those who received SOC (n=131), using a pre-specified analysis for three PRO-domains (EORTC QLQ-C30 Physical Functioning, EORTC QLQ-C30 Global Health Status/QOL, and EQ-5D-5L visual analog scale [VAS]). There was also a trend toward faster recovery to baseline QoL in the Yescarta arm versus SOC.
About Yescarta
Yescarta was first approved in Europe in 2018 and is currently indicated for five types of blood cancer: Diffuse Large B-Cell Lymphoma (DLBCL); Large B-Cell Lymphoma (LBCL); High-Grade B-Cell Lymphoma (HGBL); Primary Mediastinal Large B-Cell Lymphoma (PMBCL); and Follicular Lymphoma (FL). For the full European Prescribing Information, please visit: https://www.ema.europa.eu/en/medicines/human/EPAR/yescarta. Please see full US Prescribing Information, including BOXED WARNING and Medication Guide.
YESCARTA is a CD19-directed genetically modified autologous T cell immunotherapy indicated for the treatment of:
About Kite
Kite, a Gilead Company, is a global biopharmaceutical company based in Santa Monica, California, with manufacturing operations in North America and Europe. Kite’s singular focus is cell therapy to treat and potentially cure cancer. As the cell therapy leader, Kite has more approved CAR T indications to help more patients than any other company. For more information on Kite, please visit www.kitepharma.com. Follow Kite on social media on Twitter (@KitePharma) and LinkedIn.
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer. Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California.
Oct. 17, 2022 5:29 PM ETGilead Sciences, Inc. (GILD)
By: Dania Nadeem, SA News Editor
Home News & Views Health Canada Approves Illuccix® for Prostate Cancer Imaging
14 October 2022
Telix announces that Health Canada has approved Illuccix® [kit for the preparation of gallium (68Ga) gozetotide injection] for use in staging and re-staging intermediate and high-risk prostate cancer and localizing tumour tissue in recurrent prostate cancer.
Illuccix™ [kit for the preparation of gallium (68Ga) gozetotide injection], after radiolabeling with gallium (68Ga), is indicated for use with the positron emission tomography (PET) of prostate specific membrane antigen (PSMA) positive lesions in men with prostate cancer:
• with suspected metastasis who are suitable for initial definitive therapy
• with suspected recurrence with elevated serum prostate specific antigen (PSA) level.
“PSMA PET” (or the imaging of Prostate-Specific Membrane Antigen (PSMA) via a positron emission tomography (PET) scan) is a diagnostic tool demonstrated to detect advanced prostate cancer.
Illuccix is the first PSMA PET imaging agent to be granted regulatory approval in Canada. Health Canada is the third regulatory body worldwide to approve Illuccix, which is commercially available in Australia and the United States.
Dr. Norman Laurin, current president of the Quebec Association of Nuclear Medicine Specialists and past president of the Canadian Association of Nuclear Medicine said, “On average, 67 men in Canada are diagnosed with prostate cancer each day. Early detection, along with better understanding of the spread and stage of the disease can lead to more informed disease management decisions. Tools such as Illuccix are valuable as we look for better ways to detect prostate cancer in men.”
Kevin Richardson, CEO Telix Americas said, “PSMA PET imaging has been one of the most important developments in prostate cancer detection in recent years and we are very pleased that we can now bring this important diagnostic imaging agent to physicians and their patients across Canada.”
Illuccix will be made available in Canada to physicians and eligible patients through Telix’s partner, Isologic Innovative Radiopharmaceuticals (ISOLOGIC), whose distribution network services 265 hospitals and clinics nationwide.
To read the full ASX disclosure click here
To return to the Telix homepage please click here.
Oct. 14, 2022 4:59 AM ET
Telix Pharmaceuticals Limited (TLPPF)
By: Ravikash, SA News Editor
FDA Accepts BioMarin's Biologics License Application (BLA) for Valoctocogene Roxaparvovec AAV Gene Therapy for Adults with Severe Hemophilia A
Oct 12, 2022
If Approved, Would Be 1st Gene Therapy in U.S. for Treatment of Severe Hemophilia A
PDUFA Target Action Date is March 31, 2023
SAN RAFAEL, Calif., Oct. 12, 2022 /PRNewswire/ -- BioMarin Pharmaceutical Inc. (NASDAQ: BMRN) announced today that the U.S. Food and Drug Administration (FDA) accepted the Company's resubmission of the Biologics License Application (BLA) for its investigational AAV gene therapy, valoctocogene roxaparvovec, for adults with severe hemophilia A. The Prescription Drug User Fee Act (PDUFA) target action date is March 31, 2023. At this time, the FDA has not communicated any plans to hold an advisory committee meeting. If approved, valoctocogene roxaparvovec would be the first gene therapy in the U.S. for the treatment of severe hemophilia A.
"This BLA resubmission is an important step that brings us closer to delivering a gene therapy treatment choice to address the unmet needs of people with severe hemophilia A in the United States. We'd like to extend our sincere gratitude to the bleeding disorders community, study participants, and investigators who have been an integral part of this journey with BioMarin. We look forward to working closely with the Agency on our application for this potentially transformative therapy," said Hank Fuchs, M.D., President of Worldwide Research and Development at BioMarin. "In this application, we have provided a substantial body of evidence that supports the safe and effective use of valoctocogene roxaparvovec for the treatment of adults with severe hemophilia A. In addition, we have proposed 15 years of follow-up for all clinical study participants, as well as a post-approval registry study to follow patients dosed in a real-world setting, to further characterize long-term effects on safety and efficacy that will contribute to increasing the body of knowledge of AAV gene therapy in severe hemophilia A. While we recognize the potential for the Agency to extend the PDUFA action date to review additional long-term follow up data, we are pleased the FDA has initiated its review of the BLA without requesting additional data."
The BLA resubmission incorporates the Company's responses to all deficiencies identified in the FDA Complete Response (CR) Letter of August 18, 2020. The BLA includes a substantial amount of data from the valoctocogene roxaparvovec clinical development program, the most extensively studied gene therapy for severe hemophilia A, including two-year outcomes from all study participants in the global GENEr8-1 Phase 3 study. The GENEr8-1 Phase 3 study demonstrated stable and durable bleed control, including a reduction in the mean annualized bleeding rate (ABR) and the mean annualized Factor VIII infusion rate. In addition, the data package includes supportive evidence from five years of follow-up from the 6e13 vg/kg dose cohort in the ongoing Phase 1/2 dose escalation study. The BLA also includes a proposed long-term extension study following all clinical trial participants for up to 15 years, as well as a post-approval registry study to follow patients dosed in a real-world setting.
The FDA granted Regenerative Medicine Advanced Therapy (RMAT) designation to valoctocogene roxaparvovec in March 2021. RMAT is an expedited program intended to facilitate development and review of regenerative medicine therapies, such as valoctocogene roxaparvovec, that are expected to address an unmet medical need in patients with serious conditions. The RMAT designation is complementary to Breakthrough Therapy Designation, which the Company received for valoctocogene roxaparvovec in 2017.
In addition to the RMAT Designation and Breakthrough Therapy Designation, BioMarin's valoctocogene roxaparvovec also received orphan drug designation from the EMA and FDA for the treatment of severe hemophilia A. Orphan drug designation is reserved for medicines treating rare, life-threatening or chronically debilitating diseases. The European Commission (EC) granted conditional marketing authorization to valoctocogene roxaparvovec gene therapy under the brand name ROCTAVIAN™ on August 24, 2022.
Robust Clinical Program
BioMarin has multiple clinical studies underway in its comprehensive gene therapy program for the treatment of severe hemophilia A. In addition to the global Phase 3 study GENEr8-1 and the ongoing Phase 1/2 dose escalation study, the Company is also conducting a Phase 3, single arm, open-label study to evaluate the efficacy and safety of valoctocogene roxaparvovec at a dose of 6e13 vg/kg with prophylactic corticosteroids in people with severe hemophilia A (Study 270-303). Also ongoing are a Phase 1/2 Study with the 6e13 vg/kg dose of valoctocogene roxaparvovec in people with severe hemophilia A with pre-existing AAV5 antibodies (Study 270-203) and a Phase 1/2 Study with the 6e13 vg/kg dose of valoctocogene roxaparvovec in people with severe hemophilia A with active or prior Factor VIII inhibitors (Study 270-205).
About BioMarin
BioMarin is a global biotechnology company that develops and commercializes innovative therapies for people with serious and life-threatening genetic diseases and medical conditions. The Company selects product candidates for diseases and conditions that represent a significant unmet medical need, have well-understood biology and provide an opportunity to be first-to-market or offer a significant benefit over existing products. The Company's portfolio consists of eight commercial products and multiple clinical and preclinical product candidates for the treatment of various diseases. For additional information, please visit www.biomarin.com.
SOURCE BioMarin Pharmaceutical Inc.
Oct. 12, 2022 4:29 PM ET
BioMarin Pharmaceutical Inc. (BMRN)
By: Jonathan Block, SA News Editor3 Comments
October 14, 2022 at 7:04 AM EDT
Recommendation based on a Phase 3 Libtayo trial that was first and only to demonstrate significantly improved survival compared to chemotherapy in the second-line setting irrespective of PD-L1 expression status or tumor histology
TARRYTOWN, N.Y., Oct. 14, 2022 /PRNewswire/ -- Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) today announced the European Medicines Agency's (EMA) Committee for Medicinal Products for Human Use (CHMP) has adopted a positive opinion for Libtayo® (cemiplimab) as a monotherapy for the treatment of adult patients with recurrent or metastatic cervical cancer and disease progression on or after platinum-based chemotherapy. The European Commission is expected to make a final decision on the application in the coming months.
The positive opinion is supported by results from the Phase 3 EMPOWER-Cervical 1 trial. In the trial (n=608), the primary endpoint of overall survival was met, with Libtayo reducing the risk of death by 31% (hazard ratio [HR]: 0.69; 95% confidence interval [CI]: 0.56-0.84; one-sided p<0.001) in the total population and 27% in the squamous cell carcinoma (SCC) population (HR: 0.73; 95% CI: 0.58-0.91; one-sided p=0.003), compared to an investigator's choice of chemotherapy. No new Libtayo safety signals were observed. Among adverse events (AE) in 10% or more patients in either group, Grade 3 or higher AEs that occurred more often in the Libtayo group (n=300) than chemotherapy (n=290) included urinary tract infection (5% Libtayo, 3% chemotherapy), back pain (1% Libtayo, less than 1% chemotherapy), asthenia (2% Libtayo, 1% chemotherapy), arthralgia (less than 1% Libtayo, 0% chemotherapy) and pyrexia (less than 1% Libtayo, 0% chemotherapy). Detailed data were published in the New England Journal of Medicine in February 2022.
Libtayo is currently approved in the European Union and other countries for the treatment of certain patients with advanced basal cell carcinoma (BCC), advanced cutaneous squamous cell carcinoma (CSCC) and advanced non-small cell lung cancer (NSCLC).
About the Phase 3 Trial
EMPOWER-Cervical 1 was an open-label, multi-center Phase 3 trial that investigated Libtayo monotherapy versus an investigator's choice of commonly used chemotherapy in patients with recurrent or metastatic cervical cancer who had progressed on platinum-based chemotherapy. Patients (median age: 51 years) were randomized to receive Libtayo (350 mg every three weeks) or chemotherapy (pemetrexed, vinorelbine, topotecan, irinotecan or gemcitabine). The primary endpoint for the trial was overall survival analyzed first among patients with SCC, then in the total population.
Patients were allowed to enroll regardless of PD-L1 expression status, with 78% of patients having SCC and 22% having adenocarcinoma or adenosquamous carcinoma. The trial included women from 14 countries: Australia, Belgium, Brazil, Canada, Greece, Italy, Japan, Poland, Russia, South Korea, Spain, Taiwan, the UK and the U.S.
In March 2021, the trial was stopped early based on the highly significant effect of Libtayo on overall survival among SCC patients and following a unanimous recommendation by the Independent Data Monitoring Committee.
About Regeneron in Oncology
At Regeneron, we're applying more than three decades of scientific innovation with the goal of developing paradigm-changing therapies for patients with cancer. Our oncology portfolio is built around two foundational approaches – our approved PD-1 inhibitor Libtayo and investigational bispecific antibodies – which are being evaluated both as monotherapies and in combination with emerging therapeutic modalities. Together, they provide us with unique combinatorial flexibility to develop potentially synergistic treatments for a wide range of solid tumors and blood cancers.
If you are interested in learning more about our clinical trials, please contact us (clinicaltrials@regeneron.com or 844-734-6643) or visit our clinical trials website.
About Libtayo
Libtayo is a fully human monoclonal antibody targeting the immune checkpoint receptor PD-1 on T cells and was invented using Regeneron's proprietary VelocImmune® technology. By binding to PD-1, Libtayo has been shown to block cancer cells from using the PD-1 pathway to suppress T-cell activation. In the U.S. and other countries Libtayo is indicated in certain patients with advanced basal cell carcinoma (BCC), advanced cutaneous squamous cell carcinoma (CSCC) and advanced non-small cell lung cancer (NSCLC), as well as in advanced cervical cancer in Canada and Brazil. As of July 1, 2022, Libtayo is developed and marketed globally by Regeneron.
In the U.S., the generic name for Libtayo in its approved indications is cemiplimab-rwlc, with rwlc as the suffix designated in accordance with Nonproprietary Naming of Biological Products Guidance for Industry issued by the FDA. Outside of the U.S. the generic name of Libtayo in its approved indication is cemiplimab.
The extensive clinical program for Libtayo is focused on difficult-to-treat cancers. Libtayo is currently being investigated in trials as a monotherapy, as well as in combination with either conventional or novel therapeutic approaches for other solid tumors and blood cancers. These potential uses are investigational, and their safety and efficacy have not been evaluated by any regulatory authority.
U.S. FDA-approved Indications
Libtayo is a prescription medicine used to treat people with:
It is not known if Libtayo is safe and effective in children.
Please see full Prescribing Information, including Medication Guide.
About Regeneron
Regeneron is a leading biotechnology company that invents, develops and commercializes life-transforming medicines for people with serious diseases. Founded and led for nearly 35 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to nine FDA-approved treatments and numerous product candidates in development, almost all of which were homegrown in our laboratories. Our medicines and pipeline are designed to help patients with eye diseases, allergic and inflammatory diseases, cancer, cardiovascular and metabolic diseases, pain, hematologic conditions, infectious diseases and rare diseases.
Regeneron is accelerating and improving the traditional drug development process through our proprietary VelociSuite technologies, such as VelocImmune, which uses unique genetically humanized mice to produce optimized fully human antibodies and bispecific antibodies, and through ambitious research initiatives such as the Regeneron Genetics Center®, which is conducting one of the largest genetics sequencing efforts in the world.
For more information, please visit www.Regeneron.com or follow @Regeneron on Twitter.
Cision View original content:https://www.prnewswire.com/news-releases/libtayo-cemiplimab-receives-positive-chmp-opinion-recommending-approval-to-treat-advanced-cervical-cancer-301649532.html
SOURCE Regeneron Pharmaceuticals, Inc.
Oct. 14, 2022 9:07 AM ET
Regeneron Pharmaceuticals, Inc. (REGN), SNY
By: Ravikash, SA News Editor
Oct 14, 2022 6:30 AM
If approved, BRUKINSA would be the only BTK inhibitor for CLL in the European Union to achieve superior efficacy versus standard of care in head-to-head trials
With significantly lower rates of atrial fibrillation/flutter compared to standard of care, BRUKINSA has potential to offer a more tolerable treatment option for certain patients
CAMBRIDGE, Mass. & BASEL, Switzerland & BEIJING--(BUSINESS WIRE)-- BeiGene (NASDAQ: BGNE; HKEX: 06160; SSE: 688235), a global biotechnology company focused on developing innovative and affordable oncology medicines to improve treatment outcomes and access for patients worldwide, today announced that the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) has issued a positive opinion recommending approval of BRUKINSA® (zanubrutinib) for the treatment of adult patients with chronic lymphocytic leukemia (CLL).
“BRUKINSA was designed to overcome limitations in efficacy and safety of first-generation Bruton’s Tyrosine Kinase inhibitors (BTKi). As a result, BRUKINSA became the only BTKi to demonstrate superiority versus ibrutinib in the largest head-to-head BTKi study in relapsed/refractory (R/R) CLL, in addition to demonstrating superior Progression-Free Survival (PFS) against bendamustine plus rituximab (BR) in treatment-naïve (TN) patients regardless of age, co-morbidities, mutation, or risk status,” said Mehrdad Mobasher, M.D., M.P.H., Chief Medical Officer, Hematology at BeiGene. “This recommendation demonstrates the focus and urgency with which we are delivering on our mission to accelerate development and broaden access to innovative medicines around the globe.”
The CHMP recommendation is based on two global head-to-head Phase 3 clinical trials in which BRUKINSA demonstrated superior efficacy: ALPINE (NCT03734016) comparing BRUKINSA to ibrutinib in patients with R/R CLL and SEQUOIA (NCT03336333) comparing BRUKINSA to BR in patients with TN CLL. These two studies enrolled patients from a total of 17 countries, including the United States, China, Australia, New Zealand, and multiple countries in Europe.
Prof. Clemens Wendtner, Head of Hematology and Oncology at Munich Clinic, an academic teaching hospital of the University of Munich, Germany, commented, “BTK inhibitors have proven to be highly effective oral treatments for CLL; however, the burden of adverse events and discontinuations have a negative impact on patients’ prognosis. The findings from two large head-to-head Phase 3 trials of BRUKINSA in CLL demonstrated efficacy across lines of therapy in addition to consistently low rates of atrial fibrillation/flutter and discontinuation across trials. These clinical trial data suggest BRUKINSA has the potential to become a practice-changing treatment option for CLL.”
“We are proud of the rapid progress we have made over the past year bringing BRUKINSA to the blood cancer community in Europe,” noted Gerwin Winter, Senior Vice President, Head of Europe at BeiGene. “With this recommendation, we are looking forward to the opportunity to provide this important medicine to more people with hematologic malignancies in the European Union.”
Following the CHMP positive opinion, the European Commission will consider BeiGene’s Marketing Application, with a final decision expected within 67 days of receipt of the CHMP opinion. The decision will be applicable to all 27 member states of the European Union (EU), plus Iceland and Norway. BRUKINSA is currently approved in the EU for the treatment of adult patients with WM who have received at least one prior therapy or as the first-line treatment for patients unsuitable for chemo-immunotherapy. Last month, CHMP issued a positive opinion recommending approval of BRUKINSA for the treatment of adult patients with MZL who have received at least one prior anti-CD20-based therapy.
In Europe, BeiGene has now obtained reimbursement for BRUKINSA for the treatment of WM in Austria, Belgium, Denmark, England and Wales, Germany, Ireland, Italy, Spain, and Switzerland, while additional EU countries are currently going through the reimbursement process.
About Chronic Lymphocytic Leukemia (CLL)
A slow-growing, life-threatening and incurable cancer of adults, CLL is a type of mature B-cell malignancy in which abnormal leukemic B lymphocytes (a type of white blood cells) arise from the bone marrow and flood peripheral blood, bone marrow, and lymphoid tissues.i,ii,iii CLL is one of the most common types of leukemia, accounting for about one-quarter of new cases of leukemia.iv In Europe, the estimated incidence is 4.92/100,000 persons per year.v,vi
About BRUKINSA
BRUKINSA is a small-molecule inhibitor of Bruton’s tyrosine kinase (BTK) discovered by BeiGene scientists that is currently being evaluated globally in a broad clinical program as a monotherapy and in combination with other therapies to treat various B-cell malignancies. BRUKINSA was specifically designed to deliver targeted and sustained inhibition of the BTK protein by optimizing bioavailability, half-life, and selectivity. With differentiated pharmacokinetics compared to other approved BTK inhibitors, BRUKINSA has been demonstrated to inhibit the proliferation of malignant B cells within a number of disease-relevant tissues.
BRUKINSA is supported by a broad clinical program which includes more than 4,500 subjects in 35 trials across 28 markets. To date, BRUKINSA has received approvals covering more than 55 countries and regions, including the United States, China, the EU, Switzerland, Great Britain, Canada, Australia, and additional international markets.
BeiGene Oncology
BeiGene is committed to advancing best- and first-in-class clinical candidates internally or with like-minded partners to develop impactful and affordable medicines for patients across the globe. We have a growing R&D and medical affairs team of approximately 3,300 colleagues dedicated to advancing more than 100 clinical trials that have involved more than 16,000 subjects. Our expansive portfolio is directed predominantly by our internal colleagues supporting clinical trials in more than 45 countries and regions. Hematology-oncology, and solid tumor targeted therapies, and immuno-oncology are key focus areas for the Company, with both monotherapies and combination therapies prioritized in our research and development. BeiGene currently has three licensed medicines discovered and developed in our own labs: BTK inhibitor BRUKINSA® in the U.S., China, the European Union, Switzerland, Great Britain, Canada, Australia, and additional international markets; and the non-FC-gamma receptor binding anti-PD-1 antibody tislelizumab as well as the poly adenosine diphosphate-ribose polymerase (PARP) inhibitor pamiparib in China.
BeiGene also partners with innovative companies who share our goal of developing therapies to address global health needs. We commercialize a range of oncology medicines in China licensed from Amgen, Bristol Myers Squibb, EUSA Pharma, and Bio-Thera. We also plan to address greater areas of unmet need globally through our other collaborations including Mirati Therapeutics, Seagen, and Zymeworks.
In January 2021 BeiGene and Novartis announced a collaboration granting Novartis rights to co-develop, manufacture, and commercialize BeiGene’s anti-PD-1 antibody tislelizumab in North America, Europe, and Japan. Building upon this productive collaboration, BeiGene and Novartis announced an option, collaboration, and license agreement in December 2021 for BeiGene’s TIGIT inhibitor ociperlimab that is in Phase 3 development. Novartis and BeiGene also entered into a strategic commercial agreement through which BeiGene will promote five approved Novartis oncology products across designated regions of China.
About BeiGene
BeiGene is a global biotechnology company that is developing and commercializing innovative and affordable oncology medicines to improve treatment outcomes and access for far more patients worldwide. With a broad portfolio, we are expediting development of our diverse pipeline of novel therapeutics through our internal capabilities and collaborations. We are committed to radically improving access to medicines for far more patients who need them. Our growing global team of more than 9,000 colleagues spans five continents, with administrative offices in Beijing, China; Cambridge, U.S.; and Basel, Switzerland. To learn more about BeiGene, please visit www.beigene.com and follow us on Twitter at @BeiGeneGlobal.
View source version on businesswire.com: https://www.businesswire.com/news/home/20221014005113/en/
Source: BeiGene
Oct. 14, 2022 7:05 AM ET
By: Ravikash, SA News Editor
October 12, 2022 at 7:00 AM EDT
ROP is a leading cause of childhood blindness worldwide
TARRYTOWN, N.Y., Oct. 12, 2022 /PRNewswire/ -- Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) today announced the U.S. Food and Drug Administration (FDA) has accepted for Priority Review the supplemental Biologics License Application (sBLA) for EYLEA® (aflibercept) Injection to treat Retinopathy of Prematurity (ROP) in preterm infants. The target action date for the FDA decision is February 11, 2023.
ROP is a leading cause of childhood blindness worldwide. Each year in the U.S., between 1,100 to 1,500 infants develop disease severe enough to require medical treatment. The rare eye disease often impacts infants who are born before 31 weeks of pregnancy have been completed or who weigh less than 1,500 grams (3.3 lbs) pounds at birth. As retinal blood vessels are often only fully developed when an infant is full-term (~9 months of pregnancy), these infants are at risk of developing retinal blood vessels that are abnormal (retinal neovascularization) potentially leading to retinal detachment and irreversible vision loss. Mild cases of ROP may get better without treatment, but some cases require treatment (for which the current standard of care is laser photocoagulation) to keep ROP from causing significant visual impairment and even blindness.
The sBLA is supported by data from two randomized global Phase 3 trials – FIREFLEYE (N=113) and BUTTERFLEYE (N=120) – investigating EYLEA 0.4 mg versus laser photocoagulation (laser) in infants with ROP. In both trials, approximately 80% of EYLEA-treated infants achieved an absence of both active ROP and unfavorable structural outcomes at 52 weeks of age, although their primary endpoint of non-inferiority was not met due to laser demonstrating comparable levels of efficacy that were higher than what have been historically observed in similar ROP trials. Notably, per an exploratory analysis, the time required to complete treatment administration per patient was considerably less for EYLEA than for laser (FIREFLEYE: 4 minutes versus 122 minutes; BUTTERFLEYE: 11 minutes versus 129 minutes).
No new EYLEA safety signals were observed in either trial. Comparing EYLEA to laser, ocular adverse events (AEs) among patients occurred in 39% versus 37% in FIREFLEYE and 18% versus 26% in BUTTERFLEYE, with serious ocular AEs occurring in 8% for both groups in FIREFLEYE and 6.5% versus 11% in BUTTERFLEYE. AEs in both trials were consistent with infant prematurity or to the injection procedure, and with the AEs in similar ROP trials.
The results of FIREFLEYE were published in Journal of the American Medical Association, and data from BUTTERFLEYE were recently presented at ROP Update 2022 meeting in the U.S. Both trials were conducted pursuant to FDA Pediatric Written Request, and a Pediatric Exclusivity Determination is under review by FDA with a response expected by early 2023. A Pediatric Exclusivity Determination is granted if the specifications of the Pediatric Written Request are met, regardless of whether a pediatric indication is approved.
EYLEA was granted orphan drug designation by the FDA for the treatment of ROP in July 2019. The safety and efficacy of EYLEA for the treatment of ROP have not been fully evaluated by the FDA and other regulatory authorities.
The lead sponsors of the trials were Regeneron for BUTTERFLEYE and Bayer for FIREFLEYE. Bayer and Regeneron are collaborating on the global development of EYLEA. Regeneron maintains exclusive rights of EYLEA in the United States. Bayer has licensed the exclusive marketing rights outside the United States, where the companies share equally the profits from sales of EYLEA.
About EYLEA
EYLEA is a VEGF inhibitor formulated as an injection for the eye. It is designed to block the growth of new blood vessels and decrease the ability of fluid to pass through blood vessels (vascular permeability) in the eye by blocking VEGF-A and placental growth factor (PLGF), two growth factors involved in angiogenesis. The EYLEA safety and efficacy profile is supported by a robust body of research that includes eight pivotal Phase 3 trials, 10 years of real-world experience and more than 50 million EYLEA injections globally.
IMPORTANT EYLEA SAFETY INFORMATION AND INDICATIONS
INDICATIONS
EYLEAâ (aflibercept) Injection 2 mg (0.05 mL) is indicated for the treatment of patients with Neovascular (Wet) Age-related Macular Degeneration (AMD), Macular Edema following Retinal Vein Occlusion (RVO), Diabetic Macular Edema (DME), and Diabetic Retinopathy (DR).
CONTRAINDICATIONS
About Regeneron
Regeneron (NASDAQ: REGN) is a leading biotechnology company that invents, develops and commercializes life-transforming medicines for people with serious diseases. Founded and led for nearly 35 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to nine FDA-approved treatments and numerous product candidates in development, almost all of which were homegrown in our laboratories. Our medicines and pipeline are designed to help patients with eye diseases, allergic and inflammatory diseases, cancer, cardiovascular and metabolic diseases, pain, hematologic conditions, infectious diseases and rare diseases.
Regeneron is accelerating and improving the traditional drug development process through our proprietary VelociSuite® technologies, such as VelocImmune®, which uses unique genetically humanized mice to produce optimized fully human antibodies and bispecific antibodies, and through ambitious research initiatives such as the Regeneron Genetics Center®, which is conducting one of the largest genetics sequencing efforts in the world.
For more information, please visit www.Regeneron.com or follow @Regeneron on Twitter.
Cision View original content:https://www.prnewswire.com/news-releases/eylea-aflibercept-injection-sbla-for-treatment-of-retinopathy-of-prematurity-rop-accepted-for-fda-priority-review-301646618.html
SOURCE Regeneron Pharmaceuticals, Inc.
Oct. 12, 2022 8:53 AM ET
Regeneron Pharmaceuticals, Inc. (REGN), BAYZF, BAYRY
By: Ravikash, SA News Editor
10/11/2022
-- Supplemental Biologics License Application (sBLA) Based on Statistically Significant and Clinically Meaningful Overall Survival and Progression-Free Survival Results from the Phase 3 TROPiCS-02 Study --
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced the U.S. Food and Drug Administration (FDA) has accepted for priority review the supplemental Biologics License Application (sBLA) for Trodelvy® (sacituzumab govitecan-hziy) for the treatment of adult patients with unresectable locally advanced or metastatic hormone receptor (HR) positive, human epidermal growth factor receptor 2 (HER2) negative (IHC 0, IHC 1+ or IHC 2+/ISH–) breast cancer who have received endocrine-based therapy and at least two additional systemic therapies in the metastatic setting. The FDA grants priority review for therapies that, if approved, would be significant improvements in the safety or effectiveness of the treatment, diagnosis, or prevention of serious conditions when compared to standard applications. The Prescription Drug User Fee Act (PDUFA) target action date is currently set for February 2023.
“Trodelvy has already changed the treatment landscape in second-line metastatic triple-negative breast cancer and pre-treated metastatic urothelial cancer, and today’s news marks our third supplemental application acceptance within the last two years,” said Bill Grossman, MD, PhD, Senior Vice President, Therapeutic Area Head, Gilead Oncology. “People with pre-treated HR+/HER2- metastatic breast cancer who have progressed on endocrine-based therapies and chemotherapy have limited treatment options, and we look forward to working with the FDA to potentially make Trodelvy available to patients who need it most.”
This sBLA is based on data from the registrational Phase 3 TROPiCS-02 study, which met its primary endpoint of progression-free survival (PFS) and key secondary endpoint of overall survival (OS) over comparator chemotherapy (treatment of physician’s choice (TPC) of chemotherapy). PFS data were presented at the 2022 ASCO Annual Meeting and published in the Journal of Clinical Oncology,and OS data were recently presented at ESMO Congress 2022. In the study, Trodelvy demonstrated a 34% reduction in risk of disease progression or death (median PFS: 5.5 versus 4 months; hazard ratio [HR]: 0.66; 95% CI: 0.53-0.83; p=0.0003) and a 21% decrease in the risk of death compared to TPC (median OS: 14.4 months vs. 11.2 months; HR=0.789; 95% CI: 0.646-0.964; p=0.02).
The safety profile for Trodelvy in TROPiCS-02 was consistent with prior studies, with no new safety concerns identified in this population.
Trodelvy has not been approved by any regulatory agency for the treatment of HR+/HER2- metastatic breast cancer. Its safety and efficacy have not been established for this indication.
Sacituzumab govitecan-hziy is currently included in the National Comprehensive Cancer Network® (NCCN®) Clinical Practice Guidelines in Oncology (NCCN Guidelines®)i. This includes a Category 1 recommendation for use in adult patients with second-line metastatic triple-negative breast cancer (defined as those who received at least two prior therapies, with at least one line for metastatic disease) and a Category 2A preferred recommendation based on PFS data from TROPiCS-02 for investigational use in HR+/HER2- advanced breast cancer after prior treatment including endocrine therapy, a CDK4/6 inhibitor and at least two lines of chemotherapy. It also has a Category 2A recommendation for use in locally advanced or metastatic bladder cancer after prior treatment with platinum and a checkpoint inhibitor.ii
Trodelvy has a Boxed Warning for severe or life-threatening neutropenia and severe diarrhea; please see below for additional Important Safety Information.
About the TROPiCS-02 Study
The TROPiCS-02 study is a global, multicenter, open-label, Phase 3 study, randomized 1:1 to evaluate Trodelvy versus physicians’ choice of chemotherapy (eribulin, capecitabine, gemcitabine, or vinorelbine) in 543 patients with HR+/HER2- metastatic breast cancer who were previously treated with endocrine therapy, CDK4/6 inhibitors and two to four lines of chemotherapy for metastatic disease. The primary endpoint is progression-free survival per Response Evaluation Criteria in Solid Tumors (RECIST 1.1) as assessed by blinded independent central review (BICR) for participants treated with Trodelvy compared to those treated with chemotherapy. Secondary endpoints include overall survival, overall response rate, clinical benefit rate and duration of response, as well as assessment of safety and tolerability and quality of life measures. In the study, HER2 negativity was defined per American Society of Clinical Oncology (ASCO) and the College of American Pathologists (CAP) criteria as immunohistochemistry (IHC) score of 0, IHC 1+ or IHC 2+ with a negative in-situ hybridization (ISH) test. More information about TROPiCS-02 is available at https://clinicaltrials.gov/ct2/show/NCT03901339.
About Trodelvy
Trodelvy® (sacituzumab govitecan-hziy) is a first-in-class Trop-2 directed antibody-drug conjugate. Trop-2 is a cell surface antigen highly expressed in multiple tumor types, including in more than 90% of breast and bladder cancers. Trodelvy is intentionally designed with a proprietary hydrolyzable linker attached to SN-38, a topoisomerase I inhibitor payload. This unique combination delivers potent activity to both Trop-2 expressing cells and the microenvironment.
Trodelvy is approved in more than 35 countries, with multiple additional regulatory reviews underway worldwide, for the treatment of adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (TNBC) who have received two or more prior systemic therapies, at least one of them for metastatic disease. Trodelvy is also approved in the U.S. under the accelerated approval pathway for the treatment of adult patients with locally advanced or metastatic urothelial cancer (UC) who have previously received a platinum-containing chemotherapy and either programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor.
Trodelvy is also being developed for potential investigational use in other TNBC and metastatic UC populations, as well as a range of tumor types where Trop-2 is highly expressed, including HR+/HER2- metastatic breast cancer, metastatic non-small cell lung cancer (NSCLC), metastatic small cell lung cancer (SCLC), head and neck cancer, and endometrial cancer.
U.S. Indications for Trodelvy
In the United States, Trodelvy is indicated for the treatment of:
Please see full Prescribing Information , including BOXED WARNING.
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer. Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California.
View source version on businesswire.com: https://www.businesswire.com/news/home/20221010005766/en/
Source: Gilead Sciences, Inc.
Oct. 11, 2022 8:55 AM ET
By: Ravikash, SA News Editor1 Comment
The U.S. Food and Drug Administration (FDA) granted priority review to Gilead Sciences' (NASDAQ:GILD) application seeking expanded approval of Trodelvy to treat certain patients with a type of breast cancer.
https://seekingalpha.com/symbol/GILD
https://www.nasdaq.com/market-activity/stocks/gild/dividend-history
Oct. 12, 2022 7:05 AM ETMerck & Co., Inc. (MRK)
Approval Based on Data from the Phase 3 KEYNOTE-564 Trial
KIRKLAND, QC, Oct. 12, 2022 /CNW/ - Merck (MRK), known as MSD outside the United States and Canada, announced that Health Canada has granted approval for KEYTRUDA® (pembrolizumab), Merck's anti-PD-1 therapy, as monotherapy for the adjuvant treatment of adults with renal cell carcinoma (RCC) at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions. This approval is based on the results from the Phase 3 KEYNOTE-564 trial, which demonstrated a statistically significant improvement in disease-free survival.1

"It is estimated that in 2022 over eight thousand Canadians will be diagnosed with kidney and renal pelvis cancer, of which renal cell carcinoma is the most common type,2" said Christine Collins, Executive Director, Kidney Cancer Canada. "For people who are impacted by RCC, having a new treatment option following surgery, can offer patients and their loved ones support in managing the disease."
RCC is the most common type of kidney cancer, making up for more than 90% of cases of kidney cancer.3 It occurs most often in people over 50, and is more common in men than in women. Some of the risk factors include smoking, obesity, and high blood pressure.4 Given the impact on patients, there is active research in understanding the molecular biology of the disease and improvements in prevention, diagnosis, and availability of treatment options.5
"We are proud to be bringing another innovation in the management of renal cell carcinoma to Canadians, because this is our mission – to help improve the lives of those impacted by cancer by providing additional treatment options," said Marwan Akar, President and Managing Director of Merck Canada. "This is another great example of how scientific research, coupled with a passionate team, can help improve patient journeys and we hope to build on this momentum to continue making progress in the fight against cancer."
About KEYNOTE-564 Trial
Health Canada's approval is based on findings from KEYNOTE-564, a Phase 3 randomized, double-blind clinical trial that enrolled 994 patients.6 The eligibility criteria for this study were adult patients who have undergone nephrectomy and have intermediate-high or high risk of recurrence, or M1 no evidence of disease (M1 NED) renal cell carcinoma. Patients who had received prior systemic therapy for advanced RCC were excluded from the trial
The primary efficacy outcome measure was investigator-assessed disease-free survival (DFS). DFS was defined as the time from randomization to the first recurrence (local, regional, or distant metastasis), or death, whichever occurs first. The results of KEYNOTE-564 demonstrated a statistically significant improvement in DFS for patients randomized to receive KEYTRUDA® monotherapy compared with patients randomized to placebo.7
About KEYTRUDA®
KEYTRUDA® is an anti-PD-1 therapy that works by helping increase the ability of the body's immune system to help detect and fight tumour cells.1 KEYTRUDA® is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumour cells and healthy cells.11
KEYTRUDA® was first approved in Canada in 2015 and currently has indications in several disease areas, including advanced renal cell carcinoma, bladder cancer, non-small cell lung carcinoma, primary mediastinal B-cell lymphoma, classical Hodgkin lymphoma, colorectal cancer, endometrial carcinoma, esophageal cancer, triple-negative breast cancer, melanoma, and head and neck squamous cell carcinoma.12
About Merck
At Merck, known as MSD outside of the United States and Canada, we are unified around our purpose: We use the power of leading-edge science to save and improve lives around the world. For more than 130 years, we have brought hope to humanity through the development of important medicines and vaccines. We aspire to be the premier research-intensive biopharmaceutical company in the world – and today, we are at the forefront of research to deliver innovative health solutions that advance the prevention and treatment of diseases in people and animals. We foster a diverse and inclusive global workforce and operate responsibly every day to enable a safe, sustainable and healthy future for all people and communities. For more information about our operations in Canada, visit www.merck.ca and connect with us on YouTube and Twitter @MerckCanada.
® Merck Sharp & Dohme LLC.Used under license
Copyright © 2022 Merck & Co., Inc., Rahway, NJ, USA and its affiliates. All rights reserved.
SOURCE Merck Canada
Oct. 12, 2022 9:14 AM ET
By: Ravikash, SA News Editor1 Comment
https://seekingalpha.com/symbol/MRK
Positive new data for Roche’s Evrysdi in largest trial ever undertaken in patients with previously-treated spinal muscular atrophy (SMA)October 12, 2022
Basel, 12 October 2022- Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced new two-year data from the JEWELFISH study evaluating Evrysdi® (risdiplam) in people with Type 1, 2 or 3 SMA aged 6 months to 60 years at time of enrolment. Patients had been previously treated with other approved or investigational SMA-targeting therapies, including nusinersen (Spinraza(R)) or onasemnogene abeparvovec (Zolgensma(R)). Data showed Evrysdi improved or maintained motor function and led to rapid increases in SMN protein levels which were sustained after 2-years of treatment. These data will be presented at the 27th World Muscle Society (WMS) congress, 11-15 October 2022.
“The consistent safety profile and exploratory efficacy we have seen in the JEWELFISH study, the largest ever conducted in previously treated patients, reinforces Evrysdi as a meaningful treatment option across SMA populations,” said Dr. Claudia Chiriboga, Professor of Neurology and Pediatrics, Department of Neurology, Columbia University Medical Center, New York, USA. “The findings add to our confidence when making treatment decisions for previously-treated patients in need.”
The JEWELFISH study enrolled the broadest and most diverse patient population ever studied in an SMA trial. Of the 174 people enrolled, 36% (n=63) were adults, 63% (n=105) had a Hammersmith Functional Motor Scale Expanded (HFMSE) score of less than 10 at baseline, meaning their disease was very severe, and 83% (n=139) had scoliosis. Forty-four percent (n=76) of those enrolled had previously been treated with nusinersen (Spinraza), 41% (n=71) with olesoxime*, 8% (n=14) with onasemnogene abeparvovec (Zolgensma) and 7% (n=13) with RG7800*.
People with SMA are unable to produce enough survival motor neuron (SMN) protein, leading to debilitating and potentially fatal muscle weakness. The study showed Evrysdi led to a two-fold increase in median SMN protein levels versus baseline after 4 weeks of treatment in all patient groups, irrespective of previous treatment. The SMN protein levels achieved after 4 weeks of treatment were maintained for over two years.
Observed through exploratory efficacy endpoints, the study also suggests maintenance of motor function was sustained at two-years of treatment as measured by change from baseline in Motor Function Measure 32 (MFM-32), Revised Upper Limb Module (RULM) and HFMSE total scores compared to the natural history of SMA in untreated patients. A recent survey conducted by patient advocacy group SMA Europe, more than 96% of people with SMA viewed disease stabilization as progress in terms of their expectations of treatment.
“These important data demonstrate the safety and efficacy of Evrysdi in a broad, real-world population of people previously treated with an SMA-targeting therapy, ” said Levi Garraway, M.D, Ph.D, Roche’s Chief Medical Officer and Head of Global Products. “Those enrolled in JEWELFISH had very severe disease, with over 80% having scoliosis, so maintaining motor function–especially for a progressive disease–can be potentially life-changing.”
The overall adverse event (AE) and serious adverse event (SAE) profiles observed with Evrysdi treatment in JEWELFISH were reflective of underlying disease. The rate of AEs decreased by more than 50% between the first and second 6-month period, and then remained stable thereafter. The rate of SAEs, including pneumonia, decreased throughout the 24-month period, with a total reduction of more than 50% by the second year. The most common AEs (reported in ≥ 12% of all patients:n=173) were pyrexia (24%), upper respiratory tract infection (21%), headache (18%), nasopharyngitis (16%), diarrhoea (14%), nausea (13%) and cough (12%). The most common SAEs (reported in less than >2% of all patients) were pneumonia (3%) respiratory failure (2%), respiratory distress (2%), lower respiratory tract infection (2%) and upper respiratory tract infection (2%). The most common AEs/SAEs were consistent with those observed in treatment-naïve patients in our other three trials. Low rates of discontinuation from the study were observed, with a 5% rate per year over the 24-month period.
Roche leads the clinical development of Evrysdi as part of a collaboration with the SMA Foundation and PTC Therapeutics.
About Evrysdi® (risdiplam)
Evrysdi is a survival motor neuron 2 (SMN2) splicing modifier designed to treat SMA caused by mutations in chromosome 5q that lead to SMN protein deficiency. Evrysdi is administered daily at home in liquid form by mouth or by feeding tube.
Evrysdi is designed to treat SMA by increasing and sustaining the production of the SMN protein in the central nervous system (CNS) and peripheral tissues. SMN protein is found throughout the body and is critical for maintaining healthy motor neurons and movement.
Evrysdi was granted PRIME designation by the European Medicines Agency (EMA) in 2018 and Orphan Drug Designation by the U.S. Food and Drug Administration in 2017. In 2021 Evrysdi was awarded Drug Discovery of the Year by the British Pharmacological Society as well as the Society for Medicines Research award for Drug Discovery. Evrysdi is currently approved in 91 countries and the dossier is under review in a further 18 countries.
Evrysdi is currently being evaluated in five multicentre trials in people with SMA:
About Roche
Founded in 1896 in Basel, Switzerland, as one of the first industrial manufacturers of branded medicines, Roche has grown into the world’s largest biotechnology company and the global leader in in-vitro diagnostics. The company pursues scientific excellence to discover and develop medicines and diagnostics for improving and saving the lives of people around the world. We are a pioneer in personalised healthcare and want to further transform how healthcare is delivered to have an even greater impact. To provide the best care for each person we partner with many stakeholders and combine our strengths in Diagnostics and Pharma with data insights from the clinical practice.
In recognizing our endeavor to pursue a long-term perspective in all we do, Roche has been named one of the most sustainable companies in the pharmaceuticals industry by the Dow Jones Sustainability Indices for the thirteenth consecutive year. This distinction also reflects our efforts to improve access to healthcare together with local partners in every country we work.
Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan.
For more information, please visit www.roche.com.
Oct. 12, 2022 5:00 AM ET
Roche Holding AG (RHHBY), RHHBF, PTCT
By: Ravikash, SA News Editor

Roche (OTCQX:RHHBY) (OTCQX:RHHBF) said new two year data showed that Evrysdi helped improve or maintain motor function in people with spinal muscular atrophy (SMA).
SMA is a genetic neuromuscular disorder characterized by loss of nerve cells in the spinal cord which leads to progressive muscle weakness and muscle wasting. SMA is caused by low levels of the survival motor neuron (SMN) protein.
https://seekingalpha.com/symbol/RHHBY
https://seekingalpha.com/symbol/RHHBF

October 11, 2022 at 12:59 AM EDT Back
A majority of children on Dupixent (up to 68% on a higher dose) achieved histological disease remission at week 16
First and only Phase 3 trial to show positive results in this patient population; there are currently no approved treatments specifically indicated for children under 12 years of age with EoE
TARRYTOWN, N.Y. and PARIS, Oct. 11, 2022 /PRNewswire/ -- Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) and Sanofi today presented late-breaking positive results from a Phase 3 trial evaluating the investigational use of Dupixent® (dupilumab) in children aged 1 to 11 years with active eosinophilic esophagitis (EoE). The data will be shared at United European Gastroenterology (UEG) Week 2022 and submitted to regulatory authorities around the world, starting with the U.S. Food and Drug Administration (FDA) next year. In May 2022, Dupixent 300 mg weekly was approved by the FDA to treat EoE in people aged 12 years and older, weighing at least 40 kg.
"Eosinophilic esophagitis impacts a child's fundamental ability to eat, which is especially critical in early childhood when healthy weight gain is vital to long-term health and development," said Mirna Chehade, M.D., Mount Sinai Center for Eosinophilic Disorders, Icahn School of Medicine at Mount Sinai, New York and principal investigator of the trial. "These Phase 3 data support the potential of dupilumab to reduce esophageal damage – caused in part by underlying type 2 inflammation – and showed histological disease remission and signs of weight gain impacting the growth percentile for those children on higher dose Dupixent."
As presented at UEG Week 2022, Dupixent led to significant improvements in the primary efficacy measure for higher (n=37) and lower (n=31) dose groups at 16 weeks in the randomized, placebo-controlled Phase 3 trial. Among children treated with Dupixent, 68% of patients on higher dose and 58% of patients on lower dose achieved the primary endpoint of significant histological disease remission, compared to 3% for placebo (both p<0.0001). Children on the higher dose regimen also experienced significant improvements in abnormal endoscopic findings of their esophagus, with a reduction of 3.5 points compared to an increase of 0.3 points for placebo (p<0.0001). Symptomatically, higher dose Dupixent led to a numerical improvement in the proportion of days children experienced disease symptoms from baseline as reported by their caregivers compared to placebo, though not statistically significant. Additionally, a prespecified exploratory analysis was presented which found higher dose Dupixent led to a 3.09 percentile increase in body weight for age percentile from baseline, compared to 0.29 for placebo.
Safety results were generally consistent with the known safety profile of Dupixent in its approved EoE indication for children and adults aged 12 years and older who weigh at least 40 kg. For the 16-week treatment period, the overall rates of adverse events (AEs) were 79% (higher dose n=27/37, lower dose n=26/30) for Dupixent and 91% (n=31/34) for placebo. AEs most commonly observed with Dupixent (≥5%) compared to placebo included COVID-19 (higher dose n=5/37, lower dose n=9/30, placebo n=0/34; all cases were mild or moderate and did not lead to study discontinuation), rash (higher dose n=3/37, lower dose n=3/30, placebo n=2/34), headache (higher dose n=1/37, lower dose n=4/30, placebo n=1/34), viral gastroenteritis (higher dose n=4/37, lower dose n=0/30, placebo n=1/34), diarrhea (higher dose n=2/37, lower dose n=2/30, placebo n=1/34) and nausea (higher dose n=1/37, lower dose n=3/30, placebo n=0/34). Rates of treatment discontinuation due to AEs prior to week 16 were 0% (higher dose n=0/37, lower dose n=0/30) for Dupixent and 6% (n=2/34) for placebo.
The potential use of Dupixent in children with EoE aged 1 to 11 years is currently under clinical development, and the safety and efficacy have not been evaluated by any regulatory authority.
About Eosinophilic Esophagitis
EoE is a chronic inflammatory disease that damages the esophagus and prevents it from working properly. The results seen with Dupixent in adults and children with EoE demonstrate that IL-4 and IL-13 are key and central drivers of the type 2 inflammation underlying this disease.
In children, common symptoms of EoE include acid reflux, vomiting, abdominal discomfort, trouble swallowing, and a failure to thrive. These symptoms can impact growth, weight gain and development, and can cause food-related fear and anxiety which can persist through adulthood. Diet adjustments, which oftentimes include the elimination of many foods, are the standard treatment for EoE, as well as the use of treatments not approved for the disease in children. These include proton pump inhibitors, swallowed topical corticosteroids, or in severe cases, a feeding tube, which may be used to ensure proper caloric intake and weight gain.
Of the approximately 21,000 children under the age of 12 in the U.S. currently being treated for EoE, about 9,000 do not satisfactorily respond to their current treatment regimen and potentially require advanced therapy.
About the Dupixent Pediatric Eosinophilic Esophagitis Trial
The Phase 3, randomized, double-blind, placebo-controlled trial evaluated the efficacy and safety of Dupixent in young children aged 1 to 11 years with EoE, as determined by histological, endoscopic and patient- or caregiver-reported measures. At baseline, 98% of these patients had at least one co-existing type 2 inflammatory disease such as food allergy, allergic rhinitis, asthma and atopic dermatitis.
Patients received Dupixent subcutaneously at either a higher dose or lower dose regimen based on their weight (ranging from ≥5 kg to <60 kg) over a 16-week period, at which point all endpoints were assessed. The dosing frequency ranged between every two weeks and every four weeks, based on weight.
The primary endpoint was histological disease remission, which was defined as peak esophageal intraepithelial eosinophil count of ≤6 eosinophils (eos)/high power field (hpf). Secondary endpoints included abnormal endoscopic findings (EoE Endoscopic Reference Score [EoE-EREFS] on a 0-18 scale), as well as changes in caregiver-reported symptoms (proportion of days with one or more EoE signs [e.g., stomach pain, vomiting, food refusal] by the Pediatric EoE Sign/Symptom Questionnaire-caregiver version [PESQ-C]). The PESQ-C is a novel endpoint developed by Regeneron and Sanofi used for the first time in this trial, designed to assess symptoms in young children through their caregivers (as signs), as children may have difficulty verbalizing their symptoms themselves. An exploratory endpoint assessed change from baseline in body weight for age percentile.
The trial is ongoing with a 36-week extended active treatment period, followed by a 108-week open-label extension to evaluate long-term outcomes. These results, as well as more detailed lower dose results, will be presented or published in the future.
About Dupixent
Dupixent, which was invented using Regeneron's proprietary VelocImmune® technology, is a fully human monoclonal antibody that inhibits the signaling of the interleukin-4 (IL-4) and interleukin-13 (IL-13) pathways and is not an immunosuppressant. The Dupixent development program has shown significant clinical benefit and a decrease in type 2 inflammation in Phase 3 trials, establishing that IL-4 and IL-13 are key and central drivers of the type 2 inflammation that plays a major role in multiple related and often co-morbid diseases. These diseases include approved indications for Dupixent such as asthma, atopic dermatitis, chronic rhinosinusitis with nasal polyposis (CRSwNP), EoE and prurigo nodularis (PN).
Dupixent has received regulatory approvals in one or more countries around the world for use in certain patients with atopic dermatitis, asthma, CRSwNP, EoE or PN in different age populations. Dupixent is currently approved across these indications in the U.S. and for one or more of these indications in more than 60 countries, including in the European Union and Japan. More than 500,000 patients have been treated with Dupixent globally.
About Regeneron's VelocImmune Technology
Regeneron's VelocImmune technology utilizes a proprietary genetically engineered mouse platform endowed with a genetically humanized immune system to produce optimized fully human antibodies. When Regeneron's co-Founder, President and Chief Scientific Officer George D. Yancopoulos was a graduate student with his mentor Frederick W. Alt in 1985, they were the first to envision making such a genetically humanized mouse, and Regeneron has spent decades inventing and developing VelocImmune and related VelociSuite® technologies. Dr. Yancopoulos and his team have used VelocImmune technology to create approximately one in five of all original, FDA-approved or authorized fully human monoclonal antibodies. This includes REGEN-COV® (casirivimab and imdevimab), Dupixent, Libtayo® (cemiplimab-rwlc), Praluent® (alirocumab), Kevzara® (sarilumab), Evkeeza® (evinacumab-dgnb) and Inmazeb™ (atoltivimab, maftivimab and odesivimab-ebgn).
U.S. INDICATIONS
DUPIXENT is a prescription medicine used:
Please see accompanying full Prescribing Information including Patient Information.
About Regeneron
Regeneron is a leading biotechnology company that invents, develops and commercializes life-transforming medicines for people with serious diseases. Founded and led for nearly 35 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to nine FDA-approved treatments and numerous product candidates in development, almost all of which were homegrown in our laboratories. Our medicines and pipeline are designed to help patients with eye diseases, allergic and inflammatory diseases, cancer, cardiovascular and metabolic diseases, pain, hematologic conditions, infectious diseases and rare diseases.
Regeneron is accelerating and improving the traditional drug development process through our proprietary VelociSuite® technologies, such as VelocImmune®, which uses unique genetically humanized mice to produce optimized fully human antibodies and bispecific antibodies, and through ambitious research initiatives such as the Regeneron Genetics Center®, which is conducting one of the largest genetics sequencing efforts in the world.
For more information, please visit www.Regeneron.com or follow @Regeneron on Twitter.
About Sanofi
We are an innovative global healthcare company, driven by one purpose: we chase the miracles of science to improve people's lives. Our team, across some 100 countries, is dedicated to transforming the practice of medicine by working to turn the impossible into the possible. We provide potentially life-changing treatment options and life-saving vaccine protection to millions of people globally, while putting sustainability and social responsibility at the center of our ambitions.
Sanofi is listed on EURONEXT: SAN and NASDAQ: SNY.
Cision View original content:https://www.prnewswire.com/news-releases/dupixent-dupilumab-late-breaking-phase-3-data-presented-at-ueg-week-2022-showed-significant-histological-remission-of-eosinophilic-esophagitis-eoe-in-children-1-to-11-years-old-301645381.html
SOURCE Regeneron Pharmaceuticals, Inc.
Oct. 11, 2022 5:34 AM ET
By: Ravikash, SA News Editor
Sanofi (NASDAQ:SNY) and Regeneron Pharmaceuticals (NASDAQ:REGN) said their medicine Dupixent showed positive results in a phase 3 trial to treat children aged one to 11 years with active eosinophilic esophagitis (EoE).
https://seekingalpha.com/symbol/SNY
https://seekingalpha.com/symbol/REGN
Oct 06, 2022
- OXLUMO Now Indicated for the Treatment of Primary Hyperoxaluria Type 1 (PH1) to Lower Urinary and Plasma Oxalate Levels in Pediatric and Adult Patients -
- Approval is Based on Positive Efficacy and Safety Results of the ILLUMINATE-C Phase 3 Study of OXLUMO in PH1 Patients with Severe Renal Impairment, Including Those on Hemodialysis -
CAMBRIDGE, Mass.--(BUSINESS WIRE)--Oct. 6, 2022-- Alnylam Pharmaceuticals, Inc. (Nasdaq: ALNY), the leading RNAi therapeutics company, announced today that the U.S. Food and Drug Administration (FDA) approved a label expansion for OXLUMO® (lumasiran), an RNAi therapeutic administered via subcutaneous injection, now indicated for the treatment of primary hyperoxaluria type 1 (PH1) to lower urinary oxalate (UOx) and plasma oxalate (POx) levels in pediatric and adult patients. The approval is based on positive efficacy and safety results of the ILLUMINATE-C Phase 3 study of OXLUMO in patients with severe renal impairment, including those on hemodialysis.
PH1 is an ultra-rare genetic disease characterized by oxalate overproduction in the liver. The excess production of oxalate results in the deposition of calcium oxalate crystals in the kidneys and urinary tract and can lead to the formation of painful and recurrent kidney stones and nephrocalcinosis, which can progress to kidney failure. PH1 can also lead to oxalate deposition in multiple organs beyond the kidney, a condition known as systemic oxalosis.
The FDA approval is based on positive six-month results from the ILLUMINATE-C Phase 3 study, in which OXLUMO treatment resulted in substantial reductions in POx and demonstrated an encouraging safety and tolerability profile in patients with compromised renal function, including those with kidney failure and undergoing treatment by hemodialysis. Elevated POx is directly related to the pathophysiology of oxalosis and results in systemic deposition of oxalate in extra-renal tissues, potentially leading to bone fractures, cardiomyopathy, impaired erythropoiesis, vision loss, skin ulcers and other serious manifestations.1
The supplemental New Drug Application also included results from the open-label extensions of the ILLUMINATE-A and ILLUMINATE-B Phase 3 studies of pediatric and adult patients with PH1. The label has correspondingly been updated to highlight the maintenance of sustained reductions in UOx through Month 24 and Month 12, respectively.
“Today’s label expansion exemplifies Alnylam’s commitment to advancing research and innovation in support of the PH1 community. We also believe this expansion will strengthen prescribers’ confidence in OXLUMO for patients,” said Jorge Capapey, Vice President, Global Rare Disease Lead at Alnylam Pharmaceuticals. “Through the findings of the ILLUMINATE clinical development program, I am thrilled to see the potential benefit of OXLUMO, which remains the first and only FDA-approved PH1 treatment option, now be available for a broad range of people living with the ultra-rare disease, including those advanced PH1 patients undergoing hemodialysis. We extend our sincere gratitude to the patients, families and investigators involved in the research supporting this approval, without whom it would not be possible to bring this therapy to those living with PH1.”
The label expansion for OXLUMO is based on positive six-month results from ILLUMINATE-C, where treatment with lumasiran resulted in a substantial reduction in POx from baseline to Month Six in both dialysis-independent and -dependent patients. Patients in both cohorts had a reduction in POx as early as Month One. In Cohort A (N=6, dialysis-independent), treatment with OXLUMO led to a 33% least squares (LS) mean reduction in POx from baseline to Month Six (LS Mean; 95% CI: -82%, 15%), and in Cohort B (N=15, hemodialysis-dependent), treatment with OXLUMO led to a 42% LS mean reduction in POx from baseline to Month Six (LS Mean; 95% CI: -51%, 34%). OXLUMO demonstrated an encouraging safety and tolerability profile with no deaths or drug-related serious adverse events (SAEs) among enrolled patients. There were two treatment discontinuations due to adverse events (AEs) in the extension period of the study, neither of which was drug related. The most common AE related to lumasiran was injection-site reaction (ISR) reported in five of 21 patients (24%) which were all mild and transient.
“The significance of the label expansion of OXLUMO cannot be overstated, as this milestone provides crucial reassurance among a patient population with the highest unmet need, as well as their caregivers and loved ones, that OXLUMO is available in the US for patients living with PH1, including those with advanced disease,” said Kim Hollander, Executive Director of the Oxalosis and Hyperoxaluria Foundation. “We are grateful to Alnylam for their continued commitment to the PH1 community and for ensuring that all patients — no matter how severe their disease — have the same opportunity to potentially benefit from OXLUMO treatment.”
In November of 2020, OXLUMO was approved by the FDA for the treatment of PH1 to lower UOx levels in pediatric and adult patients. It was also approved by the European Medicines Agency (EMA) for the treatment of PH1 in all age groups. The Committee for Medicinal Products for Human Use (CHMP) of the EMA delivered a positive opinion recommending variation to the marketing authorization of OXLUMO based on ILLUMINATE-C data from patients with advanced PH1 in September 2022. Lumasiran is also being evaluated in a global Phase 2 study as an investigational treatment in patients with recurrent kidney stone disease and elevated UOx levels.
Visit OXLUMO.com for more information, including full Prescribing Information.
OXLUMO® (lumasiran) INDICATION AND IMPORTANT SAFETY INFORMATION
Indication
OXLUMO® (lumasiran) is indicated for the treatment of primary hyperoxaluria type 1 (PH1) to lower urinary and plasma oxalate levels in children and adults.
About OXLUMO (lumasiran)
OXLUMO® (lumasiran) is an RNAi therapeutic targeting hydroxyacid oxidase 1 (HAO1). HAO1 encodes glycolate oxidase (GO). Thus, by silencing HAO1 and depleting the GO enzyme, OXLUMO inhibits production of oxalate – the metabolite that directly contributes to the pathophysiology of PH1. OXLUMO utilizes Alnylam’s Enhanced Stabilization Chemistry (ESC)-GalNAc-conjugate technology, which enables subcutaneous dosing with increased potency and durability and a wide therapeutic index. OXLUMO has received regulatory approvals from the U.S. Food and Drug Administration (FDA) for the treatment of primary hyperoxaluria type 1 (PH1) to lower urinary and plasma oxalate levels in pediatric and adult patients and from the European Medicines Agency (EMA) for the treatment of PH1 in all age groups.
In the pivotal ILLUMINATE-A study, OXLUMO was shown to significantly reduce levels of urinary oxalate relative to placebo, with the majority of patients reaching normal or near-normal levels. In the ILLUMINATE-B pediatric Phase 3 study, OXLUMO demonstrated an efficacy and safety profile consistent to that observed in ILLUMINATE-A. In the ILLUMINATE-C study, OXLUMO resulted in substantial reductions in plasma oxalate in patients with advanced PH1. Across all three studies, injection site reactions (ISRs) were the most common drug-related adverse reaction.
OXLUMO is administered via subcutaneous injection once monthly for three months, then once quarterly beginning one month after the last loading dose at a dose based on actual body weight. For patients who weigh less than 10 kg, ongoing dosing remains monthly.
OXLUMO should be administered by a healthcare professional. For more information about OXLUMO, visit OXLUMO.com.
About the ILLUMINATE-C Phase 3 Study
ILLUMINATE-C (NCT04152200) is a single arm, open-label, multinational Phase 3 study with a 6-month primary analysis period and an extended 54-month dosing period to evaluate the safety and efficacy of lumasiran in PH1 patients of all ages with severe renal impairment (eGFR ≤ 45 mL/min/1.73m2 or elevated serum creatinine for patients <12 months of age). The study is being conducted at 13 study sites across 10 countries around the world. Cohort A enrolled six patients with advanced PH1 who do not yet require dialysis, and Cohort B enrolled 15 patients who are hemodialysis-dependent. The dosing regimen is based on weight with three monthly starting doses followed by ongoing monthly or quarterly doses. The primary efficacy endpoint for Cohort A was the percent change in plasma oxalate from baseline to month six, and the primary endpoint for Cohort B was the percent change in pre-dialysis plasma oxalate from baseline to month six. Key secondary endpoints are designed to evaluate additional measures of plasma oxalate and changes in urinary oxalate. Kidney function, frequency and mode of dialysis, frequency of kidney stone events and measures of systemic oxalosis, including clinical manifestations, will also be evaluated in the extension period of the study.
About Alnylam Pharmaceuticals
Alnylam (Nasdaq: ALNY) has led the translation of RNA interference (RNAi) into a whole new class of innovative medicines with the potential to transform the lives of people afflicted with rare and prevalent diseases with unmet need. Based on Nobel Prize-winning science, RNAi therapeutics represent a powerful, clinically validated approach yielding transformative medicines. Since its founding 20 years ago, Alnylam has led the RNAi Revolution and continues to deliver on a bold vision to turn scientific possibility into reality. Alnylam’s commercial RNAi therapeutic products are ONPATTRO® (patisiran), GIVLAARI® (givosiran), OXLUMO® (lumasiran), AMVUTTRA® (vutrisiran), and Leqvio® (inclisiran) being developed and commercialized by Alnylam’s partner, Novartis. Alnylam has a deep pipeline of investigational medicines, including six product candidates that are in late-stage development. Alnylam is executing on its “Alnylam P5x25” strategy to deliver transformative medicines in both rare and common diseases benefiting patients around the world through sustainable innovation and exceptional financial performance, resulting in a leading biotech profile. Alnylam is headquartered in Cambridge, MA. For more information about our people, science and pipeline, please visit www.alnylam.com and engage with us on Twitter at @Alnylam, on LinkedIn, or on Instagram.
View source version on businesswire.com: https://www.businesswire.com/news/home/20221006006075/en/
Source: Alnylam Pharmaceuticals, Inc.
Oct. 07, 2022 6:17 AM ET
Alnylam Pharmaceuticals, Inc. (ALNY)
By: Ravikash, SA News Editor
Alnylam Pharmaceuticals, Inc.
55.2 percent of induction responder patients treated with STELARA at the start of maintenance were in symptomatic remission approximately four years later (at week 200), 96.4 percent without corticosteroids
Overall, 79.1 percent of patients treated with STELARA in the long-term extension (LTE) who were receiving corticosteroids at maintenance baseline were no longer receiving corticosteroids by week 200
SPRING HOUSE, PENNSYLVANIA, October 10, 2022 – The Janssen Pharmaceutical Companies of Johnson & Johnson today announced final data from the long-term extension (LTE) of the Phase 3 UNIFI study demonstrating efficacy and safety of STELARA® (ustekinumab) through four years of treatment in adult patients with moderately to severely active ulcerative colitis (UC).1 Among all patients who had achieved clinical responsea with STELARA during induction, 64.9 percent were in symptomatic remissionb after 44 weeks of maintenance. At week 200 (four years), this proportion of patients was 55.2 percent; the majority (96.4 percent [185/192]) were not receiving corticosteroids.1 Among the 174 patients receiving STELARA as their first biologic for UC (biologic-naïve), 71.8 percent (125) of these patients were in symptomatic remissionb after 44 weeks of maintenance, and 67.2 percent (117) were in remission at week 200. A separate presentation of UNIFI LTE study data showed the majority (79.1 percent) of STELARA randomized patients who received corticosteroids at maintenance baseline and were treated with STELARA in the LTE were able to eliminate the use of corticosteroids by week 200.2 These data are being presented at the United European Gastroenterology (UEG) Week 2022 congress taking place in-person in Vienna and virtually from October 8-11.1,2
“The final LTE results of the UNIFI study demonstrated that STELARA can be an effective long-term treatment option for patients living with moderately to severely active ulcerative colitis, including in patients who are biologic-naive,” said UNIFI presenting study author Waqqas Afif, M.D., Associate Professor, Department of Medicine, Division of Experimental Medicine and Division of Gastroenterology at McGill University Health Centre in Montreal, Canada.c “Importantly, the vast majority of patients who achieved remission in the study were able to eliminate the use of steroids, which can cause significant side effects and are not a long-term treatment solution for the disease.”
UNIFI LTE Week 200 Efficacy and Safety Data (Poster PO396): 1
At week 200, among all 348 patients who had achieved clinical response to treatment with intravenous (IV) STELARA and were randomized to STELARA 90 mg every eight weeks (q8w) or every 12 weeks (q12w) at baseline of the maintenance study:
Safety was evaluated for all patients (n=588) who were treated in the LTE, including randomized and non-randomized populations.1
UNIFI LTE Week 200 Corticosteroid-Sparing Data (Poster PO395):2
Results from a separate presentation on corticosteroid-sparing effects within the UNIFI LTE study show 79.1 percent of STELARA randomized patients who received corticosteroids at maintenance baseline and were treated with STELARA in the LTE were no longer receiving corticosteroids at week 200.g Among patients who were randomized to STELARA at maintenance baseline and treated in the LTE phase of the study:
“STELARA is a well-established therapy, and the final results of the UNIFI LTE study demonstrate sustained, long-term clinical benefit in moderately to severely active ulcerative colitis,” said Jan Wehkamp, M.D., Vice President, Gastroenterology Disease Area Leader, Janssen Research & Development, LLC. “We hope this is welcome news for patients who are seeking long-term treatment options that may provide enduring relief from the debilitating symptoms of the disease.”
About UNIFI (NCT02407236)5
UNIFI was a Phase 3 protocol designed to evaluate the safety and efficacy of STELARA induction and maintenance dosing for the treatment of moderately to severely active ulcerative colitis in adults who demonstrated an inadequate response to or were unable to tolerate conventional (i.e., corticosteroids, immunomodulators) or biologic (i.e., one or more TNF blockers or vedolizumab) therapies. Both the induction and maintenance studies were randomized, double-blind, placebo-controlled, parallel group, multi-center studies.
The induction study was of at least 8 weeks duration for each participant. Participants achieving clinical responsea in the induction study were eligible for the maintenance study. The maintenance study was 44 weeks in duration. The primary endpoint of the induction study was clinical remission at week 8, and the primary endpoint for the maintenance study was clinical remission at week 44 among responders to a single intravenous (IV) STELARA infusion. 523 IV STELARA induction responders were randomized to subcutaneous (SC) maintenance therapy (175 SC placebo; 172 STELARA 90 mg q12w; 176 STELARA 90 mg q8w). 284 STELARA patients who completed week 44 entered the LTE. Randomized placebo patients were discontinued after week 44 unblinding. The long-term extension of UNIFI followed eligible participants for an additional three years upon completion of the maintenance study.
Starting at week 56, randomized patients with UC worsening could adjust to q8w dosing. Efficacy was evaluated in randomized patients using symptomatic remission.b Safety was evaluated for all 588 patients who were treated in the LTE, including the randomized and nonrandomized populations. The nonrandomized population included STELARA induction non-responders at week 8 who received SC STELARA and responded eight weeks later, and responders to placebo induction.
About STELARA® (ustekinumab)3
STELARA® (ustekinumab), a human interleukin (IL)-12 and IL-23 antagonist, is approved in the United States for the treatment of: 1) adults and children six years and older with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy; 2) adults and children six years and older with active psoriatic arthritis; 3) adult patients (18 years and older) with moderately to severely active Crohn’s disease; 4) adult patients (18 years and older) with moderately to severely active ulcerative colitis.
The Janssen Pharmaceutical Companies of Johnson & Johnson maintain exclusive worldwide marketing rights to STELARA.
https://www.stelarainfo.com/ulcerative-colitis
Please click to read the full Prescribing Information and Medication Guide for STELARA® and discuss any questions you have with your doctor.
About the Janssen Pharmaceutical Companies of Johnson & Johnson
At Janssen, we’re creating a future where disease is a thing of the past. We’re the Pharmaceutical Companies of Johnson & Johnson, working tirelessly to make that future a reality for patients everywhere by fighting sickness with science, improving access with ingenuity, and healing hopelessness with heart. We focus on areas of medicine where we can make the biggest difference: Cardiovascular, Metabolism & Retina; Immunology; Infectious Diseases & Vaccines; Neuroscience; Oncology; and Pulmonary Hypertension.
Learn more at www.janssen.com.
Follow us at www.twitter.com/JanssenGlobal.
Janssen Research & Development, LLC is a part of the Janssen Pharmaceutical Companies of Johnson & Johnson.
Oct. 10, 2022 11:21 AM ET
By: Dulan Lokuwithana, SA News Editor
The Janssen unit of Johnson & Johnson (NYSE:JNJ) announced on Monday that more than half of patients who responded to its ulcerative colitis therapy Stelara (ustekinumab) in a Phase 3 trial remained in clinical remission through four years of treatment.
https://seekingalpha.com/symbol/JNJ
https://www.stelarainfo.com/ulcerative-colitis
The China NMPA Approved CYRAMZA® (ramucirumab) for the Treatment in Patients with Hepatocellular Carcinoma
Published on: Oct 10th, 2022Back
ROCKVILLE,M.D. and SUZHOU, China, Oct 10th, 2022 — Innovent Biologics, Inc. (“Innovent”, HKEX: 01801), a world-class biopharmaceutical company that develops, manufactures and commercializes high-quality medicines for the treatment of cancer, metabolic, autoimmune, ophthalmology and other major diseases, is pleased to see that the National Medical Products Administration (NMPA) of China has approved the supplemental New Drug Application (sNDA) for CYRAMZA® (ramucirumab) in patients with hepatocellular carcinoma (HCC, also known as liver cancer), who have an alpha fetoprotein of ≥400 ng/mL and have been treated with sorafenib. In March 2022, CYRAMZA (ramucirumab) was approved by the NMPA in combination with paclitaxel for second-line treatment in patients with advanced or metastatic, gastric or gastro-esophageal junction (GEJ) adenocarcinoma, making it the first and only targeted drug approved for the second-line treatment of advanced or metastatic, gastric or gastro-esophageal junction (GEJ) adenocarcinoma in China.
CYRAMZA (ramucirumab) has been discovered and developed by Lilly. In March 2022, Innovent and Lilly expanded their strategic partnership in oncology, which includes an agreement for Innovent to obtain the sole commercialization rights of CYRAMZA (ramucirumab) once approved in China, which positions Innovent to be fully responsible for the pricing, importation, marketing, distribution and detailing of this product. With this further expanded oncology product portfolio, Innovent intends to use its experienced oncology commercial team to leverage its broad commercial coverage in hospitals and pharmacies at various tiers and to provide integrated patient solutions with strong synergies to cancer patients in China.
This new approval was based on the results of the REACH-2 study[1],[2], a global randomized, double-blind, placebo-controlled Phase 3 clinical trial. The REACH-2 study is the first Phase 3 clinical trial in HCC to obtain positive results in a biomarker-enriched population known for poor prognosis. On the primary endpoint of overall survival (OS), treatment with CYRAMZA significantly improved the OS of patients compared to placebo (HR: 0.71; 95% CI: 0.53-0.95; P=0.020). The median OS was 8.5 months with CYRAMZA, compared to 7.3 months with placebo. On the secondary endpoint of progression-free survival (PFS), median PFS was significantly improved with CYRAMZA (2.8 months vs. 1.6 months for placebo (HR: 0.45; 95% CI: 0.34-0.60; P<0.0001)). Objective response rate (ORR) was numerically higher with CYRAMZA compared to placebo (4.6% vs. 1.1%). Disease control rate (DCR) was higher with CYRAMZA than with placebo (59.9% vs. 38.9%). CYRAMZA was well-tolerated in Chinese patients and the overall patient population. The safety and efficacy profile of CYRAMZA in the Chinese population was consistent with that observed in previously reported global studies2.
Professor Shukui Qin, the principal investigator of REACH-2 in China and the Director of the Cancer Center of Nanjing Jinling Hospital stated, "Primary Liver Cancer (PLC) is the fourth most common cancer in terms of prevalence and the second leading cause of cancer-related mortality in China with only a 12.1% five-year survival rate. HCC is the main type of PLC and most HCC patients with AFP ≥ 400ng/ml experience greater disease progression on or after first-line treatment[3]. New treatment options are urgently needed to improve outcomes in these patients[4]. Globally, CYRAMZA is the first approved novel medicine specifically evaluated in the biomarker-enriched population of HCC patients with AFP ≥ 400ng/ml; the study results in Chinese patients demonstrated a consistent efficacy and safety profile with the global population. I believe the approval will ignite a positive impact on the clinical practice of liver cancer in China, bringing a new and efficacious treatment option for Chinese patients with advanced HCC.”
Dr. Yongjun Liu, President of Innovent, stated, "Hepatocellular carcinoma ranks fifth among the number of new cases in China, with approximately 410,000 new cases yearly and the number of deaths each year are nearly the same3. Most patients will experience disease progression after current first-line therapy and are left with limited treatment options. We are excited about the approval of CYRAMZA, the first innovative drug proven to have clinical benefits for a biomarker-enriched population of patients with HCC. This differentiated product will potentially be an exciting treatment option and bring new hope to patients in China with advanced HCC. The successive approvals of CYRAMZA in second-line GC/GEJ and HCC this year enable us to provide integrated patient solutions with strong portfolio synergies while enhancing our leading franchise in these two large cancer indications. Innovent is fully committed to making these new treatment options available to benefit more cancer patients in China as soon as possible.”
About CYRAMZA® (ramucirumab)
CYRAMZA® is an antiangiogenic therapy. It is a vascular endothelial growth factor (VEGF) Receptor 2 antagonist that binds specifically to VEGFR-2, thereby blocking the binding of the receptor ligands (VEGF-A, VEGF-C, and VEGF-D) – which may slow tumor growth.[6] In recent years, studies have shown that the VEGF pathway is an important signaling pathway involved in tumor angiogenesis, and it is also the main target pathway in targeted therapy of liver cancer7. From the existing research results, the single drug use of compounds targeting the VEGF pathway can bring survival benefits to patients and is a very promising treatment method in the treatment of liver cancer.[7]
In March 2022, CYRAMZA® (ramucirumab) in combination with paclitaxel was approved by the National Medical Products Administration (NMPA) for second-line treatment in patients with advanced or metastatic, gastric or gastro-esophageal junction (GEJ) adenocarcinoma.
In October 2022, CYRAMZA® (ramucirumab) was approved by the NMPA for patients with Hepatocellular Carcinoma (HCC) who have an alpha fetoprotein (AFP) of ≥400 ng/mL and have been treated with sorafenib.
About Innovent’s Strategic Cooperation with Eli Lilly and Company
Innovent entered into a strategic collaboration with Lilly focused on biological medicine in March 2015 – a groundbreaking partnership between a Chinese pharmaceutical company and a multinational pharmaceutical company. Under the agreement, Innovent and Lilly will co-develop and commercialize oncology medicines, including TYVYT® (sintilimab injection) in China. In October 2015, the two companies announced the extension of their existing collaboration to include co-development of three additional oncology antibodies targeting oncology indications. In August 2019, Innovent further entered into a licensing agreement with Lilly to develop and commercialize a potentially global best-in-class diabetes medicine in China. This collaboration with Lilly indicates that Innovent has established a comprehensive level of cooperation between China’s innovative pharmaceuticals sector and the international pharmaceuticals sector in fields such as R&D, CMC, clinical development and commercialization. In August 2020,Lilly and Innovent announced a global expansion of their strategic alliance for sintilimab, whereby Lilly obtained an exclusive license for sintilimab for geographies outside of China and plans to pursue registration of sintilimab in the U.S. and other geographies outside of China. In March 2022, Lilly and Innovent entered into a fifth agreement to expand their strategic partnership in oncology, in which Innovent obtained the sole commercialization rights to import, market, promote, distribute and detail CYRAMZA® (ramucirumab) and selpercatinib once approved in Mainland China, and a right of first negotiation for potential future commercialization of pirtobrutinib in Mainland China.
About Innovent
Inspired by the spirit of "Start with Integrity, Succeed through Action,” Innovent’s mission is to develop, manufacture and commercialize high-quality biopharmaceutical products that are affordable to ordinary people. Established in 2011, Innovent is committed to developing, manufacturing and commercializing high-quality innovative medicines for the treatment of cancer, autoimmune, metabolic, ophthalmology and other major diseases. On October 31, 2018, Innovent was listed on the Main Board of the Stock Exchange of Hong Kong Limited with the stock code: 01801.HK.
Since its inception, Innovent has developed a fully integrated multi-functional platform which includes R&D, CMC (Chemistry, Manufacturing, and Controls), clinical development and commercialization capabilities. Leveraging the platform, the company has built a robust pipeline of 34 valuable assets in the fields of cancer, metabolic, autoimmune disease and other major therapeutic areas, with 8 products approved for marketing in China – TYVYT® (sintilimab injection), BYVASDA® (bevacizumab biosimilar injection), SULINNO® (adalimumab biosimilar injection), HALPRYZA® (rituximab biosimilar injection), Pemazyre® (pemigatinib oral inhibitor), olverembatinib (BCR ABL TKI), CYRAMZA® (ramucirumab) and selpercatinib, 2 assets under NMPA NDA review, 4 assets in Phase 3 or pivotal clinical trials, and an additional 20 molecules in clinical studies.
Innovent has built an international team with advanced talent in high-end biological drug development and commercialization, including many global experts. The company has also entered into strategic collaborations with Eli Lilly and Company, Sanofi, Adimab, Incyte, MD Anderson Cancer Center and other international partners. Innovent strives to work with many collaborators to help advance China’s biopharmaceutical industry, improve drug availability and enhance the quality of the patients’ lives. For more information, please visit: www.innoventbio.com. and www.linkedin.com/company/innovent-biologics/.
Oct. 10, 2022 4:34 AM ET
Innovent Biologics, Inc. (IVBIY), IVBXF, LLY
By: Ravikash, SA News Editor1 Comment
October 6, 2022Download PDF
The Fast Track designation accelerates tirzepatide's path to U.S. FDA submission for the treatment of adults with obesity, or overweight with weight-related comorbidities
INDIANAPOLIS, Oct. 6, 2022 /PRNewswire/ -- Eli Lilly and Company (NYSE: LLY) announced today that the U.S. Food and Drug Administration (FDA) has granted Fast Track designation for the investigation of tirzepatide for the treatment of adults with obesity, or overweight with weight-related comorbidities. The FDA grants Fast Track designation to facilitate the development and expedite the review of medicines to treat serious conditions and fill an unmet medical need. Fast Track designation is intended to bring promising medicines to patients sooner.
Based on discussions with the FDA, Lilly plans to initiate a rolling submission of a new drug application (NDA) for tirzepatide in adults with obesity or overweight this year, which when complete, will be based primarily on results from two Phase 3 clinical trials: SURMOUNT-1, which is complete, and SURMOUNT-2, which is expected to complete by the end of April 2023. The rolling submission allows Lilly to submit completed sections of an application for review by FDA, rather than wait until all sections are completed.
Assuming positive SURMOUNT-2 results, Lilly aims to complete the submission shortly after SURMOUNT-2 data is available. The Fast Track designation, along with a rolling submission, accelerates tirzepatide's path to FDA submission.
"We are pleased with the FDA's decision to grant Fast Track designation for tirzepatide, and we look forward to completing our rolling submission next year," said Mike Mason, president, Lilly Diabetes. "Obesity is a chronic disease that impacts the health of nearly 100 million Americans and is a significant driver of healthcare costs. While diet and exercise are important steps, most patients don't achieve their desired treatment goals with only diet and exercise. We are dedicated to helping people living with obesity through our research and development of innovative treatments like tirzepatide, which produced significant weight reductions in patients taking tirzepatide for type 2 diabetes in SURPASS. Tirzepatide also helped nearly two-thirds of participants on the highest dose reduce their body weight by at least 20 percent in SURMOUNT-1."
About SURMOUNT-1, SURMOUNT-2 and the SURMOUNT clinical trial program1,2
SURMOUNT-1 (NCT04184622) is a multi-center, randomized, double-blind, parallel, placebo-controlled trial, which compared the efficacy and safety of tirzepatide 5 mg, 10 mg and 15 mg to placebo as an adjunct to a reduced-calorie diet and increased physical activity in adults without type 2 diabetes who have obesity, or overweight with at least one of the following comorbidities: hypertension, dyslipidemia, obstructive sleep apnea or cardiovascular disease. The trial randomized 2,539 participants across the U.S., Argentina, Brazil, China, India, Japan, Mexico, Russia and Taiwan in a 1:1:1:1 ratio to receive either tirzepatide 5 mg, 10 mg or 15 mg or placebo. The co-primary objectives of the study were to demonstrate that tirzepatide 10 mg and/or 15 mg was superior in percentage of body weight reductions from baseline and percentage of participants achieving ≥5% body weight reduction at 72 weeks compared to placebo. Participants who had pre-diabetes at study commencement will remain enrolled in SURMOUNT-1 for an additional 104 weeks of treatment following the initial 72-week completion date to evaluate the impact on body weight and potential differences in progression to type 2 diabetes at three years of treatment with tirzepatide compared to placebo.
All participants in the tirzepatide treatment arms started the study at a dose of tirzepatide 2.5 mg once-weekly and then increased the dose in a step-wise approach at four-week intervals to their final randomized maintenance dose of 5 mg (via a 2.5 mg step), 10 mg (via steps at 2.5 mg, 5 mg and 7.5 mg) or 15 mg (via steps at 2.5 mg, 5 mg, 7.5 mg, 10 mg and 12.5 mg).
SURMOUNT-2 (NCT04657003) is a multi-center, randomized, double-blind, parallel, placebo-controlled trial comparing the efficacy and safety of tirzepatide 10 mg and 15 mg to placebo as an adjunct to a reduced-calorie diet and increased physical activity in adults with type 2 diabetes who have obesity or overweight. The trial randomized 938 participants across the U.S., Argentina, Brazil, India, Japan, Puerto Rico, Russia and Taiwan in a 1:1:1 ratio to receive either tirzepatide 10 mg or 15 mg or placebo. The co-primary objectives of the study are to demonstrate that tirzepatide 10 mg and/or 15 mg is superior in percentage of body weight change from baseline and percentage of participants achieving ≥5% body weight reduction at 72 weeks compared to placebo.
The SURMOUNT Phase 3 global clinical development program for tirzepatide began in late 2019 and has enrolled more than 5,000 people with obesity or overweight across six clinical trials, four of which are global studies. Results from SURMOUNT-2, -3 and -4 are anticipated in 2023.
About tirzepatide
Tirzepatide is a once-weekly GIP (glucose-dependent insulinotropic polypeptide) receptor and GLP-1 (glucagon-like peptide-1) receptor agonist. Tirzepatide is a single novel molecule that activates the body's receptors for GIP and GLP-1, which are natural incretin hormones. GIP is a hormone that may complement the effects of GLP-1 receptor agonism. GIP has been shown to decrease food intake while blunting the metabolic adaptive responses that usually occur with calorie restriction resulting in weight reductions, and when combined with GLP-1 receptor agonism, may result in greater effects on markers of metabolic dysregulation such as body weight, glucose and lipids. Tirzepatide is in Phase 3 development for adults with obesity, or overweight with weight-related comorbidity. It is also being studied as a potential treatment for heart failure with preserved ejection fraction (HFpEF), obstructive sleep apnea (OSA), and non-alcoholic steatohepatitis (NASH). Studies of tirzepatide in chronic kidney disease and in morbidity/mortality in obesity are planned as well.
Tirzepatide was approved as Mounjaro® (tirzepatide) by the FDA on May 13, 2022. Mounjaro is a glucose-dependent insulinotropic polypeptide (GIP) receptor and glucagon-like peptide-1 (GLP-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
INDICATION AND SAFETY SUMMARY WITH WARNINGS
Mounjaro® (mown-JAHR-OH) is an injectable medicine for adults with type 2 diabetes used along with diet and exercise to improve blood sugar (glucose).
About Lilly
Lilly unites caring with discovery to create medicines that make life better for people around the world. We've been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world's most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer's disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we're motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn. P-LLY
![]()
View original content to download multimedia:https://www.prnewswire.com/news-releases/lilly-receives-us-fda-fast-track-designation-for-tirzepatide-for-the-treatment-of-adults-with-obesity-or-overweight-with-weight-related-comorbidities-301642476.html
SOURCE Eli Lilly and Company
Oct. 06, 2022 7:10 AM ET
By: Ravikash, SA News Editor1 Comment

05 October 2022
Issued: London UK For media and investors only
GSK plc (LSE/NYSE: GSK) today announced positive headline results of the PERLA phase II trial, which met its primary endpoint of objective response rate (ORR) by Response Evaluation Criteria in Solid Tumours (RECIST) criteria as determined by blinded independent central review. The trial evaluated dostarlimab in combination with chemotherapy versus pembrolizumab in combination with chemotherapy in first-line patients with metastatic non-squamous non-small cell lung cancer (NSCLC). The PERLA phase II trial is a randomised, double-blind trial of 243 patients and is the largest global head-to-head trial of programmed death receptor-1 (PD-1) inhibitors in this population.The trial was not designed to demonstrate superiority.
Full results from the PERLA phase II trial, including the primary endpoint of ORR and the key secondary endpoint of progression-free survival, with results by programmed death ligand-1 (PD-L1) expression subgroups, will be presented at an upcoming scientific meeting.
The safety and tolerability profile of dostarlimab in the PERLA phase II trial was consistent with previous clinical trials of similar regimens. The most common treatment-emergent adverse reactions were anaemia, asthenia, nausea, constipation, cough, dyspnoea, vomiting, decreased appetite, and neutropenia.
In addition, GSK is also advancing both arms of the COSTAR Lung trial into phase III. The decision follows the recommendation of the Independent Data Monitoring Committee, given that the trial met its pre-specified expansion criteria per protocol. The COSTAR Lung phase III trial is a randomized, open label 3-arm trial comparing cobolimab, an investigational selective anti–TIM-3 monoclonal antibody, plus dostarlimab plus docetaxel to dostarlimab plus docetaxel to docetaxel alone in patients with advanced NSCLC who have progressed on prior anti-PD-L1 therapy and chemotherapy.
Hesham Abdullah, Senior Vice President, Global Head of Oncology Development, GSK said: “These trials support the ambition for dostarlimab to become the backbone of our ongoing immuno-oncology-based research and development programme when used alone and in combination with standard of care and future novel cancer therapies, particularly in patients with currently limited treatment options.”
About PERLA
The PERLA phase II trial is a global, randomised, double-blind trial of 243 patients evaluating the efficacy and safety of dostarlimab plus chemotherapy compared to pembrolizumab plus chemotherapy in patients with metastatic non-squamous NSCLC without a known sensitising epidermal growth factor receptor, anaplastic lymphoma kinase, or receptor tyrosine kinase-1 mutation, V600E mutation of the BRAF gene or other genomic mutation for which an approved targeted therapy is available. The primary endpoint was objective response rate of dostarlimab plus chemotherapy versus pembrolizumab plus chemotherapy assessed by blinded independent central review per RECIST v1.1. Secondary endpoints include investigator-assessed progression-free survival per RECIST v1.1, overall survival, and safety.
About Jemperli (dostarlimab)
Jemperli is a programmed death receptor-1 (PD-1)-blocking antibody that binds to the PD-1 receptor and blocks its interaction with the PD-1 ligands PD-L1 and PD-L2. Jemperli is being investigated in registrational enabling studies, as monotherapy and as part of combination regimens, including in women with recurrent or primary advanced endometrial cancer, women with stage III or IV non-mucinous epithelial ovarian cancer, and in patients with other advanced solid tumours or metastatic cancers. Jemperli is not approved anywhere in the world in combination with chemotherapy in first-line patients with metastatic non-squamous NSCLC or in combination with other agents to treat patients with advanced NSCLC who have progressed on prior anti-PD-L1 therapy and chemotherapy.
Jemperli was discovered by AnaptysBio and licensed to TESARO, Inc., under a Collaboration and Exclusive License Agreement signed in March 2014. The collaboration has resulted in three monospecific antibody therapies that have progressed into the clinic. These are: Jemperli (GSK4057190), a PD-1 antagonist; cobolimab, (GSK4069889), a TIM-3 antagonist; and GSK4074386, a LAG-3 antagonist. GSK is responsible for the ongoing research, development, commercialization, and manufacturing of each of these Products under the Agreement.
About Cobolimab
Cobolimab is a monoclonal antibody against the inhibitory T-cell receptor, T-cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), with potential immune checkpoint inhibitory and antineoplastic activities. Cobolimab was discovered by AnaptysBio and TESARO, Inc., under a Collaboration and Exclusive License Agreement signed in March 2014. The collaboration has resulted in three monospecific antibody drugs that have progressed into the clinic. These are: dostarlimab (GSK4057190), a PD-1 antagonist; cobolimab, (GSK4069889), a TIM-3 antagonist; and GSK4074386, a LAG-3 antagonist. GSK is responsible for the ongoing research, development, commercialisation, and manufacture of each of these products under the Agreement.
Important Information for Jemperli in the EU
Indication
Jemperli is indicated as monotherapy for the treatment of adult patients with mismatch repair deficient (dMMR)/microsatellite instability‑high (MSI‑H) recurrent or advanced endometrial cancer that has progressed on or following prior treatment with a platinum‑containing regimen.
Refer to the Jemperli Prescribing Information for a full list of adverse events and the complete important safety information in the EU.
About GSK
GSK is a global biopharma company with a purpose to unite science, technology, and talent to get ahead of disease together. Find out more at gsk.com/company
Oct. 05, 2022 4:40 AM ET
By: Ravikash, SA News Editor
GSK (NYSE:GSK) said its medicine Jemperli (dostarlimab) met the main goal of showing objective response rate (ORR) in certain patients with non-small cell lung cancer (NSCLC) in a phase 2 trial.
The study, dubbed PERLA, evaluated Jemperli in combination with chemotherapy versus Merck's (NYSE:MRK) Keytruda (pembrolizumab) in combination with chemotherapy in first-line patients with metastatic NSCLC, the company noted in an Oct. 5 press release.
https://seekingalpha.com/symbol/GSK
https://www.gskusmedicalaffairs.com/patients/index.html
Tuesday, October 04, 2022 - 06:45am
NEW YORK--(BUSINESS WIRE)-- Pfizer Inc. (NYSE: PFE) today announced positive topline results from the Phase 3 TALAPRO-2 study of TALZENNA® (talazoparib), an oral poly ADP-ribose polymerase (PARP) inhibitor, in combination with XTANDI® (enzalutamide) compared to placebo plus XTANDI in men with metastatic castration-resistant prostate cancer (mCRPC), with or without homologous recombination repair (HRR) gene mutations. The study met its primary endpoint with a statistically significant and clinically meaningful improvement in radiographic progression-free survival (rPFS) compared with placebo plus XTANDI. The results of the primary endpoint exceeded the pre-specified hazard ratio of 0.696.
Results showed a trend toward improved overall survival, a key secondary endpoint, at the time of the analysis, but these data are not yet mature. Benefits were also observed in other secondary endpoints, including investigator assessed rPFS, prostate specific antigen (PSA) response, time to PSA progression, and overall response rate. Other secondary endpoints are being analyzed. At the time of topline analysis, the safety of TALZENNA plus XTANDI were generally consistent with the known safety profile of each medicine.
“XTANDI is a global standard of care, with overall survival demonstrated in mCRPC, non-metastatic CRPC, and metastatic castration-sensitive prostate cancer (mCSPC),” said Chris Boshoff, M.D., Ph.D., Chief Development Officer, Oncology and Rare Disease, Pfizer Global Product Development. “We are very pleased with the strong findings from TALAPRO-2, and although no definitive conclusions can be made across trials, the rPFS appears to be the longest observed in a randomized trial in this setting. These data highlight the potential for TALZENNA in combination with XTANDI, if approved, to become a new standard of care for mCRPC, irrespective of HRR gene mutation status. We look forward to discussing these data with global health authorities.”
“These exciting results from TALAPRO-2 underscore our long-standing commitment to men living with prostate cancer and delivering the next scientific breakthroughs,” said Suneet Varma, Global Oncology and U.S. President, Pfizer. “Based on these compelling combination data with XTANDI, we believe TALZENNA in prostate cancer may become the next potential blockbuster opportunity in our leading Pfizer Oncology portfolio, subject to regulatory approval.”
Detailed results from TALAPRO-2 will be submitted for presentation at a near-term medical congress. These data will also be shared with global regulatory authorities to potentially support a regulatory filing.
TALZENNA or the combination of TALZENNA plus XTANDI have not been approved by any regulatory agency for the treatment of mCRPC. In addition to the TALAPRO-2 trial, the combination of TALZENNA plus XTANDI is being investigated in the TALAPRO-3 trial (NCT04821622), a global, randomized, double-blind, placebo-controlled Phase 3 study in men with HRR-deficient mCSPC.
About TALAPRO-2
The Phase 3 TALAPRO-2 trial is a two-part, two-cohort, multicenter, randomized, double-blind, placebo-controlled study that enrolled 1,095 patients with mCRPC (with no systemic treatments initiated after documentation of mCRPC) at sites in the U.S., Canada, Europe, South America, and the Asia-Pacific region. The study included two patient cohorts: all-comers (n=750) and those with HRR mutations (HRRm; n=380). Patients on androgen deprivation therapy (ADT) or who had bilateral orchiectomy in the trial were randomized to receive TALZENNA 0.5 mg/day plus XTANDI 160mg/day, or placebo plus XTANDI 160 mg/day.
The primary endpoint of the trial is radiographic progression-free survival (rPFS), defined as the time from the date of randomization to first objective evidence of radiographic progression by blinded independent review, or death, whichever occurs first, in both cohort 1 (all-comers) and cohort 2 (those with HRRm). The trial is still ongoing for cohort 2. Secondary endpoints include overall survival, objective response rate, duration of response and PSA response.
For more information on the TALAPRO-2 trial (NCT03395197) go to www.clinicaltrials.gov.
About TALZENNA® (talazoparib)
TALZENNA (talazoparib) is an inhibitor of PARP enzymes, which play a role in the DNA damage repair response. Preclinical studies have demonstrated that TALZENNA blocks PARP enzyme activity and traps PARP at the site of DNA damage, leading to decreased cancer cell growth and cancer cell death. TALZENNA is being evaluated in several ongoing clinical trials in prostate cancer, as well as other novel combinations with targeted therapies in various solid tumors. TALZENNA was approved by the U.S. Food and Drug Administration in 2018 for the treatment of adult patients with deleterious or suspected deleterious germline breast cancer susceptibility gene (BRCA)-mutated (gBRCAm) human epidermal growth factor receptor 2 (HER2)-negative locally advanced or metastatic breast cancer.
Indication in the U.S.
TALZENNA (talazoparib) is indicated for the treatment of adult patients with deleterious or suspected deleterious germline breast cancer susceptibility gene (BRCA)-mutated (gBRCAm) human epidermal growth factor receptor 2 (HER2)-negative locally advanced or metastatic breast cancer. Select patients for therapy based on an FDA-approved companion diagnostic for TALZENNA.
Please see full U.S. Prescribing Information and Patient Information for TALZENNA® (talazoparib) at www.TALZENNA.com.
About XTANDI® (enzalutamide) and Important Safety Information
XTANDI (enzalutamide) is an androgen receptor inhibitor indicated in the U.S. for the treatment of patients with castration-resistant prostate cancer (CRPC) and metastatic castration-sensitive prostate cancer (mCSPC).
Overall survival benefit has been observed in patients treated with XTANDI in mCRPC, nmCRPC, and mCSPC.
Please see Full Prescribing Information for additional safety information.
About Pfizer Oncology
At Pfizer Oncology, we are committed to advancing medicines wherever we believe we can make a meaningful difference in the lives of people living with cancer. Today, we have an industry-leading portfolio of 24 approved innovative cancer medicines and biosimilars across more than 30 indications, including breast, genitourinary, colorectal, blood and lung cancers, as well as melanoma.
About the Pfizer/Astellas Collaboration
In October 2009, Medivation, Inc., which is now part of Pfizer (NYSE: PFE), and Astellas (TSE: 4503) entered into a global agreement to jointly develop and commercialize enzalutamide. The companies jointly commercialize XTANDI in the United States and Astellas has responsibility for manufacturing and all additional regulatory filings globally, as well as commercializing XTANDI outside the United States.
View source version on businesswire.com: https://www.businesswire.com/news/home/20221004005291/en/
Source: Pfizer Inc.
Oct. 04, 2022 7:17 AM ET
By: Dulan Lokuwithana, SA News Editor1 Comment
Pfizer (NYSE:PFE) announced Tuesday that its breast cancer therapy Alzenna in combination with FDA-approved prostate cancer medicine Xtandi achieved the primary endpoint in a Phase 3 trial involving patients with metastatic castration-resistant prostate cancer (mCRPC).
https://seekingalpha.com/symbol/PFE

September 28, 2022 at 5:44 PM EDT Back
Dupixent significantly reduced itch and skin lesions compared to placebo in direct-to-Phase 3 program consisting of two pivotal trials
About 75,000 adults in the U.S. living with prurigo nodularis are most in need of new treatment options
Approval represents the second dermatology indication for Dupixent and fifth disease indication overall in the U.S.
TARRYTOWN, N.Y. and PARIS, Sept. 28, 2022 /PRNewswire/ -- Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) and Sanofi today announced that the U.S. Food and Drug Administration (FDA) has approved Dupixent® (dupilumab) for the treatment of adult patients with prurigo nodularis. With this approval, Dupixent becomes the first and only medicine specifically indicated to treat prurigo nodularis in the U.S. Prurigo nodularis is a chronic, debilitating skin disease with underlying type 2 inflammation and its impact on quality of life is one of the highest among inflammatory skin diseases. The FDA evaluated the Dupixent application for prurigo nodularis under Priority Review, which is granted to therapies that have the potential to provide significant improvements in the treatment, diagnosis or prevention of serious conditions.
"Patients living with prurigo nodularis must often contend with dozens, if not hundreds, of itchy and painful nodules covering their body and have not had an approved treatment option for their disease," said George D. Yancopoulos, M.D., Ph.D., President and Chief Scientific Officer at Regeneron, and a principal inventor of Dupixent. "Dupixent has already transformed the treatment landscape of several diseases driven by type 2 inflammation – including atopic dermatitis, asthma, chronic rhinosinusitis with nasal polyposis and eosinophilic esophagitis – and been prescribed to more than half a million patients around the world for its approved indications. With this approval, those suffering with prurigo nodularis finally have a medicine to address the debilitating signs and symptoms of the disease."
"Until today, there were limited treatment options to manage the relentless itch and associated sensations of burning and stinging skin that can negatively impact the lives of patients struggling with prurigo nodularis," said Naimish Patel, M.D, Head of Global Development, Immunology and Inflammation at Sanofi. "Dupixent has the potential to transform the standard-of-care for prurigo nodularis patients by alleviating the key hallmarks of the disease, such as reducing itch and achieving clearer skin. With Dupixent now approved in two diseases in dermatology where type 2 inflammation is a central driver, we look forward to further evaluating the potential of inhibiting IL-4 and IL-13 in other chronic skin diseases."
The FDA approval is based on data from two Phase 3 trials, PRIME and PRIME2, evaluating the efficacy and safety of Dupixent in adults with prurigo nodularis. Efficacy in these trials assessed the proportion of subjects with clinically meaningful reduction in itch, clearing of skin, or both:
The safety results of the trial were generally consistent with the known safety profile of Dupixent in its approved dermatology indication. The most common adverse events (≥2%) from pooled PRIME and PRIME2 data more frequently observed with Dupixent than placebo were nasopharyngitis (5% Dupixent, 2% placebo), conjunctivitis (4% Dupixent, 1% placebo), herpes infection (3% Dupixent, 0% placebo), dizziness (3% Dupixent, 1% placebo), muscle pain (3% Dupixent, 1% placebo), and diarrhea (3% Dupixent, 1% placebo).
A regulatory filing for prurigo nodularis is under review by the European Medicines Agency, and submissions to regulatory authorities in additional countries are also planned in 2022.
About the Dupixent Prurigo Nodularis Trials
The PRIME and PRIME2 Phase 3 double-blind, placebo-controlled trials evaluated the efficacy and safety of Dupixent in 311 adults with uncontrolled prurigo nodularis.
In PRIME and PRIME2, the primary endpoint evaluated the proportion of patients with clinically meaningful improvement in itch from baseline (measured by a ≥4-point reduction in Worst-Itch Numeric Rating Scale [WI-NRS] on a 0-10 scale) at 24 and 12 weeks, respectively. Additional endpoints included the proportion of patients with clear or almost clear skin of nodules at 24 weeks (measured by a score of 0 or 1 on the Investigator's Global Assessment PN-Stage [IGA PN-S] on a 0-4 scale), and the proportion of patients who achieved a clinically meaningful response in both WI-NRS and IGA PN-S.
About Dupixent
Dupixent is administered as an injection under the skin (subcutaneous injection) at different injection sites. In patients aged 18 years and older with prurigo nodularis, Dupixent 300 mg is administered with a pre-filled syringe or pre-filled pen every two weeks following an initial loading dose. Dupixent is intended for use under the guidance of a healthcare professional and can be given in a clinic or at home by self-administration after training by a healthcare professional.
Regeneron and Sanofi are committed to helping patients in the U.S. who are prescribed Dupixent gain access to the medicine and receive the support they may need with the DUPIXENT MyWay® program. For more information, please call 1-844-DUPIXENT (1-844-387-4936) or visit www.DUPIXENT.com.
Dupixent has received regulatory approvals in one or more countries around the world for use in certain patients with atopic dermatitis, asthma, chronic rhinosinusitis with nasal polyposis (CRSwNP), eosinophilic esophagitis (EoE) or prurigo nodularis in different age populations. Dupixent is currently approved across these indications in the U.S. and for one or more of these indications in more than 60 countries, including in the European Union and Japan. More than 500,000 patients have been treated with Dupixent globally.
Dupixent, which was invented using Regeneron's proprietary VelocImmune® technology, is a fully human monoclonal antibody that inhibits the signaling of the interleukin-4 (IL-4) and interleukin-13 (IL-13) pathways and is not an immunosuppressant. The Dupixent development program has shown significant clinical benefit and a decrease in type 2 inflammation in Phase 3 trials, establishing that IL-4 and IL-13 are key and central drivers of the type 2 inflammation that plays a major role in multiple related and often co-morbid diseases. These diseases include approved indications for Dupixent such as prurigo nodularis, atopic dermatitis, asthma, CRSwNP and EoE.
About Regeneron's VelocImmune Technology
Regeneron's VelocImmune technology utilizes a proprietary genetically engineered mouse platform endowed with a genetically humanized immune system to produce optimized fully human antibodies. When Regeneron's co-Founder, President and Chief Scientific Officer George D. Yancopoulos was a graduate student with his mentor Frederick W. Alt in 1985, they were the first to envision making such a genetically humanized mouse, and Regeneron has spent decades inventing and developing VelocImmune and related VelociSuite® technologies. Dr. Yancopoulos and his team have used VelocImmune technology to create approximately one in five of all original, FDA-approved or authorized fully human monoclonal antibodies. This includes REGEN-COV® (casirivimab and imdevimab), Dupixent, Libtayo® (cemiplimab-rwlc), Praluent® (alirocumab), Kevzara® (sarilumab), Evkeeza® (evinacumab-dgnb) and Inmazeb™ (atoltivimab, maftivimab and odesivimab-ebgn).
Dupilumab Development Program
Dupilumab is being jointly developed by Regeneron and Sanofi under a global collaboration agreement. To date, dupilumab has been studied across more than 60 clinical trials involving more than 10,000 patients with various chronic diseases driven in part by type 2 inflammation.
In addition to the currently approved indications, Regeneron and Sanofi are studying dupilumab in a broad range of diseases driven by type 2 inflammation or other allergic processes in Phase 3 trials, including pediatric eosinophilic esophagitis, hand and foot atopic dermatitis, chronic inducible urticaria-cold, chronic spontaneous urticaria, chronic pruritis of unknown origin, chronic obstructive pulmonary disease with evidence of type 2 inflammation, chronic rhinosinusitis without nasal polyposis, allergic fungal rhinosinusitis, allergic bronchopulmonary aspergillosis and bullous pemphigoid. These potential uses of dupilumab are currently under clinical investigation, and the safety and efficacy in these conditions have not been fully evaluated by any regulatory authority.
INDICATIONS
DUPIXENT is a prescription medicine used:
About Regeneron Regeneron is a leading biotechnology company that invents, develops and commercializes life-transforming medicines for people with serious diseases. Founded and led for nearly 35 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to nine FDA-approved treatments and numerous product candidates in development, almost all of which were homegrown in our laboratories. Our medicines and pipeline are designed to help patients with eye diseases, allergic and inflammatory diseases, cancer, cardiovascular and metabolic diseases, pain, hematologic conditions, infectious diseases and rare diseases.
Regeneron is accelerating and improving the traditional drug development process through our proprietary VelociSuite® technologies, such as VelocImmune®, which uses unique genetically humanized mice to produce optimized fully human antibodies and bispecific antibodies, and through ambitious research initiatives such as the Regeneron Genetics Center®, which is conducting one of the largest genetics sequencing efforts in the world.
For more information, please visit www.Regeneron.com or follow @Regeneron on Twitter.
About Sanofi
We are an innovative global healthcare company, driven by one purpose: we chase the miracles of science to improve people's lives. Our team, across some 100 countries, is dedicated to transforming the practice of medicine by working to turn the impossible into the possible. We provide potentially life-changing treatment options and life-saving vaccine protection to millions of people globally, while putting sustainability and social responsibility at the center of our ambitions.
Sanofi is listed on EURONEXT: SAN and NASDAQ: SNY.
View original content:https://www.prnewswire.com/news-releases/dupixent-dupilumab-approved-by-fda-as-the-first-and-only-treatment-indicated-for-prurigo-nodularis-301636139.html
SOURCE Regeneron Pharmaceuticals, Inc.
Sep. 29, 2022 6:51 AM ET
Regeneron Pharmaceuticals, Inc. (REGN), SNYSNYNF, GCVRX
By: Dulan Lokuwithana, SA News Editor

Sep 27, 2022
SOMERSET, N.J., – SEPTEMBER 27, 2022 – Legend Biotech Corporation (NASDAQ: LEGN), a global biotechnology company developing, manufacturing and commercializing novel therapies to treat life-threatening diseases, announced today that Japan’s Ministry of Health, Labour and Welfare (MHLW) has approved CARVYKTI™ (ciltacabtagene autoleucel), a B-cell maturation antigen (BCMA)-directed chimeric antigen receptor T cell (CAR-T) therapy, for the treatment of adults with relapsed or refractory multiple myeloma, limited to cases meeting both of the following conditions:
The New Drug Application was submitted by Legend Biotech’s collaboration partner, Janssen Pharmaceuticals (Janssen). Legend entered into an exclusive worldwide license and collaboration agreement with Janssen to develop and commercialize ciltacabtagene autoleucel (cilta-cel) in December 2017.
CARVYKTI™ features two BCMA-targeting single domain antibodies, is specifically developed for each individual patient and is administered as a single infusion.
The approval is based on data from the pivotal phase 1b/2 CARTITUDE-1 study, and included patients who received a median of six prior treatment regimens (range, 3-18), and previously received a proteasome inhibitor, an immunomodulatory agent and an anti-CD38 monoclonal antibody.[1] In the study, a one-time treatment with ciltacabtagene autoleucel resulted in durable responses, with 96.9 percent (95 percent Confidence Interval [CI], 91.2-99.4) of patients with relapsed or refractory multiple myeloma (RRMM) in the non-Japanese population responding to therapy (n=97).1 Notably, 67 percent (95 percent CI, 56.7-76.2) of the patients (n=65) achieved a stringent complete response (sCR), a measure for which a physician is unable to observe any signs or symptoms of disease via imaging or other tests after treatment.[2][*] The efficacy results were also observed in Japanese patients with multiple myeloma, which were consistent with that of the non-Japanese population.[†] At a median of 18 months follow-up, the median duration of response (DOR) was 21.8 months in non-Japanese patients.[3]
The safety of cilta-cel was evaluated in 106 adult patients in the CARTITUDE-1 study, including 97 non-Japanese and 9 Japanese participants. Adverse reactions were observed in 105 (99.1%) of 106 patients treated with cilta-cel. The most common adverse reactions included cytokine release syndrome (94.3%), cytopenia (79.2%), neutropenia (75.5%), thrombocytopenia (59.4%), anemia (51.9%), neurologic events (39.6%) infections (19.8%) and hypogammaglobulinemia (11.3%).[4]
Ying Huang, Ph.D., Chief Executive Officer of Legend Biotech, said: “The approval of CARVYKTI™ by the Japanese regulatory authority is a landmark on our journey to help patients with relapsed or refractory multiple myeloma. In partnership with Janssen, we are laying the groundwork for this drug to enter the marketplace, while advancing the clinical development program for what we believe to be a vital therapeutic option for appropriate patients with multiple myeloma.”
CARVYKTI™ was approved by the U.S. Food and Drug Administration in February 2022 and was granted conditional marketing authorization by the European Commission in May 2022.
ABOUT CARVYKTI™ (CILTACABTAGENE AUTOLEUCEL; CILTA-CEL)
CARVYKTI™ is a B-cell maturation antigen (BCMA)-directed, genetically modified autologous T-cell immunotherapy, which involves reprogramming a patient’s own T-cells with a transgene encoding a chimeric antigen receptor (CAR) that identifies and eliminates cells that express BCMA. BCMA is primarily expressed on the surface of malignant multiple myeloma B-lineage cells, as well as late-stage B-cells and plasma cells. The CARVYKTI™ CAR protein features two BCMA-targeting single domain antibodies designed to confer high avidity against human BCMA. Upon binding to BCMA-expressing cells, the CAR promotes T-cell activation, expansion, and elimination of target cells.1
In December 2017, Legend Biotech Corporation entered into an exclusive worldwide license and collaboration agreement with Janssen Pharmaceuticals (Janssen) to develop and commercialize cilta-cel.
In February 2022, CARVYKTI™ was approved by the U.S. Food and Drug Administration (FDA) for the treatment of adults with relapsed or refractory multiple myeloma.[5] In May 2022, the European Commission (EC) granted conditional marketing authorization of CARVYKTI™ for the treatment of adults with relapsed and refractory multiple myeloma.[6] Cilta-cel was granted Breakthrough Therapy Designation in the U.S. in December 2019 and in China in August 2020. In addition, cilta-cel received a PRIority MEdicines (PRIME) designation from the European Commission in April 2019. Cilta-cel also received Orphan Drug Designation from the U.S. FDA in February 2019, from the European Commission in February 2020, and from the Pharmaceuticals and Medicinal Devices Agency (PMDA) in Japan in June 2020. In May 2022, the European Medicines Agency’s Committee for Orphan Medicinal Products recommended by consensus that the orphan designation for cilta-cel be maintained on the basis of clinical data demonstrating improved and sustained complete response rates following treatment.[7]
ABOUT CARTITUDE-1
CARTITUDE-1 (NCT03548207)[8] is a Phase 1b/2, open-label, single arm, multi-center trial evaluating cilta-cel for the treatment of adult patients with relapsed or refractory multiple myeloma, who previously received at least three prior lines of therapy including a proteasome inhibitor (PI), an immunomodulatory agent (IMiD) and an anti-CD38 monoclonal antibody, and who demonstrated disease progression on or after the last regimen. All patients in the study had received a median of six prior treatment regimens (range, 3-18).[9]
CARVYKTI™ U.S. FDA-APPROVED INDICATION
CARVYKTI™ (ciltacabtagene autoleucel) is a B-cell maturation antigen (BCMA)-directed genetically modified autologous T cell immunotherapy indicated for the treatment of adult patients with relapsed or refractory multiple myeloma, after four or more prior lines of therapy, including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody.
Further information is available at www.CARVYKTIrems.com or 1-844-672-0067.
Please read full Prescribing Information including Boxed Warning for CARVYKTI™.
ABOUT LEGEND BIOTECH
Legend Biotech is a global biotechnology company dedicated to treating, and one day curing, life-threatening diseases. Headquartered in Somerset, New Jersey, we are developing advanced cell therapies across a diverse array of technology platforms, including autologous and allogeneic chimeric antigen receptor T-cell, T-cell receptor (TCR-T), and natural killer (NK) cell-based immunotherapy. From our three R&D sites around the world, we apply these innovative technologies to pursue the discovery of safe, efficacious and cutting-edge therapeutics for patients worldwide.
Learn more at www.legendbiotech.com and follow us on Twitter and LinkedIn.
Sep. 27, 2022 10:29 AM ET
Legend Biotech Corporation (LEGN)JNJ
By: Jonathan Block, SA News Editor
October 03, 2022 Download this Press ReleasePDF Format (opens in new window)
– Median rPFS of 11.2 months for Rubraca vs 6.4 months for control group in the BRCA subgroup
– Median rPFS of 10.2 months for Rubraca vs 6.4 months for control group in the ITT population (inclusive of all patients with a BRCA or ATM mutation enrolled in TRITON3)
BOULDER, Colo.--(BUSINESS WIRE)-- Clovis Oncology, Inc. (NASDAQ: CLVS) today announced positive top-line data from the Phase 3, open-label, multicenter, randomized TRITON3 trial demonstrating that Rubraca monotherapy treatment achieved the primary endpoint of significantly improved radiographic progression-free survival (rPFS) by independent radiology review (IRR) compared with the control group, which consisted of physician’s choice of docetaxel, abiraterone acetate, or enzalutamide. Benefit was observed in both primary efficacy analyses of patients with chemotherapy-naïve metastatic castration-resistant prostate cancer (mCRPC): first, those who had mutations in BRCA, as well as all patients randomized in the trial, inclusive of mutations in BRCA or ATM (the overall intent-to-treat population (ITT)). The safety profile of Rubraca observed in the TRITON3 study was consistent with Rubraca labelling.
During the first quarter of 2023, the Company plans to submit a supplemental New Drug Application (sNDA) to the FDA for the BRCA subgroup of patients and intends to discuss with the FDA submitting for the broader ITT population.
“We believe that the positive results from TRITON3 further demonstrate the important role that Rubraca can play as a treatment option for men with metastatic castration-resistant prostate cancer associated with homologous recombination deficiency, and we look forward to submitting these data to the regulatory authorities in the US during Q1 2023. Not only does this provide a potential treatment option for eligible men with earlier stage disease, but it is the first and only PARP inhibitor that has demonstrated superior radiographic PFS compared to chemotherapy, which is today the standard of care for these patients,” said Patrick J. Mahaffy, President and CEO of Clovis Oncology. “Most importantly, I would like to thank the patients, physicians, and our colleagues whose commitment to this trial made these results possible, which now offer the potential to make a difference in the lives of many men with advanced prostate cancer.”
“Men with this type of metastatic prostate cancer want to get their genetically targeted therapy as early as possible, and this trial clearly shows the value of rucaparib as a treatment for these men,” said Alan H. Bryce, MD, chair of the Division of Hematology and Medical Oncology at the Mayo Clinic and principal investigator of the TRITON3 trial. “A key point is that rucaparib can replace chemotherapy in this setting. The current standard of care for these men is chemotherapy with docetaxel, and rucaparib is the only PARP inhibitor which has beaten a docetaxel-containing control arm in a clinical trial.”
“This trial demonstrates the potential for rucaparib to treat men with early-stage metastatic castration-resistant prostate cancer,” said Karim Fizazi, MD, PhD, medical oncologist at Gustave Roussy, and a full professor in Oncology at the University of Paris-Saclay and principal investigator of the TRITON3 trial. “To my knowledge, this is the first time in two decades that a potential new treatment, rucaparib, has shown in a randomized controlled trial radiographic PFS efficacy over an investigator’s choice control arm that included docetaxel chemotherapy, a long-standing standard of care for men with metastatic castration-resistant prostate cancer, and this is excellent news for patients.”
TRITON3 is a Phase 3, multicenter, open-label, randomized trial of Rubraca in patients with chemotherapy-naïve mCRPC. The study enrolled 405 patients with a mutation in BRCA or ATM who were randomized to Rubraca or the control group, which consisted of physician’s choice of docetaxel, abiraterone acetate, or enzalutamide. Approximately 55% of the patients in the control arm received docetaxel. The primary endpoint was rPFS by IRR, in patients with mutations in BRCA1, BRCA2or ATM. TRITON3 was designed as a Phase 3 trial to confirm and expand the efficacy data from TRITON2 in an earlier treatment setting against a relevant control arm.
Patients were required to have disease progression after treatment with one prior next-generation AR-targeted therapy (abiraterone acetate, enzalutamide, apalutamide or investigational agent), as well as a deleterious mutation in BRCA or ATM. Following a protocol amendment, patients were permitted to have received a qualifying AR-targeted therapy in either the hormone-sensitive or castration-resistant setting, and as a result, approximately 18% of patients in TRITON3 had received prior AR-targeted therapy in the metastatic hormone-sensitive setting only.
The primary efficacy analysis evaluated two prospectively defined molecular sub-groups in a step-down manner: 1) the BRCA subgroup, and 2) all patients randomized (ITT) in TRITON3, inclusive of those with BRCA or ATM mutations.
Following is a summary of the primary efficacy analyses of rPFS by independent radiologic review (IRR), the primary analysis of TRITON3.
Significant Improvement in rPFS in the BRCA Patient Population
The Rubraca arm (n=201) achieved statistical significance over the control arm (n=101) for the primary endpoint of rPFS with a hazard ratio of 0.50 (95% CI: 0.36-0.69). The median PFS for the population of patients with BRCA mutations treated with Rubraca was 11.2 months vs 6.4 months among those who received physician’s choice (p<0.0001).
Significant Improvement in rPFS in the ITT population, inclusive of those with BRCA or ATM mutations
Rubraca also showed statistical significance in all 405 patients randomized in TRITON3. The Rubraca arm (n=270) successfully achieved statistical significance over the control arm (n=135) for the primary endpoint of rPFS with a hazard ratio of 0.61 (95% CI: 0.47-0.80). The median PFS for all patients enrolled in TRITON3 and treated with Rubraca was 10.2 months vs 6.4 months among those who received physician’s choice (p=0.0003).
rPFS in Exploratory ATM Mutation Subgroup
In the exploratory subgroup of men with tumor ATM mutations (n=103), the hazard ratio for rPFS was 0.97 (95% CI: 0.59-1.52). Median rPFS in the Rubraca arm (n=69) was 8.1 months vs 6.8 months in the control arm (n=34) with a nominal p-value (p=0.8421).
Secondary Endpoint of Overall Survival Summary
The hazard ratio for the interim analysis of the secondary endpoint of overall survival (OS) in the BRCA subgroup and ITT population, which are not yet mature, favored Rubraca. The hazard ratio for OS in the exploratory subgroup of ATM, which is mature, favored the control arm. The 95% confidence intervals for these OS analyses included less than one for the exploratory endpoint ATM, signifying no statistical difference in outcomes between Rubraca and control.
Summary of TRITON3 Safety
The safety profile of Rubraca observed in TRITON3 was consistent with Rubraca labelling.The most common (≥5%) treatment-emergent grade 3 or higher adverse events (TEAEs) among all patients treated with Rubraca in the TRITON3 study were anemia/decreased hemoglobin (23.7%), neutropenia/decreased neutrophil count (7.4%), asthenia/fatigue (7.0%), thrombocytopenia/decreased platelet count (5.9%) and increased ALT/AST (5.2%). The discontinuation rate for TEAEs was 14.8% for Rubraca-treated patients and 21.5% for the control arm.
Clovis Oncology will submit an expanded description of the TRITON3 results for presentation in a scientific session at the Prostate Cancer Foundation Annual Scientific Retreat later this month and plans to submit additional data to a medical meeting in early 2023.
Rubraca is not currently approved in the chemotherapy-naïve mCRPC setting.
About the TRITON3 Clinical Trial
TRITON3 (NCT02975934) is a Phase 3, multicenter, open-label, randomized trial of Rubraca in patients with chemotherapy-naïve mCRPC. Patients with a mutation in BRCA or ATM were randomized to Rubraca or the control group, which consisted of physician’s choice of docetaxel, abiraterone acetate, or enzalutamide. The primary objective was efficacy, as analysed by independent radiology review (IRR) of radiographic progression-free survival (rPFS) in patients with mutations in BRCA1, BRCA2 or ATM. TRITON3 was designed as a Phase 3 trial to confirm and expand the efficacy data from TRITON2 in an earlier treatment setting against a relevant control arm.
About Rubraca (rucaparib)
Rubraca is an oral, small molecule inhibitor of PARP1, PARP2 and PARP3 being developed in multiple tumor types, including ovarian and metastatic castration-resistant prostate cancers, as monotherapy, and in combination with other anti-cancer agents. Exploratory studies in other tumor types are also underway.
Rubraca is an unlicensed medical product outside of the US and Europe.
Rubraca US FDA Approved Indications
About Clovis Oncology
Clovis Oncology, Inc. is a biopharmaceutical company focused on acquiring, developing, and commercializing innovative anti-cancer agents in the US, Europe, and additional international markets. Clovis Oncology targets development programs at specific subsets of cancer populations, and simultaneously develops, with partners, for those indications that require them, diagnostic tools intended to direct a compound in development to the population that is most likely to benefit from its use. Clovis Oncology is headquartered in Boulder, Colorado, with additional office locations in the US and Europe. Please visit www.clovisoncology.com for more information.
View source version on businesswire.com: https://www.businesswire.com/news/home/20221003005303/en/
Source: Clovis
![]()
Oct. 03, 2022 8:33 AM ET
Clovis Oncology, Inc. (CLVS)JNJ, PFE, ALPMF, ALPMY
By: Ravikash, SA News Editor
Clovis Oncology (NASDAQ:CLVS) said its medicine Rubraca helped extend the lives of patients with a type of prostate cancer without their disease getting worse, achieving the main goal of a phase 3 trial.
https://seekingalpha.com/symbol/CLVS
September 27, 2022 6:45 am ET
KEYTRUDA now approved for 23 uses in 13 different types of cancer in Japan
RAHWAY, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside of the United States and Canada, today announced that KEYTRUDA, Merck’s anti-PD-1 therapy, received four new approvals from Japan’s Ministry of Health, Labor and Welfare (MHLW):
“Based on compelling data from our clinical trial program, KEYTRUDA has become an important treatment option in Japan and now has 23 approved uses across 13 different types of cancer,” said Dr. Eliav Barr, senior vice president, head of global clinical development and chief medical officer, Merck Research Laboratories. “These four new approvals provide certain patients with advanced or recurrent cervical cancer, high-risk early-stage triple-negative breast cancer, renal cell carcinoma and completely resected stage IIB and IIC melanoma the opportunity to be treated with KEYTRUDA.”
“In Japan, the cancer mortality rate continues to rise annually, and patients are in need of new treatment options – particularly those with breast cancer, which is the most prevalent cancer among women in the country,” said Kyle Tattle, Representative Director and President, MSD Japan. “These new approvals for KEYTRUDA are the result of strong collaboration with the MHLW and provide additional treatment options for appropriate patients with difficult-to-treat cancers.”
Approval as neoadjuvant and adjuvant treatment regimen for triple-negative breast cancer
The approval of KEYTRUDA plus chemotherapy as neoadjuvant treatment, and then continued as monotherapy as adjuvant treatment after surgery for patients with hormone receptor-negative and HER2-negative breast cancer at high risk of recurrence is based on results from the Phase 3 KEYNOTE-522 trial, in which KEYTRUDA in combination with chemotherapy before surgery and continued as a single agent after surgery resulted in a statistically significant and clinically meaningful improvement in event-free survival (EFS), reducing the risk of disease progression that precludes curative surgery, local or distant recurrence, second primary malignancy or death by 37% (HR=0.63 [95% CI, 0.48-0.82]; p=0.00031) compared to neoadjuvant placebo in combination with chemotherapy and adjuvant placebo alone after surgery in these patients.
The Japanese package insert states that in KEYNOTE-522, adverse reactions were observed in 774 patients (98.9%) out of the safety analysis set of 783 patients (including 45/45 Japanese patients) receiving KEYTRUDA at a dose of 200 mg every three weeks in combination with chemotherapy as neoadjuvant treatment, and then continued as monotherapy as adjuvant treatment after surgery. The most common adverse reactions (≥20%) were nausea (63.2%), alopecia (60.2%), anemia (54.8%), neutropenia (46.9%), fatigue (42.1%), diarrhea (30.4%), elevated alanine aminotransferase (26.1%), vomiting (25.5%), asthenia (25.3%), rash (25.0%), constipation (24.0%), decreased neutrophil count (23.6%) and elevated aspartate aminotransferase (20.1%).
Triple-negative breast cancer (TNBC) is the most aggressive type of breast cancer, which has the highest risk of recurrence within the first five years after diagnosis and is associated with worse outcomes compared to other forms of breast cancer. While some breast cancers may test positive for estrogen receptors, progesterone receptors or overexpression of HER2, TNBC tests negative for all three. Triple-negative breast cancer is known to be prevalent in Japan, as approximately 15% of patients with breast cancer in Japan are diagnosed with TNBC. Breast cancer is the most commonly diagnosed cancer in women in Japan, with more than 94,000 people diagnosed in 2020.
Approval in renal cell carcinoma
The approval of KEYTRUDA for the adjuvant treatment of certain patients with RCC at increased risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions is based on results from the Phase 3 KEYNOTE-564 trial, in which KEYTRUDA as adjuvant treatment demonstrated a statistically significant improvement in disease-free survival, reducing the risk of disease recurrence or death by 32% (HR=0.68 [95% CI, 0.53-0.87]; p=0.0010) compared to placebo.
The Japanese package insert states that in KEYNOTE-564, adverse reactions were observed in 386 patients (79.1%) out of the safety analysis set of 488 patients receiving KEYTRUDA at a dose of 200 mg every three weeks or 400 mg every six weeks for up to 12 months. The most common adverse reactions (≥10%) were fatigue (20.3%), pruritus (18.6%), hypothyroidism (17.6%), diarrhea (15.8%), rash (15.0%) and hyperthyroidism (10.2%).
Renal cell carcinoma is by far the most common type of kidney cancer; about nine out of 10 kidney cancer diagnoses are RCCs. Renal cell carcinoma is about twice as common in men than in women. In Japan, it is estimated there were more than 25,000 new cases of kidney cancer diagnosed and more than 8,000 deaths from the disease in 2020.
Approval in advanced cervical cancer
The approval for KEYTRUDA plus chemotherapy, with or without bevacizumab, for the treatment of patients with advanced or recurrent cervical cancer with no prior chemotherapy who are not amenable to curative treatment is based on results from the Phase 3 KEYNOTE-826 trial, in which KEYTRUDA plus chemotherapy, with or without bevacizumab, demonstrated a statistically significant improvement in overall survival, reducing the risk of death by 33% (HR=0.67 [95% CI, 0.54-0.84]; p=0.0003), and progression-free survival, reducing the risk of disease progression or death by 35% (HR=0.65 [95% CI, 0.53-0.79]; p<0.0001), compared to chemotherapy with or without bevacizumab in these patients.
The Japanese package insert states that in KEYNOTE-826, adverse reactions were observed in 298 patients (97.1%) out of the safety analysis set of 307 patients (including 35/35 Japanese patients) receiving KEYTRUDA at a dose of 200 mg every three weeks in combination with investigator’s choice of anti-cancer regimens. The most common adverse reactions (≥20%) were alopecia (55.7%), anemia (48.5%), nausea (33.9%), diarrhea (24.8%), peripheral neuropathy (24.4%), fatigue (22.8%), peripheral sensory neuropathy (22.5%), neutropenia (22.1%) and vomiting (20.5%).
Cervical cancer forms in the lower part of the uterus and in the cells lining the cervix. All women are at risk for cervical cancer, and the disease is most frequently diagnosed between the ages of 35 to 44. In Japan, it is estimated there were more than 12,700 new cases of cervical cancer diagnosed and nearly 4,200 deaths from the disease in 2020.
Expanded indication in the adjuvant treatment of melanoma
The expanded indication for KEYTRUDA as monotherapy for the adjuvant treatment of patients with stage IIB or IIC melanoma after complete resection is based on results of the Phase 3 KEYNOTE-716 trial, in which KEYTRUDA as adjuvant treatment significantly prolonged recurrence-free survival, reducing the risk of disease recurrence or death by 35% (HR=0.65 [95% CI, 0.46-0.92]; p=0.00658) compared to placebo in these patients. With this expansion, Keytruda is now approved for the adjuvant treatment of resected stage IIB, IIC and stage III melanoma.
The Japanese package insert states that in KEYNOTE-716, adverse reactions were observed in 400 patients (82.8%) out of the safety analysis set of 483 patients (including 2/2 Japanese patients) receiving KEYTRUDA at a dose of 200 mg every three weeks. The most common adverse reactions (≥10%) were pruritus (24.2%), fatigue (21.1%), diarrhea (18.6%), arthralgia (16.1%), rash (15.7%) and hypothyroidism (15.5%).
Melanoma, the most serious form of skin cancer, is characterized by the uncontrolled growth of pigment-producing cells. In Japan, the rates of skin cancer have been rapidly increasing, and it is estimated there were more than 1,500 new cases of melanoma diagnosed and nearly 700 deaths from the disease in Japan in 2020.
About KEYTRUDA® (pembrolizumab) injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of adult and pediatric (12 years and older) patients with stage IIB, IIC, or III melanoma following complete resection.
Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [tumor proportion score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [Combined Positive Score (CPS) ≥1] as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy.
KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Carcinoma
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
Non-muscle Invasive Bladder Cancer
KEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC) as determined by an FDA-approved test.
Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or gastroesophageal junction (GEJ) (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
KEYTRUDA, in combination with chemotherapy, with or without bevacizumab, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
KEYTRUDA is indicated for the adjuvant treatment of patients with RCC at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions.
Endometrial Carcinoma
KEYTRUDA, as a single agent, is indicated for the treatment of patients with advanced endometrial carcinoma that is MSI-H or dMMR, as determined by an FDA-approved test, who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation.
Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
Merck’s focus on cancer
Our goal is to translate breakthrough science into innovative oncology medicines to help people with cancer worldwide. At Merck, the potential to bring new hope to people with cancer drives our purpose and supporting accessibility to our cancer medicines is our commitment. As part of our focus on cancer, Merck is committed to exploring the potential of immuno-oncology with one of the largest development programs in the industry across more than 30 tumor types. We also continue to strengthen our portfolio through strategic acquisitions and are prioritizing the development of several promising oncology candidates with the potential to improve the treatment of advanced cancers. For more information about our oncology clinical trials, visit www.merck.com/clinicaltrials.
About Merck
At Merck, known as MSD outside of the United States and Canada, we are unified around our purpose: We use the power of leading-edge science to save and improve lives around the world. For more than 130 years, we have brought hope to humanity through the development of important medicines and vaccines. We aspire to be the premier research-intensive biopharmaceutical company in the world – and today, we are at the forefront of research to deliver innovative health solutions that advance the prevention and treatment of diseases in people and animals. We foster a diverse and inclusive global workforce and operate responsibly every day to enable a safe, sustainable and healthy future for all people and communities. For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf .
Source: Merck & Co., Inc.
Sep. 27, 2022 7:16 AM ET
By: Dulan Lokuwithana, SA News Editor3 Comments
PUBLISHED27 September 2022 27 September 2022 07:05 BST
Koselugo (selumetinib) has been approved in Japan for the treatment of paediatric patients three years of age and older with plexiform neurofibromas (PNs) in neurofibromatosis type 1 (NF1) with clinical symptoms, such as pain and disfigurement, and PNs which cannot be completely removed by surgery without risk of substantial morbidity.1
The approval by the Japanese Ministry of Health, Labour and Welfare (MHLW) is based on positive results from the SPRINT Stratum 1 Phase II trial sponsored by the National Institutes of Health's National Cancer Institute (NCI) Cancer Therapy Evaluation Program (CTEP). The trial showed Koselugo, an oral treatment option, reduced the size of inoperable tumours in children.1,2 Additionally, a Phase I trial in Japanese paediatric NF1 patients with symptomatic and inoperable PNs was also evaluated as a basis for the approval, with the trial showing tumour reduction.
NF1 is a debilitating genetic condition affecting one in 3,000 individuals worldwide, most commonly diagnosed in children under 10.3,4 In 30-50% of patients, tumours develop on the nerve sheaths (plexiform neurofibromas) and can cause clinical issues such as disfigurement, motor dysfunction, pain, airway dysfunction, visual impairment and bladder or bowel dysfunction.2,5-8
Professor Yoshihiro Nishida, MD, PhD, Department of Rehabilitation Medicine at Nagoya University, Nagoya, Japan, and Japan Phase I trial investigator said: “People living with plexiform neurofibromas caused by neurofibromatosis type 1 often face painful physical, emotional and social burdens. This approval marks a major step forward in addressing the debilitating impact these plexiform neurofibromas have on paediatric patients living with neurofibromatosis type 1 in Japan. Koselugo provides a suitable intervention to treat symptomatic plexiform neurofibromas, which may improve long-term patient activities of daily living and quality of life.”
Marc Dunoyer, Chief Executive Officer, Alexion, said: “As the first medicine approved in Japan for paediatric patients with symptomatic, inoperable plexiform neurofibromas in neurofibromatosis type 1, Koselugo offers new hope for patients and families affected by this incurable genetic disease, whose only previous treatment option was repeated surgery. This approval is a testament to our longstanding commitment to rare disease research and we are energised by the opportunity to further accelerate innovation and care for the neurofibromatosis type 1 community.”
The SPRINT Stratum 1 Phase II trial showed Koselugo demonstrated an objective response rate (ORR) of 66% (33 of 50 patients, confirmed partial responses) in paediatric patients with PNs in NF1 when treated with Koselugo as twice-daily oral monotherapy.1 ORR is defined as the percentage of patients with confirmed complete (disappearance of PNs) or partial response (at least 20% reduction in tumour volume).1 The most common adverse reactions in the SPRINT trial were vomiting, blood creatine phosphokinase increase, diarrhoea and nausea.1
Results from the SPRINT Stratum 1 Phase II trial were published online in The New England Journal of Medicine.2
In addition to Japan, Koselugo is also approved in the US and EU for the treatment of paediatric patients with NF1 and symptomatic, inoperable PNs. Further regulatory submissions are underway.
SPRINT
The SPRINT Stratum 1 Phase II trial was designed to evaluate the objective response rate and impact on patient-reported and functional outcomes in paediatric patients with NF1-related inoperable PNs treated with Koselugo (selumetinib) monotherapy.2 This trial sponsored by NCI CTEP was conducted under a Cooperative Research and Development Agreement between NCI and AstraZeneca with additional support from Neurofibromatosis Therapeutic Acceleration Program (NTAP).
Koselugo
Koselugo (selumetinib) is the first and only approved therapy by the Japanese MHLW for the treatment of paediatric patients three years of age and older with plexiform neurofibromas (PNs) in neurofibromatosis type 1 (NF1) with clinical symptoms, such as pain and disfigurement, and PNs which cannot be completely removed by surgery without risk of substantial morbidity.1 Koselugo blocks specific enzymes (MEK1 and MEK2), which are involved in stimulating cells to grow.1 In NF1, these enzymes are overactive, causing tumour cells to grow in an unregulated way. By blocking these enzymes, Koselugo slows down the growth of tumour cells.1
Koselugo is approved for use in the US, EU and Japan and has received Orphan Drug Designation in Russia, Switzerland, South Korea, Taiwan and Australia, and health authorities worldwide are reviewing regulatory submissions.
AstraZeneca and MSD Strategic Collaboration
In July 2017, AstraZeneca and Merck & Co., Inc., Rahway, NJ, US, known as MSD outside the US and Canada, announced a global strategic collaboration to co-develop and co-commercialise Lynparza and Koselugo (selumetinib), a mitogen-activated protein kinase (MEK) inhibitor, for multiple cancer types. Working together, the companies will develop Lynparza and Koselugo in combination with other potential new medicines and as monotherapies. Independently, the companies will develop Lynparza and Koselugo in combination with their respective PD-L1 and PD-1 medicines.
Alexion
Alexion, AstraZeneca Rare Disease, is the group within AstraZeneca focused on rare diseases, created following the 2021 acquisition of Alexion Pharmaceuticals, Inc. As a leader in rare diseases for 30 years, Alexion is focused on serving patients and families affected by rare diseases and devastating conditions through the discovery, development and commercialisation of life-changing medicines. Alexion focuses its research efforts on novel molecules and targets in the complement cascade and its development efforts on haematology, nephrology, neurology, metabolic disorders, cardiology and ophthalmology. Headquartered in Boston, Massachusetts, Alexion has offices around the globe and serves patients in more than 50 countries.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Sep. 27, 2022 4:34 AM ET
By: Ravikash, SA News Editor
AstraZeneca (NASDAQ:AZN) and Merck's (NYSE:MRK) Koselugo was approved in Japan to treat patients three years of age and older with plexiform neurofibromas (PNs) in neurofibromatosis type 1 (NF1) with clinical symptoms, such as pain and disfigurement, and PNs which cannot be completely removed by surgery without risk of substantial morbidity.
https://seekingalpha.com/symbol/AZN

PUBLISHED27 September 2022 27 September 2022 07:00 BST
AstraZeneca’s Tezspire (tezepelumab) has been approved in Japan for the treatment of bronchial asthma in patients with severe or refractory disease in whom asthma symptoms cannot be controlled with mid- or high-dose inhaled corticosteroids and other long-term maintenance therapies.1
The approval by the Japanese Ministry of Health, Labour and Welfare (MHLW) was based on efficacy and safety results from the PATHFINDER clinical trial programme. The application included results from the pivotal NAVIGATOR Phase III trial in which Tezspire demonstrated superiority across every primary and key secondary endpoint in patients with severe asthma, compared to placebo, when added to standard therapy.2
Tezspire is the first and only biologic for severe asthma that acts at the top of the inflammatory cascade by blocking thymic stromal lymphopoietin (TSLP), an epithelial cytokine.2-5 Tezspire consistently and significantly reduced asthma exacerbations across the PATHWAY Phase II and NAVIGATOR Phase III clinical trials which included a broad population of severe asthma patients irrespective of key biomarkers, including blood eosinophil counts, allergic status and fractional exhaled nitric oxide (FeNO).2,3
Hironori Sagara, Professor and Chairman of the Department of Medicine Division of Respiratory Medicine & Allergology, Showa University School of Medicine, Tokyo, Japan, said: “Severe asthma is a debilitating disease, and many patients continue to experience frequent exacerbations and a significantly reduced quality of life despite recent advances in treatment. Across clinical trials, Tezspire has demonstrated significant results in a broad population of severe asthma patients, and with this approval, physicians can now offer patients in Japan a meaningful new treatment option.”
Mene Pangalos, Executive Vice President, BioPharmaceuticals R&D, AstraZeneca, said: “Tezspire is the first and only biologic approved by the Japanese Ministry of Health, Labour and Welfare that has been shown to consistently and significantly reduce attacks in exacerbation trials in a broad population of severe asthma patients irrespective of biomarker levels. Tezspire has the potential to improve outcomes for many patients with severe asthma and we are working to make this important medicine available in Japan as quickly as possible.”
In clinical studies, the most common adverse reactions in patients who received Tezspire were pharyngitis, arthralgia, rash and injection site reactions.1 The results from the NOZOMI Phase III long-term safety trial of Tezspire in Japanese patients were published in The Journal of Asthma in June 2022.
Results from the NAVIGATOR Phase III trial were published in The New England Journal of Medicine in May 2021.
Tezspire is approved in the US, the EU and other countries for the treatment of severe asthma. Other regulatory reviews evaluating Tezspire are ongoing in several markets around the world.
Clinical trials
In addition to the Phase IIb PATHWAY trial, the PATHFINDER programme included two Phase III trials, NAVIGATOR2,17 and SOURCE.18,19 The programme also includes additional mechanistic and long-term safety trials.20,21
NAVIGATOR is a Phase III, randomised, double-blinded, placebo-controlled trial in adults (18–80 years old) and adolescents (12–17 years old) with severe, uncontrolled asthma, who were receiving standard of care (SoC). SoC was treatment with medium- or high-dose inhaled corticosteroids plus at least one additional controller medication with or without daily OCS treatment. The trial population included approximately equal proportions of patients with high (≥300 cells per microlitre) and low (<300 cells per microlitre) blood eosinophil counts. The trial comprised a five-to-six-week screening period, a 52-week treatment period and a 12-week post-treatment follow-up period. All patients received their prescribed controller medications without change throughout the trial.2
The primary efficacy endpoint was the annualised asthma exacerbation rate (AAER) during the 52-week treatment period. Key secondary endpoints included the effect of Tezspire on lung function, asthma control and health-related quality of life.2
As part of prespecified analyses, the AAER over 52 weeks was also assessed in patients grouped by baseline blood eosinophil count, FeNO level and serum specific immunoglobin E (IgE) status (perennial aeroallergen sensitivity positive or negative).2 These are inflammatory biomarkers used by clinicians to inform treatment options and involve tests analysing a patient’s blood (eosinophils/IgE) and exhaled air (FeNO).
There were no clinically meaningful differences in safety results between the Tezspire and placebo groups in the NAVIGATOR trial.2 The most frequently reported adverse events for Tezspire were nasopharyngitis, upper respiratory tract infection and headache.2
NAVIGATOR is the first Phase III trial to show benefit in severe asthma irrespective of eosinophils by targeting TSLP.2 These results support the FDA Breakthrough Therapy Designation granted to Tezspire in September 2018 for patients with severe asthma, without an eosinophilic phenotype. In July 2021, Tezspire was the first and only biologic to be granted Priority Review in the US for the treatment of asthma by the FDA.22
Tezspire
Tezspire (tezepelumab) is being developed by AstraZeneca in collaboration with Amgen as a first-in-class human monoclonal antibody that inhibits the action of TSLP, a key epithelial cytokine that sits at the top of multiple inflammatory cascades and is critical in the initiation and persistence of allergic, eosinophilic and other types of airway inflammation associated with severe asthma, including airway hyperresponsiveness.3,4 TSLP is released in response to multiple triggers associated with asthma exacerbations, including allergens, viruses and other airborne particles.3,4 Expression of TSLP is increased in the airways of patients with asthma and has been correlated with disease severity.3,5 Blocking TSLP may prevent the release of pro-inflammatory cytokines by immune cells, resulting in the prevention of asthma exacerbations and improved asthma control.2,3,5 Tezspire acts at the top of the inflammation cascade and has the potential to help address a broad population of severe asthma patients irrespective of biomarker levels.2,3
Tezspire is approved in the US for the add-on maintenance treatment of adult and paediatric patients aged 12 years and older with severe asthma.27 Tezspire is also in development for other potential indications including chronic obstructive pulmonary disease, chronic rhinosinusitis with nasal polyps, chronic spontaneous urticaria and eosinophilic esophagitis (EoE). In October 2021, tezepelumab was granted Orphan Drug Designation by the FDA for the treatment of EoE.
Amgen collaboration
In 2020, Amgen and AstraZeneca updated a 2012 collaboration agreement for Tezspire. Both companies will continue to share costs and profits equally after payment by AstraZeneca of a mid single-digit inventor royalty to Amgen. AstraZeneca continues to lead development and Amgen continues to lead manufacturing. All aspects of the collaboration are under the oversight of joint governing bodies. Under the amended agreement, Amgen and AstraZeneca will jointly commercialise Tezspire in North America. Amgen will record product sales in the US, with AZ recording its share of US profits as Collaboration Revenue. Outside of the US, AstraZeneca will record product sales, with Amgen recording profit share as Other/Collaboration revenue.
AstraZeneca in Respiratory & Immunology
Respiratory & Immunology, part of BioPharmaceuticals, is one of AstraZeneca’s main disease areas and is a key growth driver for the Company.
AstraZeneca is an established leader in respiratory care with a 50-year heritage. The Company aims to transform the treatment of asthma and COPD by focusing on earlier biology-led treatment, eliminating preventable asthma attacks, and removing COPD as a top-three leading cause of death. The Company’s early respiratory research is focused on emerging science involving immune mechanisms, lung damage and abnormal cell-repair processes in disease and neuronal dysfunction.
With common pathways and underlying disease drivers across respiratory and immunology, AstraZeneca is following the science from chronic lung diseases to immunology-driven disease areas. The Company’s growing presence in immunology is focused on five mid- to late-stage franchises with multi-disease potential, in areas including rheumatology (including systemic lupus erythematosus), dermatology, gastroenterology, and systemic eosinophilic-driven diseases. AstraZeneca’s ambition in Respiratory & Immunology is to achieve disease modification and durable remission for millions of patients worldwide.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca
Sep. 27, 2022 4:47 AM ET
By: Ravikash, SA News Editor
Amgen (NASDAQ:AMGN) and AstraZeneca's (NASDAQ:AZN) Tezspire was approved in Japan for a broad population of patients with severe asthma with no phenotype or biomarker limitations.
https://seekingalpha.com/symbol/AZN
https://seekingalpha.com/symbol/AMGN
September 21, 2022Download PDF
Tumor-agnostic data supporting approval demonstrated an overall response rate (ORR) of 44% across multiple tumor types
FDA simultaneously grants traditional approval in adults with locally advanced or metastatic non-small cell lung cancer (NSCLC) with a RET gene fusion, as detected by an FDA-approved test
INDIANAPOLIS, Sept. 21, 2022 /PRNewswire/ -- Eli Lilly and Company (NYSE: LLY) today announced the U.S. Food and Drug Administration (FDA) has granted approval to Retevmo® (selpercatinib, 40 mg & 80 mg capsules) for adult patients with locally advanced or metastatic solid tumors with a rearranged during transfection (RET) gene fusion that have progressed on or following prior systemic treatment or who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on ORR and duration of response (DOR). Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trial.
"In the LIBRETTO-001 trial, selpercatinib demonstrated clinically meaningful and durable responses across a variety of tumor types in patients with RET-driven cancers, including pancreatic, colon and other cancers in need of new treatment options," said Vivek Subbiah, M.D., associate professor of Investigational Cancer Therapeutics at The University of Texas MD Anderson Cancer Center and co-investigator for LIBRETTO-001. "These data and FDA approval of the tumor-agnostic indication underscore the importance of routine, comprehensive genomic testing for patients across a wide variety of tumor types."
In addition to the tumor-agnostic approval, the FDA has granted traditional approval for Retevmo in adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with a RET gene fusion, as detected by an FDA-approved test. This FDA action broadens the Retevmo label to include patients with locally advanced disease and converts the May 2020 accelerated approval for NSCLC to a traditional approval.
The labeling for Retevmo contains warnings and precautions for hepatotoxicity (evidence of liver dysfunction), interstitial lung disease (ILD)/pneumonitis, hypertension, QT interval prolongation, hemorrhagic events, hypersensitivity, tumor lysis syndrome, risk of impaired wound healing, hypothyroidism, and embryo-fetal toxicity.
"Since its initial accelerated approval, Retevmo has shifted the treatment paradigm for patients with RET-altered cancers," said David Hyman, M.D., chief medical officer, Loxo@Lilly. "Retevmo is the first and only RET inhibitor to receive both tumor-agnostic accelerated approval and traditional approval in NSCLC, further supporting its ability to deliver meaningful clinical benefit for patients across diverse tumor types."
The two approvals are supported by data from the pivotal LIBRETTO-001 trial, which is the largest clinical trial of patients with RET-driven cancers treated with a RET inhibitor. The multicenter, open-label, multi-cohort study enrolled patients with locally advanced or metastatic RET-driven solid tumors, including NSCLC. Major efficacy outcomes were ORR and DOR, assessed by a blinded independent review committee (BIRC). Prespecified secondary endpoints included central nervous system (CNS) ORR and CNS DOR.
RET Fusion-Positive Solid Tumors
Among the 41 patients in the tumor-agnostic data set, the most common cancers were pancreatic adenocarcinoma (27%), colorectal (24%), salivary (10%), and unknown primary (7%). Thirty-seven patients (90%) received prior systemic therapy (median 2 [range 0 – 9]; 32% received 3 or more). Efficacy results are summarized below:
The activity of Retevmo in patients with CNS metastases was also evaluated. Among the 247 patients with previously treated RET fusion-positive NSCLC, 16 had measurable CNS metastases at baseline as assessed by BIRC. One patient received radiation therapy (RT) to the brain within two months prior to study entry. Responses in intracranial lesions were observed in 87.5% (14 of 16) of patients; 39% of responders had an intracranial DOR of 12 months or greater. Among the 69 patients with treatment-naïve RET fusion-positive NSCLC, five had measurable CNS metastases at baseline as assessed by BIRC. Two patients received RT to the brain within two months prior to study entry. Responses in intracranial lesions were observed in four of these five patients; 38% of responders had an intracranial DOR of 12 months or greater.
"Retevmo's accelerated approval played an important role in providing earlier access for patients who needed new treatment options. We are now pleased to see the conversion from an accelerated approval to a traditional approval," said Andrea Ferris, president and chief executive officer, LUNGevity Foundation. "As a targeted treatment, this traditional approval further reinforces the need for comprehensive biomarker testing for lung cancer patients, with the hope that as many patients as possible can benefit from receiving treatments tailored to their specific tumor mutations."
In the full LIBRETTO-001 safety population (n=796) with advanced solid tumors, the most common adverse reactions (≥25%) were edema, diarrhea, fatigue, dry mouth, hypertension, abdominal pain, constipation, rash, nausea, and headache. The most common Grade 3 or 4 laboratory abnormalities (≥5%) were decreased lymphocytes, increased alanine aminotransferase (ALT), increased aspartate aminotransferase (AST), decreased sodium, and decreased calcium. For more information, see "IMPORTANT SAFETY INFORMATION FOR RETEVMO® (selpercatinib)" below.
About LIBRETTO-001
The Phase 1/2 LIBRETTO-001 trial is the largest clinical trial of patients with RET-driven cancers treated with a RET inhibitor. The trial, which spans 16 countries and 85 sites, included a dose escalation phase (Phase 1) and a dose expansion phase (Phase 2). The primary objective was to determine ORR by blinded independent review committee (BIRC) and other objectives included DOR, CNS ORR & DOR, safety and PFS.
About Retevmo® (selpercatinib, 40 mg & 80 mg capsules)
Retevmo (selpercatinib, formerly known as LOXO-292) (pronounced ret-tév-mo) is a selective and potent RET kinase inhibitor. Retevmo may affect both tumor cells and healthy cells, which can result in side effects. RET-driver alterations are predominantly mutually exclusive from other oncogenic drivers. Retevmo is a U.S. FDA-approved oral prescription medicine, 120 mg or 160 mg dependent on weight (<50 kg or ≥50 kg, respectively), taken twice daily until disease progression or unacceptable toxicity.
Please see full Prescribing Information for Retevmo.
About Lilly
Lilly unites caring with discovery to create medicines that make life better for people around the world. We've been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world's most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer's disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we're motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn. P-LLY
Retevmo® is a registered trademark owned by or licensed to Eli Lilly and Company, its subsidiaries, or affiliates.
PP-SE-US-1166 09/2022
©Lilly USA, LLC 2022. ALL RIGHTS RESERVED.
![]()
View original content to download multimedia:https://www.prnewswire.com/news-releases/fda-approves-lillys-retevmo-selpercatinib-the-first-and-only-ret-inhibitor-for-adults-with-advanced-or-metastatic-solid-tumors-with-a-ret-gene-fusion-regardless-of-type-301630358.html
SOURCE Eli Lilly and Company
Sep. 22, 2022 6:58 AM ET
By: Dulan Lokuwithana, SA News Editor1 Comment
Eli Lilly and Company (NYSE:LLY) announced that the FDA greenlighted its oral lung cancer medication Retevmo under the agency’s accelerated approval for certain adult patients with solid tumors with a specific genetic makeup.
https://seekingalpha.com/news/3885170-lly-stock-gains-after-fda-approval-for-solid-tumor-therapy
https://seekingalpha.com/symbol/LLY
PUBLISHED23 September 2022
Ultomiris (ravulizumab) has been approved in Europe as an add-on to standard therapy for the treatment of adult patients with generalised myasthenia gravis (gMG) who are anti-acetylcholine receptor (AChR) antibody-positive.
This decision marks the first and only approval for a long-acting C5 complement inhibitor for the treatment of gMG in Europe. gMG is a rare, debilitating, chronic, autoimmune neuromuscular disease that leads to a loss of muscle function and severe weakness.1 The diagnosed prevalence of gMG in the EU is estimated at approximately 89,000.2-8
The approval by the European Commission follows the positive opinion of the Committee for Medicinal Products for Human Use and is based on results from the CHAMPION-MG Phase III trial, which were published online in NEJM Evidence. In the trial, Ultomiris was superior to placebo in the primary endpoint of change from baseline in the Myasthenia Gravis-Activities of Daily Living Profile (MG-ADL) total score at Week 26, a patient-reported scale that assesses patients’ abilities to perform daily activities.9 Additionally, in prolonged follow-up results from the open-label extension, clinical benefit of Ultomiris was observed through 60 weeks.9
Renato Mantegazza, Professor at the Department of Neuroimmunology and Neuromuscular Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy, and CHAMPION-MG trial investigator, said: “As physicians, we see first-hand how gMG can have a debilitating impact on quality of life. Today’s approval is a major advancement for treating gMG in Europe, offering patients and physicians a new, long-acting treatment option which has shown reliable efficacy and sustained improvements in activities of daily living.”
Marc Dunoyer, Chief Executive Officer, Alexion, said: “This approval in Europe of the first and only long-acting C5 inhibitor is an important step towards realising our vision of improving the lives of people living with gMG and increasing access to Ultomiris worldwide. Alexion’s pioneering leadership in complement science has affirmed C5 inhibition as a proven approach for managing this debilitating disease. We’re proud to offer a new treatment option that provides more convenience in dosing and has shown clinical benefit in a broader range of patients, including those who remain symptomatic despite their initial standard of care treatment.”
In CHAMPION-MG, the safety profile of Ultomiris was comparable to placebo and consistent with that observed in Phase III trials of Ultomiris in paroxysmal nocturnal haemoglobinuria (PNH) and atypical haemolytic uraemic syndrome (aHUS). The most common adverse reactions in patients receiving Ultomiris were diarrhoea, upper respiratory tract infection, nasopharyngitis and headache.9
Ultomiris was approved in the US in April 2022 and Japan in August 2022 for certain adults with gMG. Regulatory reviews are ongoing in additional countries.
CHAMPION-MG
The global Phase III randomised, double-blind, placebo-controlled, multicentre 26-week trial evaluated the safety and efficacy of Ultomiris in adults with gMG. The trial enrolled 175 patients across North America, Europe, Asia-Pacific, and Japan. Participants were required to have a confirmed myasthenia gravis diagnosis at least six months prior to the screening visit with a positive serologic test for anti-AChR antibodies, MG-ADL total score of at least 6 at trial entry and Myasthenia Gravis Foundation of America Clinical Classification Class II to IV at screening. Patients could stay on stable standard of care medicines, with a few exceptions, for the duration of the randomised control period.17
Patients were randomised 1:1 to receive Ultomiris or placebo for a total of 26 weeks. Patients received a single weight-based loading dose on Day 1, followed by regular weight-based maintenance dosing beginning on Day 15, every eight weeks. The primary endpoint of change from baseline in the MG-ADL total score at Week 26 was assessed along with multiple secondary endpoints evaluating improvement in disease-related and quality-of-life measures.
Patients who completed the randomised control period were eligible to continue into an open-label extension period evaluating the safety and efficacy of Ultomiris, which is ongoing.
Ultomiris
Ultomiris (ravulizumab), the first and only long-acting C5 complement inhibitor, offers immediate, complete and sustained complement inhibition. The medication works by inhibiting the C5 protein in the terminal complement cascade, a part of the body’s immune system. When activated in an uncontrolled manner, the complement cascade over-responds, leading the body to attack its own healthy cells. Ultomiris is administered intravenously every eight weeks in adult patients, following a loading dose.
Ultomiris is approved in the US, EU and Japan for the treatment of certain adults with gMG.
Ultomiris is also approved in the US, EU and Japan for the treatment of certain adults with PNH and for certain children with PNH in the US and EU.
Additionally, Ultomiris is approved in the US, EU and Japan for certain adults and children with aHUS to inhibit complement-mediated thrombotic microangiopathy.
As part of a broad development programme, Ultomiris is being assessed for the treatment of additional haematology and neurology indications.
Alexion
Alexion, AstraZeneca Rare Disease, is the group within AstraZeneca focused on rare diseases, created following the 2021 acquisition of Alexion Pharmaceuticals, Inc. As a leader in rare diseases for nearly 30 years, Alexion is focused on serving patients and families affected by rare diseases and devastating conditions through the discovery, development, and commercialisation of life-changing medicines. Alexion focuses its research efforts on novel molecules and targets in the complement cascade and its development efforts on haematology, nephrology, neurology, metabolic disorders, cardiology, and ophthalmology. Headquartered in Boston, Massachusetts, Alexion has offices around the globe and serves patients in more than 50 countries.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Sep. 23, 2022 4:29 AM ET
By: Ravikash, SA News Editor
The European Commission (EC) approved the expanded use of AstraZeneca's (NASDAQ:AZN) Ultomiris (ravulizumab) as an add-on to standard therapy for adult patients with generalized myasthenia gravis (gMG) who are anti-acetylcholine receptor (AChR) antibody-positive.
https://seekingalpha.com/symbol/AZN
PUBLISHED21 September 2022 21 September 2022 07:00 BST
AstraZeneca’s Tezspire (tezepelumab) has been approved in the European Union (EU) as an add-on maintenance treatment in patients 12 years and older with severe asthma who are inadequately controlled with high dose inhaled corticosteroids plus another medicinal product.
The approval by the European Commission was based on results from the PATHFINDER clinical trial programme, which included the pivotal NAVIGATOR Phase III trial in which Tezspire demonstrated superiority across every primary and key secondary endpoint in patients with severe asthma, compared to placebo, when added to standard therapy.1 The approval follows the recommendation by The Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency in July 2022.
Tezspire is the first and only biologic approved in Europe for severe asthma that acts at the top of the inflammatory cascade by blocking thymic stromal lymphopoietin (TSLP), an epithelial cytokine.1-4 Tezspire consistently and significantly reduced asthma exacerbations across the PATHWAY Phase II and the NAVIGATOR Phase III clinical trials, which included a broad population of severe asthma patients irrespective of key biomarkers, including blood eosinophil counts, allergic status and fractional exhaled nitric oxide (FeNO).1,2
Professor Guy Brusselle, Department of Respiratory Medicine, Ghent University Hospital, Ghent, Belgium, said: “Severe asthma is a complex disease given approximately 60% of patients have multiple drivers of inflammation. With the European approval of Tezspire, a first-in-class biologic acting at the top of the inflammation cascade, we have an opportunity to treat a broader population of patients with severe asthma, fulfilling a high unmet need in this disease."
Mene Pangalos, Executive Vice President, BioPharmaceuticals R&D, AstraZeneca, said: “Severe asthma continues to have a debilitating impact for people living with the disease, with many patients experiencing frequent exacerbations, an increased risk of hospitalisation and a significantly reduced quality of life. Tezspire is now the first and only biologic approved in Europe for patients with severe asthma with no phenotype or biomarker limitation and we look forward to bringing this important medicine to patients as quickly as possible.”
In clinical trials, the most common adverse events in patients who received Tezspire were pharyngitis, rash, arthralgia and injection site reactions.5
Results from the NAVIGATOR Phase III trial were published in The New England Journal of Medicine in May 2021.
Tezspire has been approved in the US and other countries for the treatment of severe asthma, and regulatory reviews are ongoing in additional countries around the world.6
Clinical trials
In addition to the Phase IIb PATHWAY trial, the PATHFINDER programme included two Phase III trials, NAVIGATOR1,19 and SOURCE.20,21 The programme also includes additional mechanistic and long-term safety trials.22,23
NAVIGATOR is a Phase III, randomised, double-blinded, placebo-controlled trial in adults (18–80 years old) and adolescents (12–17 years old) with severe, uncontrolled asthma, who were receiving standard of care (SoC). SoC was treatment with medium- or high-dose inhaled corticosteroids plus at least one additional controller medication with or without daily OCS treatment. The trial population included approximately equal proportions of patients with high (≥300 cells per microlitre) and low (<300 cells per microlitre) blood eosinophil counts. The trial comprised a five-to-six-week screening period, a 52-week treatment period and a 12-week post-treatment follow-up period. All patients received their prescribed controller medications without change throughout the trial.1
The primary efficacy endpoint was the annualised asthma exacerbation rate (AAER) during the 52-week treatment period. Key secondary endpoints included the effect of Tezspire on lung function, asthma control and health-related quality of life.1
As part of prespecified analyses, the AAER over 52 weeks was also assessed in patients grouped by baseline blood eosinophil count, FeNO level and serum specific immunoglobin E (IgE) status (perennial aeroallergen sensitivity positive or negative).1 These are inflammatory biomarkers used by clinicians to inform treatment options and involve tests analysing a patient’s blood (eosinophils/IgE) and exhaled air (FeNO).
There were no clinically meaningful differences in safety results between the Tezspire and placebo groups in the NAVIGATOR trial.1 The most frequently reported adverse events for Tezspire were nasopharyngitis, upper respiratory tract infection and headache.1
NAVIGATOR is the first Phase III trial to show benefit in severe asthma irrespective of eosinophils by targeting TSLP.1 These results support the FDA Breakthrough Therapy Designation granted to Tezspire in September 2018 for patients with severe asthma, without an eosinophilic phenotype. In July 2021, Tezspire was the first and only biologic to be granted Priority Review in the US for the treatment of asthma by the FDA.
Tezspire
Tezspire (tezepelumab) is being developed by AstraZeneca in collaboration with Amgen as a first-in-class human monoclonal antibody that inhibits the action of TSLP, a key epithelial cytokine that sits at the top of multiple inflammatory cascades and is critical in the initiation and persistence of allergic, eosinophilic and other types of airway inflammation associated with severe asthma, including airway hyperresponsiveness.2,3 TSLP is released in response to multiple triggers associated with asthma exacerbations, including allergens, viruses and other airborne particles.2,3 Expression of TSLP is increased in the airways of patients with asthma and has been correlated with disease severity.2,4 Blocking TSLP may prevent the release of pro-inflammatory cytokines by immune cells, resulting in the prevention of asthma exacerbations and improved asthma control.1,2,4 Tezspire acts at the top of the inflammation cascade and has the potential to help address a broad population of severe asthma patients irrespective of biomarker levels.1,2
Tezspire is approved in the US for the add-on maintenance treatment of adult and paediatric patients aged 12 years and older with severe asthma.5 Tezspire is also in development for other potential indications including chronic obstructive pulmonary disease (COPD), chronic rhinosinusitis with nasal polyps, chronic spontaneous urticaria and eosinophilic esophagitis (EoE). In October 2021, tezepelumab was granted Orphan Drug Designation by the FDA for the treatment of EoE.
Amgen collaboration
In 2020, Amgen and AstraZeneca updated a 2012 collaboration agreement for Tezspire. Both companies will continue to share costs and profits equally after payment by AstraZeneca of a mid single-digit inventor royalty to Amgen. AstraZeneca continues to lead development and Amgen continues to lead manufacturing. All aspects of the collaboration are under the oversight of joint governing bodies. Under the amended agreement, Amgen and AstraZeneca will jointly commercialise Tezspire in North America. Amgen will record product sales in the US, with AZ recording its share of US profits as Collaboration Revenue. Outside of the US, AstraZeneca will record product sales, with Amgen recording profit share as Other/Collaboration revenue.
AstraZeneca in Respiratory & Immunology
Respiratory & Immunology, part of BioPharmaceuticals, is one of AstraZeneca’s main disease areas and is a key growth driver for the Company.
AstraZeneca is an established leader in respiratory care with a 50-year heritage. The Company aims to transform the treatment of asthma and COPD by focusing on earlier biology-led treatment, eliminating preventable asthma attacks, and removing COPD as a top-three leading cause of death. The Company’s early respiratory research is focused on emerging science involving immune mechanisms, lung damage and abnormal cell-repair processes in disease and neuronal dysfunction.
With common pathways and underlying disease drivers across respiratory and immunology, AstraZeneca is following the science from chronic lung diseases to immunology-driven disease areas. The Company’s growing presence in immunology is focused on five mid- to late-stage franchises with multi-disease potential, in areas including rheumatology (including systemic lupus erythematosus), dermatology, gastroenterology, and systemic eosinophilic-driven diseases. AstraZeneca’s ambition in Respiratory & Immunology is to achieve disease modification and durable remission for millions of patients worldwide.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca
Sep. 21, 2022 4:27 AM ET
By: Ravikash, SA News Editor
22 September 2022
Issued: London UK
For media and investors only
GSK plc (LSE/NYSE: GSK) today announced that the US Food and Drug Administration (FDA) will convene a meeting of the Oncologic Drugs Advisory Committee (ODAC) to discuss overall survival (OS) data from the ENGOT-OV16/NOVA phase III clinical trial. NOVA is a randomised, double-blind, placebo-controlled phase III trial of Zejula (niraparib), an oral, once-daily poly (ADP-ribose) polymerase (PARP) inhibitor for the maintenance treatment of women with platinum-sensitive recurrent ovarian cancer.
The phase III NOVA trial met the primary endpoint of progression-free survival (PFS) in both the gBRCAm and non-gBRCAm cohorts, demonstrating a statistically significant and clinically meaningful treatment effect of Zejula in this patient population, regardless of biomarker status. These PFS results served as the primary basis for the US FDA approval for the maintenance treatment of women with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in complete or partial response to platinum-based chemotherapy. Overall survival was a secondary endpoint. Updated final overall survival data was recently shared with the FDA.
Hesham Abdullah, SVP, Global Head of Oncology Development, GSK said: “We believe PARP inhibitors, including Zejula, are important options for the maintenance treatment of patients with recurrent ovarian cancer, across all biomarker subgroups, who are in complete or partial response to platinum-based chemotherapy. We look forward to continuing our ongoing discussions with the FDA.”
The ODAC meeting is scheduled for 22 November 2022. This is not related to the niraparib indication in the maintenance treatment of adult patients with advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to first-line platinum-based chemotherapy.
About ovarian cancer
Ovarian cancer is the eighth most common cancer in women worldwide. [1] Despite high response rates to platinum-based chemotherapy in the front-line setting, approximately 85% of patients will experience disease recurrence.[2] Once the disease recurs, it is rarely curable, with decreasing time intervals to each subsequent recurrence.
About Zejula (niraparib)
Zejula is an oral, once-daily PARP inhibitor currently being evaluated in multiple pivotal trials. GSK is building a robust clinical development programme by assessing activity across multiple tumour types and evaluating several potential combinations of Zejula with other therapeutics. The ongoing development programme includes several combination studies.
ZEJULA is indicated:
Please see accompanying US Prescribing Information.
About GSK
GSK is a global biopharma company with a purpose to unite science, technology, and talent to get ahead of disease together. Find out more at gsk.com/company.
Sep. 23, 2022 5:04 AM ET
By: Ravikash, SA News Editor
GSK (NYSE:GSK) said on Sept. 22 that a panel of the U.S. Food and Drug Administration (FDA) is scheduled to hold a meeting on Nov. 22 to discuss overall survival (OS) data from a phase 3 trial of Zejula (niraparib) for recurrent ovarian cancer.
https://seekingalpha.com/symbol/GSK
Sep 20, 2022
– AMVUTTRA Demonstrated Halting or Reversal in Neuropathy Impairment with Subcutaneous Administration Once Every Three Months –
– Decision Follows Positive Opinion from the European Medicines Agency (EMA) Committee for Medicinal Products for Human Use (CHMP) in July 2022 –
CAMBRIDGE, Mass.--(BUSINESS WIRE)--Sep. 20, 2022-- Alnylam Pharmaceuticals, Inc. (Nasdaq: ALNY), the leading RNAi therapeutics company, today announced that the European Commission (EC) has granted marketing authorization for AMVUTTRA® (vutrisiran), an RNAi therapeutic for the treatment of hereditary transthyretin-mediated (hATTR) amyloidosis in adult patients with stage 1 or stage 2 polyneuropathy. The EC approval is based on positive 18-month results from the HELIOS-A Phase 3 study, where AMVUTTRA significantly improved the signs and symptoms of hATTR amyloidosis, with more than 50 percent of patients experiencing halting or reversal of their polyneuropathy manifestations.
“On behalf of patients across Europe, we could not be prouder to announce the approval of AMVUTTRA, our second RNAi therapeutic for hATTR amyloidosis. We are committed to delivering continued innovation to people affected by rare, life-limiting conditions where high unmet need still exists. Today’s decision now means we can progress working with health authorities across Europe to achieve responsible and sustainable access arrangements that allow us to bring AMVUTTRA to patients as quickly as possible. Our thanks go to the patients, families, investigators and study staff who have enabled us to reach this significant milestone,” said Kasha Witkos, SVP, Head International Region at Alnylam.
“Although the considerable research into hATTR amyloidosis over the past few years has resulted in a more positive outlook for those diagnosed with the condition, there remain unmet needs in treatment for patients living with this rapidly progressive, multi-system disease”, said Professor David Adams, Head of the Neurology department at Bicêtre hospital AP-HP, University of Paris-Saclay and Lead Investigator for the HELIOS-A Study. “RNAi therapeutics are changing the future of medicine and I am honored to have contributed to these research efforts that have enabled us to bring an innovative new medicine to patients. Results from the HELIOS-A study have shown the potential AMVUTTRA has to benefit patients with hATTR amyloidosis with stage 1 or stage 2 polyneuropathy, whilst also helping reduce treatment burden through subcutaneous dosing once every three months.”
HELIOS-A was a global, randomized, open-label, multicenter, Phase 3 study that evaluated the efficacy and safety of AMVUTTRA across a diverse group of patients with hATTR amyloidosis with stage 1 or stage 2 polyneuropathy. Results of the HELIOS-A study were published in Amyloid in July, 2022.
In the HELIOS-A study, AMVUTTRA met the primary and all secondary endpoints of the study at both 9 months and 18 months, demonstrating reversal in neuropathy impairment and an encouraging safety and tolerability profile. AMVUTTRA demonstrated improvement in the mean change from baseline in modified Neuropathy Impairment Score + 7 (mNIS+7) at 18 months (the primary endpoint for the EU), as compared to external placebo data from the landmark APOLLO Phase 3 study of patisiran.
After 18 months of dosing, the most frequently occurring adverse reactions in AMVUTTRA-treated patients were arthralgia (joint stiffness) and pain in extremity (pain in arms and legs). Other less frequent adverse reactions reported with AMVUTTRA were dyspnea (shortness of breath), injection site reaction and an increase in blood alkaline phosphatase (a liver enzyme).
Vutrisiran was previously granted Orphan Drug Designation in the European Union (EU) and U.S. for the treatment of ATTR amyloidosis and in Japan for transthyretin type familial amyloidosis with polyneuropathy. In June 2022, AMVUTTRA was approved by the U.S. Food and Drug Administration (FDA) for the treatment of the polyneuropathy of hATTR amyloidosis in adults. Vutrisiran is under review by the Brazilian Health Regulatory Agency (ANVISA) and the Japanese Pharmaceuticals and Medical Devices Agency (PMDA).
About AMVUTTRA® (vutrisiran)
AMVUTTRA is an RNAi therapeutic, which is designed to target and silence specific messenger RNA (mRNA), blocking the production of wild-type and variant transthyretin (TTR) protein before it is made. AMVUTTRA utilizes Alnylam’s Enhanced Stabilization Chemistry (ESC)-GalNAc-conjugate delivery platform, designed for increased potency and high metabolic stability to allow for quarterly, and potentially biannual, subcutaneous administration. Vutrisiran is also in development for the treatment of ATTR amyloidosis with cardiomyopathy, which encompasses both hereditary ATTR (hATTR) and wild-type ATTR (wtATTR) amyloidosis.
About Alnylam Pharmaceuticals
Alnylam (Nasdaq: ALNY) is leading the translation of RNA interference (RNAi) into a whole new class of innovative medicines with the potential to transform the lives of people afflicted with rare genetic, cardio-metabolic, hepatic infectious, and central nervous system (CNS)/ocular diseases. Based on Nobel Prize-winning science, RNAi therapeutics represent a powerful, clinically validated approach for the potential treatment of a wide range of severe and debilitating diseases. Founded in 2002, Alnylam is delivering on a bold vision to turn scientific possibility into reality, with a robust RNAi therapeutics platform. Alnylam’s has developed RNAi therapeutic products which are licensed for the treatment of hATTR amyloidosis, acute hepatic porphyria, primary hyperoxaluria type 1 and primary hypercholesterolemia / mixed dyslipidemia. Alnylam has a deep pipeline of investigational medicines, including six product candidates that are in late-stage development. Alnylam is executing on its “Alnylam P5x25” strategy to deliver transformative medicines in both rare and common diseases benefiting patients around the world through sustainable innovation and exceptional financial performance, resulting in a leading biotech profile. Alnylam is headquartered in Cambridge, MA.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220920005241/en/
Source: Alnylam Pharmaceuticals, Inc.
Sep. 20, 2022 7:20 AM ET
Alnylam Pharmaceuticals, Inc. (ALNY)
By: Ravikash, SA News Editor
Sep 20, 2022 at 12:45 PM EDTDownload PDF
Combined clinical trial and real-world data with six years of follow-up provides important evidence of efficacy
Analysis compares findings from the Phase 3 POISE trial and open-label extension to natural history data from the Global PBC and UK-PBC patient registries
MORRISTOWN, N.J., Sept. 20, 2022 (GLOBE NEWSWIRE) -- Intercept Pharmaceuticals, Inc. (Nasdaq: ICPT), a biopharmaceutical company focused on the development and commercialization of novel therapeutics to treat progressive non-viral liver diseases, today announced a key publication in Gastroenterology showing that people receiving OCA for primary biliary cholangitis (PBC) in a clinical trial setting had greater transplant-free survival compared to patients with PBC selected from “real-world” patient registries who did not receive OCA.
Key findings include:
“This important study is the first to demonstrate that initiating treatment with OCA in appropriate patients with PBC appears to have a meaningful impact on clinical outcomes,” said Professor Gideon Hirschfield, FRCP, Ph.D., Lily and Terry Horner Chair in Autoimmune Liver Disease at the University of Toronto. “I am proud of this innovative analysis, and further, the continued value we are deriving from large, academic-led, prospective databases like the Global PBC and UK-PBC study groups. Leveraging these registries helps advance our collective understanding of this disease to ultimately influence clinical decision-making and benefit people living with PBC.”
“Given the known challenges associated with conducting blinded, placebo-controlled trials when the treatment has been approved and is available, this study provides evidence that a well-matched external comparator group can be a credible, alternate approach to evaluating clinical efficacy,” said M. Michelle Berrey, M.D., M.P.H., President of Research & Development and Chief Medical Officer of Intercept. “We look forward to further leveraging real-world databases to continue generating insights on the clinical benefit of Ocaliva in PBC.”
This study compared patients with PBC who received up to six years of OCA in the Phase 3 POISE study and its open-label extension (n=209) to external controls who were extracted from two large, academic-led patient registries. External controls met POISE entry criteria but were not treated with OCA. Treatment with ursodeoxycholic acid (UDCA) was permitted.
The primary endpoint of this study was time to first occurrence of liver transplant or death. Over the six-year follow-up, there were five deaths/liver transplants in 209 subjects in the POISE study, 135 in 1,381 patients in the Global PBC control, and 281 in 2,135 patients in the UK-PBC control. Preliminary results from this analysis were first presented at the 2021 Annual Meeting of the American Association for the Study of Liver Diseases (AASLD).
Data Sources for this Study:
The Global PBC registry included 6,484 patients with PBC from eight countries in Europe and North America during the study period who were not treated with OCA. Of these, 1,381 met the entry criteria for this study.
The UK-PBC registry included over 6,900 patients with PBC from the UK during the study period who were not treated with OCA. Of these, 2,135 met the entry criteria for this study.
The POISE trial studied the safety and efficacy of once-daily treatment with Ocaliva in PBC patients with an inadequate therapeutic response to, or who were unable to tolerate, ursodeoxycholic acid (UDCA). There were 217 patients randomized to one of three groups in the trial: placebo, OCA 10 mg, or OCA 5 mg for six months titrated to 10 mg based on clinical response. Seven subjects did not participate in the open-label extension and were not included in the current study. Patients completing the double-blind phase had the option to continue in an open-label extension (OLE) phase for a maximum of five additional years, during which all patients received treatment with OCA 5-10 mg once daily. Of the 198 patients who completed the double-blind phase, more than 95 percent continued in the long-term safety extension phase of the trial. Additional information regarding the POISE trial can be found on the NIH clinical study listing website: http://clinicaltrials.gov/ct2/show/NCT01473524.
About Primary Biliary Cholangitis
Primary biliary cholangitis (PBC) is a rare, progressive and chronic autoimmune disease that affects the bile ducts in the liver and is most prevalent (approximately 1 in 10,000) in women over the age of 40. PBC causes bile acids to build up in the liver, resulting in inflammation and scarring (fibrosis), which, if left untreated, can lead to cirrhosis, liver transplant, or death.
About Intercept
Intercept is a biopharmaceutical company focused on the development and commercialization of novel therapeutics to treat progressive non-viral liver diseases, including primary biliary cholangitis (PBC) and nonalcoholic steatohepatitis (NASH). For more information, please visit www.interceptpharma.com or connect with the company on Twitter and LinkedIn.
About Ocaliva® (obeticholic acid)
OCALIVA, a farnesoid X receptor (FXR) agonist, is indicated for the treatment of adult patients with primary biliary cholangitis (PBC).
This indication is approved under accelerated approval based on a reduction in alkaline phosphatase (ALP). An improvement in survival or disease-related symptoms has not been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
Source: Intercept Pharmaceuticals, Inc.
Sep. 20, 2022 2:10 PM ET
Intercept Pharmaceuticals, Inc. (ICPT)
By: Jonathan Block, SA News Editor
PUBLISHED22 September 2022 22 September 2022 07:00 BST
AstraZeneca and MSD’s Lynparza (olaparib) has been approved in China for the maintenance treatment of adult patients with advanced epithelial ovarian, fallopian tube or primary peritoneal cancer who are in complete or partial response to 1st-line platinum-based chemotherapy in combination with bevacizumab, and whose cancer is associated with homologous recombination deficiency (HRD)-positive status.
In China, ovarian cancer is the third most common gynaecologic cancer, with a five-year survival rate of approximately 39%, largely because more than 70% of women are diagnosed with advanced disease (Stage III or IV).1,2 In 2020, there were over 55,000 new cases of ovarian cancer in China.3
The approval by China's National Medical Products Administration was based on an HRD-positive subgroup exploratory analysis of the PAOLA-1 Phase III trial which showed Lynparza plus bevacizumab demonstrated a substantial progression-free survival (PFS) improvement versus bevacizumab alone for patients with HRD-positive advanced ovarian cancer. During European Society for Medical Oncology Congress (ESMO) 2022, the final overall survival (OS) results were presented from the PAOLA-1 Phase III trial demonstrating that Lynparza plus bevacizumab provided a clinically meaningful improvement in overall survival in HRD-positive advanced ovarian cancer.
Professor Ding Ma, Member of the Chinese Academy of Engineering, said: “Ovarian cancer has the highest fatality rate among gynaecologic cancers in China. The emergence of PARP inhibitors and their application in the 1st-line treatment of ovarian cancer could help patients delay disease progression and achieve long-term remission. In the PAOLA-1 trial, the combination of olaparib and bevacizumab demonstrated clinically meaningful improvements in overall survival. This approval provides HRD-positive patients with a new option for 1st-line maintenance therapy.”
Professor Beihua Kong, Chairman of the Gynaecological Oncology Branch of the Chinese Medical Association, said: "Ovarian cancer has entered the era of precision medicine, and HRD detection (including BRCA1/2 mutations) has important clinical value for newly diagnosed patients with advanced ovarian cancer, to help guide first-line treatment decisions. The approval of the combination of olaparib and bevacizumab brings a clinically meaningful survival benefit to HRD-positive patients, and further reflects the importance of a precision approach to help guide treatment decisions in ovarian cancer."
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “The maintenance treatment of Lynparza in combination with bevacizumab has shown to both improve progression-free survival and provide a clinically meaningful improvement in overall survival in patients with HRD-positive advanced ovarian cancer following response to platinum-based chemotherapy. I am thrilled we can now bring this targeted treatment option to these patients in China.”
Dr Eliav Barr, Senior Vice President, Head of Global Clinical Development and Chief Medical Officer, MSD Research Laboratories, said: “This approval is an important milestone for patients with newly diagnosed advanced ovarian cancer in China and underscores the critical importance of HRD testing for all women with advanced ovarian cancer at the point of diagnosis.”
The initial results from the PAOLA-1 Phase III trial showed that Lynparza plus bevacizumab reduced the risk of disease progression or death by 67% in the subgroup of patients with HRD-positive advanced ovarian cancer (based on a hazard ratio [HR] of 0.33; 95% confidence interval [CI] 0.25-0.45 from the pre-specified exploratory analysis). The primary endpoint in the intent-to-treat population was a statistically significant and clinically meaningful improvement in PFS. The data from the PAOLA-1 trial was published in The New England Journal of Medicine in 2019.
Further results from the five-year analysis of the PAOLA-1 trial recently presented at the ESMO 2022 showed Lynparza plus bevacizumab increased median overall survival to 56.5 months versus 51.6 months with bevacizumab alone, in patients with newly diagnosed advanced ovarian cancer irrespective of HRD status. This increase was not statistically significant. In HRD-positive patients, Lynparza plus bevacizumab provided a clinically meaningful improvement in overall survival, reducing the risk of death by 38% versus bevacizumab (based on a HR of 0.62; 95% CI 0.45-0.85 from the pre-specified exploratory sub-group analysis) despite PAOLA-1 having 30% Stage IV patients. The safety and tolerability profile of Lynparza in this trial was in line with that observed in prior clinical trials, with no new safety signals.
Lynparza in combination with bevacizumab is approved in the US, and several other countries as a 1st-line maintenance treatment for patients with HRD-positive advanced ovarian cancer and is currently under regulatory review in other countries around the world. In China, Lynparza is approved for the treatment of BRCA-mutated metastatic castration-resistant prostate cancer as well as a 1st-line maintenance therapy in BRCA-mutated advanced ovarian cancer.
PAOLA-1
PAOLA-1 is a double-blinded Phase III trial testing the efficacy and safety of Lynparza added to standard of care bevacizumab versus bevacizumab alone, as a 1st-line maintenance treatment for newly diagnosed advanced FIGO Stage III-IV high-grade serous or endometroid ovarian, fallopian tube, or peritoneal cancer patients who had a complete or partial response to 1st-line treatment with platinum-based chemotherapy and bevacizumab. AstraZeneca and MSD announced in August 2019 that the trial met its primary endpoint of PFS in the overall trial population.
The primary analysis of the PAOLA-1 Phase III trial showed that Lynparza, in combination with bevacizumab maintenance treatment, reduced the risk of disease progression or death by 67% (based on a HR of 0.33; 95% CI 0.25-0.45) in the subgroup of patients with HRD-positive advanced ovarian cancer. The addition of Lynparza improved PFS to a median of 46.8 months versus 17.6 with bevacizumab alone in this patient population.
Updated results from the five-year analysis of the PAOLA-1 Phase III trial demonstrate that in a pre-specified exploratory subgroup analysis of HRD-positive patients, Lynparza plus bevacizumab provided a clinically meaningful improvement in overall survival, reducing the risk of death by 38% versus bevacizumab (based on a HR of 0.62; 95% CI 0.45-0.85). In addition, 65.5% of patients treated with Lynparza plus bevacizumab were still alive at five years versus 48.4% of those treated with bevacizumab alone. Lynparza plus bevacizumab also improved median PFS to almost four years (46.8 months) versus 17.6 months with bevacizumab plus placebo, and 46.1% of patients treated with Lynparza plus bevacizumab remain progression free at five years versus 19.2% of patients treated with bevacizumab alone.
PAOLA-1 is an ENGOT (European Network of Gynaecological Oncological Trial groups) trial, sponsored by ARCAGY Research (Association de Recherche sur les CAncers dont GYnécologiques) on behalf of GINECO (Groupe d’Investigateurs National des Etudes des Cancers Ovariens et du sein), lead group for the PAOLA-1 trial. ARCAGY-GINECO is an academic group specialising in clinical and translational research in patients’ gynaecological cancers, labelled by the French National Cancer Institute (INCa), and a member of the GCIG (Gynecologic Cancer InterGroup).
Homologous recombination deficiency
HRD, which defines a subgroup of ovarian cancer, encompasses a wide range of genetic abnormalities, including BRCA mutations and beyond. As with BRCA gene mutations, HRD interferes with normal cell DNA repair mechanisms and confers sensitivity to PARP inhibitors including Lynparza.13
Lynparza
Lynparza (olaparib) is a 1st-in-class PARP inhibitor and the first targeted treatment to block DNA damage response (DDR) in cells/tumours harbouring a deficiency in homologous recombination repair (HRR), such as mutations in BRCA1 and/or BRCA2. Inhibition of PARP with Lynparza leads to the trapping of PARP bound to DNA single-strand breaks, stalling of replication forks, their collapse and the generation of DNA double-strand breaks and cancer cell death. Lynparza is being tested in a range of PARP-dependent tumour types with defects and dependencies in the DDR pathway.
Lynparza is currently approved in a number of countries across multiple tumour types including maintenance treatment of platinum-sensitive relapsed ovarian cancer and as both monotherapy and in combination with bevacizumab for the 1st-line maintenance treatment of BRCA-mutated (BRCAm) and HRD-positive advanced ovarian cancer, respectively; for gBRCAm, HER2-negative metastatic breast cancer (in the EU and Japan this includes locally advanced breast cancer); for gBRCAm, HER2-negative high-risk early breast cancer (in Japan this includes all BRCAm HER2-negative high-risk early breast cancer); for gBRCAm metastatic pancreatic cancer; and HRR gene-mutated metastatic castration-resistant prostate cancer (BRCAm only in the EU and Japan). In China, Lynparza is approved for the treatment of BRCA-mutated metastatic castration-resistant prostate cancer as well as a 1st-line maintenance therapy in BRCA-mutated advanced ovarian cancer.
Lynparza, which is being jointly developed and commercialised by AstraZeneca and MSD, has been used to treat over 75,000 patients worldwide. Lynparza has a broad clinical trial development programme, and AstraZeneca and MSD are working together to understand how it may affect multiple PARP-dependent tumours as a monotherapy and in combination across multiple cancer types. Lynparza is the foundation of AstraZeneca's industry-leading portfolio of potential new medicines targeting DDR mechanisms in cancer cells.
The AstraZeneca and MSD strategic oncology collaboration
In July 2017, AstraZeneca and Merck & Co., Inc., Kenilworth, NJ, US, known as MSD outside the US and Canada, announced a global strategic oncology collaboration to co-develop and co-commercialise Lynparza, the world’s first PARP inhibitor, and Koselugo (selumetinib), a MEK inhibitor, for multiple cancer types. Working together, the companies will develop Lynparza and Koselugo in combination with other potential new medicines and as monotherapies. Independently, the companies will develop Lynparza and Koselugo in combination with their respective PD-L1 and PD-1 medicines.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
By harnessing the power of six scientific platforms – Immuno-Oncology, Tumour Drivers and Resistance, DNA Damage Response, Antibody Drug Conjugates, Epigenetics, and Cell Therapies – and by championing the development of personalised combinations, AstraZeneca has the vision to redefine cancer treatment and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development and commercialisation of prescription medicines, primarily for the treatment of diseases in three therapy areas - Oncology, Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Sep. 22, 2022 4:23 AM ET
By: Ravikash, SA News Editor1 Comment
Sep 20, 2022
ACTON, Mass.--(BUSINESS WIRE)--Sep. 20, 2022-- Insulet Corporation (NASDAQ: PODD) (Insulet or the Company), the global leader in tubeless insulin pump technology with its Omnipod® brand of products, today announced it has received CE marking under the European Medical Device Regulation for its Omnipod 5 Automated Insulin Delivery System (Omnipod 5) for individuals aged two years and older with type 1 diabetes.
“There is tremendous enthusiasm for Omnipod 5 from the global diabetes community, and we are thrilled to have achieved this latest milestone,” said Jim Hollingshead, President and CEO of Insulet. “With Omnipod 5, customers can enjoy the lifestyle benefits of a tubeless, wearable innovation and achieve positive clinical outcomes while managing their diabetes.”
Omnipod 5 is the first CE marked tubeless hybrid closed loop system (also known as automated insulin delivery) that integrates with the Dexcom G6 Continuous Glucose Monitoring (CGM) System to automatically adjust insulin and help protect against high and low glucose levels 1. The system2 consists of the tubeless Pod enhanced with SmartAdjustTM technology, the Omnipod 5 Controller with its integrated SmartBolus Calculator, and the Dexcom G6 CGM.
Every five minutes, SmartAdjust technology receives a CGM value and trend, and predicts where glucose will be 60 minutes into the future. The system then increases, decreases, or pauses insulin delivery based on the user’s desired and customized glucose target.
"Today’s announcement is a significant step forward for people in Europe living with diabetes,” said Kevin Sayer, Chairman, President and CEO of Dexcom. “We’re proud to partner with Insulet, combining our industry-leading Dexcom G6 CGM technology with their tubeless, wearable insulin delivery system, to improve clinical outcomes and reduce the burden of glucose management for people with diabetes.”
New clinical data demonstrating the safety and effectiveness of Omnipod 5 on recently diagnosed people with type 1 diabetes will be presented at the upcoming European Association for the Study of Diabetes (EASD) meeting in Stockholm, Sweden on Thursday, September 22. To learn more and to register for EASD, visit the conference website.
Insulet expects Omnipod 5 to be available in select countries starting mid-2023. To learn more about Omnipod 5, visit the Omnipod website.
About Insulet Corporation:
Insulet Corporation (NASDAQ: PODD), headquartered in Massachusetts, is an innovative medical device company dedicated to simplifying life for people with diabetes and other conditions through its Omnipod product platform. The Omnipod Insulin Management System provides a unique alternative to traditional insulin delivery methods. With its simple, wearable design, the disposable Pod provides up to three days of non-stop insulin delivery, without the need to see or handle a needle. Insulet’s latest innovation, the Omnipod 5 Automated Insulin Delivery System, is a tubeless automated insulin delivery system, integrated with a continuous glucose monitor to manage blood sugar with no multiple daily injections and zero fingersticks3. Insulet also leverages the unique design of its Pod by tailoring its Omnipod technology platform for the delivery of non-insulin subcutaneous drugs across other therapeutic areas. For more information, please visit: insulet.com and omnipod.com.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220920005138/en/
Source: Insulet Corporation
Sep. 20, 2022 9:52 AM ET
By: Ravikash, SA News Editor1 Comment

SEP 20, 2022
The LINQ II ICM is the first continuous, long-term cardiac monitor cleared by FDA for the pediatric patient population
DUBLIN, Sept. 20, 2022 /PRNewswire(opens new window)/ -- Medtronic plc (NYSE:MDT), a global leader in healthcare technology, today announced the LINQ II™ Insertable Cardiac Monitor (ICM) system is the first-and-only ICM to receive 510(k) clearance by the U.S. Food and Drug Administration (FDA) for use in pediatric patients over the age of 2 who have heart rhythm abnormalities and require long-term, continuous monitoring.
"For pediatric cardiologists who see many young patients needing continuous, long-term monitoring for infrequent or unknown heart rhythm conditions, this expanded indication for the LINQ II ICM is critically important," said Jennifer Silva, M.D., director of pediatric cardiac electrophysiology at Washington University in St. Louis and St. Louis Children's Hospital. "The data generated from these small monitors can help us better tailor treatment decisions and ongoing management for our patients."
The LINQ II system is a small (one-third the size of a AAA battery), wireless ICM for patients with abnormal heart rhythms who experience infrequent symptoms including dizziness, palpitations, syncope (fainting) and chest pain, thereby requiring long-term monitoring or ongoing management. The LINQ II ICM, which has a battery life of up to 4.5 years1, allows patients to undergo magnetic resonance imaging (MRI) when needed, and, as an implantable device, does not interfere with daily activities such as showering, bathing, or swimming. The latest-generation device has been implanted in thousands of patients globally since it was first commercialized in 2020.
"As a result of this milestone, physicians will be able to provide actionable data to help diagnose underlying heart conditions and define treatment protocols in our younger patients with abnormal heart rhythms," said Julie Brewer, president of the Cardiovascular Diagnostics and Services business, which is part of the Cardiovascular Portfolio at Medtronic. "And parents can have peace of mind knowing their child's heart is being monitored continuously, and their doctor will be notified of abnormal heartbeats."
The LINQ II ICM system also includes the recently launched AccuRhythm™ AI algorithms, which applies artificial intelligence (AI) to heart rhythm event data collected by the LINQ II ICM, improving the accuracy of information physicians receive so they can better diagnose and treat abnormal heart rhythms. The two AI algorithms have shown to reduce the number of false alerts specific to the most common ICM false alerts — atrial fibrillation (AF) and pause (asystole) — by 74.1% and 97.4% respectively,2, 3 while preserving more than 99% of true alerts.4, 5
With integrated remote patient management, patients or their caregivers can choose to use their smartphones with the LINQ II ICM to automatically transfer device data via the MyCareLink Heart™ mobile app using BlueSync™ technology that enables secure communication via Bluetooth.
Medtronic has been a pioneer in the development of insertable cardiac monitors for more than 20 years; to date, more than 1.7 million patients have received a Medtronic ICM.6
About Medtronic
Bold thinking. Bolder actions. We are Medtronic. Medtronic plc, headquartered in Dublin, Ireland, is the leading global healthcare technology company that boldly attacks the most challenging health problems facing humanity by searching out and finding solutions. Our Mission — to alleviate pain, restore health, and extend life — unites a global team of 95,000+ passionate people across 150 countries. Our technologies and therapies treat 70 health conditions and include cardiac devices, surgical robotics, insulin pumps, surgical tools, patient monitoring systems, and more. Powered by our diverse knowledge, insatiable curiosity, and desire to help all those who need it, we deliver innovative technologies that transform the lives of two people every second, every hour, every day. Expect more from us as we empower insight-driven care, experiences that put people first, and better outcomes for our world. In everything we do, we are engineering the extraordinary. For more information on Medtronic (NYSE:MDT), visit www.Medtronic.com(opens new window) and follow @Medtronic(opens new window) on Twitter and LinkedIn(opens new window).
SOURCE Medtronic plc
Sep. 20, 2022 10:06 AM ET
By: Anuron Mitra, SA News Editor

PUBLISHED20 September 2022 20 September 2022 07:00 BST
AstraZeneca's Evusheld (tixagevimab and cilgavimab, formerly AZD7442), a long-acting antibody combination, has been approved in the European Union (EU) for the treatment of adults and adolescents (aged 12 years and older weighing at least 40 kg) with COVID‑19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID‑19.
The approval by the European Commission was based on results from the TACKLE Phase III COVID-19 treatment trial which showed one intramuscular (IM) dose of Evusheld provided clinically and statistically significant protection against progression to severe COVID-19 or death from any cause compared to placebo. Evusheld treatment earlier in the disease course led to more favourable outcomes. TACKLE was conducted in non-hospitalised adults with mild-to-moderate COVID-19 who were symptomatic for seven days or less. 90% of trial participants were at high risk of progression to severe COVID-19 due to co-morbidities or age. Evusheld was generally well tolerated in the trial.1
Michel Goldman, M.D., Ph.D., Professor, Institute for Interdisciplinary Innovation in Healthcare, Université Libre de Bruxelles, and former Executive Director of the European Innovative Medicines Initiative, said: “Many people, including those who are immunocompromised, older adults and those with underlying health conditions, are at high risk of severe disease, hospitalisation and death if they become infected. Evusheld, delivered in a convenient intramuscular formulation, is now a much-needed new COVID-19 treatment option for these vulnerable populations.”
Iskra Reic, Executive Vice President, Vaccines and Immune Therapies, AstraZeneca, said: “COVID-19 remains an ongoing health concern for millions of Europeans and around the world, especially for those who may not be well-protected against the virus from vaccination. With this approval, Evusheld is now the only long-acting antibody combination available for both prevention and treatment of COVID-19 in Europe, allowing us to protect even more people from this devastating disease.”
The recommended dose of Evusheld for treatment in Europe is 300mg of tixagevimab and 300mg of cilgavimab, administered as two separate, sequential IM injections.
Evusheld has been shown to retain in vitro neutralisation of Omicron BA.5, which is currently the dominant SARS-CoV-2 variant in Europe.2 Real-world evidence generated to date has demonstrated significantly lower rates of symptomatic COVID-19 and/or hospitalisation/death for immunocompromised patients receiving Evusheld compared to control arms. This includes real-world evidence collected while Omicron BA.5, BA.4, BA.2, BA.1 and BA.1.1 were circulating.3-6
Evusheld was granted marketing authorisation in the EU for pre-exposure prophylaxis (prevention) of COVID-19 in a broad population of adults and adolescents earlier this year and is already available in a majority of countries in Europe.
TACKLE
TACKLE is a Phase III, randomised, double-blind, placebo-controlled, multi-centre trial assessing the safety and efficacy of a single 600mg IM dose of Evusheld (300mg each of cilgavimab and tixagevimab) compared to placebo for the treatment of mild-to-moderate COVID-19. The trial was conducted in 95 sites in the US, Latin America, Europe and Japan. 903 participants were randomised (1:1) to receive either Evusheld (n = 452) or saline placebo (n = 451), administered in two separate, sequential IM injections.
Participants were adults 18 years-old and over who had mild-to-moderate COVID-19 and were symptomatic for seven days or less. Participants had a documented laboratory-confirmed SARS-CoV-2 infection, as determined by a molecular test (antigen or nucleic acid) from any respiratory tract specimen (e.g. oropharyngeal, nasopharyngeal, or nasal swab or saliva) collected no more than three days prior to day 1. Participants were not vaccinated against COVID-19 at the time of screening.
Detailed results from TACKLE, published in The Lancet Respiratory Medicine, showed Evusheld significantly reduced the relative risk of progressing to severe COVID-19 or death (from any cause) by 50% (95% confidence interval [CI] 15, 71; p=0.010) through day 29 compared to placebo in non-hospitalised patients with mild-to-moderate COVID-19 who were symptomatic for seven days or less, the trial’s primary endpoint. In pre-specified analyses of participants who received treatment within three days of symptom onset, Evusheld reduced the risk of developing severe COVID-19 or death (from any cause) by 88% compared to placebo (95% CI 9, 98), and the risk reduction was 67% (95% CI 31, 84) compared with placebo when participants received Evusheld within five days of symptom onset.1
Evusheld was generally well tolerated in the trial. Adverse events (AEs) occurred more frequently in the placebo group (163/451; 36%) than the Evusheld group (132/452; 29%). The most common AE was COVID-19 pneumonia, occurring in 49 participants (11%) in the placebo group and 26 participants (6%) in the Evusheld group. Serious AEs occurred in 54 participants (12%) in the placebo group and 33 participants (7%) in the Evusheld group. There were six COVID-19-reported deaths in the placebo group and three in the Evusheld group.1
Evusheld
Evusheld, formerly known as AZD7442, is a combination of two long-acting antibodies - tixagevimab (AZD8895) and cilgavimab (AZD1061) - derived from B-cells donated by convalescent patients after SARS-CoV-2 infection. Discovered by Vanderbilt University Medical Center and licensed to AstraZeneca in June 2020, the human monoclonal antibodies bind to distinct sites on the SARS-CoV-2 spike protein7 and were optimised by AstraZeneca with half-life extension and reduction of Fc effector function and complement C1q binding.8 The half-life extension more than triples the durability of its action compared to conventional antibodies;9-11 data from the PROVENT Phase III trial show protection lasting six months.12 The reduced Fc effector function aims to minimise the risk of antibody-dependent enhancement of disease - a phenomenon in which virus-specific antibodies promote, rather than inhibit, infection and/or disease.13
Evusheld is authorised for use for pre-exposure prophylaxis (prevention) of COVID-19 in the US (emergency use), EU, Japan and many other countries. Evusheld is approved for treatment of those with risk factors for severe SARS-CoV-2 infection in Japan. Regulatory submissions are progressing for both prevention and treatment indications around the world.
Evusheld is being developed with support from the US government, including federal funds from the Department of Health and Human Services; Office of the Assistant Secretary for Preparedness and Response; Biomedical Advanced Research and Development Authority in partnership with the Department of Defense; Joint Program Executive Office for Chemical, Biological, Radiological and Nuclear Defense, under Contract No. W911QY-21-9-0001.
Under the terms of the licensing agreement with Vanderbilt, AstraZeneca will pay single-digit royalties on future net sales.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Sep. 20, 2022 4:52 AM ET
By: Ravikash, SA News Editor
The European Commission (EC) approved AstraZeneca's (NASDAQ:AZN) Evusheld to treat people aged 12 years and older with COVID‑19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID‑19.
https://seekingalpha.com/news/3884207-astrazenecas-evusheld-gets-approval-in-eu-to-treat-covid-19
https://seekingalpha.com/symbol/AZN
https://www.evusheld.com/en/patient
Sep. 20, 2022 8:00 AM ET
Takeda Pharmaceutical Company Limited (TAK)
TORONTO, Sept. 20, 2022 /CNW/ - Takeda Canada Inc. ("Takeda") is pleased to announce that Health Canada has authorized (Notice of Compliance) LIVTENCITY™ (maribavir) for the treatment of adults with post-transplant cytomegalovirus (CMV) infection/disease who (FREEW) are refractory (with or without genotypic resistance) to one or more prior antiviral therapies.1 Compared to conventionally used antivirals to treat post-transplant CMV, LIVTENCITY offers twice the efficacy1 and more than 10 times less toxicity.1*

LIVTENCITY was evaluated using the Health Canada priority review process which recognizes the significant unmet need for this patient population. The Canadian approval of LIVTENCITY represents the second market globally to receive authorization for this important CMV treatment.
"Health Canada's authorization of LIVTENCITY represents a much-needed treatment option for post-transplant patients with refractory/resistant CMV infection or disease," said Dr. Shariq Haider, Professor of Medicine McMaster University Division of Infectious Disease and General Medicine and Research Lead Transplant Malignant ID Juravinski Hospital and Cancer Center, Hamilton ON. "Until now, physicians have relied upon conventional non-indicated antiviral therapies as the only option to treat post-transplant CMV which can be difficult to manage post-transplant. The addition of an effective oral treatment for CMV, with a better safety profile, is welcome news for Canadian patients, physicians and the transplant community."
The approval of LIVTENCITY is based on the SOLSTICE Study which assessed the efficacy and safety of LIVTENCITY treatment compared to Investigator Assigned Treatment (IAT) in 350 Hematopoietic Stem Cell Transplant (HSCT) and Solid Organ Transplant (SOT) recipients with CMV infections that were refractory to treatment with ganciclovir, valganciclovir, foscarnet, or cidofovir, including CMV infections with or without confirmed resistance to 1 or more anti-CMV agents.1 Beneficial treatment effect was consistent regardless of transplant type, baseline CMV DNA viral load, genotypic resistance, CMV syndrome at baseline and age.1
"A diagnosis of CMV infection can be particularly unsettling for patients given the complex challenges associated with receiving a life-saving transplant," said Rute Fernandes, General Manager, Takeda, Canada. "Providing patients with the first indicated post-transplant CMV treatment underscores Takeda's commitment to finding solutions for patients where there is an unmet need, and we look forward to working with the appropriate agencies to bring LIVTENCITY to Canadians."
Current antiviral treatment options for post-transplant cytomegalovirus (CMV) infections have unfavorable toxicities, such as neutropenia or nephrotoxicity, that may impact patient care and transplant outcomes.2
About LIVITENCITY
LIVTENCITY, is a selective orally bioavailable benzimidazole riboside antiviral drug with a novel mechanism of action against human CMV (HCMV). LIVTENCITY attaches to the UL97 encoded kinase at the adenosine triphosphate (ATP) binding site, abolishing phosphotransferase needed in processes such as DNA replication, encapsidation, and nuclear egress of viral capsids.1
The recommended dose of LIVTENCITY is 400 mg (two 200 mg tablets) twice daily resulting in a daily dose of 800 mg.1 LIVTENCITY is intended for oral use only and can be taken with or without food.1
The primary efficacy endpoint was confirmed CMV viremia clearance (plasma CMV DNA concentration below the lower limit of quantification (<LLOQ; i.e., <137 IU/mL)) at Week 8. The key secondary endpoint was CMV viremia clearance and CMV infection symptom control at the end of Study Week 8 with maintaining this treatment effect through Study Week 16.1
For the primary endpoint, LIVTENCITY was superior to IAT (56% vs. 24%, respectively, p<0.001). For the key secondary endpoint, 19% vs. 10% achieved both CMV viremia clearance and CMV infection symptom control in the LIVTENCITY and IAT group, respectively (p=0.013).1
Most common adverse reactions with LIVTENCITY were: taste disturbance (44.8%), nausea (8.5%), vomiting (7.7%), immunosuppressant drug concentration level increased (6.4%), diarrhea (3.8%), abdominal pain (2.1%), neutropenia (1.7%), acute kidney injury (1.7%), anemia (1.3%), and decreased appetite (1.3%).1
About Takeda Canada Inc.
Takeda Canada Inc. is the Canadian organization of Takeda Pharmaceutical Company Limited (TKPHF) (TSE: 4502) (NYSE: TAK), a global, values-based, R&D-driven biopharmaceutical leader headquartered in Japan, committed to discovering and delivering life-transforming treatments, guided by our commitment to patients, our people and the planet. Takeda focuses its R&D efforts on four therapeutic areas: Oncology, Rare Genetics and Hematology, Neuroscience, and Gastroenterology (GI). We also make targeted R&D investments in Plasma-Derived Therapies and Vaccines. We are focusing on developing highly innovative medicines that contribute to making a difference in people's lives by advancing the frontier of new treatment options and leveraging our enhanced collaborative R&D engine and capabilities to create a robust, modality-diverse pipeline. Our employees are committed to improving quality of life for patients and to working with our partners in health care in approximately 80 countries and regions. For more information, visit: takeda.com/en-ca
SOURCE Takeda Canada Inc.
Sep. 20, 2022 9:21 AM ET
Takeda Pharmaceutical Company Limited (TAK)
By: Anuron Mitra, SA News Editor
https://seekingalpha.com/symbol/TAK
DOWNLOAD AS PDFSEPTEMBER 19, 2022
Approval follows the positive opinion granted by European Committee for Medicinal Products for Human Use (CHMP) in July 2022
LUPKYNIS is the first oral medicine approved in both the U.S. and Europe for the treatment of adults living with active lupus nephritis
Approval triggers $30.0 million milestone payment, to be recognized as revenue in the 3rd quarter
VICTORIA, British Columbia--(BUSINESS WIRE)-- Aurinia Pharmaceuticals Inc. (NASDAQ:AUPH) (Aurinia or the Company), a biopharmaceutical company committed to delivering therapeutics that change the trajectory of autoimmune disease, announced that the European Commission (EC) has granted marketing authorization of LUPKYNIS® (voclosporin) to treat adults with active lupus nephritis (LN), a serious complication of systemic lupus erythematosus (SLE). The U.S. Food and Drug Administration (FDA) approved LUPKYNIS on January 22, 2021, in combination with a background immunosuppressive therapy regimen to treat adult patients with active LN.
The centralized marketing authorization is valid in all European Union (EU) member states as well as in Iceland, Liechtenstein, Norway and Northern Ireland.
“Today marks the first approved oral treatment for lupus nephritis in both the European Union and provides adults across Europe living with this potentially life-threatening disease a new treatment option,” said Peter Greenleaf, President and Chief Executive Officer, Aurinia. “People with lupus nephritis and their physicians have long been challenged by the lack of treatments available. In partnership with Otsuka, we’re excited to reach patients across Europe with a meaningful therapy that can help enable positive long-term kidney outcomes.”
Aurinia and Otsuka Pharmaceutical Co., Ltd., (Otsuka) entered a collaboration and licensing agreement in December 2020 for the development and commercialization of voclosporin for the treatment of LN in the EU, Japan, the United Kingdom, Russia, Switzerland, Norway, Belarus, Iceland, Liechtenstein, and Ukraine. As part of the agreement, Aurinia will receive a $30.0 million EC approval-related milestone payment to be recognized as revenue in the quarter, with receipt of cash to follow within 30 days of invoicing. In addition to the milestone payment, Aurinia is eligible to receive further payments tied to additional regulatory and reimbursement milestones, low double-digit royalties on future net sales, as well as revenues for the supply of product to Otsuka under a cost-plus arrangement.
A decision on marketing authorization for LUPKYNIS in Great Britain is expected from the UK Medicines and Healthcare products Regulatory Agency in the coming weeks. In addition, a marketing authorization application (MAA) for LUPKYNIS was submitted to the Swiss Agency for Therapeutic Products (Swissmedic) and is currently under review. Swissmedic previously granted orphan drug status to voclosporin in LN in February 2022.
The EC approval of LUPKYNIS is based on the results of the pivotal Phase 3 AURORA 1 study and the recent AURORA 2 continuation study, which demonstrated voclosporin, in combination with mycophenolate mofetil (MMF) and low-dose corticosteroids, led to statistically superior complete renal response rates at 52 weeks compared to MMF and low-dose corticosteroids alone. The safety profile of voclosporin and MMF and low-dose corticosteroids was generally comparable to MMF and low-dose corticosteroids alone.
About Lupus Nephritis
LN is a serious manifestation of SLE, a chronic and complex autoimmune disease. About 200,000-300,000 people live with SLE in the U.S. and about one-third of these people are diagnosed with lupus nephritis at the time of their SLE diagnosis. About 50 percent of all people with SLE may develop lupus nephritis. If poorly controlled, LN can lead to permanent and irreversible tissue damage within the kidney. Black and Asian individuals with SLE are four times more likely to develop LN and individuals of Hispanic ancestry are approximately twice as likely to develop the disease when compared with Caucasian individuals. Black and Hispanic individuals with SLE also tend to develop LN earlier and have poorer outcomes when compared to Caucasian individuals.
About LUPKYNIS
LUPKYNIS® is the first U.S. FDA-approved and EC-approved oral medicine for the treatment of adult patients with active lupus nephritis (LN). LUPKYNIS is a novel, structurally modified calcineurin inhibitor (CNI) with a dual mechanism of action, acting as an immunosuppressant through inhibition of T-cell activation and cytokine production and promoting podocyte stability in the kidney. The recommended starting dose of LUPKYNIS is three capsules twice daily with no requirement for serum drug monitoring. Dose modifications can be made based on Aurinia’s proprietary personalized eGFR-based dosing protocol. Boxed Warning, warnings, and precautions for LUPKYNIS are consistent with those of other CNI-immunosuppressive treatments.
About Aurinia
Aurinia Pharmaceuticals is a fully integrated biopharmaceutical company focused on delivering therapies to treat targeted patient populations that are impacted by serious diseases with a high unmet medical need. In January 2021, the Company introduced LUPKYNIS® (voclosporin), the first FDA-approved oral therapy dedicated for the treatment of adult patients with active lupus nephritis. The Company’s head office is in Victoria, British Columbia, its U.S. commercial office is in Rockville, Maryland. The Company focuses its development efforts globally.
INDICATION AND IMPORTANT SAFETY INFORMATION
INDICATIONS
LUPKYNIS is indicated in combination with a background immunosuppressive therapy regimen for the treatment of adult patients with active LN. Limitations of Use: Safety and efficacy of LUPKYNIS have not been established in combination with cyclophosphamide. Use of LUPKYNIS is not recommended in this situation.
Please see Prescribing Information, including Boxed Warning, and Medication Guide for LUPKYNIS.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220919005617/en/
Source: Aurinia Pharmaceuticals Inc.
Released September 19, 2022
Sep. 19, 2022 12:16 PM ET
Aurinia Pharmaceuticals Inc. (AUPH)OTSKF, OTSKY
By: Jonathan Block, SA News Editor2 Comments
09/19/2022
- FDA Has Set Action Date of January 19, 2023 -
BOTHELL, Wash.--(BUSINESS WIRE)-- Seagen Inc. (Nasdaq: SGEN) today announced that the U.S. Food and Drug Administration (FDA) has accepted for Priority Review the supplemental New Drug Application (sNDA) seeking accelerated approval for TUKYSA® (tucatinib) in combination with trastuzumab for adult patients with HER2-positive colorectal cancer who have received at least one prior treatment regimen for unresectable or metastatic disease.
The sNDA submission is based on the results of the pivotal phase 2 MOUNTAINEER trial. These data were presented at the European Society for Medical Oncology (ESMO) World Congress on Gastrointestinal Cancer in July 2022.
“There are currently no FDA-approved therapies for metastatic colorectal cancer that specifically target HER2,” said Marjorie Green, M.D., Senior Vice President and Head of Late-Stage Development, Seagen. “The FDA’s prioritization of our application for tucatinib in combination with trastuzumab supports our belief in its significant potential to benefit people with previously treated HER2-positive metastatic colorectal cancer.”
The FDA grants Priority Review to applications for medicines that, if approved, would provide significant improvements in safety or effectiveness of the treatment, diagnosis or prevention of serious diseases. Under the Prescription Drug User Fee Act (PDUFA), the FDA has set a target action date of January 19, 2023. In July 2022, the FDA granted Breakthrough Therapy Designation for tucatinib in combination with trastuzumab for the treatment of adult patients with unresectable or metastatic HER2-positive colorectal cancer who have previously received fluoropyrimidine-, oxaliplatin- and irinotecan-containing chemotherapy based on results from the MOUNTAINEER trial.
In February 2022, Seagen initiated the randomized, global phase 3 MOUNTAINEER-03 clinical trial, which is evaluating tucatinib in combination with trastuzumab and standard chemotherapy versus chemotherapy given with or without cetuximab or bevacizumab in first-line HER2-positive metastatic colorectal cancer. MOUNTAINEER-03 is intended to serve as a confirmatory trial in the U.S. and to support global filings. Merck, known as MSD outside of the U.S. and Canada, is commercializing TUKYSA in regions outside of the U.S., Canada and Europe.
About MOUNTAINEER
MOUNTAINEER is a U.S. and European open-label, multicenter phase 2 clinical trial of tucatinib in combination with trastuzumab or as a single agent that enrolled 117 patients with HER2-positive unresectable or metastatic colorectal cancer following previous standard-of-care therapies. Patients evaluated in MOUNTAINEER had not received prior anti-HER2 therapy. Patients received tucatinib (300 mg) twice per day orally in combination with trastuzumab intravenously (8 mg/kg loading dose, then 6 mg/kg every three weeks thereafter). The primary endpoint of the trial is confirmed objective response rate by RECIST (Response Evaluation Criteria in Solid Tumors) version 1.1 criteria per blinded independent central review. Duration of response, progression-free survival, overall survival and safety and tolerability of the combination regimen are secondary objectives.
About TUKYSA (tucatinib)
TUKYSA is an oral medicine that is a tyrosine kinase inhibitor of the HER2 protein. In vitro (in lab studies), TUKYSA inhibited phosphorylation of HER2 and HER3, resulting in inhibition of downstream MAPK and AKT signaling and cell growth (proliferation), and showed anti-tumor activity in HER2-expressing tumor cells. In vivo (in living organisms), TUKYSA inhibited the growth of HER2-expressing tumors. The combination of TUKYSA and the anti-HER2 antibody trastuzumab showed increased anti-tumor activity in vitro and in vivo compared to either medicine alone.
TUKYSA is approved in 38 countries. It was approved by the U.S. FDA in April 2020 and by the European Medicines Agency and the UK Medicines and Healthcare Products Regulatory Agency in February 2021.
U.S. Indication and Important Safety Information
TUKYSA is indicated in combination with trastuzumab and capecitabine for treatment of adult patients with advanced unresectable or metastatic HER2-positive breast cancer, including patients with brain metastases, who have received one or more prior anti-HER2-based regimens in the metastatic setting.
About Seagen
Seagen is a global biotechnology company that discovers, develops and commercializes transformative cancer medicines to make a meaningful difference in people’s lives. Seagen is headquartered in the Seattle, Washington area, and has locations in California, Canada, Switzerland and the European Union. For more information on the company’s marketed products and robust pipeline, visit www.seagen.com and follow @SeagenGlobal on Twitter.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220919005223/en/
Source: Seagen Inc.
Sep. 19, 2022 9:01 AM ET
By: Anuron Mitra, SA News Editor
European Commission approves Roche’s Vabysmo, the first bispecific antibody for the eye, for two leading causes of vision loss
September 18, 2022
Basel, 19 September 2022 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the European Commission (EC) approved Vabysmo® (faricimab) for the treatment of neovascular or ‘wet’ age-related macular degeneration (nAMD) and visual impairment due to diabetic macular edema (DME). These retinal conditions are two of the leading causes of vision loss worldwide, affecting more than 40 million people 1,2,3,4
“Many people with nAMD and DME struggle to keep up with the monthly eye injections and physician visits, often associated with current standards of care, and unfortunately their vision may suffer as a result of undertreatment,” said Prof Ramin Tadayoni, head of the ophthalmology department, Lariboisière, Saint-Louis and Rothschild Hospitals, Paris, France, and European Society of Retina Specialists (EURETINA) president elect. “For people in Europe living with these conditions, today’s approval offers the first new mechanism of action in over a decade; one which could improve and protect their vision with fewer injections over time.”
Vabysmo is the only injectable eye medicine approved in Europe with phase III studies supporting treatment at intervals of up to four months for people living with nAMD and DME.5,6,7 With the potential to require fewer eye injections over time, while also improving and maintaining vision and anatomy, Vabysmo could offer a less burdensome treatment schedule for individuals, their caregivers and healthcare systems 6,7,8,9
“The approval of Vabysmo in Europe is the result of years of pioneering research from Roche ophthalmologists and scientists, who are deeply committed to improving outcomes for people with retinal conditions,” said Levi Garraway, M.D., PhD., Roche’s Chief Medical Officer and Head of Global Product Development. “We are delighted to offer people in Europe this first-of-its-kind treatment option and are working to bring Vabysmo to people with nAMD and DME as soon as possible.”
Today’s approval is based on results across four phase III studies in two indications, involving 3,220 patients: TENAYA and LUCERNE in nAMD at year one, and YOSEMITE and RHINE in DME up to two years. The studies showed that people treated with Vabysmo, given at intervals of up to four months, achieved similar vision gains and anatomical improvements compared to aflibercept given every two months.6,7,8 The totality of data across all four studies at two years showed that more than 60% of people treated with Vabysmo were able to extend treatment to every four months, while improving and maintaining vision. Additionally, up to two years, people with nAMD and DME treated with Vabysmo received 33% (10 vs. 15) and 21% (11 vs. 14) fewer median number of injections compared to aflibercept, respectively.6,9
Vabysmo, a bispecific antibody, is uniquely engineered to target and inhibit two disease pathways, linked to a number of vision-threatening retinal conditions, by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A), to restore vascular stability. By independently blocking both pathways involving Ang-2 and VEGF-A, Vabysmo is designed to stabilise blood vessels and thereby reduce inflammation, leakage and abnormal vessel growth (neovascularisation) more than inhibition of VEGF-A alone.7 This sustained blood vessel stabilisation may improve disease control, vision and anatomical outcomes for longer.7,8
Vabysmo is now approved in the European Union and nine other countries around the world, including the US, Japan, and the UK, for people living with nAMD and DME, and submissions to other regulatory authorities are ongoing.5,10,11,12 Globally, more than 100,000 Vabysmo doses have been distributed for treatment of these conditions to date.13 Roche also continues to explore areas where Vabysmo has the potential to deliver additional benefits to patients, including retinal vein occlusion.
About the Vabysmo® (faricimab) clinical development programme
Roche has a robust phase III clinical development programme for Vabysmo. The programme includes AVONELLE-X, an extension study of TENAYA and LUCERNE, evaluating the long-term safety and tolerability of Vabysmo in neovascular or ‘wet’ age-related macular degeneration (nAMD), and RHONE-X, an extension study of YOSEMITE and RHINE, evaluating the long-term safety and tolerability of Vabysmo in diabetic macular edema (DME).14,15 Additionally, the BALATON and COMINO trials are underway, evaluating the efficacy and safety of Vabysmo in people with macular edema following retinal vein occlusion.16,17 Roche has also initiated the phase IV ELEVATUM study of Vabysmo in underrepresented patient populations with DME.18
About the TENAYA and LUCERNE studies7,9
TENAYA (NCT03823287) and LUCERNE (NCT03823300) were two identical, randomised, multicentre, double-masked, global phase III studies evaluating the efficacy and safety of Vabysmo® (faricimab) compared to aflibercept in 1,329 people living with neovascular or ‘wet’ age-related macular degeneration (671 in TENAYA and 658 in LUCERNE).
Both studies met their primary endpoint, with Vabysmo given at intervals of up to every four months consistently shown to offer visual acuity gains and anatomical improvements that were non-inferior to aflibercept given every two months. A secondary endpoint in both studies measured the proportion of people in the Vabysmo arm that were treated on dosing schedules of every three or four months during the first year. Importantly, 46% (n=144/315) of those receiving Vabysmo in TENAYA and 45% (n=142/316) in LUCERNE were able to be treated every four months in the first year, and an additional 34% (n=107/315) and 33% (n=104/316), respectively, were able to be treated every three months. Combined, nearly 80% of people receiving Vabysmo were able to go three months or longer between treatments during the first year.
At two years, vision improvements were comparable across both treatment arms. In TENAYA, the average vision gains from baseline at two years were +3.7 eye chart letters in the Vabysmo arm and +3.3 letters in the aflibercept arm. In LUCERNE, the average vision gains from baseline at two years were +5.0 letters in the Vabysmo arm and +5.2 letters in the aflibercept arm. Furthermore, 59% (n=160/271) of Vabysmo patients in TENAYA and 67% (n=192/287) in LUCERNE achieved four-month dosing at two years. This is an increase over one-year results, which showed 46% (n=144/315) of Vabysmo patients in TENAYA and 45% (n=142/316) in LUCERNE achieved four-month dosing. An additional 15% (n=41/271) of Vabysmo patients in TENAYA and 14% (n=41/287) in LUCERNE achieved three-month dosing at two years. Combined, approximately 80% of Vabysmo patients were able to go three months or longer between treatments at the end of the second year.
Vabysmo was generally well tolerated in both studies, with a favourable benefit-risk profile. In TENAYA and LUCERNE, the most common adverse reactions (≥3% of people) included cataract, conjunctival haemorrhage, vitreous floaters, retinal pigment epithelial tears, increase of intraocular pressure and eye pain. Safety results were consistent across study arms.
Two-year data from TENAYA and LUCERNE were presented at the 2022 American Society of Retina Specialists Annual Scientific Meeting. These data will be submitted to the European Medicines Agency in due course.
About the YOSEMITE and RHINE studies6,8
YOSEMITE (NCT03622580) and RHINE (NCT03622593) were two identical, randomised, multicentre, double-masked, global phase III studies evaluating the efficacy and safety of Vabysmo® (faricimab) compared to aflibercept in 1,891 people with visual impairment due to diabetic macular edema (940 in YOSEMITE and 951 in RHINE).
Both studies met their primary endpoint, with Vabysmo given at intervals of up to every four months consistently shown to offer visual acuity gains and anatomical improvements that were non-inferior to aflibercept given every two months. A secondary endpoint in both studies measured the proportion of people in the Vabysmo treat-and-extend arm that achieved dosing schedules of every three or four months. Importantly, 53% (n=151/286) of those in the Vabysmo treat-and-extend arm in YOSEMITE and 51% (n=157/308) in RHINE achieved four-month dosing at the end of the first year, and an additional 21% (n=60/286) and 20% (n=63/308), respectively, achieved three-month dosing. At two years, the number of people in the Vabysmo treat-and-extend arm achieving four-month dosing increased to 60% (n=162/270) in YOSEMITE and 64% (n=185/287) in RHINE. An additional 18% (n=49/270) of people in YOSEMITE and 14% (n=39/287) in RHINE achieved three-month dosing. Combined, almost 80% of people in the Vabysmo treat-and-extend arm were able to go three months or longer between treatments at the end of the second year.
Vabysmo was generally well tolerated in both studies, with a favourable benefit-risk profile. In YOSEMITE and RHINE, the most common adverse reactions (≥3% of people) included cataract, conjunctival haemorrhage, vitreous floaters, increase of intraocular pressure and eye pain. Safety results were consistent across study arms.
About Vabysmo® (faricimab)8
Vabysmo is the first bispecific antibody approved for the eye. It targets and inhibits two disease pathways linked to a number of vision-threatening retinal conditions, by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A). Ang-2 and VEGF-A contribute to vision loss by destabilising blood vessels, causing new leaky blood vessels to form and increasing inflammation. By blocking pathways involving Ang-2 and VEGF-A, Vabysmo is designed to stabilise blood vessels.
About Roche
Founded in 1896 in Basel, Switzerland, as one of the first industrial manufacturers of branded medicines, Roche has grown into the world’s largest biotechnology company and the global leader in in-vitro diagnostics. The company pursues scientific excellence to discover and develop medicines and diagnostics for improving and saving the lives of people around the world. We are a pioneer in personalised healthcare and want to further transform how healthcare is delivered to have an even greater impact. To provide the best care for each person we partner with many stakeholders and combine our strengths in Diagnostics and Pharma with data insights from the clinical practice.
In recognising our endeavour to pursue a long-term perspective in all we do, Roche has been named one of the most sustainable companies in the pharmaceuticals industry by the Dow Jones Sustainability Indices for the thirteenth consecutive year. This distinction also reflects our efforts to improve access to healthcare together with local partners in every country we work.
Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan.
For more information, please visit www.roche.com.
Sep. 19, 2022 7:39 AM ET
Roche Holding AG (RHHBF), RHHBYREGN
By: Jonathan Block, SA News Editor
PUBLISHED16 September 2022
AstraZeneca's Evusheld (tixagevimab and cilgavimab, formerly AZD7442), a long-acting antibody combination, has been recommended for marketing authorisation in the European Union (EU) for the treatment of adults and adolescents (aged 12 years and older weighing at least 40 kg) with COVID‑19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID‑19.
The Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency based its positive opinion on results from the TACKLE Phase III COVID-19 treatment trial which showed one intramuscular (IM) dose of Evusheld provided clinically and statistically significant protection against progression to severe COVID-19 or death from any cause compared to placebo. Evusheld treatment earlier in the disease course led to more favourable outcomes. TACKLE was conducted in non-hospitalised adults with mild-to-moderate COVID-19 who were symptomatic for seven days or less. 90% of trial participants were at high risk of progression to severe COVID-19 due to co-morbidities or age. Evusheld was generally well tolerated in the trial.1
Michel Goldman, M.D., Ph.D., Professor, Institute for Interdisciplinary Innovation in Healthcare, Université Libre de Bruxelles, and former Executive Director of the European Innovative Medicines Initiative, said: “Despite the remarkable efficacy of COVID-19 vaccines in the overall population, patients with an impaired immune system remain at very high risk to develop severe COVID-19 requiring hospitalisation. For high-risk patients, there is a major need for tolerable and effective therapies that can be used to block COVID-19 progression and prevent poor outcomes. As we head into the autumn and winter months when COVID-19 infections may rise, Evusheld represents an important new treatment option to protect patients infected with the SARS-CoV-2 virus against severe disease and death.”
Iskra Reic, Executive Vice President, Vaccines and Immune Therapies, AstraZeneca, said: “Evusheld has already made an important difference around the world helping prevent COVID-19 infections in vulnerable populations who can’t mount an adequate response to COVID-19 vaccination. This positive CHMP opinion underscores Evusheld’s potential as a COVID-19 treatment for patients at increased risk of progressing to severe disease.”
The recommended dose of Evusheld for treatment in the EU is 300mg of tixagevimab and 300mg of cilgavimab, administered as two separate, sequential IM injections.
Evusheld been shown to retain in vitro neutralisation of Omicron BA.5, which is currently the dominant SARS-CoV-2 variant in Europe.2 Real-world evidence generated to date has demonstrated significantly lower rates of symptomatic COVID-19 and/or hospitalisation/death for immunocompromised patients receiving Evusheld compared to control arms. This includes real-world evidence collected while Omicron BA.5, BA.4, BA.2, BA.1 and BA.1.1 were circulating.3-6
Evusheld was granted marketing authorisation in the EU for pre-exposure prophylaxis (prevention) of COVID-19 in a broad population of adults and adolescents earlier this year and is already available in a majority of countries in Europe.
AstraZeneca anticipates that the European Commission will shortly complete its review of the CHMP positive opinion to determine whether to grant marketing authorisation for treatment of COVID-19 in appropriate populations.
Evusheld
Evusheld, formerly known as AZD7442, is a combination of two long-acting antibodies - tixagevimab (AZD8895) and cilgavimab (AZD1061) - derived from B-cells donated by convalescent patients after SARS-CoV-2 infection. Discovered by Vanderbilt University Medical Center and licensed to AstraZeneca in June 2020, the human monoclonal antibodies bind to distinct sites on the SARS-CoV-2 spike protein7 and were optimised by AstraZeneca with half-life extension and reduction of Fc effector function and complement C1q binding.8 The half-life extension more than triples the durability of its action compared to conventional antibodies;9-11 data from the PROVENT Phase III trial show protection lasting six months.12 The reduced Fc effector function aims to minimise the risk of antibody-dependent enhancement of disease - a phenomenon in which virus-specific antibodies promote, rather than inhibit, infection and/or disease.13
Evusheld is authorised for use for pre-exposure prophylaxis (prevention) of COVID-19 in the US (emergency use), EU, Japan and many other countries. Evusheld is approved for treatment of those with risk factors for severe SARS-CoV-2 infection in Japan. Regulatory submissions are progressing for both prevention and treatment indications around the world.
Evusheld is being developed with support from the US government, including federal funds from the Department of Health and Human Services; Office of the Assistant Secretary for Preparedness and Response; Biomedical Advanced Research and Development Authority in partnership with the Department of Defense; Joint Program Executive Office for Chemical, Biological, Radiological and Nuclear Defense, under Contract No. W911QY-21-9-0001.
Under the terms of the licensing agreement with Vanderbilt, AstraZeneca will pay single-digit royalties on future net sales.
https://www.evusheld.com/en/patient
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Sep. 16, 2022 4:35 AM ET
By: Ravikash, SA News Editor
September 16, 2022
– Positive Opinion Based on Landmark ZUMA-7 Study in Which 41% of Patients Demonstrated Event-Free Survival at Two Years versus 16% for Standard of Care -
SANTA MONICA, Calif.--(BUSINESS WIRE)-- Kite, a Gilead Company (Nasdaq: GILD), today announces that the European Medicines Agency (EMA) Committee for Medicinal Products for Human Use (CHMP) has issued a positive opinion for Yescarta® (axicabtagene ciloleucel) for adult patients with diffuse large B-cell lymphoma (DLBCL) and high-grade B-cell lymphoma (HGBL) that relapses within 12 months from completion of, or is refractory to, first-line chemoimmunotherapy. If approved, Yescarta will be the first Chimeric Antigen Receptor (CAR) T-cell therapy approved for patients in Europe who do not respond to first-line treatment. Although 60% of newly diagnosed LBCL patients will respond to their initial treatment, 40% will relapse or will not respond and need 2nd line treatment.
“At Kite, we are committed to bringing the curative potential of cell therapy to the world, and changing the way cancer is treated,” said Christi Shaw, CEO, Kite. “Today’s positive CHMP opinion brings us a step closer to utilizing cell therapy earlier in the treatment journey, potentially transforming the standard of care for the most common and aggressive form of non-Hodgkin lymphoma.”
The European Commission will review the CHMP opinion, and a final decision on the marketing authorization is expected in the coming months.
“For people with DLBCL and HGBL who do not respond to first-line treatment or have an early relapse, outcomes are often poor and there are limited curative treatment options for these patients,” said Marie José Kersten, Professor of Hematology at Amsterdam University Medical Centers, Amsterdam. “If approved, axicabtagene ciloleucel may offer a new standard of care for patients with relapsed or refractory DLBCL and HGBL. Importantly, in a randomized trial of axicabtagene ciloleucel versus the current standard of care, quality of life also showed greater improvement in the experimental arm.”
The positive opinion for Yescarta is based on the primary results of the landmark Phase 3 ZUMA-7 study, the largest and longest trial of a CAR T-cell therapy versus standard of care (SOC) in second-line LBCL. Results demonstrated that at a median follow-up of two years, Yescarta-treated patients had a four-fold greater improvement in the primary endpoint of event-free survival (EFS; hazard ratio 0.40; 95% CI: 0.31-0.51, P<0.001) over the current SOC (8.3 months v 2.0 months). Additionally, Yescarta demonstrated a 2.5 fold increase in patients who were alive at two years without disease progression or need for additional cancer treatment vs SOC (41% v 16%). Improvements in EFS with Yescarta were consistent across key patient subgroups, including elderly patients (HR: 0.28 [95% CI: 0.16-0.46]), primary refractory patients (HR: 0.43 [95% CI: 0.32- 0.57]), high-grade B cell lymphoma including double-hit and triple-hit lymphoma patients (HGBL; HR: 0.28 [95% CI: 0.14-0.59]), and double expressor lymphoma patients (HR: 0.42 [95% CI: 0.27-0.67]).
In a separate, secondary analysis of Patient-Reported Outcomes (PROs) published in Blood patients receiving Yescarta and eligible for the PROs portion of the study (n=165) showed statistically significant improvements in Quality of Life (QoL) at Day 100 compared with those who received SOC (n=131), using a pre-specified analysis for three PRO-domains (EORTC QLQ-C30 Physical Functioning, EORTC QLQ-C30 Global Health Status/QOL, and EQ-5D-5L visual analog scale [VAS]). There was also a trend toward faster recovery to baseline QoL in the Yescarta arm versus SOC.
In the ZUMA-7 trial, Yescarta had a manageable safety profile that was consistent with previous studies. Among the 170 Yescarta-treated patients evaluable for safety, Grade ≥3 cytokine release syndrome (CRS) and neurologic events were observed in 6% and 21% of patients, respectively. No Grade 5 CRS or neurologic events occurred. In the SOC arm, 83% of patients had high-grade events, mostly cytopenias (low blood counts).
About ZUMA-7
ZUMA-7 is an ongoing, randomized, open-label, global, multicenter (US, Australia, Canada, Europe, Israel) Phase 3 study of 359 patients at 77 centers, evaluating the safety and efficacy of a single-infusion of Yescarta versus current SOC for second-line therapy (platinum-based salvage combination chemotherapy regimen followed by high-dose chemotherapy and autologous stem cell transplant in those who respond to salvage chemotherapy) in adult patients with relapsed or refractory LBCL within 12 months of first-line therapy. The primary endpoint is event free survival (EFS) as determined by blinded central review, and defined as the time from randomization to the earliest date of disease progression per Lugano Classification, commencement of new lymphoma therapy, or death from any cause. Key secondary endpoints include objective response rate (ORR) and overall survival (OS). Additional secondary endpoints include patient reported outcomes (PROs) and safety.
About Yescarta
Yescarta was first approved in Europe in 2018 and is currently indicated for three types of blood cancer: Diffuse Large B-Cell Lymphoma (DLBCL); Primary Mediastinal Large B-Cell Lymphoma (PMBCL); and Follicular Lymphoma (FL). For the full European Prescribing Information, please visit: https://www.ema.europa.eu/en/medicines/human/EPAR/yescarta
Please see full US Prescribing Information, including BOXED WARNING and Medication Guide.
YESCARTA is a CD19-directed genetically modified autologous T cell immunotherapy indicated for the treatment of:
About Kite
Kite, a Gilead Company, is a global biopharmaceutical company based in Santa Monica, California, with manufacturing operations in North America and Europe. Kite’s singular focus is cell therapy to treat and potentially cure cancer. As the cell therapy leader, Kite has more approved CAR T indications to help more patients than any other company. For more information on Kite, please visit www.kitepharma.com. Follow Kite on social media on Twitter (@KitePharma) and LinkedIn.
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer. Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220916005209/en/
Source: Gilead Sciences, Inc.
Sep. 16, 2022 7:38 AM ETGilead Sciences, Inc. (GILD)By: Ravikash, SA News Editor1 Comment
https://seekingalpha.com/symbol/GILD
Download PDFSeptember 16, 2022
DANVERS, Mass. – September 16, 2022 – Abiomed (Nasdaq: ABMD) announces two approvals from the U.S. Food and Drug Administration (FDA) related to clinical research of Impella® heart pumps in acute myocardial infarction (AMI) cardiogenic shock patients.
The FDA has approved the on-label RECOVER IV randomized controlled trial (RCT) for AMI cardiogenic shock patients. RECOVER IV is a two-arm trial that will assess whether percutaneous coronary intervention (PCI), with Impella support initiated prior to the PCI, is superior to PCI without Impella support.
“This landmark trial will be the culmination of over 20 years of research in the interventional therapy of AMI and will apply all the clinical advancements we have made to improve survival and heart recovery for AMI patients with cardiogenic shock as demonstrated in multiple prospective studies,” said William O’Neill, MD, medical director of the Center for Structural Heart Disease at Henry Ford Health and a RECOVER IV national co-principal investigator.
The primary endpoint of RECOVER IV is all-cause mortality at 30 days. Secondary endpoints include major adverse cardiovascular and cerebrovascular events (MACCE) at 30 days, days alive out of the hospital at six months, recovery of left ventricular (LV) function, need for durable ventricular assist device (VAD) or heart transplant, and health-related quality of life as measured by responses to the Kansas City Cardiomyopathy Questionnaire (KCCQ) at one year. Abiomed’s goal in conducting the trial is to achieve a global AMI cardiogenic shock Class I guideline recommendation for Impella and related best practice protocols, including Impella implantation pre-PCI (see figure 1).
“I am optimistic that RECOVER IV will further demonstrate the benefits of hemodynamic support and best practice protocols. These benefits include ventricular unloading using Impella pre-PCI, reduced LV wall stress, reduced pulmonary congestion, increased collateral coronary blood flow, and enhanced cardio protection so that more AMI cardiogenic shock patients can survive and achieve native heart recovery. The heart team and field have evolved and understand how important myocardial recovery is for both AMI and AMI cardiogenic shock to reduce the growing epidemic of heart failure,” said Navin K. Kapur, MD, executive director of the Cardiovascular Center for Research and Innovation (CVCRI) at Tufts Medical Center and a national co-principal investigator for RECOVER IV.
FDA Approves and Closes RECOVER III Post-Approval Study
Additionally, the FDA has approved and closed Impella’s prospective AMI cardiogenic shock post-approval study (PAS), RECOVER III. This study gathered real world evidence on AMI cardiogenic shock patients treated with Impella between 2017 – 2019, collecting detailed data including stages of cardiogenic shock, cardiac output, and timing of implantation. RECOVER III fulfills Abiomed’s PAS requirement and the FDA’s approval and closure of RECOVER III further validates Impella as a safe and effective therapy for AMI cardiogenic shock.
Impella remains the only mechanical circulatory support (MCS) device that has received the FDA’s highest level of pre-market approval (PMA) and PAS regulatory approval for AMI cardiogenic shock. Based on RECOVER III, Impella’s label for AMI cardiogenic shock will be updated to reflect data for up to one-year post-procedure.
AMI Cardiogenic Shock Clinical History
AMI cardiogenic shock has one of the highest mortality rates in the field of medicine. The survival rate has remained approximately 50% for cardiogenic shock patients in SCAI stages D and E without Impella support and associated best practices. Survival alone in cardiogenic shock is no longer the gold standard. Multiple Impella best practice studies demonstrate greater than 70% survival with greater than 90% native heart recovery (see figure 2). Impella best practices, such as pre-PCI implantation (see figure 3), have been developed by the recognized physician experts in the field of circulatory support and published over the last decade in multiple clinical studies from the U.S., Germany, Italy and Japan. Heart recovery after AMI cardiogenic shock improves patient quality of life and makes Impella one of the most cost-effective therapies in the CMS Medicare population and in private insurance. In the U.S. alone, more than 200,000 patients are admitted to the hospital every year in cardiogenic shock.
FDA Regulatory History of AMI Cardiogenic Shock
Impella is the most studied heart pump in the history of the FDA (see figures 4 & 5)and has exclusive FDA PMA as a safe and effective therapy for cardiogenic shock, high-risk PCI and right heart failure. Since 2004, more than 1,200 peer-reviewed studies, including real-world evidence analyses, prospective clinical studies and RCTs have published about the clinical benefits of Impella.
Impella has been used to treat more than 235,000 patients globally and is included in 13 clinical society guidelines. In 2021, the European Society of Cardiology upgraded Impella to a Class IIa recommendation for the treatment of cardiogenic shock. The intra-aortic balloon (IAB) is currently Class III (harmful) for routine use in cardiogenic shock in Europe and Japan based on the IABP-SHOCK II RCT, which demonstrated IAB compared to inotropic therapy provided no benefit to survival or hemodynamic augmentation. In 2020, IAB became Class III (harmful) in the U.S. guideline recommendations for post-cardiotomy cardiogenic shock.
All MCS and ventricular assist devices (VADs) since 1992 were approved with single arm studies compared to historical survival rates judged on objective performance criteria (OPC) due to the ethical and logistical challenges of randomizing critically ill patients requiring immediate hemodynamic augmentation. In 2008 and 2009, Abiomed attempted the FDA RECOVER II RCT, which compared Impella to IAB in AMI cardiogenic shock. RECOVER II enrolled only 1 patient in 15 months at more than 30 sites before being halted for logistical and ethical consent challenges and lack of enrollment.
The FDA granted Impella 510(k) clearance in 2008 and after multiple FDA and prospective physician-initiated studies, the FDA approved high-risk PCI in 2015, AMI cardiogenic shock in 2016 (see figure 6), and other forms of heart failure with cardiogenic shock in 2018. With the fulfillment of RECOVER III and approval of RECOVER IV RCT, Abiomed is pursuing an on-label study to strengthen the global guidelines and improve outcomes for patients.
Abiomed has sponsored and funded several AMI cardiogenic shock studies since 2006 (see figure 7) including the only FDA studies in this space. The difficulty randomizing AMI cardiogenic shock patients in execution has been demonstrated in multiple studies including IMPRESS in STEMI (n=18), IMPRESS in Cardiac Arrest (n=48), Seyfarth, et al. (n=26) and the Abiomed-sponsored FDA RECOVER II RCT (n=1). All these studies failed to randomize and were halted early for failure to enroll their designated numbers based on logistical and ethical consent challenges.
Recent FDA Regulatory Changes for Exception from Informed Consent (EFIC):
Randomizing patients in AMICS has been challenging because they require emergent care and are too sick to provide traditional informed consent to enroll in a trial. In 1996, the FDA created the exception from informed consent (EFIC) pathway for emergency clinical research. This pathway allows investigators to broadly educate a community about a trial, then enroll patients without consent from patients, their family or their legally authorized representatives.
In 2022, after engaging with the FDA on the RECOVER IV RCT study design, the FDA approved the RECOVER IV RCT study protocol, which includes the use of EFIC. This community awareness process is rare and only used when the patients being studied are experiencing a life-threatening medical condition causing serious deficiency of mental function. This is a significant milestone for the field and physician leaders. The next steps include local hospital Institutional Review Board (IRB) approvals and a commitment from physicians to randomize.
“This pivotal randomized trial is historic as the first to use EFIC community consent to enroll cardiogenic shock patients. I applaud the FDA for its partnership to help solve the consent challenges in cardiogenic shock RCTs and call on the physician community to enroll and randomize patients in RECOVER IV,” said Gregg W. Stone, MD, professor of medicine and director of academic affairs for the Mount Sinai Heart Health System in New York and study chair for RECOVER IV.
For more information on current best practices in treating AMI cardiogenic shock patients please click here.
For more information on the RECOVER IV RCT study please click here.
Figure 1.
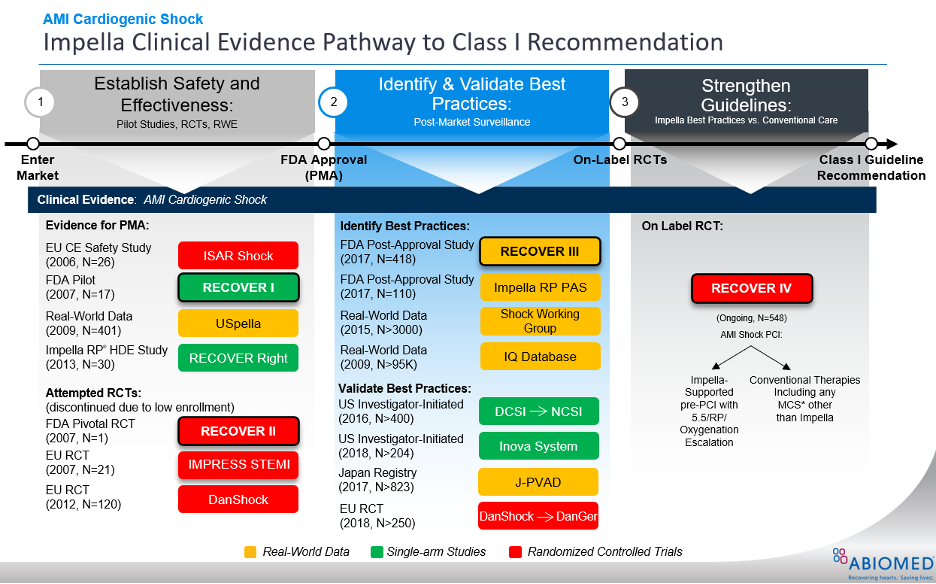
RECOVER IV is an on-label, two arm RCT that is designed to provide the clinical evidence needed to achieve a Class I guideline recommendation for Impella use in AMI cardiogenic shock.
Figure 2.
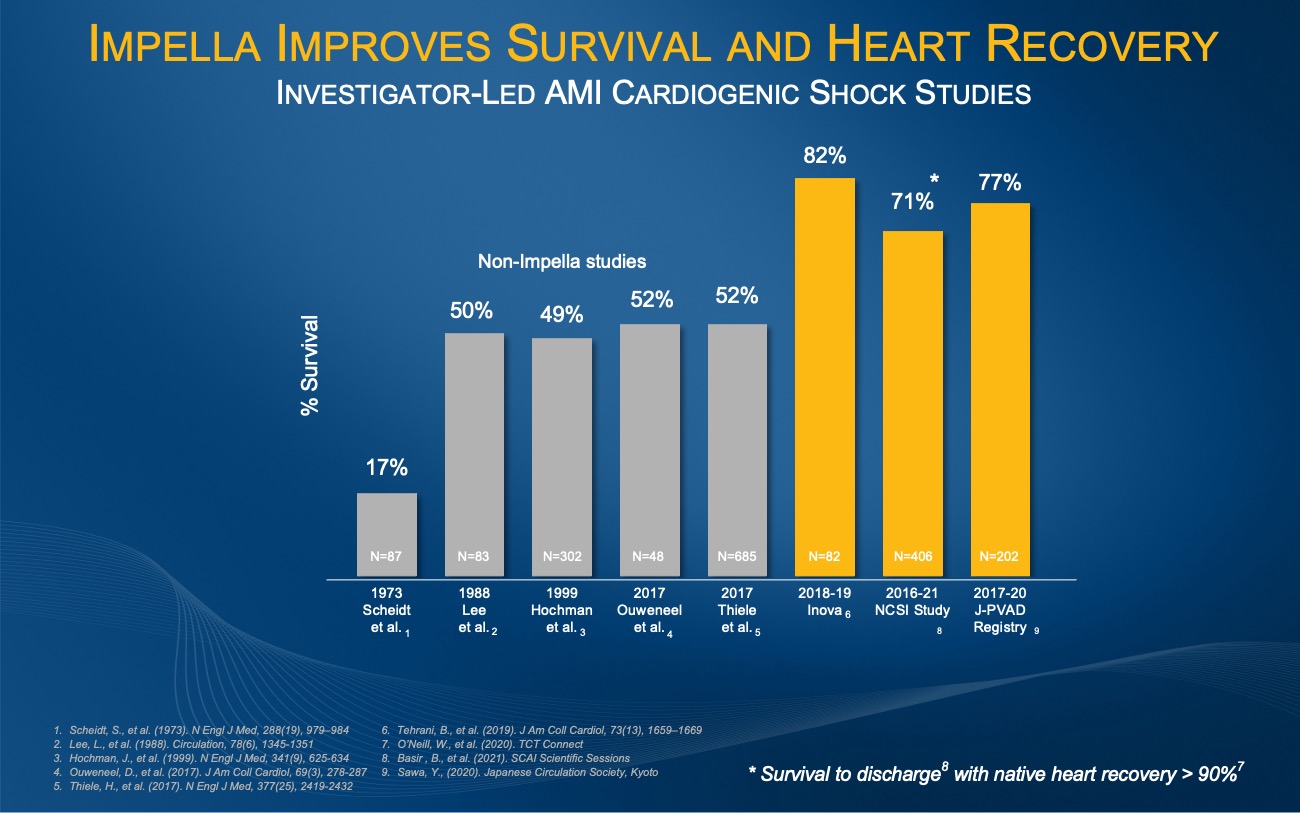
The investigator-led Inova, NCSI and J-PVAD studies all demonstrate an improvement from the historical AMI cardiogenic shock survival rate of approximately 50% when patients are treated with best practices including Impella.
Figure 3.
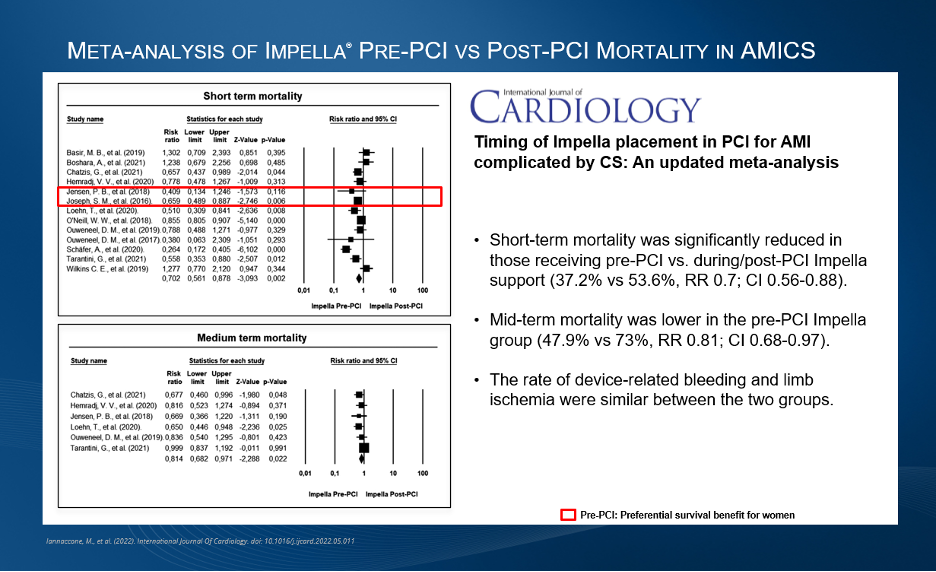
A 2022 meta-analysis demonstrated a reduction in short-term mortality and lower mid-term mortality for patients receiving Impella pre-PCI vs. post-PCI in AMICS.
Figure 4.
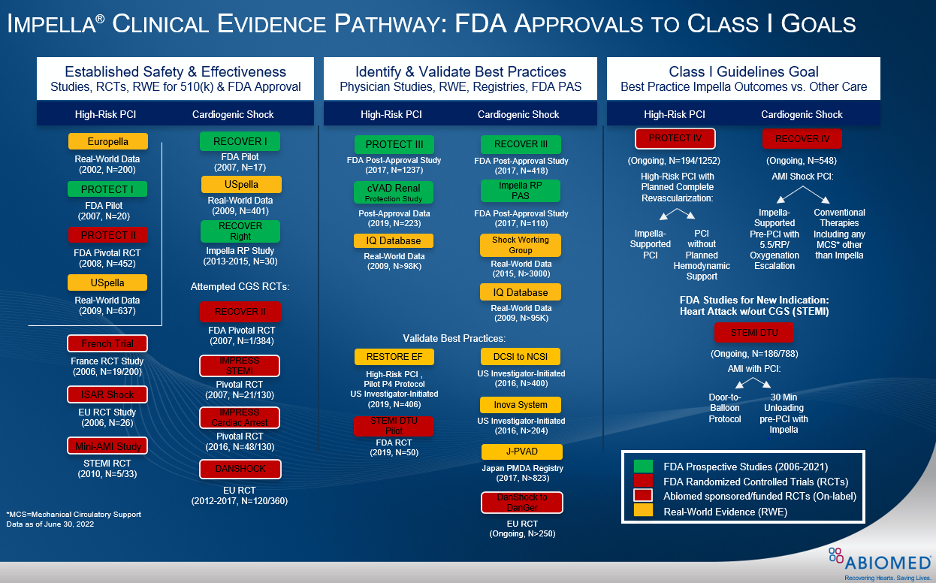
Impella is the most studied MCS device in the history of the FDA. Since 2004, more than 1,200 peer-reviewed studies, including real-world evidence analyses, prospective clinical studies and RCTs have published about the clinical benefits and cost effectiveness of Impella.
Figure 5.

Impella remains the most studied heart pump in the history of the FDA from 2006-2022, fueled by rigorous levels of evidence and high-quality data sources.
Figure 6.
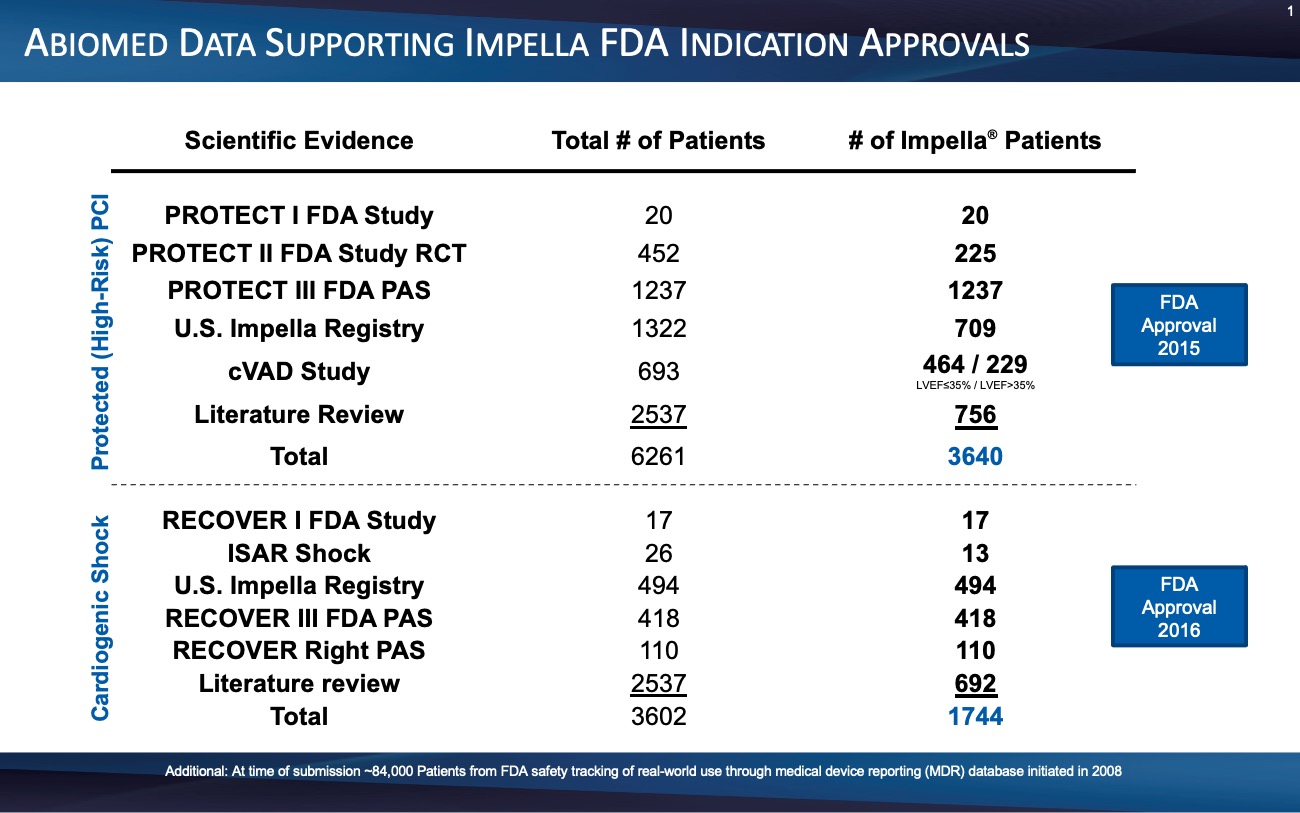
After multiple FDA and prospective physician-initiated studies, the FDA granted Impella a PMA for AMI cardiogenic shock in 2016.
Figure 7.
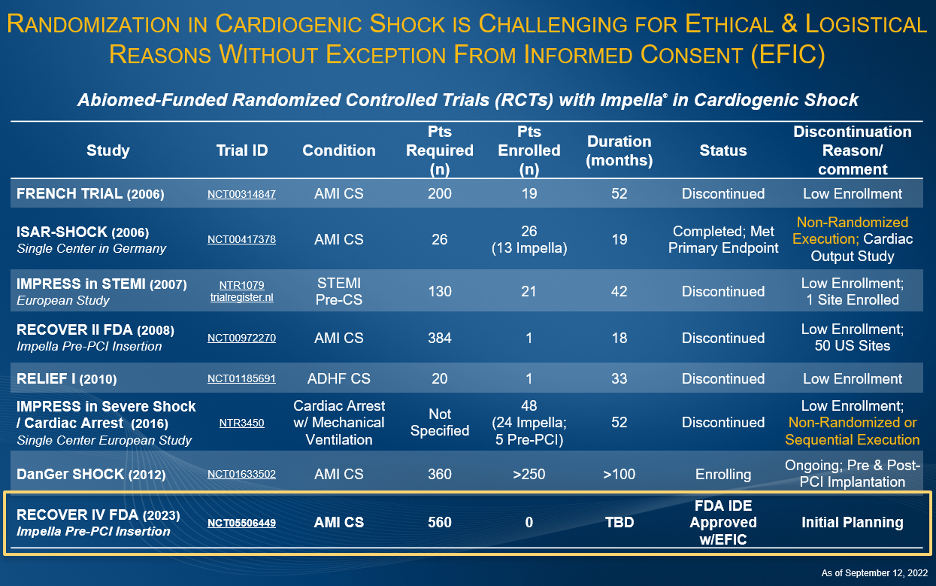
Abiomed has sponsored and funded several AMI cardiogenic shock studies since 2006 including the only FDA studies. Difficulty in randomization has been demonstrated in multiple studies including IMPRESS in STEMI (n=18), IMPRESS in Cardiac Arrest (n=48), Seyfarth, et al. (n=26) and the Abiomed-sponsored FDA RECOVER II RCT (n=1). All these studies failed to randomize in execution and were halted early for failure to enroll their designated numbers.
ABOUT IMPELLA HEART PUMPS
Impella 2.5® and Impella CP® with SmartAssist® are U.S. FDA approved to treat certain advanced heart failure patients undergoing elective and urgent percutaneous coronary interventions (PCI), such as stenting or balloon angioplasty, to reopen blocked coronary arteries.
Impella 2.5, Impella CP, Impella CP with SmartAssist®, Impella 5.0®, Impella LD®, and Impella 5.5® with SmartAssist® are U.S. FDA approved to treat heart attack or cardiomyopathy patients in cardiogenic shock and have the unique ability to enable native heart recovery, allowing patients to return home with their own heart.
ABOUT ABIOMED
Based in Danvers, Massachusetts, U.S.A., Abiomed is a leading provider of medical technology that provides circulatory support and oxygenation. Our products are designed to enable the heart to rest by improving blood flow and/or provide sufficient oxygenation to those in respiratory failure. For additional information, please visit: http://www.abiomed.com/.
Sep. 16, 2022 9:23 AM ET
By: Dania Nadeem, SA News Editor
PUBLISHED16 September 2022
A prespecified interim analysis of the ALPHA Phase III trial evaluating danicopan (ALXN2040), an investigational, oral factor D inhibitor, as an add-on to C5 inhibitor therapy Ultomiris (ravulizumab) or Soliris (eculizumab) showed positive high-level results in patients with paroxysmal nocturnal haemoglobinuria (PNH) who experience clinically significant extravascular haemolysis (EVH).
The trial met its primary endpoint of change in haemoglobin from baseline at 12 weeks and key secondary endpoints, including transfusion avoidance and change in Functional Assessment of Chronic Illness Therapy (FACIT) Fatigue score. Danicopan plus Ultomiris or Soliris demonstrated superiority compared to placebo plus Ultomiris or Soliris for this specific patient population, with statistically significant and clinically meaningful improvements in haemoglobin levels, transfusion avoidance and FACIT Fatigue scores from baseline.
PNH is a rare and severe blood disorder characterised by the destruction of red blood cells, known as intravascular haemolysis (IVH), and white blood cell and platelet activation that can cause thrombosis (blood clots) and result in organ damage and potentially premature death.1-3
Marc Dunoyer, Chief Executive Officer, Alexion, said: “Alexion has relentlessly innovated for the PNH community, pioneering with Soliris, the first treatment for PNH, and establishing Ultomiris as a standard of care. We are proud of our continued innovation to advance new ways of targeting the complement cascade to help address the needs of patients living with this debilitating disease. These are the first positive Phase III results for an oral factor D inhibitor and demonstrate the potential for danicopan add-on therapy to improve signs and symptoms and reduce the need for transfusions for the limited proportion of people living with PNH who experience clinically significant EVH.”
Professor Jong-Wook Lee, MD, PhD, Department of Haematology at Seoul St. Mary's Hospital of The Catholic University of Korea, and investigator in the ALPHA trial, said: “C5 inhibitors are a proven treatment option for patients living with PNH, yet a small percentage may continue to experience anaemia and burden of transfusion due to clinically significant EVH, however it is not life-threatening. These data show that danicopan has the potential to resolve clinically significant EVH while allowing patients to remain on standard of care treatment with Ultomiris or Soliris.”
Danicopan was generally well tolerated and there were no clinically meaningful differences in safety results observed between the danicopan plus C5 inhibitor group and control group.
Alexion, AstraZeneca Rare Disease, will present these data at a forthcoming medical meeting and intends to proceed with regulatory submissions in the coming months.
ALPHA
ALPHA is a pivotal, global Phase III trial designed to evaluate the efficacy of danicopan as an add-on to C5 inhibitor therapy Ultomiris (ravulizumab) or Soliris (eculizumab) in patients with PNH who experience clinically significant EVH. In the double-blind, placebo controlled, multiple-dose trial, patients were enrolled and randomised to receive danicopan or placebo (2:1) in addition to their ongoing C5 inhibitor therapy for 12 weeks. Prespecified interim analysis for efficacy was planned once 75 percent (N~63) of participants completed 12 weeks of treatment period 1. At 12 weeks, patients on placebo plus C5 inhibitor are switched to danicopan plus a C5 inhibitor and patients on danicopan plus a C5 inhibitor remain on treatment for an additional 12 weeks.
Patients who complete both treatment periods (24 weeks) have the option to participate in a long-term extension period and continue to receive danicopan in addition to C5 inhibitor therapy.
Danicopan (ALXN2040)
Danicopan is an investigational oral medicine in development as an add-on to C5 inhibitor therapy Ultomiris (ravulizumab) or Soliris (eculizumab) for patients with PNH who experience clinically significant EVH. It is designed to selectively inhibit factor D, a complement system protein that plays a key role in the amplification of the complement system response. Danicopan has been granted Breakthrough Therapy designation by the US Food and Drug Administration and PRIority MEdicines (PRIME) status by the European Medicines Agency. Danicopan has also been granted Orphan Drug Designation in the US and orphan designation in the EU for the treatment of PNH. Alexion is also evaluating danicopan as a potential monotherapy for geographic atrophy in a Phase II clinical trial.
Alexion
Alexion, AstraZeneca Rare Disease, is the group within AstraZeneca focused on rare diseases, created following the 2021 acquisition of Alexion Pharmaceuticals, Inc. As a leader in rare diseases for 30 years, Alexion is focused on serving patients and families affected by rare diseases and devastating conditions through the discovery, development and commercialisation of life-changing medicines. Alexion focuses its research efforts on novel molecules and targets in the complement cascade and its development efforts on haematology, nephrology, neurology, metabolic disorders, cardiology and ophthalmology. Headquartered in Boston, Massachusetts, Alexion has offices around the globe and serves patients in more than 50 countries.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Sep. 16, 2022 5:29 AM ET
By: Ravikash, SA News Editor
AstraZeneca (NASDAQ:AZN) said its investigational drug danicopan as an add-on to Ultomiris or Soliris showed positive high-level results in patients with paroxysmal nocturnal haemoglobinuria (PNH) who experience clinically significant extravascular haemolysis (EVH) in a phase 3 trial.
https://seekingalpha.com/symbol/AZN
September 16, 2022
-- If Granted by the European Commission, Veklury Will Become the First and Only Authorized Antiviral Treatment for Pediatric Patients Under 12 Years of Age in the European Union --
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion to extend the indication of Veklury® (remdesivir) for the treatment of pediatric patients (weighing at least 40 kg) who do not require supplemental oxygen and are at increased risk of progressing to severe COVID-19 and pediatric patients (4 weeks of age and older and weighing at least 3 kg) with SARS-CoV-2 with pneumonia who require supplemental oxygen (low- or high-flow oxygen or other non-invasive ventilation at the start of treatment). The European Commission (EC) will review the CHMP recommendation, and if adopted, Veklury will be the only authorized COVID-19 treatment for adolescents at high risk of progressing to severe COVID-19 and pediatric patients with COVID-19 requiring supplemental oxygen.
“As the pandemic persists, there remains a critical need for proven and effective antiviral therapies like Veklury that can treat some of the most vulnerable in our society,” said Merdad Parsey, MD, PhD, Chief Medical Officer, Gilead Sciences. “We are proud of today’s CHMP opinion that represents a positive step toward bridging the treatment gap for children and helping them recover from COVID-19 more quickly.”
This positive opinion was based on results from the ongoing CARAVAN Phase 2/3 study, which demonstrated Veklury was generally well-tolerated among pediatric patients hospitalized with COVID-19, with a high proportion of participants showing clinical improvement and recovery, as well as data from trials in adults. Of the 53 pediatric patients enrolled in the CARAVAN study, no new safety signals were apparent for patients treated with Veklury. Overall, 75% and 85% showed clinical improvement (≥2 point increase on an ordinal scale) at Day 10 and last assessment, respectively, while 60% and 83% were discharged by Day 10 and Day 30, respectively. In the study, 38 patients (72%) experienced adverse events (AEs), with 11 patients (21%) experiencing serious adverse events (SAEs) that were determined not to be study-drug related, including three participant deaths, which were consistent with the patients’ underlying medical conditions prior to study entry or with COVID-19 during hospitalization.
“As the COVID-19 pandemic evolves, it’s important to have effective treatments with well-established safety profiles, including for the vulnerable groups like children to help them recover faster from COVID-19,” said Pablo Rojo, MD, PhD, Pediatric Infectious Diseases Specialist of Hospital 12 de Octubre, Madrid, Spain and Associate Professor of Complutense University, Madrid, Spain. “Therefore, the medical community welcomes this CHMP positive opinion for Veklury in pediatric patients and looks forward to the EC decision.”
In the European Economic Area (EEA), Veklury is the only antiviral indicated for both the treatment of COVID-19 in adult patients who do not require supplemental oxygen and are at increased risk of developing severe COVID-19, as well as adults and adolescents (aged 12 to less than 18 years and weighing at least 40 kg) with pneumonia requiring supplemental oxygen (low- or high-flow oxygen or other non-invasive ventilation at start of treatment).
About Veklury
Veklury (remdesivir) is a nucleotide analog invented by Gilead, building on more than a decade of the company’s antiviral research. Veklury is a foundation for the treatment of hospitalized patients with COVID-19 and is a recommended treatment for reducing disease progression in non-hospitalized patients at high risk of disease progression. Veklury has an established safety profile and minimal known drug interactions in diverse populations. It can help reduce disease progression across a spectrum of disease severity and enable patients to recover faster, freeing up limited hospital resources and saving healthcare systems money.
Veklury directly inhibits viral replication inside of the cell by targeting the SARS-CoV-2 viral RNA polymerase. Based on in vitro analyses, Veklury retains antiviral activity against Omicron subvariants BA.2.12.2, BA.4 and BA.5, which are currently the most common circulating variants. Data continue to confirm that Veklury retains antiviral activity against all Omicron subvariants analyzed to date. Gilead continuously evaluates the activity of Veklury against new SARS-CoV-2 variants of concern as they emerge around the world.
Veklury is approved in more than 50 countries worldwide. To date, Veklury and generic remdesivir have been made available to more than 11 million patients around the world, including more than 7 million people in 127 middle- and low-income countries through Gilead’s voluntary licensing program. These licenses currently remain royalty-free, reflecting Gilead’s existing commitment to enabling broad patient access to remdesivir.
U.S. Indication for Veklury
Veklury® (remdesivir 100 mg for injection) is indicated for the treatment of COVID-19 in adults and pediatric patients (≥28 days old and weighing ≥3 kg) with positive results of SARS-CoV-2 viral testing, who are:
For more information, please see the U.S. full Prescribing Information available at www.gilead.com.
U.S. Important Safety Information for Veklury
View source version on businesswire.com: https://www.businesswire.com/news/home/20220915006144/en/
Source: Gilead Sciences, Inc.
September 15, 2022
WHO Expands Recommendation for Veklury® (Remdesivir) to Patients With Severe Disease in Latest Update to COVID-19 Guideline
Sep. 16, 2022 7:38 AM ET
By: Ravikash, SA News Editor1 Comment
A committee of the European Medicines Agency (EMA) recommended the approval of expanded use of Gilead Sciences' (NASDAQ:GILD) Veklury (remdesivir) to treat pediatric patients who do not require supplemental oxygen and are at increased risk of progressing to severe COVID-19.
https://seekingalpha.com/symbol/GILD

Sep. 15, 2022 9:00 AM ETIncyte Corporation (INCY)
SINGAPORE, Sept. 15, 2022 /PRNewswire/ -- A NEW targeted therapy to treat a rare bile duct cancer called cholangiocarcinoma has been approved for use in Australia.
PEMAZYRE® (pemigatinib) has been provisionally approved by the Therapeutic Goods Administration (TGA) "for the treatment of adult patients with locally advanced or metastatic cholangiocarcinoma with a fibroblast growth factor receptor 2 (FGFR2) fusion or rearrangement that has progressed after at least one prior line of systemic therapy. The decision to approve this indication has been made on the basis of overall response rate (ORR) and duration of response (DOR). Continued approval of this indication depends on verification and description of benefit in confirmatory trial(s)." 1
This means it will be available to Australian patients with cholangiocarcinoma who have been tested for an abnormal gene change in their tumour called an FGFR2 fusion or rearrangement – a defect that can drive cancer growth.
This defect is estimated to be present in around 15% of patients diagnosed with cholangiocarcinoma.2
Australian oncologist Dr David Goldstein said the approval of PEMAZYRE was "a significant step forward" for Australian patients who are diagnosed with this rare and aggressive cancer.
He commented: "This TGA approval of a targeted therapy heralds an era of precision medicine in this rare cancer.
"The study underpinning this approval, FIGHT 202, saw clinical outcomes you would not expect from previous experience after initial treatment ceases to be effective.
"This is one of only a few second-line therapies to offer serious real benefits to patients and will be very focused upon the right tumour type.
"Up until now, we have had some chemotherapies that were effective, but only in a minority of patients.
"This TGA approval is a positive first step. Subsequent reimbursement via the Pharmaceutical Benefits Scheme is really the only way forward to translate this scientific knowledge of targeting a specific tumour type into everyday clinical practice."
PEMAZYRE, developed by Incyte (INCY) and partnered with independent pharmaceutical company Specialised Therapeutics (ST) for commercialisation in Australia, New Zealand and Singapore, belongs to a class of drugs called kinase inhibitors and works by blocking the abnormal FGFR2 protein in bile duct tumour cells and preventing cell growth.
The TGA approval follows PEMAZYRE's approval in the United States, Europe, Great Britain, Canada and Japan.
While PEMAZYRE is not yet reimbursed in Australia, it is currently being made available to eligible patients via a co-pay access program.
ST CEO Carlo Montagner said PEMAZYRE was a "wonderful inclusion" to the company's portfolio of rare cancer therapies and was synergistic with its mission of providing therapies to patient populations where there is a high unmet medical need.
He said: "We look forward to providing this new highly-targeted treatment to eligible patients with cholangiocarcinoma who have limited therapy options."
PEMAZYRE's approval is based on a clinical trial known as the FIGHT-202 study – a Phase 2 investigation evaluating the safety and efficacy of PEMZYRE in adult patients with previously treated, locally advanced or metastatic cholangiocarcinoma with documented FGFR2 fusion or rearrangement. 2
In the primary analysis of 108 patients enrolled in the trial, treatment with PEMAZYRE resulted in an objective response rate of 37% with 3.7% of patients having a confirmed complete response and 33.3% having a confirmed partial response. The disease control rate was 82.2%.1
The safety analysis, including 147 patients, demonstrated that PEMAZYRE was generally well tolerated.
The most common adverse reactions were hyperphosphataemia: includes hyperphosphataemia and increased blood phosphorous (60.5%), alopecia (49.7%), diarrhoea (46.9%), nail toxicity (44.9%), fatigue (43.5%), nausea (41.5%), dysgeusia (40.8%), stomatitis (37.4%), constipation (36.7%), dry mouth (34.0%), dry eye (27.9%), arthralgia (25.9%), hypophosphataemia (23.1%), dry skin (21.8%) and palmar-plantar erythrodysaesthesia syndrome (16.3%).1
Ends.
About PEMAZYRE
PEMAZYRE (pemigatinib) a fibroblast growth factor receptor (FGFR) inhibitor, has provisional approval in Australia for the treatment of adult patients with locally advanced or metastatic cholangiocarcinoma with a fibroblast growth factor receptor 2 (FGFR2) fusion or rearrangement that has progressed after at least one prior line of systemic therapy. The decision to approve this indication has been made on the basis of overall response rate (ORR) and duration of response (DOR). Continued approval of this indication depends on verification and description of benefit in confirmatory trial(s).
PEMAZYRE is marketed by Incyte in the United States, Europe and Japan. Incyte has established various license or distribution agreements for Pemazyre in certain geographies and retains all other rights to develop and commercialize pemigatinib outside of the United States.
Pemazyre is a trademark of Incyte Corporation.
References:
1. PEMAZYRE (pemigatinib) Product Information Australia
View original content:https://www.prnewswire.com/news-releases/new-treatment-for-rare-cancer-cholangiocarcinoma-approved-in-australia-301625157.html
Sep. 15, 2022 10:24 AM ETIncyte Corporation (INCY)By: Dania Nadeem, SA News Editor
https://seekingalpha.com/symbol/INCY
The committee recommended 12 extensions of indication for medicines that are already authorised in the EU: Adtralza , Biktarvy, Brukinsa, Evusheld , Exparel liposomal, Revolade, Skyrizi, Vaxneuvance, Veklury (includes two extensions of indication for two paediatric populations, see the COVID-19 update below), Xalkori and Yescarta.
Sep. 16, 2022 9:31 AM ET
By: Anuron Mitra, SA News Editor
09/15/2022CATEGORY:
Positive results from CheckMate -76K reinforce the potential benefit of Opdivo in earlier stages of melanoma
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) today announced that the Phase 3 CheckMate -76K trial evaluating Opdivo (nivolumab) as a single agent in the adjuvant settingin patients with completely resected stage IIB/C melanoma met its primary endpoint and demonstrated a statistically significant and clinically meaningful benefit in recurrence-free survival (RFS) versus placebo at a pre-specified interim analysis. No new safety signals were observed at the time of the analysis.
CheckMate -76K is part of BMS’ development program studying Opdivo and Opdivo-based combinations in earlier stages of cancer, which currently spans seven tumor types.
“Stage IIB/C melanoma patients are at high risk of disease recurrence, with approximately one third of stage IIB and half of stage IIC patients experiencing recurrence within five years after surgery. The results of the CheckMate -76K study represent a significant advancement for patients with stage IIB/C melanoma and an extension of our legacy in the treatment of melanoma,” said Gina Fusaro, PhD, development program lead, melanoma, Bristol Myers Squibb. “Recurrence represents a life-altering event for people living with cancer. Treating with Opdivo in earlier stages of cancer, when the immune system may be more responsive, has the potential to help prevent recurrence – a critical goal of improving patient outcomes.”
The company will complete a full evaluation of the CheckMate -76K data and looks forward to sharing the results at an upcoming medical conference, as well as with health authorities. Bristol Myers Squibb thanks the patients and investigators who were involved in the CheckMate -76K clinical trial.
About CheckMate -76K
CheckMate -76K is a randomized Phase 3, double-blind study evaluating adjuvant Opdivo (nivolumab) 480 mg Q4W for up to 12 months versus placebo in patients with completely resected stage IIB/C melanoma.
The primary endpoint of the trial is recurrence-free survival (RFS). Secondary endpoints of the trial include overall survival (OS), distant metastases-free survival (DMFS), progression-free survival on next-line therapy (PFS2), and safety endpoints.
About Melanoma
Melanoma is a form of skin cancer characterized by the uncontrolled growth of pigment-producing cells (melanocytes) located in the skin. Metastatic melanoma is the deadliest form of the disease and occurs when cancer spreads beyond the surface of the skin to other organs. The incidence of melanoma has been increasing steadily for the last 30 years. In the United States, 99,780 new diagnoses of melanoma and about 7,650 related deaths are estimated for 2022. Globally, the World Health Organization estimates that by 2035, melanoma incidence will reach 424,102, with 94,308 related deaths. Melanomas can be mostly treatable when caught in very early stages; however, survival rates can decrease as the disease progresses.
Bristol Myers Squibb: Creating a Better Future for People with Cancer
Bristol Myers Squibb is inspired by a single vision — transforming patients’ lives through science. The goal of the company’s cancer research is to deliver medicines that offer each patient a better, healthier life and to make cure a possibility. Building on a legacy across a broad range of cancers that have changed survival expectations for many, Bristol Myers Squibb researchers are exploring new frontiers in personalized medicine, and through innovative digital platforms, are turning data into insights that sharpen their focus. Deep scientific expertise, cutting-edge capabilities and discovery platforms enable the company to look at cancer from every angle. Cancer can have a relentless grasp on many parts of a patient’s life, and Bristol Myers Squibb is committed to taking actions to address all aspects of care, from diagnosis to survivorship. Because as a leader in cancer care, Bristol Myers Squibb is working to empower all people with cancer to have a better future.
About Opdivo
Opdivo is a programmed death-1 (PD-1) immune checkpoint inhibitor that is designed to uniquely harness the body’s own immune system to help restore anti-tumor immune response. By harnessing the body’s own immune system to fight cancer, Opdivo has become an important treatment option across multiple cancers.
Opdivo’s leading global development program is based on Bristol Myers Squibb’s scientific expertise in the field of Immuno-Oncology, and includes a broad range of clinical trials across all phases, including Phase 3, in a variety of tumor types. To date, the Opdivo clinical development program has treated more than 35,000 patients. The Opdivo trials have contributed to gaining a deeper understanding of the potential role of biomarkers in patient care, particularly regarding how patients may benefit from Opdivo across the continuum of PD-L1 expression.
In July 2014, Opdivo was the first PD-1 immune checkpoint inhibitor to receive regulatory approval anywhere in the world. Opdivo is currently approved in more than 65 countries, including the United States, the European Union, Japan and China. In October 2015, the Company’s Opdivo and Yervoy combination regimen was the first Immuno-Oncology combination to receive regulatory approval for the treatment of metastatic melanoma and is currently approved in more than 50 countries, including the United States and the European Union.
OPDIVO U.S. INDICATIONS
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of adult patients with unresectable or metastatic melanoma.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adult patients with unresectable or metastatic melanoma.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of adult patients with melanoma with involvement of lymph nodes or metastatic disease who have undergone complete resection.
OPDIVO® (nivolumab), in combination with platinum-doublet chemotherapy, is indicated as neoadjuvant treatment of adult patients with resectable (tumors ≥4 cm or node positive) non-small cell lung cancer (NSCLC).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) whose tumors express PD-L1 (≥1%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab) and 2 cycles of platinum-doublet chemotherapy, is indicated for the first-line treatment of adult patients with metastatic or recurrent non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) with progression on or after platinum-based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving OPDIVO.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with unresectable malignant pleural mesothelioma (MPM).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with intermediate or poor risk advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab), in combination with cabozantinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with advanced renal cell carcinoma (RCC) who have received prior anti-angiogenic therapy.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with classical Hodgkin lymphoma (cHL) that has relapsed or progressed after autologous hematopoietic stem cell transplantation (HSCT) and brentuximab vedotin or after 3 or more lines of systemic therapy that includes autologous HSCT. This indication is approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN) with disease progression on or after platinum-based therapy.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
OPDIVO® (nivolumab), as a single agent, is indicated for the adjuvant treatment of adult patients with urothelial carcinoma (UC) who are at high risk of recurrence after undergoing radical resection of UC.
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of adult and pediatric (12 years and older) patients with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adults and pediatric patients 12 years and older with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adult patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with unresectable advanced, recurrent or metastatic esophageal squamous cell carcinoma (ESCC) after prior fluoropyrimidine- and platinum-based chemotherapy.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of completely resected esophageal or gastroesophageal junction cancer with residual pathologic disease in adult patients who have received neoadjuvant chemoradiotherapy (CRT).
OPDIVO® (nivolumab), in combination with fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of adult patients with unresectable advanced or metastatic esophageal squamous cell carcinoma (ESCC).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with unresectable advanced or metastatic esophageal squamous cell carcinoma (ESCC).
OPDIVO® (nivolumab), in combination with fluoropyrimidine- and platinum- containing chemotherapy, is indicated for the treatment of adult patients with advanced or metastatic gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma.
Please see US Full Prescribing Information for OPDIVO and YERVOY.
About Bristol Myers Squibb
Bristol Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information about Bristol Myers Squibb, visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
Source: Bristol Myers Squibb
Sep. 15, 2022 8:10 AM ET
Bristol-Myers Squibb Company (BMY)
By: Ravikash, SA News Editor
On 15 September 2022, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion recommending a change to the terms of the marketing authorisation for the medicinal product Xalkori. The marketing authorisation holder for this medicinal product is Pfizer Europe MA EEIG.
The CHMP adopted 2 new indications for the treatment of paediatric patients with anaplastic large cell lymphoma (ALCL) or inflammatory myofibroblastic tumour (IMT). For information, the full indications for Xalkori will therefore be as follows:1
Xalkori as monotherapy is indicated for:
Detailed recommendations for the use of this product will be described in the updated summary of product characteristics (SmPC), which will be published in the revised European public assessment report (EPAR), and will be available in all official European Union languages after a decision on this change to the marketing authorisation has been granted by the European Commission.
First published: 16/09/2022
EMA/CHMP/757723/2022
Indications
XALKORI® (crizotinib) is a prescription medicine used to treat people with non-small cell lung cancer (NSCLC) that has spread to other parts of the body and is caused by a defect in either a gene called ALK (anaplastic lymphoma kinase) or a gene called ROS1. It is not known if XALKORI is safe and effective in children.
Sep. 16, 2022 9:47 AM ET
By: Ravikash, SA News Editor1 Comment

September 16, 2022 7:30 am ET
RAHWAY, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside of the United States and Canada, announced today that the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended the approval of VAXNEUVANCE™(Pneumococcal 15-valent Conjugate Vaccine) (pronounced VAKS-noo-vans) for active immunization for the prevention of invasive disease, pneumonia and acute otitis media caused by Streptococcus pneumoniae in infants, children and adolescents from 6 weeks to less than 18 years of age. VAXNEUVANCE is currently authorized for use in the European Union (EU) for individuals 18 years of age and older.
The CHMP opinion will now be considered by the European Commission (EC) for amending the marketing authorization in the EU, and a final decision is expected by the end of the year.
“We are committed to advancing protection for those at increased risk for pneumococcal disease, which includes those under the age of 2 years and children of any age who have certain underlying conditions,” said Dr. Eliav Barr, senior vice president, head of global clinical development and chief medical officer, Merck Research Laboratories. “We are pleased with the CHMP’s positive opinion as it brings us one step closer to our goal of helping to protect against pneumococcal strains that pose substantial risk to infants and children in Europe.”
Pneumococcal disease is an infection caused by the bacterium Streptococcus pneumoniae, or pneumococcus. While there are more than 100 different types of S. pneumoniae, called serotypes, a selected number of serotypes are responsible for the majority of pneumococcal infections. Invasive pneumococcal disease (IPD) can cause serious and potentially life-threatening infections such as bacteremia (infection in the bloodstream); bacteremic pneumonia (pneumonia with bacteremia); and meningitis (infection of the coverings of the brain and spinal cord).
The CHMP opinion was based on data from eight randomized, double-blind clinical studies that enrolled approximately 8,400 individuals from a variety of pediatric populations and clinical circumstances; of these, approximately 5,400 received VAXNEUVANCE.
In July 2021, VAXNEUVANCE received approval from the U.S. Food and Drug Administration (FDA) for active immunization for the prevention of invasive disease caused by Streptococcus pneumoniae serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, 22F, 23F and 33F in adults 18 years of age and older, and in June 2022, the FDA approved an expanded indication for VAXNEUVANCE to include individuals 6 weeks through 17 years of age.
About VAXNEUVANCE (Pneumococcal 15-valent Conjugate Vaccine)
VAXNEUVANCE, Merck’s 15-valent pneumococcal conjugate vaccine, consists of purified capsular polysaccharides from S.pneumoniae serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, 22F, 23F and 33F individually conjugated to CRM197 carrier protein.
VAXNEUVANCE is indicated in the EU for active immunization for the prevention of invasive pneumococcal disease and pneumonia caused by S. pneumoniae in individuals 18 years of age and older.
VAXNEUVANCE is indicated in the U.S. for active immunization of individuals 6 weeks of age and older for the prevention of invasive disease caused by the S. pneumoniae serotypes contained in the vaccine.
About Merck
At Merck, known as MSD outside of the United States and Canada, we are unified around our purpose: We use the power of leading-edge science to save and improve lives around the world. For more than 130 years, we have brought hope to humanity through the development of important medicines and vaccines. We aspire to be the premier research-intensive biopharmaceutical company in the world – and today, we are at the forefront of research to deliver innovative health solutions that advance the prevention and treatment of diseases in people and animals. We foster a diverse and inclusive global workforce and operate responsibly every day to enable a safe, sustainable and healthy future for all people and communities. For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Please see Prescribing Information for VAXNEUVANCE (Pneumococcal 15-valent Conjugate Vaccine) at https://www.merck.com/product/usa/pi_circulars/v/vaxneuvance/vaxneuvance_pi.pdf and Patient Information/Medication Guide for VAXNEUVANCE at https://www.merck.com/product/usa/pi_circulars/v/vaxneuvance/vaxneuvance_ppi.pdf .
Source: Merck & Co., Inc.
Sep. 16, 2022 8:13 AM ET
By: Ravikash, SA News Editor
Sep. 13, 2022 7:05 AM ETMerck & Co., Inc. (MRK)
Approval is based on the Phase 3 KEYNOTE-716 Trial
Melanoma is the most serious of all skin cancers, with over 5,000 Canadians diagnosed each year.1,2
KIRKLAND, QC, Sept. 13, 2022 /CNW/ - Merck (MRK), known as MSD outside the United States and Canada, announced that Health Canada has granted approval for KEYTRUDA® (pembrolizumab), Merck's anti-PD-1 therapy, for the adjuvant treatment of adult and pediatric (12 years and older) patients with stage IIB or IIC melanoma following complete resection.3 This approval is based on the results from the Phase 3 KEYNOTE-716 trial, which demonstrated a statistically significant improvement in recurrence-free survival (RFS).3

In 2022, an estimated 9,000 Canadians will be diagnosed with melanoma,4 a form of cancer that takes place when melanocytes, the cells responsible for melanin production, start to grow uncontrollably and develop into a tumour.5 Although it is the least common of all skin cancers, melanoma is the most serious type and early diagnosis and treatment are critical.1,5
"We welcome the news of a new treatment option for Canadians living with this disease as the incidence of melanoma continues to rise across the country,"6,8 said Kathy Barnard, Founder/President of Save Your Skin Foundation. "Having options available right after surgery can help take action against a disease that moves quickly if not caught."
"Melanoma can affect anyone, including children, for whom, although rare, this is the most common amongst pediatric skin cancer types,"7 said Falyn Katz, Executive Director, Melanoma Canada. "Having options available, like this one, can help to make a difference for Canadians facing this particular type of skin cancer, helping them navigate the disease."
About KEYNOTE-716 Trial
Health Canada's approval is based on findings from KEYNOTE-716, a multicenter, randomized, double-blind, placebo-controlled clinical trial in patients (n=976) with completely resected stage IIB or IIC melanoma.3 Only patients that had not been previously treated for melanoma beyond complete surgical resection for their melanoma prior to study entry were included in the trial.3 Patients were randomized to KEYTRUDA® 200 mg or the pediatric (≥12 years old) dose of KEYTRUDA® 2 mg/kg intravenously (up to a maximum of 200 mg) every three weeks (n=487) or placebo (n=489) for up to one year until disease recurrence or unacceptable toxicity.3 The primary efficacy outcome was investigator-assessed recurrence-free survival (RFS). RFS was defined as the time between the date of randomization and the date of first recurrence (local, regional, or distant metastasis) or death, whichever occurs first.3
The results of KEYNOTE-716 demonstrated a statistically significant improvement in RFS for patients randomized to receive KEYTRUDA® compared with patients randomized to placebo at the first pre-specified interim analysis.3 At the time of median follow-up (14.3 months), 11% (n=54/487) of patients who received KEYTRUDA® had a recurrence or died compared to 17% (n=82/489) of patients who received placebo.
In the study, the safety profile of KEYTRUDA® was consistent with previously reported studies in patients with solid tumors. The most common treatment-related adverse events (reported in at least 15% of patients) were pruritis, fatigue, diarrhea, and rash. KEYTRUDA® was discontinued for treatment-related adverse events in 15% of patients in KEYNOTE-716. Refer to the product monograph for complete information.3
"This approval is important for people living with melanoma and for all of us at Merck," said Marwan Akar, President, and Managing Director, Merck Canada. "With this new indication, KEYTRUDA® is the first checkpoint inhibitor approved in Canada in the adjuvant space in Stage IIB or IIC melanoma, meaning it can be considered for patients earlier in their journey. We are proud to continue to pursue our passion to save and improve lives as well as reinforce our commitment to finding innovative and effective options for more patients with melanoma."
About KEYTRUDA®
KEYTRUDA® is an anti-PD-1 therapy that works by helping increase the ability of the body's immune system to help detect and fight tumour cells.3 KEYTRUDA® is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumour cells and healthy cells.3
KEYTRUDA® was first approved in Canada in 2015 and currently has indications in several disease areas, including advanced renal cell carcinoma, bladder cancer, non-small cell lung carcinoma, primary mediastinal B-cell lymphoma, classical Hodgkin lymphoma, colorectal cancer, endometrial carcinoma, esophageal cancer, triple-negative breast cancer, melanoma, and head and neck squamous cell carcinoma.3
About Merck
At Merck, known as MSD outside of the United States and Canada, we are unified around our purpose: We use the power of leading-edge science to save and improve lives around the world. For more than 130 years, we have brought hope to humanity through the development of important medicines and vaccines. We aspire to be the premier research-intensive biopharmaceutical company in the world – and today, we are at the forefront of research to deliver innovative health solutions that advance the prevention and treatment of diseases in people and animals. We foster a diverse and inclusive global workforce and operate responsibly every day to enable a safe, sustainable and healthy future for all people and communities. For more information about our operations in Canada, visit www.merck.ca and connect with us on YouTube and Twitter @MerckCanada.
SOURCE Merck Canada
Sep. 13, 2022 8:10 AM ET
By: Ravikash, SA News Editor2 Comments


PUBLISHED11 September 2022 11 September 2022 9:20 BST
Updated results after approximately four years of follow-up of the POSEIDON Phase III trial showed a limited course of tremelimumab when added to AstraZeneca’s Imfinzi (durvalumab) plus four cycles of chemotherapy demonstrated a sustained improvement in overall survival (OS) compared to chemotherapy alone in the 1st-line treatment of patients with Stage IV (metastatic) non-small cell lung cancer (NSCLC). Additional post-hoc, exploratory analyses continued to show a trend for OS improvement with this combination in patients with STK11, KEAP1 and KRAS-mutated NSCLC, as well as in patients whose tumours were PD-L1-negative (less than 1% tumour cell expression).
These late-breaking results were presented today at the European Society of Medical Oncology (ESMO) Congress 2022 (Abstract #LBA59).
KRAS mutations are the most common tumour growth driver in NSCLC, occurring in approximately 25% of patients. NSCLC tumours with STK11 and KEAP1 mutations are often associated with poor outcomes and classified as immunologically “cold.” KRAS-mutated NSCLC can be responsive to immunotherapy but also can have poor outcomes, particularly when associated with STK11 or KEAP1 co-mutations.1-6
Melissa Johnson, MD, Director of Lung Cancer Research, Sarah Cannon Research Institute at Tennessee Oncology in Nashville, Tennessee, and a lead investigator in the POSEIDON Phase III trial, said: "Metastatic non-small cell lung cancer is a devastating diagnosis, particularly for patients whose cancers are less responsive to standard treatments such as chemotherapy and immune therapy. These results support the addition of a limited course of tremelimumab to durvalumab plus chemotherapy as a potential new treatment option for patients with these harder-to-treat forms of lung cancer.”
Susan Galbraith, Executive Vice President, Oncology R&D, AstraZeneca, said: "These updated POSEIDON results at nearly four years of follow-up show further evidence that the addition of a limited course of tremelimumab to Imfinzi plus chemotherapy improves outcomes for metastatic non-small cell lung cancer patients, including those with specific tumour mutations where a high unmet need for effective, well tolerated treatments remains. We look forward to bringing this potential new treatment option to patients as quickly as possible."
These latest data show that a limited course of five cycles of tremelimumab added to Imfinzi plus platinum-based chemotherapy improved overall survival by 25% compared to chemotherapy alone (hazard ratio [HR] 0.75; 95% CI 0.63-0.88). Updated median OS was 14 months for the combination versus 11.7 for chemotherapy alone. An estimated 25% of patients treated with the combination were alive at three years versus 13.6% for those treated with chemotherapy alone.
A trend for OS improvement continued to be observed in patients with STK11, KEAP1, and KRAS-mutated mNSCLC when treated with the combination, which reduced the risk of death by 38% (based on a HR of 0.62; 95% CI 0.34-1.12), 57% (based on a HR of 0.43; 95% CI 0.16-1.25), and 45% (based on a HR of 0.55; 95% CI 0.36-0.85), respectively. The combination also extended sustained OS benefit to patients with less than 1% PD-L1 tumour cell expression. These post hoc, exploratory subgroup analyses should be interpreted with caution due to the small sample sizes.
Consistent with previous POSEIDON Phase III trial readouts, OS benefit in these updated results appeared more pronounced with tremelimumab plus Imfinzi and chemotherapy in patients with non-squamous NSCLC histology. A 32% improvement in OS compared to chemotherapy alone (HR 0.68; 95% CI 0.55–0.85) was reported for patients with non-squamous histology, with an updated median OS of 17.2 months for the combination versus 13.1 months for chemotherapy. An estimated 31.4% of patients with non-squamous NSCLC treated with the combination were alive at three years versus 17.3% for those receiving chemotherapy alone.
Tremelimumab plus Imfinzi and chemotherapy continued to be well-tolerated, with no new safety signals identified based on the collection of serious adverse events (AEs) during the approximately four years of follow-up. Serious treatment-related AEs of any grade occurred in 27.6% of patients in the Imfinzi, tremelimumab and chemotherapy arm versus 17.7% in the chemotherapy alone arm as assessed by investigators. Treatment-related AEs leading to death occurred in 3.3% of patients in the combination arm versus 2.4% in the chemotherapy arm.
These updated results build on the primary progression-free survival (PFS) and OS results presented at the 2021 World Conference on Lung Cancer (WCLC) in September 2021 as well as post-hoc exploratory results in patients with STK11, KEAP1 or KRAS-mutated metastatic NSCLC recently presented at WCLC 2022 in August.
Tremelimumab is under review by global regulatory authorities in combination with Imfinzi and chemotherapy in 1st-line metastatic NSCLC based on the results of the POSEIDON trial.
POSEIDON
The POSEIDON trial was a randomised, open-label, multi-centre, global, Phase III trial of Imfinzi plus platinum-based chemotherapy or Imfinzi, tremelimumab and chemotherapy versus chemotherapy alone in the 1st-line treatment of 1,013 patients with metastatic NSCLC. The trial population included patients with either non-squamous or squamous disease and the full range of PD-L1 expression levels. POSEIDON excluded patients with certain epidermal growth factor receptor (EGFR) mutations or anaplastic lymphoma kinase (ALK) fusions.
In the experimental arms, patients were treated with a flat dose of 1,500mg of Imfinzi, or Imfinzi and 75mg of tremelimumab with up to four cycles of chemotherapy every three weeks, followed by maintenance treatment with Imfinzi once every four weeks, or Imfinzi and a fifth dose of 75mg of tremelimumab given at week 16. In comparison, the control arm allowed up to six cycles of chemotherapy. Pemetrexed maintenance treatment was allowed in all arms in patients with non-squamous disease if given during the induction phase. Nearly all patients with non-squamous disease (95.5%) had pemetrexed and platinum, while the majority of patients with squamous disease receiving chemotherapy (88.3%) received gemcitabine and platinum.
Primary endpoints included PFS and OS for the Imfinzi plus chemotherapy arm. Key secondary endpoints included PFS and OS in the Imfinzi plus tremelimumab and chemotherapy arm. As both PFS endpoints were met for Imfinzi plus chemotherapy and Imfinzi, tremelimumab and chemotherapy, the prespecified statistical analysis plan allowed for testing OS in the Imfinzi plus tremelimumab and chemotherapy arm. The trial was conducted in more than 150 centres across 18 countries, including the US, Europe, South America, Asia and South Africa.
Imfinzi
Imfinzi (durvalumab) is a human monoclonal antibody that binds to the PD-L1 protein and blocks the interaction of PD-L1 with the PD-1 and CD80 proteins, countering the tumour's immune-evading tactics and releasing the inhibition of immune responses.
Imfinzi is the only approved immunotherapy in the curative-intent setting of unresectable, Stage III non-small cell lung cancer (NSCLC) in patients whose disease has not progressed after chemoradiotherapy, and is the global standard of care in this setting based on the PACIFIC Phase III trial.
Imfinzi is currently approved in a number of countries in multiple tumour types including for the treatment of extensive-stage small cell lung cancer (ES-SCLC); for previously treated patients with advanced bladder cancer and for locally advanced or metastatic BTC in combination with chemotherapy (gemcitabine plus cisplatin).
Since the first approval in May 2017, more than 100,000 patients have been treated with Imfinzi.
Imfinzi combinations have demonstrated clinical benefit in multiple additional cancer settings with positive Phase III trials in unresectable advanced liver cancer (HIMALAYA), metastatic NSCLC (POSEIDON) and resectable NSCLC (AEGEAN). The data from HIMALAYA and POSEIDON are under review with global health authorities.
As part of a broad development programme, Imfinzi is being tested as a single treatment and in combinations with other anti-cancer treatments for patients with SCLC, NSCLC, bladder cancer, several GI cancers, ovarian cancer, endometrial cancer and other solid tumours.
Tremelimumab
Tremelimumab is a human monoclonal antibody and potential new medicine that targets the activity of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). Tremelimumab blocks the activity of CTLA-4, contributing to T-cell activation, priming the immune response to cancer and fostering cancer cell death.
Beyond POSEIDON, tremelimumab is being tested in combination with Imfinzi across multiple tumour types including in bladder cancer (VOLGA and NILE), locoregional HCC (EMERALD-3) and SCLC (ADRIATIC).
Tremelimumab is also under review by global regulatory authorities in combination with Imfinzi in unresectable advanced liver cancer based on the results of the HIMALAYA Phase III trial.
AstraZeneca in lung cancer
AstraZeneca is working to bring patients with lung cancer closer to cure through the detection and treatment of early-stage disease, while also pushing the boundaries of science to improve outcomes in the resistant and advanced settings. By defining new therapeutic targets and investigating innovative approaches, the Company aims to match medicines to the patients who can benefit most.
The Company's comprehensive portfolio includes leading lung cancer medicines and the next wave of innovations, including Tagrisso (osimertinib) and Iressa (gefitinib); Imfinzi (durvalumab) and tremelimumab; Enhertu (trastuzumab deruxtecan) and datopotamab deruxtecan in collaboration with Daiichi Sankyo; Orpathys (savolitinib) in collaboration with HUTCHMED; as well as a pipeline of potential new medicines and combinations across diverse mechanisms of action.
AstraZeneca is a founding member of the Lung Ambition Alliance, a global coalition working to accelerate innovation and deliver meaningful improvements for people with lung cancer, including and beyond treatment.
AstraZeneca in immuno-oncology (IO)
Immunotherapy is a therapeutic approach designed to stimulate the body’s immune system to attack tumours. The Company’s Immuno-Oncology (IO) portfolio is anchored in immunotherapies that have been designed to overcome evasion of the anti-tumour immune response. AstraZeneca is invested in using IO approaches that deliver long-term survival for new groups of patients across tumour types.
The Company is pursuing a comprehensive clinical-trial programme that includes Imfinzi as a single treatment and in combination with tremelimumab and other novel antibodies in multiple tumour types, stages of disease, and lines of treatment, and where relevant using the PD-L1 biomarker as a decision-making tool to define the best potential treatment path for a patient.
In addition, the ability to combine the IO portfolio with radiation, chemotherapy, and targeted small molecules from across AstraZeneca’s oncology pipeline, and from research partners, may provide new treatment options across a broad range of tumours.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Sep. 12, 2022 7:17 AM ET AstraZeneca PLC (AZN)
By: Ravikash, SA News Editor
AstraZeneca (NASDAQ:AZN) said ~4 years of follow-up data from a phase 3 trial suggested that a limited five cycles of tremelimumab added to Imfinzi (durvalumab) plus four cycles of chemotherapy improved overall survival (OS) by 25% compared to chemotherapy alone as initial treatment of patients with stage IV (metastatic) non-small cell lung cancer (NSCLC).
https://seekingalpha.com/symbol/AZN
PUBLISHED12 September 2022 12 September 2022 07:00 BST
Updated results from the TOPAZ-1 Phase III trial showed AstraZeneca’s Imfinzi (durvalumab), in combination with standard-of-care chemotherapy demonstrated a clinically meaningful and durable overall survival (OS) benefit as a treatment for patients with advanced biliary tract cancer (BTC).
These results from TOPAZ-1, the first Phase III trial to show improved OS with an immunotherapy combination in this setting, will be presented today at the European Society for Medical Oncology (ESMO) Congress 2022 in Paris (abstract #56P).
Sep. 12, 2022 5:30 AM ET AstraZeneca PLC (AZN)
By: Ravikash, SA News Editor
AstraZeneca (NASDAQ:AZN) said Imfinzi, in combination with standard-of-care chemotherapy, showed a clinically meaningful and durable overall survival (OS) benefit for patients with advanced biliary tract cancer (BTC) in a phase 3 trial called TOPAZ-1.
https://seekingalpha.com/symbol/AZN
Phase 1b Results Demonstrate Encouraging Median Progression-Free Survival of 5.7 Months in Difficult-to-Treat Patient Population
THOUSAND OAKS, Calif., Sept. 12, 2022 /PRNewswire/ -- Amgen (NASDAQ: AMGN) today announced updated data from its Phase 1b CodeBreaK 101 study, one of the most comprehensive global clinical development programs in patients with KRAS G12C-mutated colorectal cancer (CRC). These data show that combining LUMAKRAS®/LUMYKRAS® (sotorasib) with Vectibix® (panitumumab), Amgen's monoclonal anti-epidermal growth factor receptor (anti-EGFR) antibody, demonstrated encouraging efficacy and safety. Overall, the confirmed objective response rate (ORR) was 30% in patients with chemo-refractory metastatic colorectal cancer (mCRC). These data were presented today during an oral session at the European Society for Medical Oncology (ESMO) Annual Meeting in Paris, France.
"We are thrilled to see these CodeBreaK 101 results, which show that LUMAKRAS plus Vectibix achieved a 30% confirmed objective response rate in patients with KRAS G12C-mutated metastatic colorectal cancer. Treatment response rates can be as low as 2% in this patient population, and the current standard of care offers a median progression-free survival benefit of two months, so developing new treatment options is critically important for patients," said David M. Reese, M.D., executive vice president of Research and Development at Amgen. "These data are encouraging as we continue to focus on combination approaches in colorectal cancer, including advancing CodeBreaK 300, the Phase 3 LUMAKRAS plus Vectibix trial in the chemotherapy-refractory patient population."
In total, 40 patients with heavily pre-treated (median of two prior lines of therapy; range 1-7) KRAS G12C-mutated chemo-refractory mCRC were enrolled in the dose expansion cohort for the combination of LUMAKRAS and Vectibix. Disease control was seen in 37 patients for a total of 92.5% with a median progression free survival (PFS) of 5.7 months. There were no apparent differences found in the efficacy between left-sided and right-sided tumors. Tumor shrinkage of any magnitude was observed in 88% of patients, based on Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, and the median Duration of Treatment (DoT) was 5.9 months with 25% of patients remaining on treatment at the time of data cutoff. With a median follow up of 8.8 months, median overall survival (OS) was not yet reached. Treatment-related adverse events (TRAEs) reported with the combination were consistent with known safety profiles of the individual medicines, and no TRAEs resulted in discontinuation of either drug.
"These data are positive news for patients because they demonstrate encouraging efficacy and safety for sotorasib and panitumumab, as current treatments for chemo-refractory colorectal cancer provide limited survival benefits and can have notable safety issues," said Yasutoshi Kuboki, chief, Department of Experimental Therapeutics, National Cancer Center Hospital East, Japan. "There is a significant treatment gap for patients with KRAS G12C-mutated colorectal cancer and it is imperative that we continue to explore and develop precision therapies, like sotorasib, for these patients."
KRAS is a commonly mutated oncogene in CRC, with mutations in approximately 40% of all cases, with the KRAS G12C mutation present in approximately 3-5% of colorectal cancers. These patients face a dismal prognosis and have very limited treatment options.
*LUMAKRAS is marketed as LUMYKRAS® (sotorasib) in the European Union, the United Kingdom and Switzerland.
About LUMAKRAS®/LUMYKRAS® (sotorasib)
Amgen took on one of the toughest challenges of the last 40 years in cancer research by developing LUMAKRAS/LUMYKRAS, a KRASG12C inhibitor.1 LUMAKRAS/LUMYKRAS has demonstrated a positive benefit-risk profile with rapid, deep, and durable anticancer activity in patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) harboring the KRAS G12C mutation with a once daily oral formulation.2
Amgen is progressing the largest and broadest global KRASG12C inhibitor development program with unparalleled speed and exploring more than 10 sotorasib combination regimens, with clinical trial sites spanning five continents. To date, over 6,500 patients around the world have received LUMAKRAS/LUMYKRAS through the clinical development program and commercial use.
In May 2021, LUMAKRAS was the first KRASG12C inhibitor to receive regulatory approval with its approval in the U.S., under accelerated approval. LUMAKRAS/LUMYKRAS is also approved in the European Union, Japan, United Arab Emirates, South Korea, Hong Kong, Switzerland, Taiwan, Qatar, and in Australia, Brazil, Canada, Great Britain, Singapore, and Israel under the FDA's Project Orbis. Additionally, Amgen has submitted MAAs in Argentina, Colombia, Kuwait, Macao, Malaysia, Mexico, Russia, Saudi Arabia, Thailand and Turkey.
LUMAKRAS/LUMYKRAS is also being studied in multiple other solid tumors.3
About CodeBreaK
The CodeBreaK clinical development program for Amgen's drug sotorasib is designed to study patients with an advanced solid tumor with the KRAS G12C mutation and address the longstanding unmet medical need for these cancers.
CodeBreaK 100, the Phase 1 and 2, first-in-human, open-label multicenter study, enrolled patients with KRAS G12C-mutant solid tumors.11 Eligible patients must have received a prior line of systemic anticancer therapy, consistent with their tumor type and stage of disease. The primary endpoint for the Phase 2 study was centrally assessed objective response rate. The Phase 2 trial in NSCLC enrolled 126 patients, 124 of whom had centrally evaluable lesions by RECIST at baseline.2 The Phase 2 trial in colorectal cancer (CRC) is fully enrolled and results have been published.12
CodeBreaK 200, the global Phase 3 randomized active-controlled study comparing sotorasib to docetaxel in KRAS G12C-mutated NSCLC completed enrollment of 345 patients. Eligible patients had previously treated, locally advanced and unresectable or metastatic KRAS G12C-mutated NSCLC. The primary endpoint is progression-free survival and key secondary endpoints include overall survival, objective response rate, and patient-reported outcomes.13
Amgen also has several Phase 1b studies investigating sotorasib monotherapy and sotorasib combination therapy across various advanced solid tumors (CodeBreaK 101) open for enrollment.14 A Phase 2 randomized study will evaluate sotorasib in patients with stage IV KRAS G12C-mutated NSCLC in need of first-line treatment (CodeBreaK 201).15
For information, please visit www.hcp.codebreaktrials.com.
LUMAKRAS® (sotorasib) U.S. Indication
LUMAKRAS is indicated for the treatment of adult patients with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.
This indication is approved under accelerated approval based on overall response rate (ORR) and duration of response (DOR). Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
Please see LUMAKRAS full Prescribing Information.
About Vectibix® (panitumumab)
Vectibix is the first fully human monoclonal anti-EGFR antibody approved by the FDA for the treatment of mCRC. Vectibix was approved in the U.S. in September 2006 as a monotherapy for the treatment of patients with EGFR-expressing mCRC after disease progression after prior treatment with fluoropyrimidine-, oxaliplatin-, and irinotecan-containing chemotherapy.
In May 2014, the FDA approved Vectibix for use in combination with FOLFOX, as first-line treatment in patients with wild-type KRAS (exon 2) mCRC. With this approval, Vectibix became the first-and-only biologic therapy indicated for use with FOLFOX, one of the most commonly used chemotherapy regimens, in the first-line treatment of mCRC for patients with wild-type KRAS mCRC.
In June 2017, the FDA approved a refined indication for Vectibix for use in in patients with wild-type RAS (defined as wild-type in both KRAS and NRAS as determined by an FDA-approved test for this use) mCRC.
INDICATION AND LIMITATION OF USE
Vectibix® is indicated for the treatment of patients with wild-type RAS (defined as wild-type in both KRAS and NRAS as determined by an FDA-approved test for this use) metastatic colorectal cancer (mCRC): as first-line therapy in combination with FOLFOX, and as monotherapy following disease progression after prior treatment with fluoropyrimidine-, oxaliplatin-, and irinotecan-containing chemotherapy.
Limitation of Use: Vectibix® is not indicated for the treatment of patients with RAS mutant mCRC or for whom RAS mutation status is unknown.
To see the Vectibix® Prescribing Information, including Boxed Warning visit www.vectibix.com.
About Amgen Oncology
At Amgen Oncology, our mission to serve patients drives all that we do. That's why we're relentlessly focused on accelerating the delivery of medicines that have the potential to empower all angles of care and transform lives of people with cancer.
For the last four decades, we have been dedicated to discovering the firsts that matter in oncology and to finding ways to reduce the burden of cancer. Building on our heritage, Amgen continues to advance the largest pipeline in the Company's history, moving with great speed to advance those innovations for the patients who need them.
For more information, follow us on www.twitter.com/amgenoncology.
About Amgen
Amgen is committed to unlocking the potential of biology for patients suffering from serious illnesses by discovering, developing, manufacturing and delivering innovative human therapeutics. This approach begins by using tools like advanced human genetics to unravel the complexities of disease and understand the fundamentals of human biology.
Amgen focuses on areas of high unmet medical need and leverages its expertise to strive for solutions that improve health outcomes and dramatically improve people's lives. A biotechnology pioneer since 1980, Amgen has grown to be one of the world's leading independent biotechnology companies, has reached millions of patients around the world and is developing a pipeline of medicines with breakaway potential.
Amgen is one of the 30 companies that comprise the Dow Jones Industrial Average and is also part of the Nasdaq-100 index. In 2021, Amgen was named one of the 25 World's Best Workplaces™ by Fortune and Great Place to Work™ and one of the 100 most sustainable companies in the world by Barron's.
For more information, visit www.amgen.com and follow us on www.twitter.com/amgen.
View original content to download multimedia:https://www.prnewswire.com/news-releases/lumakras-sotorasib-combined-with-vectibix-panitumumab-shows-confirmed-30-objective-response-rate-in-patients-with-kras-g12c-mutated-metastatic-colorectal-cancer-301621681.html
SOURCE Amgen
Sep. 12, 2022 5:53 AM ETAmgen Inc. (AMGN)By: Ravikash, SA News Editor
Sep 09, 2022
Basel, September 9, 2022 — Novartis today announced results from a new pooled exploratory analysis across the entire MONALEESA Phase III program, confirming nearly one year of additional overall survival (OS) benefit in a subgroup of patients with aggressive forms of hormone receptor-positive, human epidermal growth factor receptor-2 negative (HR+/HER2-) advanced breast cancer (aBC)1. This subgroup analysis found that patients with visceral metastases—including liver metastases and multiple metastatic sites, which are typically associated with a poor prognosis—who were treated with Kisqali® (ribociclib) plus endocrine therapy in the first-line setting, achieved a median OS of 62.7 months compared to 52.1 months for those treated with endocrine therapy alone (HR=0.79; 95% CI: 0.65-0.97)1. Data from this analysis will be presented at the European Society of Medical Oncology (ESMO) Congress in Paris, France.
“Patients who have visceral metastases typically have a worse prognosis and often demonstrate resistance to treatment, so as a clinician it is encouraging to see significant survival benefit with ribociclib in the first-line setting in patients with more aggressive disease,” said Denise A. Yardley, MD, Senior Investigator, Breast Cancer Research Program, Sarah Cannon Research Institute at Tennessee Oncology, USA. “Ribociclib is the only CDK4/6 inhibitor to show a consistent overall survival benefit in combination with endocrine therapy, while also maintaining quality of life across the Phase III program.”
Those with liver metastases on Kisqali plus endocrine therapy in the first-line achieved 44.2 months median OS compared to 38.1 months for those on endocrine therapy alone (HR=0.77; 95% CI: 0.55-1.07). For patients with visceral metastases in three or more organs, first-line
treatment with Kisqali-endocrine therapy achieved 57.7 months median OS compared to 49.3 months for those on endocrine therapy alone (HR=0.81; 95% CI: 0.63-1.03)1.
“The goal for advanced breast cancer treatment is to help people live longer, and we are proud that Kisqali continues to deliver a significant survival benefit while also maintaining quality of life, even for those with harder-to-treat disease,” said Jeff Legos, Executive Vice President, Global Head of Oncology and Hematology at Novartis. “We are committed to demonstrating what makes Kisqali a unique CDK4/6 inhibitor, thus providing patients and oncologists confidence in this therapeutic option.”
HARMONIA head-to-head CDK4/6 inhibitor trial design
Also at ESMO, the trial design will be presented for HARMONIA, the first prospective, head-to-head Phase III trial of CDK4/6 inhibitors being conducted in collaboration with SOLTI Innovative Cancer Research, to evaluate Kisqali vs. Ibrance®* (palbociclib) for patients with advanced HR+/HER2-, HER2-enriched subtype, ultimately exploring what makes Kisqali unique at a molecular level13. HARMONIA seeks to test if Kisqali improves the course of HR+/HER2- aBC by changing tumor biology to enable a better response to endocrine therapy as compared to Ibrance*, and could further substantiate differences seen among these CDK4/6 inhibitors. HER2-enriched is an intrinsic subtype associated with a very poor prognosis and endocrine-resistance, as compared to luminal disease. The global, multicenter, randomized, open-label, Phase III study has a primary outcome of progression-free survival (PFS), and secondary outcomes include OS and PFS2. HARMONIA is currently ongoing with an anticipated enrollment of 456 patients.
About Kisqali® (ribociclib)
Kisqali is the only CDK4/6 inhibitor with proven overall survival benefit across all its three pivotal Phase III advanced breast cancer trials2-12, and is recognized by the National Comprehensive Cancer Network (NCCN) guidelines as the only CDK4/6 inhibitor with overall survival benefit in first-line HR+/HER2- advanced breast cancer14. Additionally, Kisqali has the highest rating of any CDK4/6 inhibitor on the ESMO Magnitude of Clinical Benefit Scale, achieving a score of five out of five for first-line premenopausal patients with HR+/HER2- advanced breast cancer15. Further, Kisqali in combination with either letrozole or fulvestrant has uniquely, among other CDK4/6i, received a score of four out of five for postmenopausal patients with HR+/HER2- advanced breast cancer treated in the first line16.
Kisqali has been approved in more than 95 countries worldwide, including by the United States Food and Drug Administration (FDA) and the European Commission, for the treatment of women with HR+/HER2- advanced or metastatic breast cancer in combination either with an aromatase inhibitor or with fulvestrant as initial endocrine-based therapy or following disease progression on endocrine therapy13,17. Kisqali in combination with fulvestrant is approved as initial endocrine-based therapy or following disease progression on endocrine therapy in men by the FDA17.
Novartis is committed to continuing to study Kisqali in breast cancer. NATALEE is a large Phase III clinical trial of Kisqali with endocrine therapy in the adjuvant treatment of HR+/HER2- early breast cancer being conducted in collaboration with Translational Research In Oncology (TRIO)18. Additionally, Novartis is collaborating with the Akershus University Hospital in Norway on the NEOLETRIB trial, a neoadjuvant Phase II trial studying the effects of Kisqali in HR+/HER2- early breast cancer and to discover the potentially unique underlying mechanism of action19.
Kisqali was developed by the Novartis Institutes for BioMedical Research (NIBR) under a research collaboration with Astex Pharmaceuticals.
Please see full Prescribing Information for Kisqali, available at www.Kisqali.com.
About Novartis in Advanced Breast Cancer
Novartis tackles breast cancer with superior science, collaboration and a passion for transforming patient care. We've taken a bold approach to our research by including patient populations often neglected in clinical trials, identifying new pathways or mutations that may play a role in disease progression and developing therapies that not only maintain, but also improve, quality of life for patients. Our priority over the past 30 years and today is to deliver treatments proven to improve and extend lives for those diagnosed with metastatic breast cancer.
About Novartis
Novartis is reimagining medicine to improve and extend people’s lives. As a leading global medicines company, we use innovative science and digital technologies to create transformative treatments in areas of great medical need. In our quest to find new medicines, we consistently rank among the world’s top companies investing in research and development. Novartis products reach nearly 800 million people globally and we are finding innovative ways to expand access to our latest treatments. About 108,000 people of more than 140 nationalities work at Novartis around the world. Find out more at
https://www.novartis.com.
Novartis is on Twitter. Sign up to follow @Novartis at https://twitter.com/novartisnews
For Novartis multimedia content, please visit https://www.novartis.com/news/media-library
For questions about the site or required registration, please contact media.relations@novartis.com
Sep. 09, 2022 4:34 AM ET
By: Ravikash, SA News Editor
Novartis (NYSE:NVS) said its drug Kisqali showed about one year of additional overall survival (OS) benefit in a subgroup of patients with aggressive forms of hormone receptor-positive, human epidermal growth factor receptor-2 negative (HR+/HER2-) advanced breast cancer, as per a new pooled analysis.
https://seekingalpha.com/symbol/NVS
https://www.us.kisqali.com/metastatic-breast-cancer/
September 12, 2022 8:55 am ET
Results demonstrated a 64.5% confirmed objective response rate in patients treated with investigational combination of enfortumab vedotin and pembrolizumab
Data highlighted in late-breaking presentation at the European Society for Medical Oncology Congress 2022
BOTHELL, W.A., TOKYO and RAHWAY, N.J., Sept.12, 2022 – Seagen Inc. (Nasdaq:SGEN), Astellas Pharma Inc. (TSE:4503, President and CEO: Kenji Yasukawa, Ph.D., “Astellas”) and Merck (NYSE: MRK), known as MSD outside of the United States and Canada, today announced results from the Phase 1b/2 EV-103 clinical trial (also known as KEYNOTE-869) Cohort K investigating PADCEV® (enfortumab vedotin-ejfv) in combination with Merck’s anti-PD-1 therapy KEYTRUDA® (pembrolizumab) and PADCEV alone as first-line treatment in patients with unresectable locally advanced or metastatic urothelial cancer (la/mUC) who are ineligible to receive cisplatin-based chemotherapy. The findings were presented today at the European Society for Medical Oncology (ESMO) Congress as part of a late-breaking abstract presentation (Abstract #LBA73).
In patients treated with enfortumab vedotin and pembrolizumab (n=76), results demonstrated a 64.5% confirmed objective response rate (ORR) (95% CI, 52.7-75.1) per RECIST v1.1 by blinded independent central review (BICR), the primary endpoint of Cohort K, with 10.5% of patients experiencing a complete response and 53.9% of patients experiencing a partial response. The median duration of response (DOR) per BICR was not reached (95% CI, 10.25-NR). All-grade treatment-related adverse events (TRAEs) of special interest for enfortumab vedotin in combination with pembrolizumab were skin reactions (67.1%), peripheral neuropathy (60.5%), ocular disorders (dry eye, blurred vision and corneal disorders) (26.3%), hyperglycemia (14.5%) and infusion-related reactions (3.9%). Pembrolizumab adverse events of special interest were consistent with previously observed safety data from monotherapy with the exception of severe skin reactions. Overall, the results were generally consistent with previously reported efficacy and safety results of the EV-103/KEYNOTE-869 dose-escalation cohort and expansion Cohort A.
Please see Important Safety Information at the end of this press release for both drugs, including BOXED WARNING for enfortumab vedotin and immune mediated adverse reactions for pembrolizumab.
Cohort K also included a monotherapy arm in which patients were treated with enfortumab vedotin alone (n=73), though this study was not designed to support a formal comparison between the two arms. Results showed a 45.2% confirmed ORR (95% CI, 33.5-57.3) per RECIST v1.1 by BICR, with 4.1% of patients experiencing a complete response and 41.1% of patients experiencing a partial response. The median DOR was 13.2 months (95% CI, 6.14-15.97) per RECIST v1.1 by BICR. All-grade TRAEs of special interest for enfortumab vedotin were peripheral neuropathy (54.8%), skin reactions (45.2%), ocular disorders (dry eye, blurred vision and corneal disorders) (28.8%), hyperglycemia (11.0%) and infusion-related reactions (5.5%).
Additional secondary endpoints in the EV-103 Cohort K trial included progression-free survival (PFS) and overall survival (OS). Among patients treated with enfortumab vedotin and pembrolizumab, median PFS was not reached (95% CI, 8.31-NR). Median OS was 22.3 months (95% CI, 19.09-NR). Among patients treated with enfortumab vedotin, median PFS was 8.0 months (95% CI, 6.05-10.35), and median OS was 21.7 months (95% CI, 15.21-NR).
The TRAEs of any grade that occurred in more than 20% of patients treated with enfortumab vedotin alone or in combination with pembrolizumab were fatigue, peripheral sensory neuropathy, alopecia, rash maculo-papular, pruritus, dysgeusia, weight decreased, diarrhea, decreased appetite, nausea and dry eye.
“Results from EV-103/KEYNOTE-869 Cohort K support the ongoing investigation of enfortumab vedotin and pembrolizumab in cisplatin-ineligible patients with locally advanced or metastatic urothelial cancer who are in need of treatment options, and this combination may be an important therapeutic option for these patients,” said Dr. Jonathan E. Rosenberg, M.D., chief, genitourinary medical oncology service, division of solid tumor oncology and Enno W. Ercklentz chair, Memorial Sloan Kettering Cancer Center and EV-103/KEYNOTE-869 Cohort K primary investigator. Dr. Rosenberg has consulting relationships with Seagen, Astellas and Merck.
“Nearly 65% of patients who were treated with enfortumab vedotin and pembrolizumab responded to this combination, with almost 11% showing no detectable cancer following treatment. These study results represent an encouraging finding for people with advanced urothelial cancer who are not eligible for cisplatin treatment,” said Margorie Green, senior vice president and head of late stage development, Seagen.
“We’re encouraged by these positive findings from the combination of enfortumab vedotin and pembrolizumab in people with advanced urothelial cancer who historically have had limited treatment options in the first-line setting, and we intend to discuss these results with regulatory authorities,” said Ahsan Arozullah, M.D., M.P.H., senior vice president and head of development therapeutic areas, Astellas.
“We’re pleased that this combination provided a meaningful benefit to this group of advanced bladder cancer patients in this study, and we will continue to investigate enfortumab vedotin plus pembrolizumab through our collaboration,” said Eliav Barr, M.D., senior vice president and head of global clinical development, chief medical officer, Merck Research Laboratories.
In February 2020, the U.S. Food and Drug Administration granted Breakthrough Therapy Designation for enfortumab vedotin in combination with pembrolizumab for patients with unresectable la/mUC who are ineligible to receive cisplatin-based chemotherapy in the first-line setting. The designation is based on results from the dose-escalation cohort and expansion Cohort A of the phase 1b/2 trial, EV-103/KEYNOTE-869 (NCT03288545), evaluating patients with la/mUC who are ineligible to receive cisplatin-based chemotherapy treated in the first-line setting with enfortumab vedotin in combination with pembrolizumab.
Seagen, Astellas and Merck are further investigating enfortumab vedotin plus pembrolizumab in Phase 3 studies, including EV-302/KEYNOTE-A39 (NCT04223856), which is intended to confirm these results for the investigational treatment combination in previously untreated la/mUC, and in muscle-invasive bladder cancer in EV-304/KEYNOTE-B15 (NCT04700124) and EV-303/KEYNOTE-905 (NCT03924895).
About the EV-103/KEYNOTE-869 trial (Cohort K)
The EV-103/KEYNOTE-869 trial (NCT03288545) is an ongoing, multi-cohort, open-label, multicenter Phase 1b/2 trial of enfortumab vedotin alone or in combination with pembrolizumab and/or chemotherapy in first- or second-line settings in patients with la/mUC and in patients with muscle-invasive bladder cancer.
Cohort K of the EV-103/KEYNOTE-869 trial is a randomized 1:1 cohort investigating enfortumab vedotin alone (n=73) or in combination with pembrolizumab (n=76) in adult patients with unresectable la/mUC who are ineligible for cisplatin-based chemotherapy and have received no prior treatment for la/mUC. The enfortumab vedotin monotherapy study arm is intended to characterize the activity of enfortumab vedotin alone in this patient population. The key outcome measure of EV-103/KEYNOTE-869 Cohort K is ORR per BICR using RECIST 1.1. Secondary endpoints include ORR per investigator assessment; DOR, disease control rate and progression-free survival per BICR and investigator assessment; overall survival; and assessment of safety.
About PADCEV
PADCEV (enfortumab vedotin-ejfv) is a first-in-class antibody-drug conjugate (ADC) that is directed against Nectin-4, a protein located on the surface of cells and highly expressed in bladder cancer. Nonclinical data suggest the anticancer activity of PADCEV is due to its binding to Nectin-4 expressing cells followed by the internalization and release of the anti-tumor agent monomethyl auristatin E (MMAE) into the cell, which result in the cell not reproducing (cell cycle arrest) and in programmed cell death (apoptosis).
PADCEV (enfortumab vedotin-ejfv) U.S. Indication & Important Safety Information
Indication
PADCEV® is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer (mUC) who:
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf.
About the Seagen, Astellas and Merck collaboration
Seagen and Astellas entered a clinical collaboration agreement with Merck to evaluate the combination of Seagen’s and Astellas’ PADCEV® (enfortumab vedotin-ejfv) and Merck’s KEYTRUDA® (pembrolizumab) in patients with previously untreated metastatic urothelial cancer. KEYTRUDA is a registered trademark of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Rahway, N.J., USA.
About Seagen
Seagen Inc. is a global biotechnology company that discovers, develops and commercializes transformative cancer medicines to make a meaningful difference in people’s lives. Seagen is headquartered in the Seattle, Washington area, and has locations in California, Canada, Switzerland and the European Union. For more information on the company’s marketed products and robust pipeline, visit www.seagen.com and follow @SeagenGlobal on Twitter.
About Astellas
Astellas Pharma Inc. is a pharmaceutical company conducting business in more than 70 countries around the world. We are promoting the Focus Area Approach that is designed to identify opportunities for the continuous creation of new drugs to address diseases with high unmet medical needs by focusing on Biology and Modality. Furthermore, we are also looking beyond our foundational Rx focus to create Rx+® healthcare solutions that combine our expertise and knowledge with cutting-edge technology in different fields of external partners. Through these efforts, Astellas stands on the forefront of healthcare change to turn innovative science into value for patients. For more information, please visit our website at https://www.astellas.com/en.
Merck’s focus on cancer
Our goal is to translate breakthrough science into innovative oncology medicines to help people with cancer worldwide. At Merck, the potential to bring new hope to people with cancer drives our purpose and supporting accessibility to our cancer medicines is our commitment. As part of our focus on cancer, Merck is committed to exploring the potential of immuno-oncology with one of the largest development programs in the industry across more than 30 tumor types. We also continue to strengthen our portfolio through strategic acquisitions and are prioritizing the development of several promising oncology candidates with the potential to improve the treatment of advanced cancers. For more information about our oncology clinical trials, visit www.merck.com/clinicaltrials.
About Merck
At Merck, known as MSD outside of the United States and Canada, we are unified around our purpose: We use the power of leading-edge science to save and improve lives around the world. For more than 130 years, we have brought hope to humanity through the development of important medicines and vaccines. We aspire to be the premier research-intensive biopharmaceutical company in the world – and today, we are at the forefront of research to deliver innovative health solutions that advance the prevention and treatment of diseases in people and animals. We foster a diverse and inclusive global workforce and operate responsibly every day to enable a safe, sustainable and healthy future for all people and communities. For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
09/12/2022
– Results demonstrated a 64.5% confirmed objective response rate in patients treated with investigational combination of enfortumab vedotin and pembrolizumab –
– Data highlighted in late-breaking presentation at the European Society for Medical Oncology (ESMO) Congress 2022 –
BOTHELL, Wash. & TOKYO & RAHWAY, N.J.--(BUSINESS WIRE)-- Seagen Inc. (Nasdaq:SGEN), Astellas Pharma Inc. (TSE:4503, President and CEO: Kenji Yasukawa, Ph.D., “Astellas”) and Merck (NYSE: MRK), known as MSD outside of the United States and Canada, today announced results from the phase 1b/2 EV-103 clinical trial (also known as KEYNOTE-869) Cohort K investigating PADCEV® (enfortumab vedotin-ejfv) in combination with Merck’s anti-PD-1 therapy KEYTRUDA® (pembrolizumab) and PADCEV alone as first-line treatment in patients with unresectable locally advanced or metastatic urothelial cancer (la/mUC) who are ineligible to receive cisplatin-based chemotherapy. The findings were presented today at the European Society for Medical Oncology (ESMO) Congress as part of a late-breaking abstract presentation (Abstract #LBA73).
Sep. 12, 2022 9:51 AM ET
Merck & Co., Inc. (MRK), SGEN, ALPMY, ALPMF
By: Ravikash, SA News Editor
Astellas Pharma (OTCPK:ALPMF) (OTCPK:ALPMY), Seagen (NASDAQ:SGEN) and Merck (NYSE:MRK) said Padcev and Keytruda showed a 64.5% confirmed objective response rate (ORR) in patients with a type of urothelial cancer.
https://seekingalpha.com/symbol/ALPMF
https://seekingalpha.com/symbol/ALPMY
https://seekingalpha.com/symbol/SGEN
https://seekingalpha.com/symbol/MRK
Sep 08, 2022
– Study Results Validate the Hypothesis That TTR Silencing by an RNAi Therapeutic has the Potential to be an Effective Approach for Treating Cardiomyopathy of ATTR Amyloidosis –
– Patisiran met Primary Endpoint, Demonstrating Significant Clinical Benefit on Functional Capacity (6-MWT) Compared to Placebo at Month 12 –
– Patisiran Also met the First Secondary Endpoint, Demonstrating Significant Clinical Benefit on Health Status and Quality of Life, as Measured by the Kansas City Cardiomyopathy Questionnaire, Compared to Placebo at Month 12 –
– Patisiran Demonstrated Encouraging Safety and Tolerability Profile in Patients with ATTR Amyloidosis with Cardiomyopathy –
– Alnylam to Host Conference Call Today at 8:00 a.m. ET –
CAMBRIDGE, Mass.--(BUSINESS WIRE)--Sep. 8, 2022-- Alnylam Pharmaceuticals, Inc. (Nasdaq: ALNY), the leading RNAi therapeutics company, today announced positive results from the APOLLO-B Phase 3 study of patisiran, an investigational RNAi therapeutic in development for the treatment of transthyretin-mediated (ATTR) amyloidosis with cardiomyopathy. The results were presented today in a late-breaker session at the 18th International Symposium on Amyloidosis (ISA). The Company previously announced positive topline results from the APOLLO-B study in August 2022.
The 12-month study achieved its primary endpoint, with patisiran demonstrating a statistically significant and clinically meaningful benefit on functional capacity, as measured by the 6-Minute Walk Test (6-MWT), compared to placebo, with a median difference of 14.7 meters (p-value 0.0162) favoring patisiran. The study also met its first secondary endpoint, demonstrating a statistically significant and clinically meaningful benefit on health status and quality of life, as measured by the Kansas City Cardiomyopathy Questionnaire Overall Summary (KCCQ-OS) score, compared to placebo with least squares (LS) mean difference of 3.7 points (p-value 0.0397) favoring patisiran. The study also included additional secondary composite outcomes endpoints. A non-significant result (p-value 0.0574) was found on the composite endpoint of all-cause mortality, frequency of cardiovascular events, and change from baseline in 6-MWT over 12 months compared to placebo. As a result, formal statistical testing was not performed on the final two composite endpoints. Patisiran also demonstrated an encouraging safety and tolerability profile in patients with ATTR amyloidosis with cardiomyopathy.
“The results of the APOLLO-B Phase 3 study are impressive, as I believe they underscore the potential for patisiran to provide a benefit on functional capacity and quality of life in patients living with ATTR amyloidosis with cardiomyopathy. Furthermore, these results were seen after only 12 months of treatment,” said Mathew Maurer, M.D., Arnold and Arlene Goldstein Professor of Cardiology at Columbia University Irving Medical Center. “The cardiac manifestations associated with ATTR amyloidosis can have a devastating impact on patients’ lives and current treatment options are limited. With the rapidly progressive nature of the disease, there is a significant need for treatments like patisiran, which has the potential to be a new option for patients and physicians to treat the cardiomyopathy of ATTR amyloidosis.”
APOLLO-B Study Results
APOLLO-B is a Phase 3, randomized, double-blind, placebo-controlled multicenter global study designed and powered to evaluate the effects of patisiran on functional capacity and quality of life in patients with ATTR amyloidosis with cardiomyopathy. The study enrolled 360 adult patients with ATTR amyloidosis (hereditary or wild-type) with cardiomyopathy at 69 sites in 21 countries. Patients were randomized 1:1 to receive 0.3 mg/kg of patisiran or placebo intravenously administered every three weeks over a 12-month treatment period. After 12 months, all patients will receive patisiran in an open-label extension.
At 12 months, the study results presented today are as follows:
Patisiran also demonstrated an encouraging safety and tolerability profile, including no cardiac safety concerns relative to placebo, during the 12-month treatment period. The majority of adverse events (AEs) were mild or moderate in severity. Treatment emergent AEs occurring in 5% or more patients in the patisiran group and observed at least 3% more commonly than in the placebo group included infusion-related reactions (12.2% vs 9.0%), arthralgia (7.7% vs 4.5%), and muscle spasms (6.6% vs 2.2%). In the safety analysis there were 5 deaths (2.8%) observed in patisiran-treated patients and 8 deaths (4.5%) observed in the placebo group.
“We believe the totality of the APOLLO-B study results provides a compelling clinical profile of patisiran for patients and families living with ATTR amyloidosis with cardiomyopathy, a fatal, multi-system disease. It was encouraging to see the impact of patisiran on functional capacity in the study, as the change from baseline in 6-MWT for patisiran-treated patients was in the range of the approximately 5 meter decline typically seen in healthy older adults over a 12-month period.1 Furthermore, health status and quality of life in patisiran-treated patients was maintained relative to baseline, which is another important aspect of the potential benefit that patisiran treatment may provide to patients,” said Pushkal Garg, M.D., Chief Medical Officer at Alnylam. “Importantly, we believe these data validate the therapeutic hypothesis that TTR silencing by an RNAi therapeutic may be an effective approach to treating cardiomyopathy of both wild-type and hereditary ATTR amyloidosis. ATTR amyloidosis with cardiomyopathy is an increasingly recognized cause of heart failure, affecting greater than 250,000 patients around the world, and with these results, we can take another important step forward towards delivering a potential new treatment to the patients who need it, assuming favorable regulatory review.”
Alnylam plans to file a supplemental new drug application (sNDA) for patisiran as a potential treatment for ATTR amyloidosis with cardiomyopathy in the U.S. in late 2022. Additional results from the APOLLO-B study will be presented at the upcoming Heart Failure Society of America (HFSA) annual meeting being held in Washington DC, September 30 to October 3, 2022.
To view the APOLLO-B data presented at ISA, please visit, Capella.
Other Patisiran Data: 36-Month Global Open Label Extension (OLE) and Phase 4 Observational Study Results
Alnylam also presented 36-month results from the ongoing Global OLE study of patisiran in eligible patients who completed the APOLLO Phase 3 and Phase 2 OLE studies, evaluating the effect of long-term patisiran treatment on mortality and ambulatory function.
Findings demonstrate that patients who initiated treatment with patisiran earlier in the Phase 3 APOLLO and Phase 2 OLE studies experienced greater survival as compared with the APOLLO placebo patients who initiated treatment with patisiran later during the Global OLE study. Further, those same patients who initiated patisiran earlier showed stabilized or improved ambulation versus patients in the APOLLO placebo group as assessed by polyneuropathy disability (PND) score. The long-term safety profile of patisiran was consistent with prior analyses.
These results highlight the substantial impact of earlier diagnosis and treatment with patisiran in patients with hereditary transthyretin-mediated (hATTR) amyloidosis with polyneuropathy.
Additional data were also presented from a Phase 4 observational study to evaluate the effectiveness of patisiran on ambulatory status in patients with hATTR amyloidosis with polyneuropathy with a V122I or T60A variant. These variants are historically associated with cardiomyopathy, thus further evidence that patients with these variants also experience polyneuropathy is emerging.
After 12 months of treatment with patisiran, 93.3% of patients (42/45) experienced stabilization or improvement in PND score from baseline. Treatment with patisiran also resulted in clinically meaningful improvements in QOL and autonomic symptoms after 12 months. Patisiran demonstrated an acceptable safety profile, consistent with existing data.
About ONPATTRO® (patisiran)
ONPATTRO is an RNAi therapeutic that is approved in the United States and Canada for the treatment of the polyneuropathy of hATTR amyloidosis in adults. ONPATTRO is also approved in the European Union, Switzerland and Brazil for the treatment of hATTR amyloidosis in adults with Stage 1 or Stage 2 polyneuropathy, and in Japan for the treatment of hATTR amyloidosis with polyneuropathy. ONPATTRO is an intravenously administered RNAi therapeutic targeting transthyretin (TTR). It is designed to target and silence TTR messenger RNA, thereby reducing the production of TTR protein before it is made. Reducing the pathogenic protein leads to a reduction in amyloid deposits in tissues. For more information about ONPATTRO, including full Prescribing Information, visit ONPATTRO.com.
ONPATTRO Indication and Important Safety Information
Indication
ONPATTRO is indicated for the treatment of the polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults.
About Alnylam Pharmaceuticals
Alnylam (Nasdaq: ALNY) has led the translation of RNA interference (RNAi) into a whole new class of innovative medicines with the potential to transform the lives of people afflicted with rare and prevalent diseases with unmet need. Based on Nobel Prize-winning science, RNAi therapeutics represent a powerful, clinically validated approach yielding transformative medicines. Since its founding 20 years ago, Alnylam has led the RNAi Revolution and continues to deliver on a bold vision to turn scientific possibility into reality. Alnylam’s commercial RNAi therapeutic products are ONPATTRO® (patisiran), GIVLAARI® (givosiran), OXLUMO® (lumasiran), AMVUTTRA™ (vutrisiran) and Leqvio® (inclisiran) being developed and commercialized by Alnylam’s partner, Novartis. Alnylam has a deep pipeline of investigational medicines, including multiple product candidates that are in late-stage development. Alnylam is executing on its “Alnylam P5x25” strategy to deliver transformative medicines in both rare and common diseases benefiting patients around the world through sustainable innovation and exceptional financial performance, resulting in a leading biotech profile. Alnylam is headquartered in Cambridge, MA. For more information about our people, science and pipeline, please visit www.alnylam.com and engage with us on Twitter at @Alnylam, on LinkedIn, or on Instagram.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220907006229/en/
Alnylam Pharmaceuticals, Inc.
Source: Alnylam Pharmaceuticals, Inc.
Sep. 08, 2022 6:31 AM ET
Alnylam Pharmaceuticals, Inc. (ALNY)
By: Dulan Lokuwithana, SA News Editor
RNAi therapeutics company Alnylam Pharmaceuticals (NASDAQ:ALNY) announced full results from its APOLLO-B Phase 3 study of patisiran, an investigational medication for transthyretin-mediated (ATTR) amyloidosis with cardiomyopathy.
September 11, 2022 Download this Press ReleasePDF Format (opens in new window)
BOULDER, Colo.--(BUSINESS WIRE)-- Clovis Oncology, Inc. (NASDAQ: CLVS), today announced results from a subgroup analysis of data from the monotherapy comparison of the randomized, Phase 3 ATHENA (GOG-3020/ENGOT-ov45) trial (ATHENA-MONO). These data showed that Rubraca as first-line maintenance treatment improved progression-free survival (PFS) versus placebo across disease risk subgroups including surgical outcome, response to first-line chemotherapy, and additional analyses in other subgroups. The data were presented by Rebecca S. Kristeleit, MD, PhD, of Guy’s and St. Thomas’ NHS Foundation Trust in London and lead ENGOT/NCRI National Cancer Research Institute (https://www.ncri.org.uk/) investigator of the ATHENA trial as a Mini Oral abstract at the European Society of Medical Oncology (ESMO) Congress 2022 in Paris.
“These additional results from the ATHENA-MONO analysis of the Phase 3 ATHENA trial demonstrate that rucaparib should be considered a new first-line maintenance treatment option for women with advanced ovarian cancer,” Dr. Kristeleit said. “In this analysis, rucaparib prolonged progression-free survival for patients with or without high risk factors for progression, irrespective of molecular characteristics, adding to our understanding of the efficacy of rucaparib in the broadest population of patients assessed in a clinical trial for first-line PARP inhibitor monotherapy.”
ATHENA is a double-blind, placebo-controlled, Phase 3 trial of Rubraca in first-line ovarian cancer maintenance treatment. It has two parts which are statistically independent. The results presented at ESMO are from the ATHENA-MONO part (Rubraca versus placebo), with results from the ATHENA-COMBO part (Rubraca plus nivolumab versus Rubraca) expected in Q1 2023.
“As further demonstrated by the additional data presented at ESMO, the ATHENO-MONO analysis continues to reinforce the potential of Rubraca as a first-line maintenance therapy for women with advanced ovarian cancer,” said Patrick J. Mahaffy, President and CEO of Clovis Oncology. “We remain grateful to the patients who participated in the trial and for the support of the clinical community familiar with these results.”
ATHENA-MONO enrolled 538 women with high-grade ovarian, fallopian tube, or primary peritoneal cancer. The primary efficacy analysis evaluated two prospectively defined molecular subgroups in a step-down manner: 1) homologous recombination deficiency (HRD)-positive (inclusive of BRCAm tumors and BRCAwt/LOH high tumors), and 2) all patients randomized (overall intent-to-treat population [ITT]) in ATHENA-MONO. The following exploratory subgroup analyses are included in the ESMO presentation:
PFS by Surgical Outcome
Patients who received Rubraca as maintenance therapy showed benefit regardless of surgical outcome, whether there was complete resection (R0) during cytoreductive surgery or not.
In HRD-positive patients:
Among the ITT population:
PFS by First-Line Chemotherapy Response
Similarly, patients treated with Rubraca as maintenance therapy showed benefit among all subgroups when evaluated against response per RECIST v1.1 at any time during first-line chemotherapy.
Among HRD-positive patients:
Among the ITT population:
Additional Analyses in the ITT Population by Subgroup
Additional analyses in other subgroups based on baseline clinical characteristics, including FIGO stage, timing of surgery, and CA-125 levels, also demonstrated that patients treated with Rubraca experienced a progression-free survival benefit compared to those treated with placebo. Safety was similar between subgroups analyzed.
Dr. Kristeleit’s presentation, as well as other company-sponsored Rubraca data presented at ESMO, can be viewed at the time of presentation at https://www.clovisoncology.com/pipeline/scientific-presentations/.
Rubraca is not currently approved in the first-line ovarian cancer maintenance setting.
About Rubraca (rucaparib)
Rucaparib is an oral, small molecule inhibitor of PARP1, PARP2 and PARP3 being developed in multiple tumor types, including ovarian and metastatic castration-resistant prostate cancers, as monotherapy, and in combination with other anti-cancer agents. Exploratory studies in other tumor types are also underway.
Rubraca is an unlicensed medical product outside of the US and Europe.
Rubraca Ovarian Cancer US FDA Approved Indication
Rubraca is indicated for the maintenance treatment of adult women with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to platinum-based chemotherapy.
Please Click here for full Prescribing Information for Rubraca.
About Clovis Oncology
Clovis Oncology, Inc. is a biopharmaceutical company focused on acquiring, developing, and commercializing innovative anti-cancer agents in the US, Europe, and additional international markets. Clovis Oncology targets development programs at specific subsets of cancer populations, and simultaneously develops, with partners, for those indications that require them, diagnostic tools intended to direct a compound in development to the population that is most likely to benefit from its use. Clovis Oncology is headquartered in Boulder, Colorado, with additional office locations in the US and Europe. Please visit www.clovisoncology.com for more information.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220911005019/en/
Source: Clovis Oncology, Inc.
Sep. 12, 2022 6:48 AM ET Clovis Oncology, Inc. (CLVS)
By: Ravikash, SA News Editor
PUBLISHED11 September 2022
Detailed positive results from an interim analysis of the DESTINY-Lung02 Phase II trial showed Enhertu (trastuzumab deruxtecan) demonstrated clinically meaningful tumour responses in previously-treated patients with HER2-mutant (HER2m) unresectable and/or metastatic non-squamous non-small cell lung cancer (NSCLC). Results will be presented today as a late-breaking presentation at the European Society for Medical Oncology (ESMO) Congress 2022.
Enhertu is a specifically engineered HER2-directed antibody drug conjugate (ADC) being jointly developed and commercialised by AstraZeneca and Daiichi Sankyo.
At a pre-specified interim analysis of DESTINY-Lung02, patients receiving Enhertu at a dose of 5.4mg/kg or 6.4mg/kg demonstrated clinically meaningful activity. The safety profile for both doses was also consistent with the overall safety profile of Enhertu, with the 5.4mg/kg dose demonstrating a favourable safety profile in this patient population. A confirmed objective response rate (ORR) of 53.8% (95% confidence interval [CI] 39.5-67.8) and 42.9% (95% CI 24.5-62.8) was seen in the 5.4mg/kg and 6.4mg/kg arms respectively, as assessed by blinded independent central review (BICR). One complete response (CR) was observed in each arm (5.4mg/kg: 1.9%, 6.4mg/kg: 3.6%), with 27 (51.9%) partial responses (PR) observed in the 5.4mg/kg arm and 11 (39.3%) PRs observed in the 6.4mg/kg arm.
Koichi Goto, MD, Medical Oncologist and Investigator at National Cancer Center Hospital East, Kashiwa, Japan, said: “DESTINY-Lung02 reinforces HER2 as an actionable mutation in patients with metastatic non-small cell lung cancer and further demonstrates that Enhertu provides a clinically meaningful tumour response for these patients who have historically had limited treatment options. The response seen in this trial, along with the disease control observed support Enhertu as a potential treatment option in this type of non-small cell lung cancer.”
Cristian Massacesi, Chief Medical Officer & Oncology Chief Development Officer, AstraZeneca, said: “The clinically meaningful activity, together with the favourable safety profile seen in the DESTINY-Lung02 trial helps establish the optimal dose of Enhertu at 5.4 milligrams per kilogram in previously-treated HER2-mutant non-small cell lung cancer. As we continue to explore the potential of this important medicine across multiple HER2-targetable tumour types, these data reaffirm the need to undertake HER2 testing in patients diagnosed with lung cancer.”
Ken Takeshita, MD, Global Head, R&D, Daiichi Sankyo, said: “The DESTINY-Lung02 results are consistent with the data previously seen with Enhertu in non-small cell lung cancer and the efficacy demonstrated in this interim analysis, which supported the recent US FDA accelerated approval of Enhertu in patients with HER2-mutant non-small cell lung cancer, reinforces the potential to establish this medicine as a treatment option for these patients. These data will help inform future regulatory submissions worldwide with the goal of continuing to offer this innovative medicine to as many patients as possible.”
DESTINY-Lung01 updated results
Updated results from the DESTINY-Lung01 Phase II trial, which evaluated Enhertu in HER2m (cohort 2) or HER2-over-expressing (cohort 1 and cohort 1a) NSCLC, were also presented at ESMO and showed that Enhertu continues to demonstrate consistent efficacy, safety and survival with longer follow-up.
After a median follow-up of 16.7 months, results of previously treated patients with HER2m NSCLC (cohort 2) showed the median DoR for Enhertu in the overall patient population increased to 10.6 months (95% CI 5.6-18.3), with median overall survival (OS) increasing to 18.6 months (95% CI 13.8-25.8). Subgroup analyses of patients with or without a presence of baseline asymptomatic brain metastases showed that treatment with Enhertu resulted in a median PFS of 7.1 months (95% CI 5.5-9.8) and 9.7 months (95% CI 4.5-16.9) respectively, and a median OS of 14.0 months (95% CI: 9.8-19.5) and 27.0 months (95% CI: 15.3-NE), respectively. The subgroup analysis of patients who had received either two or fewer prior therapies or more than two prior therapies showed a median PFS of 8.3 months (95% CI: 5.8-15.2) and 6.8 months (95% CI: 4.4-9.8) respectively, and a median OS of 22.1 months (95% CI: 14.0-31.3) and 13.8 months (95% CI: 7.1-18.6), respectively.
Additionally, updated results from cohort 1 (Enhertu 6.4mg/kg) and cohort 1a (Enhertu 5.4mg/kg), which evaluated patients with previously-treated metastatic HER2-overexpressing NSCLC, highlight encouraging anti-tumour activity. In cohort 1, a confirmed ORR of 26.5% (95% CI 15.0-41.1) was seen in patients receiving Enhertu 6.4mg/kg, with a median progression-free survival (PFS) of 5.7 months (95% CI 2.8-7.2) and a median OS of 12.4 months (95% CI 7.8-17.2). In cohort 1a, a confirmed ORR of 34.1% (95% CI 20.1-50.6) was seen in patients receiving Enhertu 5.4mg/kg, with a median PFS of 6.7 months (95% CI 4.2-8.4), and a median OS of 11.2 months (95% CI 8.4-NE).
The overall safety profile of Enhertu in DESTINY-Lung01 was consistent with previous data, with no new safety signals identified with the longer follow-up. In the HER2m NSCLC patient cohort, there was one additional case of treatment-related ILD or pneumonitis observed, as determined by an independent adjudication committee. ILD has been observed in 27.5% of patients treated with Enhertu 6.4mg/kg in the HER2m cohort, with the majority identified as low Grade, and two Grade 5 events reported. In the HER2-overexpressing NSCLC patient cohorts, there were two additional cases of treatment-related ILD or pneumonitis observed in the 6.4mg/kg dose cohort, and two cases observed in the 5.4mg/kg dose cohort, as determined by an independent adjudication committee. ILD has been observed in 20.4% and 4.9% of patients treated with Enhertu at the 6.4mg/kg and 5.4mg/kg doses respectively in the HER2-overexpressing cohort, with the majority identified as low Grade, and four Grade 5 events (three in the 6.4mg/kg dose cohort and one in the 5.4mg/kg dose cohort) reported. Data from the DESTINY-Lung01 Phase II trial were previously published in The New England Journal of Medicine.1
DESTINY-Lung02
DESTINY-Lung02 is a global, randomised, Phase II trial evaluating the safety and efficacy of Enhertu in patients with HER2m metastatic NSCLC with disease recurrence or progression during or after at least one regimen of prior anticancer therapy that must have contained a platinum-based chemotherapy. Patients were randomised 2:1 to receive Enhertu 5.4mg/kg (n=102) or Enhertu 6.4mg/kg (n=50).The primary endpoint of the trial is confirmed ORR as assessed by BICR. Secondary endpoints include confirmed disease control rate (DCR), DoR and PFS assessed by investigator and BICR, investigator-assessed OS and safety. DESTINY-Lung02 enrolled 152 patients at multiple sites, including Asia, Europe and North America. For more information about the trial, visit ClinicalTrials.gov.
DESTINY-Lung01
DESTINY-Lung01 is a global Phase II, open-label, two-cohort trial evaluating the efficacy and safety of Enhertu (5.4mg/kg or 6.4mg/kg) in patients with HER2m (cohort 2, n=91) or HER2-overexpressing (defined as IHC 3+ or IHC 2+) [cohort 1 and 1a, n=90] unresectable or metastatic non-squamous NSCLC who had progressed after one or more systemic therapies. The primary endpoint is confirmed ORR by independent central review. Key secondary endpoints include DoR, DCR, PFS, OS and safety. DESTINY-Lung01 enrolled 181 patients at multiple sites, including Asia, Europe and North America. For more information about the trial, visit ClinicalTrials.gov.
Enhertu
Enhertu is a HER2-directed ADC. Designed using Daiichi Sankyo’s proprietary DXd ADC technology, Enhertu is the lead ADC in the oncology portfolio of Daiichi Sankyo and the most advanced programme in AstraZeneca’s ADC scientific platform. Enhertu consists of a HER2 monoclonal antibody attached to a topoisomerase I inhibitor payload, an exatecan derivative, via a stable tetrapeptide-based cleavable linker.
Enhertu (5.4mg/kg) is approved in more than 30 countries for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received a (or one or more) prior anti-HER2-based regimen either in the metastatic setting, or in the neoadjuvant or adjuvant setting and have developed disease recurrence during or within six months of completing therapy based on the results from the DESTINY-Breast03 trial.
Enhertu (5.4mg/kg) is approved in several countries for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received two or more prior anti-HER2-based regimens based on the results from the DESTINY-Breast01 trial.
Enhertu (5.4mg/kg) is approved in the US for the treatment of adult patients with unresectable or metastatic HER2-low (IHC 1+ or IHC 2+/ISH-) breast cancer who have received a prior chemotherapy in the metastatic setting or developed disease recurrence during or within six months of completing adjuvant chemotherapy based on the results from the DESTINY-Breast04 trial.
Enhertu (5.4mg/kg) is approved under accelerated approval in the US for the treatment of adult patients with unresectable or metastatic NSCLC whose tumours have activating HER2 (ERBB2) mutations, as detected by a FDA-approved test, and who have received a prior systemic therapy based on the results from the DESTINY-Lung02 trial. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
Enhertu (6.4mg/kg) is approved in several countries for the treatment of adult patients with locally advanced or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma who have received a prior trastuzumab-based regimen based on the results from the DESTINY-Gastric01 trial.
Enhertu development programme
A comprehensive development programme is underway globally, evaluating the efficacy and safety of Enhertu monotherapy across multiple HER2-targetable cancers, including breast, gastric, lung and colorectal cancers. Trials in combination with other anticancer treatments, such as immunotherapy, are also underway.
Regulatory applications for Enhertu in breast and gastric cancer are currently under review in several countries based on the DESTINY-Breast01, DESTINY-Breast03, DESTINY-Breast04, DESTINY-Gastric01 and DESTINY-Gastric02 trials, respectively.
Daiichi Sankyo collaboration
Daiichi Sankyo Company, Limited (TSE: 4568) [referred to as Daiichi Sankyo] and AstraZeneca entered into a global collaboration to jointly develop and commercialise Enhertu (a HER2-directed ADC) in March 2019 and datopotamab deruxtecan (a TROP2-directed ADC) in July 2020, except in Japan where Daiichi Sankyo maintains exclusive rights. Daiichi Sankyo is responsible for the manufacturing and supply of Enhertu and datopotamab deruxtecan.
AstraZeneca in lung cancer
AstraZeneca is working to bring patients with lung cancer closer to cure through the detection and treatment of early-stage disease, while also pushing the boundaries of science to improve outcomes in the resistant and advanced settings. By defining new therapeutic targets and investigating innovative approaches, the Company aims to match medicines to the patients who can benefit most.
The Company’s comprehensive portfolio includes leading lung cancer medicines and the next wave of innovations, including Tagrisso (osimertinib) and Iressa (gefitinib); Imfinzi (durvalumab) and tremelimumab; Enhertu (trastuzumab deruxtecan) and datopotamab deruxtecan in collaboration with Daiichi Sankyo; Orpathys (savolitinib) in collaboration with HUTCHMED; as well as a pipeline of potential new medicines and combinations across diverse mechanisms of action.
AstraZeneca is a founding member of the Lung Ambition Alliance, a global coalition working to accelerate innovation and deliver meaningful improvements for people with lung cancer, including and beyond treatment.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Sep. 12, 2022 7:49 AM ET
AstraZeneca PLC (AZN), DSKYF, DSNKY
By: Ravikash, SA News Editor
AstraZeneca (NASDAQ:AZN) and Daiichi Sankyo (OTCPK:DSKYF) (OTCPK:DSNKY) said their drug Enhertu showed clinically meaningful tumor responses in previously-treated patients with HER2-mutant (HER2m) unresectable and/or metastatic non-squamous non-small cell lung cancer (NSCLC) in a phase 2 trial.
https://seekingalpha.com/symbol/DSKYF
https://seekingalpha.com/symbol/DSNKY
https://seekingalpha.com/symbol/AZN
September 8, 2022 at 9:00 AM EDT Back
91% and 89% of diabetic macular edema (DME) patients were rapidly initiated and maintained on 12- and 16-week dosing intervals (without need for regimen modification) through week 48, respectively
79% and 77% of wet age-related macular degeneration (wAMD) patients were rapidly initiated and maintained on 12- and 16-week dosing intervals (without need for regimen modification) through week 48, respectively
Safety of aflibercept 8 mg consistent with the established safety profile of EYLEA® (aflibercept) Injection
TARRYTOWN, N.Y., Sept. 8, 2022 /PRNewswire/ -- Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) today announced that the primary endpoints were met in two pivotal trials investigating novel aflibercept 8 mg with 12- and 16-week dosing regimens in patients with diabetic macular edema (DME) and wet age-related macular degeneration (wAMD). The PHOTON trial in DME and the PULSAR trial in wAMD both demonstrated that aflibercept 8 mg 12- and 16-week dosing regimens achieved non-inferiority in vision gains compared to the EYLEA 8-week dosing regimen. In these trials, the safety of aflibercept 8 mg was consistent with the established safety profile of EYLEA. Regeneron and Bayer will submit these data to regulatory authorities in countries around the world.
"These pivotal aflibercept 8 mg trials demonstrated that nearly 90% of patients with diabetic macular edema and almost 80% of patients with wet age-related macular degeneration were able to maintain a 16-week dosing regimen," said David Brown, M.D., FACS, Director of Research at Retina Consultants of Texas in the U.S. and a trial investigator. "These unprecedented durability data coupled with a safety profile consistent with that of EYLEA support aflibercept 8 mg as a potential new standard-of-care in these diseases."
"These groundbreaking results are excellent news for patients. These outcomes have shown that aflibercept 8 mg not only improved vision with less frequent injections, but also demonstrated a similar safety profile to EYLEA," said Jean-François Korobelnik, M.D., Ph.D., Professor of Ophthalmology and Head of the Department of Ophthalmology at University Hospital of Bordeaux in France and a trial investigator.
PHOTON (N=658) and PULSAR (N=1,009) are double-masked, active-controlled pivotal trials that are being conducted in multiple centers globally. At 48 weeks, >90% of patients in all dosing groups in both trials completed the treatment period with efficacy results as follows:
Over the last decade, EYLEA has become the standard-of-care for diabetic macular edema and wet age-related macular degeneration," said George D. Yancopoulos, M.D., Ph.D., President and Chief Scientific Officer at Regeneron, and a principal inventor of aflibercept. "The results of these trials with our novel aflibercept 8 mg formulation demonstrated that a remarkably high percentage of patients were maintained on 12- and 16-week dosing intervals through week 48, suggesting aflibercept 8 mg has the potential to be as paradigm-changing as EYLEA."
Detailed efficacy and safety data from PHOTON and PULSAR are planned for presentation at an upcoming medical meeting.
Aflibercept 8 mg is being jointly developed by Regeneron and Bayer AG. In the U.S., Regeneron maintains exclusive rights to EYLEA and aflibercept 8 mg. Bayer has licensed the exclusive marketing rights outside of the U.S., where the companies share equally the profits from sales of EYLEA.
Aflibercept 8 mg is investigational, and its safety and efficacy have not been evaluated by any regulatory authority.
About the Aflibercept 8 mg Trial Program
In both PHOTON and PULSAR, patients with DME and wAMD were randomized into three treatment groups to receive either: aflibercept 8 mg every 12 weeks, aflibercept 8 mg every 16 weeks, or EYLEA every 8 weeks. Patients treated with aflibercept 8 mg in both trials had 3 initial monthly doses, and patients treated with EYLEA received 5 initial monthly doses in PHOTON and 3 in PULSAR. In the first year, patients in the aflibercept 8 mg groups could have their dosing intervals shortened down to an every 8-week interval if protocol-defined criteria for disease progression was observed. Intervals could not be extended until the second year of the study, with those results still to be assessed. Patients in all EYLEA groups maintained a fixed 8-week dosing regimen throughout their participation in the trials. The lead sponsors of the trials were Regeneron for PHOTON and Bayer for PULSAR.
INDICATIONS
EYLEA® (aflibercept) Injection 2 mg (0.05 mL) is indicated for the treatment of patients with Neovascular (Wet) Age-related Macular Degeneration (AMD), Macular Edema following Retinal Vein Occlusion (RVO), Diabetic Macular Edema (DME), and Diabetic Retinopathy (DR).
CONTRAINDICATIONS
About Regeneron
Regeneron (NASDAQ: REGN) is a leading biotechnology company that invents, develops and commercializes life-transforming medicines for people with serious diseases. Founded and led for nearly 35 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to nine FDA-approved treatments and numerous product candidates in development, almost all of which were homegrown in our laboratories. Our medicines and pipeline are designed to help patients with eye diseases, allergic and inflammatory diseases, cancer, cardiovascular and metabolic diseases, pain, hematologic conditions, infectious diseases and rare diseases.
Regeneron is accelerating and improving the traditional drug development process through our proprietary VelociSuite® technologies, such as VelocImmune®, which uses unique genetically humanized mice to produce optimized fully human antibodies and bispecific antibodies, and through ambitious research initiatives such as the Regeneron Genetics Center®, which is conducting one of the largest genetics sequencing efforts in the world.
For more information, please visit www.Regeneron.com or follow @Regeneron on Twitter.
SOURCE Regeneron Pharmaceuticals, Inc.
Sep. 08, 2022 9:53 AM ET
Regeneron Pharmaceuticals, Inc. (REGN)BAYRY, BAYZF
By: Dulan Lokuwithana, SA News Editor9 Comments
Regeneron (NASDAQ:REGN) announced Thursday that its blockbuster eye medication Eylea reached the primary endpoints in two pivotal trials designed to investigate a less frequent dosing regimen in patients with diabetic macular edema (DME) and wet age-related macular degeneration (wAMD).
https://seekingalpha.com/symbol/REGN
PUBLISHED9 September 2022 09 September 2022 13:10 BST
Positive long-term follow-up results from the PAOLA-1 and SOLO-1 Phase III trials of AstraZeneca and MSD’s Lynparza (olaparib) with or without bevacizumab demonstrated clinically meaningful improvements in overall survival (OS). Further results showed class-leading progression-free survival (PFS) in combination with bevacizumab for homologous recombination deficiency (HRD)-positive patients, versus active comparator, bevacizumab, and as monotherapy for patients with BRCA mutations, versus placebo, respectively.
Both trials which were conducted in biomarker-selected, newly diagnosed patients with advanced ovarian cancer in the 1st-line maintenance setting also demonstrated a consistent safety profile.1,2
The results for PAOLA-1 (Abstract #LBA29) and SOLO-1 (Abstract #517O) were presented on 9 September at the 2022 European Society of Medical Oncology (ESMO) and SOLO-1 results were published in Journal of Clinical Oncology.
Ovarian cancer is one of the most common gynaecologic cancers, with a poor prognosis and a high mortality rate.3 Over two thirds of patients are diagnosed with advanced disease, and approximately 50-70% of these patients die within five years.4,5 Up to one in five women with advanced ovarian cancer have a BRCA mutation, and roughly half of women have HRD-positive tumours (which includes those with a BRCA mutation).6-8
Professor Isabelle Ray-Coquard, principal investigator from the PAOLA-1 trial and President of the Gineco group, said: “For women facing an advanced ovarian cancer diagnosis who are HRD-positive, a targeted treatment in the 1st-line maintenance setting is critical to helping them live longer. These latest results at the five-year landmark demonstrate that olaparib with bevacizumab reduces the risk of death by 38% in HRD-positive patients compared to bevacizumab alone, further reinforcing the clinically meaningful long-term survival benefit of this combination. This should be promising news for both clinicians and patients, as we see these additional data show that this combination may allow patients more time with family and loved ones. These results also highlight the importance of biomarker testing as part of a precision medicine approach to guide treatment decisions in ovarian cancer patients.”
Professor Paul DiSilvestro, investigator from the SOLO-1 trial and Director of the Program in Women’s Oncology at Women and Infants Hospital in Providence, Rhode Island, said: “The long-term results from SOLO-1 confirm that olaparib continues to elicit a clinically meaningful improvement in overall survival in the 1st-line maintenance setting for more than seven years. Achieving long-term survival for patients with newly diagnosed advanced ovarian cancer is critical because the 1st-line setting offers the greatest potential to impact patient survival.”
Susan Galbraith, Executive Vice President, Oncology R&D, AstraZeneca, said: “Historically the five year survival rate of newly diagnosed patients with advanced ovarian cancer is 30-50%. In that context, it is phenomenal to share the long term overall survival data from both PAOLA-1 and SOLO-1, with two out of three patients still alive in these trials. We continue to believe in Lynparza’s ability to help biomarker-selected patients with advanced ovarian cancer to achieve better outcomes.”
Dr Eliav Barr, Senior Vice President, Head of Global Clinical Development and Chief Medical Officer, MSD Research Laboratories, said: “These latest data from the PAOLA-1 and SOLO-1 trials further highlight the importance of HRD testing, including for BRCA1/2 mutations, for all newly diagnosed advanced ovarian cancer patients at the point of diagnosis. Maintenance therapy with Lynparza may provide certain patients with HRD-positive and/or BRCA-mutated advanced ovarian cancer the opportunity to live longer.”
Updated results from the PAOLA-1 Phase III trial
Updated results from the PAOLA-1 Phase III trial demonstrate that Lynparza plus bevacizumab increased median overall survival to 56.5 months versus 51.6 months with bevacizumab alone, in patients with newly diagnosed advanced ovarian cancer irrespective of HRD status. This increase was not statistically significant.
In HRD-positive patients, Lynparza plus bevacizumab provided a clinically meaningful improvement in overall survival, reducing the risk of death by 38% versus bevacizumab (based on a HR of 0.62; 95% CI 0.45-0.85) despite PAOLA-1 having 30% Stage IV patients. 65.5% of patients treated with Lynparza plus bevacizumab were still alive at five years versus 48.4% of those treated with bevacizumab alone. Lynparza plus bevacizumab also improved median PFS to almost four years (46.8 months) versus 17.6 months with bevacizumab plus placebo and 46.1% of patients treated with Lynparza plus bevacizumab remain progression free at five years versus 19.2% of patients treated with bevacizumab alone. The safety and tolerability profile of Lynparza in this trial was in line with that observed in prior clinical trials, with no new safety signals.
Updated results from the SOLO-1 Phase III trial
Updated results from the SOLO-1 Phase III trial demonstrate that Lynparza provided a clinically meaningful improvement in overall survival (OS) versus placebo in patients with BRCA-mutated (BRCAm) newly diagnosed advanced ovarian cancer, reducing the risk of death by 45% (based on an HR of 0.55; 95% CI 0.40-0.76; nominal p=0.0004 [not statistically significant]). Median OS was still not reached with Lynparza versus 75.2 months with placebo. At the seven-year descriptive OS analysis, 67% of Lynparza patients were alive versus 47% of placebo patients (44% of whom had a subsequent PARP inhibitor) and 45% of Lynparza patients versus 21% of placebo patients were alive and had not received a first subsequent treatment.
Additional data showed median time to first subsequent therapy was 64 months with Lynparza versus 15.1 months with placebo. The safety and tolerability profile of Lynparza in this trial was in line with that observed in prior clinical trials, with no new safety signals.
PAOLA-1
PAOLA-1 is a double-blinded Phase III trial testing the efficacy and safety of Lynparza added to standard-of-care bevacizumab versus bevacizumab alone, as a 1st-line maintenance treatment for newly diagnosed advanced FIGO Stage III-IV high-grade serous or endometroid ovarian, fallopian tube, or peritoneal cancer patients who had a complete or partial response to 1st-line treatment with platinum-based chemotherapy and bevacizumab. AstraZeneca and MSD announced in August 2019 that the trial met its primary endpoint of PFS in the overall trial population.
PAOLA-1 is an ENGOT (European Network of Gynaecological Oncological Trial groups) trial, sponsored by ARCAGY Research (Association de Recherche sur les CAncers dont GYnécologiques) on behalf of GINECO (Groupe d’Investigateurs National des Etudes des Cancers Ovariens et du sein). ARCAGY-GINECO is an academic group specialising in clinical and translational research in patients’ cancers and a member of the GCIG (Gynecologic Cancer InterGroup).
SOLO-1
SOLO-1 is a Phase III randomised, double-blinded, placebo-controlled, multicentre trial to evaluate the efficacy and safety of Lynparza tablets (300 mg twice daily) as maintenance monotherapy compared with placebo, in newly-diagnosed patients with advanced BRCAm ovarian cancer following platinum-based chemotherapy. The trial randomised 391 patients with a deleterious or suspected deleterious BRCA1 or BRCA2 mutation who were in clinical complete or partial response following platinum-based chemotherapy. Patients were randomised (2:1) to receive Lynparza or placebo for up to two years or until disease progression (at the investigator’s discretion). The primary endpoint was PFS and key secondary endpoints included time to second disease progression or death, time to first subsequent treatment and overall survival. AstraZeneca and MSD announced in June 2018 that the trial met its primary endpoint of PFS in the overall trial population.
BRCA
BRCA1 and BRCA2 are human genes that produce proteins responsible for repairing damaged DNA and play an important role maintaining the genetic stability of cells.13 When either of these genes is mutated or altered such that its protein product either is not made or does not function correctly, DNA damage may not be repaired properly, and cells become unstable. As a result, cells are more likely to develop additional alterations that can lead to cancer. Cancers with BRCA mutations are more likely to be sensitive to PARP inhibitors including Lynparza.13-16
Homologous recombination deficiency
HRD, which defines a subgroup of ovarian cancer, encompasses a wide range of genetic abnormalities, including BRCA mutations and beyond. As with BRCA gene mutations, HRD interferes with normal cell DNA repair mechanisms and confers sensitivity to PARP inhibitors including Lynparza.2
Lynparza
Lynparza (olaparib) is a first-in-class PARP inhibitor and the first targeted treatment to block DNA damage response (DDR) in cells/tumours harbouring a deficiency in homologous recombination repair (HRR), such as those with mutations in BRCA1 and/or BRCA2, or those where deficiency is induced by other agents (such as new hormonal agents – NHAs).
Inhibition of PARP proteins with Lynparza leads to the trapping of PARP bound to DNA single-strand breaks, stalling of replication forks, their collapse and the generation of DNA double-strand breaks and cancer cell death.
Lynparza is currently approved in a number of countries across multiple tumour types including maintenance treatment of platinum-sensitive relapsed ovarian cancer and as both monotherapy and in combination with bevacizumab for the 1st-line maintenance treatment of BRCA-mutated (BRCAm) and homologous recombination repair deficient (HRD)-positive advanced ovarian cancer, respectively; for gBRCAm, HER2-negative metastatic breast cancer (in the EU and Japan this includes locally advanced breast cancer); for gBRCAm, HER2-negative high-risk early breast cancer (in Japan this includes all BRCAm HER2-negative high-risk early breast cancer); for gBRCAm metastatic pancreatic cancer; and HRR gene-mutated metastatic castration-resistant prostate cancer (BRCAm only in the EU and Japan).
Lynparza, which is being jointly developed and commercialised by AstraZeneca and MSD, is the foundation of AstraZeneca's industry-leading portfolio of potential new medicines targeting DDR mechanisms in cancer cells.
The AstraZeneca and MSD strategic oncology collaboration
In July 2017, AstraZeneca and Merck & Co., Inc., Kenilworth, NJ, US, known as MSD outside the US and Canada, announced a global strategic oncology collaboration to co-develop and co-commercialise Lynparza, the world’s first PARP inhibitor, and Koselugo (selumetinib), a mitogen-activated protein kinase (MEK) inhibitor, for multiple cancer types.
Working together, the companies will develop Lynparza and Koselugo in combination with other potential new medicines and as monotherapies. The companies will develop Lynparza and Koselugo in combination with their respective PD-L1 and PD-1 medicines independently.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Sep. 09, 2022 9:25 AM ET
AstraZeneca PLC (AZN), MRKRHHBY, RHHBF
By: Ravikash, SA News Editor
AstraZeneca (NASDAQ:AZN) and Merck's (NYSE:MRK) Lynparza showed clinically meaningful survival benefit as first line therapy in certain patients with ovarian cancer as per long-term data from two phase 3 trials.
https://seekingalpha.com/symbol/AZN
https://seekingalpha.com/symbol/MRK
September 07, 2022
-- 3.2 Month Survival Benefit Demonstrated in Patients who had Already Received Prior Endocrine-based Therapy and at Least Two Prior Chemotherapies --
-- Trodelvy Now Shows a Survival Benefit in both Pre-treated HR+/HER2- Metastatic Breast Cancer and Second-Line Metastatic Triple-Negative Breast Cancer --
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced the positive overall survival (OS) results from the Phase 3 TROPiCS-02 study evaluating Trodelvy® (sacituzumab govitecan-hziy) versus comparator chemotherapy (physicians’ choice of chemotherapy, TPC) in patients with HR+/HER2- metastatic breast cancer who received endocrine-based therapies and at least two chemotherapies. In the study, Trodelvy demonstrated a statistically significant and clinically meaningful improvement of 3.2 months in OS compared to TPC (median OS: 14.4 months vs. 11.2 months; hazard ratio [HR]=0.79; [95% confidence interval [CI]: 0.65-0.96]; p=0.02). OS was a key secondary endpoint of the trial.
These findings will be presented on Friday, September 9 at 4:20pm CEST during the European Society for Medical Oncology (ESMO) Congress 2022 as a late-breaking oral presentation (#LBA76) in the Brest Auditorium, Paris Expo Porte de Versailles.
Other key secondary endpoints including objective response rate (ORR) demonstrated statistically significant improvement in favoring Trodelvy versus TPC. Time to deterioration (TTD) of Global Health Status/Quality of Life (QoL) and Fatigue scale per EORTC-QLQ-C30 also favored Trodelvy versus TPC (QoL: 4.3 months vs. 3.0 months, p=0.006; Fatigue: 2.2 months vs. 1.4 months, p=0.002). No statistically significant difference in TTD on the Pain Scale was observed.
“It is outstanding to see a clinically meaningful survival benefit of over three months for patients with pre-treated HR+/HER2- metastatic breast cancer,” said Hope S. Rugo, MD, Professor of Medicine and Director, Breast Oncology and Clinical Trials Education at the University of California San Francisco Comprehensive Cancer Center, U.S. “Nearly all patients with HR+/HER2- metastatic breast cancer will develop resistance to endocrine-based therapies even in combination with targeted agents, so these data are welcome news for the breast cancer community. The results of TROPiCS-02 highlight the potential for sacituzumab govitecan in patients with pre-treated HR+/HER2- metastatic breast cancer.”
The safety profile for Trodelvy was consistent with prior studies, with no new safety signals identified in this patient population.
“With these data from TROPiCS-02, Trodelvy has now demonstrated a survival benefit in both pre-treated HR+/HER2- metastatic breast cancer and second-line metastatic TNBC – two difficult-to-treat forms of breast cancer,” said Bill Grossman, MD, PhD, Senior Vice President, Therapeutic Area Head, Gilead Oncology. “Our Gilead Oncology ambition is to transform care for people with cancer, and the meaningful improvement in survival benefit seen in the TROPiCS-02 study with Trodelvy is another step forward in pursuing this ambition for patients.”
The TROPiCS-02 study met its primary endpoint of progression-free survival earlier this year; detailed results were presented during the 2022 American Society of Clinical Oncology (ASCO) Annual Meeting.
Trodelvy has not been approved by any regulatory agency for the treatment of HR+/HER2- metastatic breast cancer. Its safety and efficacy have not been established for this indication. Gilead has submitted a supplemental Biologics License Application (sBLA) to the U.S. Food and Drug Administration (FDA) based on data from TROPiCS-02; these data will also be shared with health authorities outside the U.S.
Sacituzumab govitecan-hziy is currently included in the National Comprehensive Cancer Network® (NCCN®) Clinical Practice Guidelines in Oncology (NCCN Guidelines®)i. This includes a Category 1 recommendation for use in adult patients with second-line metastatic triple-negative breast cancer (defined as those who received at least two prior therapies, with at least one line for metastatic disease). It also has a Category 2A preferred recommendation for investigational use in HR+/HER2- advanced breast cancer after prior treatment including endocrine therapy, a CDK4/6 inhibitor and at least two lines of chemotherapy.
Trodelvy has a Boxed Warning for severe or life-threatening neutropenia and severe diarrhea; please see below for additional Important Safety Information.
About the TROPiCS-02 Study
The TROPiCS-02 study is a global, multicenter, open-label, Phase 3 study, randomized 1:1 to evaluate Trodelvy versus physicians’ choice of chemotherapy (eribulin, capecitabine, gemcitabine, or vinorelbine) in 543 patients with HR+/HER2- metastatic breast cancer who were previously treated with endocrine therapy, CDK4/6 inhibitors and two to four lines of chemotherapy for metastatic disease. The primary endpoint is progression-free survival per Response Evaluation Criteria in Solid Tumors (RECIST 1.1) as assessed by blinded independent central review (BICR) for participants treated with Trodelvy compared to those treated with chemotherapy. Secondary endpoints include overall survival, overall response rate, clinical benefit rate and duration of response, as well as assessment of safety and tolerability and quality of life measures. In the study, HER2 negativity was defined per American Society of Clinical Oncology (ASCO) and the College of American Pathologists (CAP) criteria as immunohistochemistry (IHC) score of 0, IHC 1+ or IHC 2+ with a negative in-situ hybridization (ISH) test. More information about TROPiCS-02 is available at https://clinicaltrials.gov/ct2/show/NCT03901339.
About Trodelvy
Trodelvy® (sacituzumab govitecan-hziy) is a first-in-class Trop-2 directed antibody-drug conjugate. Trop-2 is a cell surface antigen highly expressed in multiple tumor types, including in more than 90% of breast and bladder cancers. Trodelvy is intentionally designed with a proprietary hydrolyzable linker attached to SN-38, a topoisomerase I inhibitor payload. This unique combination delivers potent activity to both Trop-2 expressing cells and the microenvironment.
Trodelvy is approved in more than 35 countries, with multiple additional regulatory reviews underway worldwide, for the treatment of adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (TNBC) who have received two or more prior systemic therapies, at least one of them for metastatic disease. Trodelvy is also approved in the U.S. under the accelerated approval pathway for the treatment of adult patients with locally advanced or metastatic urothelial cancer (UC) who have previously received a platinum-containing chemotherapy and either programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor.
Trodelvy is also being developed for potential investigational use in other TNBC and metastatic UC populations, as well as a range of tumor types where Trop-2 is highly expressed, including hormone receptor-positive/human epidermal growth factor receptor 2-negative (HR+/HER2-) metastatic breast cancer, metastatic non-small cell lung cancer (NSCLC), metastatic small cell lung cancer (SCLC), head and neck cancer, and endometrial cancer.
U.S. Indications for Trodelvy
In the United States, Trodelvy is indicated for the treatment of:
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer. Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220906006036/en/
Source: Gilead Sciences, Inc.
Sep. 08, 2022 6:53 AM ET
By: Dulan Lokuwithana, SA News Editor1 Comment
Gilead Sciences (NASDAQ:GILD) announced that its antibody-drug conjugate Trodelvy indicated a statistically significant and clinically meaningful improvement of 3.2 months in overall survival (OS) versus chemotherapy in certain patients with HR+/HER2- metastatic breast cancer.
https://seekingalpha.com/news/3881135-gild-stock-on-watch-after-new-breast-cancer-data-for-trodelvy
https://seekingalpha.com/symbol/GILD
September 06, 2022
– Study met primary and secondary endpoints –
– Results add to body of data supporting the safety of linaclotide for this patient population –
– There are currently no FDA approved prescription pediatric therapies for Functional Constipation (FC); FC affects an estimated 4 to 6 million children ages 6 to 17 in the United States 1 –
– Ironwood and its partner, AbbVie, to evaluate a potential supplemental New Drug Application (sNDA) submission –
BOSTON, Mass.--(BUSINESS WIRE)-- Ironwood Pharmaceuticals, Inc. (Nasdaq: IRWD), today announced positive topline data from a Phase III clinical trial evaluating LINZESS (linaclotide) 72 mcg in pediatric patients aged 6-17 with functional constipation (FC). The trial met its primary and secondary endpoints, demonstrating that linaclotide (72 mcg) improved frequency of spontaneous bowl movements (SBM) and stool consistency. Linaclotide was generally well-tolerated, and the safety profile is consistent with previously reported studies with linaclotide in FC and irritable bowel syndrome (IBS) in pediatric patients.
“Functional constipation is one of the most common gastrointestinal complaints in pediatric patients, one that significantly impacts young patients’ lives. Despite its high prevalence, FC remains challenging to treat because there are no FDA approved prescription treatments for children,” said Jeffrey S. Hyams, MD, Head, Division of Digestive Diseases, Hepatology, and Nutrition, Connecticut Children’s Medical Center, Professor of Pediatrics, University of Connecticut School of Medicine. “These strong data further our understanding of the safety profile of linaclotide in pediatric patients aged 6-17 with FC and demonstrate evidence of its potential to provide therapeutic benefit to these patients suffering from FC.”
In this study, a total of 330 patients were randomized in a 1:1 ratio between linaclotide or placebo. Topline data indicate that linaclotide showed a statistically significant and clinically meaningful improvement compared to placebo in 12-week SBM frequency rate (SBMs/week), the primary endpoint. Linaclotide-treated patients demonstrated a greater than two-fold least squares mean change from baseline in SBMs/week (2.220) compared to placebo (1.050) (p<0.0001). Stool consistency, as assessed by Bristol Stool Form Scale (BSFS) scores, which was the secondary endpoint, also showed an improvement at weeks 12 with linaclotide compared to placebo. The least squares mean change from baseline at week 12 was 1.108 and 0.685 points, respectively (p=0.0001). The BSFS is a 7-point scale ranging from 1 (separate hard, difficult-to-pass lumps) to 7 (liquid stools).
Overall, linaclotide (72 mcg) was well tolerated. The most frequently reported treatment-emergent adverse event was diarrhea, which occurred in 4.3% of linaclotide-treated participants versus 1.8% in the placebo group.
“We are excited by the results of this Phase III trial and are focused on identifying an expeditious regulatory path forward, with our partner AbbVie, to support delivering this potential first functional constipation therapeutic to pediatric patients ages 6-17 in need,” said Mike Shetzline, M.D., Ph.D., chief medical officer, senior vice president and head of research and drug development at Ironwood Pharmaceuticals.
“Our vision for LINZESS has always been simple but powerful – keep patients at the center of everything we do,” said Tom McCourt, chief executive officer at Ironwood Pharmaceuticals. “We have seen the positive impacts that LINZESS has had with more than 4 million unique patients with IBS-C or chronic idiopathic constipation (CIC) treated since launch in 2012, and our team now looks forward to potentially expanding its clinical utility to the treatment of functional constipation in an underserved population.”
These data are from the FC cohort of the Phase III, multicenter, randomized, double-blind, parallel-group, safety, and efficacy study of linaclotide vs. placebo in children ages 6-17 years with FC. Participants in the FC cohort must have fulfilled modified Rome III Criteria for child/adolescent FC.
Ironwood expects to share these Phase III trial data at upcoming scientific meetings and via peer-reviewed publications.
LINZESS is developed and marketed by Ironwood and AbbVie in the United States and is indicated for the treatment of adults with IBS-C or CIC. It is not approved for use in patients less than 18 years of age.
About Linaclotide
Linaclotide is a guanylate cyclase-C (GC-C) agonist that is thought to work in two ways based on nonclinical studies. Linaclotide binds to the GC-C receptor locally, within the intestinal epithelium. Activation of GC-C results in increased intestinal fluid secretion and accelerated transit and a decrease in the activity of pain-sensing nerves in the intestine. The clinical relevance of the effect on pain fibers, which is based on nonclinical studies, has not been established. In the United States, Ironwood and AbbVie co-develop and co-commercialize LINZESS® for the treatment of adults with IBS-C or CIC. In Europe, AbbVie markets linaclotide under the brand name CONSTELLA® for the treatment of adults with moderate to severe IBS-C. In Japan, Ironwood's partner Astellas markets linaclotide under the brand name LINZESS for the treatment of adults with IBS-C or chronic constipation. In China, (including Hong Kong and Macau) Ironwood’s partner Astra Zeneca markets linaclotide under the brand name LINZESS for the treatment of adults with IBS-C. Ironwood is also partnered with AbbVie for development and commercialization of linaclotide in all other territories worldwide. LINZESS® and CONSTELLA® are registered trademarks of Ironwood Pharmaceuticals, Inc. Any other trademarks referred to in this press release are the property of their respective owners. All rights reserved.
LINZESS Important Safety Information
INDICATIONS AND USAGE
LINZESS (linaclotide) is indicated in adults for the treatment of both irritable bowel syndrome with constipation (IBS-C) and chronic idiopathic constipation (CIC).
Please see full Prescribing Information including Boxed Warning: http://www.allergan.com/assets/pdf/linzess_pi
About Ironwood Pharmaceuticals
Ironwood Pharmaceuticals (Nasdaq: IRWD), an S&P SmallCap 600® company, is a leading gastrointestinal (GI) healthcare company on a mission to advance the treatment of GI diseases and redefine the standard of care for GI patients. We are pioneers in the development of LINZESS® (linaclotide), the U.S. branded prescription market leader for adults with irritable bowel syndrome with constipation (IBS-C) or chronic idiopathic constipation (CIC). Under the guidance of our seasoned industry leaders, we continue to build upon our history of GI innovation and challenge what has been done before to shape what the future holds. We keep patients at the heart of our R&D and commercialization efforts to reduce the burden of GI diseases and address significant unmet needs.
Founded in 1998, Ironwood Pharmaceuticals is headquartered in Boston, Massachusetts.
We routinely post information that may be important to investors on our website at www.ironwoodpharma.com. In addition, follow us on Twitter and on LinkedIn.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220906005224/en/
Source: Ironwood Pharmaceuticals, Inc.
Sep. 06, 2022 8:41 AM ET
Ironwood Pharmaceuticals, Inc. (IRWD), ABBV
By: Ravikash, SA News Editor4 Comments
AbbVie (NYSE:ABBV) and Ironwood Pharmaceuticals' medicine (NASDAQ:IRWD) Linzess met the main and secondary goals of a phase 3 trial in pediatric patients aged six years to 17 years with functional constipation (FC).
https://seekingalpha.com/symbol/IRWD
https://seekingalpha.com/symbol/ABBV
September 5, 2022 at 7:29 AM EDT Back
Results from the longest global Phase 3 open-label extension trial in this age group in asthma show sustained improvement in lung function, low rate of asthma attacks and a consistent safety profile for up to two years
TARRYTOWN, N.Y. and PARIS, Sept. 5, 2022 /PRNewswire/ -- Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) and Sanofi today announced results from a Phase 3 open-label extension trial demonstrating the efficacy and safety profile of Dupixent® (dupilumab) as a maintenance therapy when added to other asthma medications was consistent for up to two years in children aged 6 to 11 years with uncontrolled moderate-to-severe asthma with evidence of type 2 inflammation. These results were presented today in a late-breaking session at the 2022 European Respiratory Society (ERS) International Congress, which coincides with the milestone that more than 500,000 people around the world have been treated with Dupixent across all of its approved indications.
"Children with uncontrolled moderate-to-severe asthma may experience long-term persistent coughing, difficulty breathing, unpredictable asthma attacks and impaired lung function, which can lead to complications later in life as they grow and develop," said Leonard B. Bacharier, M.D., Professor of Pediatrics and Director of the Center for Pediatric Asthma Research, Monroe Carell Jr. Children's Hospital at Vanderbilt University Medical Center in Nashville, Tennessee, and principal investigator of the trial. "An established safety profile balanced with efficacy is always a priority when treating children with a chronic disease, such as those with uncontrolled moderate-to-severe asthma with an eosinophilic phenotype or oral corticosteroid dependent asthma. These new data further support the consistent safety profile of long-term Dupixent - which is indicated for the treatment of uncontrolled moderate-to-severe asthma with an eosinophilic phenotype or oral corticosteroid dependent asthma - and its ability to provide sustained improvements in lung function and reductions in asthma exacerbations in children as young as 6 years old."
The results are from data in children who entered the extension trial after finishing active treatment or placebo in the Phase 3 trial (pivotal trial). Children in the extension trial were treated for up to an additional year with Dupixent, providing up to two years of data in total. Children treated with Dupixent in the extension trial experienced a:
The safety results of the trial were generally consistent with the known safety profile of Dupixent in its approved respiratory indications. Over the 52-week treatment period, the overall rates of adverse events (AEs) were 61-68%. The most common AEs (≥5%) were nasopharyngitis (9-10%), pharyngitis (6-10%), upper respiratory tract infection (4-8%), influenza (5-6%), eosinophilia (3-6%), allergic rhinitis (3-7%), diarrhea (4-6%) and injection site reactions (3-7%).
About the LIBERTY ASTHMA EXCURSION Trial
The Phase 3, multicenter, open-label extension trial evaluated the long-term safety and efficacy of Dupixent in 365 children with uncontrolled moderate-to-severe asthma who had previously participated in the placebo-controlled VOYAGE trial (the pivotal trial) when they were 6 to 11 years of age. Based on body weight, patients in the open-label extension trial received Dupixent 100 mg or 200 mg every two weeks or Dupixent 300 mg every four weeks, for 52 weeks.
The primary endpoint assessed the number of patients experiencing any treatment emergent adverse event. Secondary endpoints included the annualized rate of severe asthma exacerbations over one year and change from pivotal trial baseline in FEV1pp.
About Dupixent
Dupixent, which was invented using Regeneron's proprietary VelocImmune® technology, is a fully human monoclonal antibody that inhibits the signaling of the interleukin-4 (IL-4) and interleukin-13 (IL-13) pathways and is not an immunosuppressant. The Dupixent development program has shown significant clinical benefit and a decrease in type 2 inflammation in Phase 3 trials, establishing that IL-4 and IL-13 are key and central drivers of the type 2 inflammation that plays a major role in multiple related and often co-morbid diseases. These diseases include approved indications for Dupixent such as asthma, atopic dermatitis, chronic rhinosinusitis with nasal polyposis (CRSwNP) and eosinophilic esophagitis (EoE), as well as investigational diseases such as prurigo nodularis.
In the EU, Dupixent is approved in children aged 6 to 11 years as an add-on maintenance treatment for severe asthma with type 2 inflammation characterized by raised blood eosinophils and/or raised FeNO, who are inadequately controlled with medium to high dose inhaled corticosteroids (ICS) plus another medicinal product for maintenance treatment. For adolescents and adults 12 years and older with severe asthma with type 2 inflammation, patients must be inadequately controlled with high dose ICS plus another medicinal product for maintenance treatment.
Dupixent has received regulatory approvals around the world for use in in certain patients with atopic dermatitis, asthma, CRSwNP or EoE in different age populations. Dupixent is currently approved across these indications in the U.S. and for one or more of these indications in more than 60 countries, including in the European Union and Japan.
About Regeneron's VelocImmune Technology
Regeneron's VelocImmune technology utilizes a proprietary genetically engineered mouse platform endowed with a genetically humanized immune system to produce optimized fully human antibodies. When Regeneron's co-Founder, President and Chief Scientific Officer George D. Yancopoulos was a graduate student with his mentor Frederick W. Alt in 1985, they were the first to envision making such a genetically humanized mouse, and Regeneron has spent decades inventing and developing VelocImmune and related VelociSuite® technologies. Dr. Yancopoulos and his team have used VelocImmune technology to create approximately one in five of all original, FDA-approved or authorized fully human monoclonal antibodies. This includes REGEN-COV® (casirivimab and imdevimab), Dupixent, Libtayo® (cemiplimab-rwlc), Praluent® (alirocumab), Kevzara® (sarilumab), Evkeeza® (evinacumab-dgnb) and Inmazeb™ (atoltivimab, maftivimab and odesivimab-ebgn).
Dupilumab Development Program
Dupilumab is being jointly developed by Regeneron and Sanofi under a global collaboration agreement. To date, dupilumab has been studied across more than 60 clinical trials involving more than 10,000 patients with various chronic diseases driven in part by type 2 inflammation.
In addition to the currently approved indications, Regeneron and Sanofi are studying dupilumab in a broad range of diseases driven by type 2 inflammation or other allergic processes in Phase 3 trials, including prurigo nodularis, pediatric eosinophilic esophagitis, hand and foot atopic dermatitis, chronic inducible urticaria-cold, chronic spontaneous urticaria, chronic pruritis of unknown origin, chronic obstructive pulmonary disease with evidence of type 2 inflammation, chronic rhinosinusitis without nasal polyposis, allergic fungal rhinosinusitis, allergic bronchopulmonary aspergillosis and bullous pemphigoid. These potential uses of dupilumab are currently under clinical investigation, and the safety and efficacy in these conditions have not been fully evaluated by any regulatory authority.
U.S. Indications
DUPIXENT is a prescription medicine used:
About Regeneron
Regeneron is a leading biotechnology company that invents, develops and commercializes life-transforming medicines for people with serious diseases. Founded and led for nearly 35 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to numerous FDA-approved treatments and product candidates in development, almost all of which were homegrown in our laboratories. Our medicines and pipeline are designed to help patients with eye diseases, allergic and inflammatory diseases, cancer, cardiovascular and metabolic diseases, pain, hematologic conditions, infectious diseases and rare diseases.
Regeneron is accelerating and improving the traditional drug development process through our proprietary VelociSuite® technologies, such as VelocImmune®, which uses unique genetically humanized mice to produce optimized fully human antibodies and bispecific antibodies, and through ambitious research initiatives such as the Regeneron Genetics Center, which is conducting one of the largest genetics sequencing efforts in the world.
For more information, please visit www.Regeneron.com or follow @Regeneron on Twitter.
About Sanofi
We are an innovative global healthcare company, driven by one purpose: we chase the miracles of science to improve people's lives. Our team, across some 100 countries, is dedicated to transforming the practice of medicine by working to turn the impossible into the possible. We provide potentially life-changing treatment options and life-saving vaccine protection to millions of people globally, while putting sustainability and social responsibility at the center of our ambitions.
Sanofi is listed on EURONEXT: SAN and NASDAQ: SNY.
SOURCE Regeneron Pharmaceuticals, Inc.
Sep. 05, 2022 6:18 AM ET
By: Ravikash, SA News Editor
Sanofi (NASDAQ:SNY) and Regeneron Pharmaceuticals (NASDAQ:REGN) said their medicine Dupixent showed efficacy and safety of up to two years in children aged six to 11 years with asthma.
https://seekingalpha.com/symbol/SNY
https://seekingalpha.com/symbol/REGN
September 04, 2022
-- Progression-Free Survival Efficacy of Trodelvy Consistent with That Observed in the TROPiCS-02 Intention-to-Treat Population --
-- Results Presented at ESMO 2022 Highlight Trodelvy as a Potential Treatment Option in HR+/HER2-Low and IHC0 Status Metastatic Breast Cancer --
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced new data from a post hoc subgroup analysis from the Phase 3 TROPiCS-02 study evaluating Trodelvy® (sacituzumab govitecan-hziy) versus comparator chemotherapies (physicians’ choice of chemotherapy, TPC) in patients with HR+/HER2- metastatic breast cancer who progressed on endocrine-based therapies and at least two chemotherapies. The analysis examined progression-free survival (PFS) in the intention-to-treat population by HER2-immunohistochemistry (IHC) status, and the results demonstrated that Trodelvy improved median PFS vs. TPC in both HER2-low (IHC1+ and IHC2+/ISH-negative) and IHC0 groups.
Detailed findings will be presented at a mini-oral session (Abstract #1362) during the European Society for Medical Oncology (ESMO) Congress 2022 in the Évry Auditorium, Paris Expo Porte de Versailles, on September 10.
“These data demonstrate Trodelvy’s efficacy across HER2-low and IHC0 status in pre-treated metastatic breast cancer patients in the TROPiCS-02 trial,” said Professor Peter Schmid, Professor of Cancer Medicine; Centre Lead, Centre of Experimental Cancer Medicine; Director, Barts Breast Cancer Centre. “Once patients have developed resistance to endocrine-based therapies, their prognosis is extremely poor. The results highlight the potential for Trodelvy as a treatment option for people living with pre-treated HR+/HER2- metastatic breast cancer, regardless of their HER2-negative status.”
“These results show Trodelvy improved progression-free survival regardless of HER2 status in this pre-treated patient population and reinforce the strength of clinical activity in a population where need is highest,” said Bill Grossman, MD, PhD, Senior Vice President, Therapeutic Area Head, Gilead Oncology. “Trodelvy is already transforming the standard of care in second-line metastatic triple-negative breast cancer, and we’re excited about its potential in other breast cancers where there is significant need for new treatment options.”
In the study, HER2 negativity was defined per American Society of Clinical Oncology (ASCO) and the College of American Pathologists (CAP) criteria as immunohistochemistry (IHC) score of 0, IHC 1+ or IHC 2+ with a negative in-situ hybridization (ISH) test.
Trodelvy has not been approved by any regulatory agency for the treatment of HR+/HER2- metastatic breast cancer. Its safety and efficacy have not been established for this indication. Gilead has submitted a supplemental Biologics License Application (sBLA) to the U.S. Food and Drug Administration (FDA) based on data from TROPiCS-02; these data will also be shared with health authorities outside the U.S.
Sacituzumab govitecan is currently included in the National Comprehensive Cancer Network® (NCCN®) Clinical Practice Guidelines in Oncology (NCCN Guidelines®)i. This includes a Category 1 recommendation for use in adult patients with second-line metastatic triple-negative breast cancer (defined as those who received at least two prior therapies, with at least one line for metastatic disease). It also has a Category 2A preferred recommendation for investigational use in HR+/HER2- advanced breast cancer after prior treatment including endocrine therapy, a CDK4/6 inhibitor and at least two lines of chemotherapy.
Trodelvy has a Boxed Warning for severe or life-threatening neutropenia and severe diarrhea; please see below for additional Important Safety Information.
About the TROPiCS-02 Study
The TROPiCS-02 study is a global, multicenter, open-label, Phase 3 study, randomized 1:1 to evaluate Trodelvy versus physicians’ choice of chemotherapy (eribulin, capecitabine, gemcitabine, or vinorelbine) in 543 patients with HR+/HER2- metastatic breast cancer who were previously treated with endocrine therapy, CDK4/6 inhibitors and two to four lines of chemotherapy for metastatic disease. The primary endpoint is progression-free survival per Response Evaluation Criteria in Solid Tumors (RECIST 1.1) as assessed by blinded independent central review (BICR) for participants treated with Trodelvy compared to those treated with chemotherapy. Secondary endpoints include overall survival, overall response rate, clinical benefit rate and duration of response, as well as assessment of safety and tolerability and quality of life measures. More information about TROPiCS-02 is available at https://clinicaltrials.gov/ct2/show/NCT03901339.
About Trodelvy
Trodelvy® (sacituzumab govitecan-hziy) is a first-in-class Trop-2 directed antibody-drug conjugate. Trop-2 is a cell surface antigen highly expressed in multiple tumor types, including in more than 90% of breast and bladder cancers. Trodelvy is intentionally designed with a proprietary hydrolyzable linker attached to SN-38, a topoisomerase I inhibitor payload. This unique combination delivers potent activity to both Trop-2 expressing cells and the microenvironment.
Trodelvy is approved in more than 35 countries, with multiple additional regulatory reviews underway worldwide, for the treatment of adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (TNBC) who have received two or more prior systemic therapies, at least one of them for metastatic disease. Trodelvy is also approved in the U.S. under the accelerated approval pathway for the treatment of adult patients with locally advanced or metastatic urothelial cancer (UC) who have previously received a platinum-containing chemotherapy and either programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor.
Trodelvy is also being developed for potential investigational use in other TNBC and metastatic UC populations, as well as a range of tumor types where Trop-2 is highly expressed, including hormone receptor-positive/human epidermal growth factor receptor 2-negative (HR+/HER2-) metastatic breast cancer, metastatic non-small cell lung cancer (NSCLC), metastatic small cell lung cancer (SCLC), head and neck cancer, and endometrial cancer.
U.S. Indications for Trodelvy
In the United States, Trodelvy is indicated for the treatment of:
Please see full Prescribing Information , including BOXED WARNING.
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer. Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California.
U.S. Prescribing Information for Trodelvy including BOXED WARNING, is available at www.gilead.com.
Trodelvy, Gilead and the Gilead logo are trademarks of Gilead Sciences, Inc., or its related companies.
For more information about Gilead, please visit the company’s website at www.gilead.com, follow Gilead on Twitter (@GileadSciences) or call Gilead Public Affairs at 1-800-GILEAD-5 or 1-650-574-3000.
i Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Breast Cancer Version 4.2022. © National Comprehensive Cancer Network, Inc. 2022. All rights reserved. Accessed August 2022. To view the most recent and complete version of the guideline, go online to NCCN.org. NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220902005309/en/
Source: Gilead Sciences, Inc.
Sep. 05, 2022 6:44 AM ET
By: Ravikash, SA News Editor7 Comments
Gilead Sciences (NASDAQ:GILD) said Trodelvy showed progression-free survival (PFS) benefit in patients with breast cancer patients regardless of the HER2 mutation status.
https://seekingalpha.com/symbol/GILD
PUBLISHED5 September 2022 5 September 2022 07:05 BST
AstraZeneca’s Forxiga (dapagliflozin), a sodium-glucose cotransporter 2 (SGLT2) inhibitor, has been approved in China to reduce the risk of sustained estimated glomerular filtration rate (eGFR) decline, end-stage kidney disease (ESKD), cardiovascular (CV) death and hospitalisation for heart failure (hHF) in adults with chronic kidney disease (CKD) at risk of progression with and without type-2 diabetes (T2D).
The approval by China’s National Medical Products Administration (NMPA) is based on positive results from the DAPA-CKD Phase III trial1.
CKD is a serious, progressive condition defined by decreased kidney function and is often associated with an increased risk of heart disease or stroke2-4. The condition affects 850 million people worldwide5. However, diagnosis rates remain low and up to 90% of patients are unaware they have the disease4.
Member of the DAPA-CKD Executive Committee, Fan Fan Hou, Southern Medical University, Guangzhou, China, said: “With unprecedented results of DAPA-CKD, dapagliflozin becomes the first SGLT2 inhibitor approved in China for the treatment of chronic kidney disease. This transformational milestone brings great hope to the 120 million subjects suffering from chronic kidney disease in China.”
Sir Mene Pangalos, Executive Vice President, BioPharmaceuticals R&D, AstraZeneca said: “In addition to AstraZeneca’s commitment to drive increased awareness, prevention and earlier diagnoses, this approval marks another important step forward in our ambition to stop, reverse and ultimately cure chronic kidney disease globally. We are thrilled at the opportunity to bring Forxiga to millions of patients across the country and improve patient outcomes.”
The DAPA-CKD Phase III trial demonstrated that Forxiga, on top of standard-of-care (SoC) treatment with an angiotensin-converting enzyme inhibitor or an angiotensin receptor blocker, reduced the relative risk of worsening of renal function, onset of ESKD, or risk of CV or renal death by 39%, the primary composite endpoint, compared to placebo (absolute risk reduction [ARR]=5.3%, p<0.0001) in patients with CKD Stages 2-4 and elevated urinary albumin excretion. Forxiga also significantly reduced the relative risk of death from any cause by 31% (ARR=2.1%, p=0.0035) compared to placebo6. The safety and tolerability of Forxiga were consistent with the well-established safety profile of the medicine.
Forxiga (known as Farxiga in the US) is now approved in 100 countries around the world including the US, the European Union and Japan for the treatment of CKD in adults with and without T2D.
DAPA-CKD
DAPA-CKD was an international, multi-centre, randomised, double-blinded Phase III trial in 4,304 patients designed to evaluate the efficacy of Forxiga 10mg, compared with placebo, in patients with CKD Stage 2-4 and elevated urinary albumin excretion, with and without T2D. Forxiga was given once daily in addition to SoC. The primary composite endpoint was worsening of renal function or risk of death (defined as a composite of an eGFR decline ≥50%, onset of ESKD or death from CV or renal cause). The secondary endpoints included the time to first occurrence of the renal composite (sustained ≥50% eGFR decline, ESKD or renal death), the composite of CV death or hospitalisation for HF (hHF), and death from any cause. The trial was conducted in 21 countries1. Detailed results from the trial were published in The New England Journal of Medicine1.
Forxiga
Forxiga (dapagliflozin) is a first-in-class, oral, once-daily SGLT2 inhibitor. Research has shown Forxiga’s efficacy in preventing and delaying cardiorenal disease, while also protecting the organs – important findings given the underlying links between the heart, kidneys and pancreas1,10,11. Damage to one of these organs can cause the other organs to fail, contributing to leading causes of death worldwide, including T2D, HF and CKD12-15.
Forxiga is approved as an adjunct to diet and exercise to improve glycaemic control in adults with T2D and in T2D to reduce the risk of hHF or CV death when added to SoC based on the findings of the DECLARE-TIMI 58 Phase III CV outcomes trial11. Forxiga is also approved for the treatment of HFrEF and the treatment of CKD based on the findings of the DAPA-HF and DAPA-CKD Phase III trials6,10.
DapaCare is a robust programme of clinical trials to evaluate the potential CV, renal and organ protection benefits of Forxiga. It includes more than 35 completed and ongoing Phase IIb/III trials in more than 35,000 patients, as well as more than 2.5 million patient-years’ experience. Forxiga is currently being tested in patients without T2D following an acute myocardial infarction (MI) or heart attack in the DAPA-MI Phase III trial - a first of its kind, indication-seeking registry-based randomised controlled trial16.
AstraZeneca in CVRM
Cardiovascular, Renal and Metabolism (CVRM), part of BioPharmaceuticals, forms one of AstraZeneca’s main disease areas and is a key growth driver for the Company. By following the science to understand more clearly the underlying links between the heart, kidneys and pancreas, AstraZeneca is investing in a portfolio of medicines for organ protection and improving outcomes by slowing disease progression, reducing risks and tackling co-morbidities. The Company’s ambition is to modify or halt the natural course of CVRM diseases and potentially regenerate organs and restore function, by continuing to deliver transformative science that improves treatment practices and CV health for millions of patients worldwide.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Sep. 05, 2022 6:28 AM ET
By: Ravikash, SA News Editor
LUMAKRAS Met Primary Endpoint of Progression-Free Survival, Demonstrating Superiority Over Standard of Care Docetaxel Chemotherapy, in KRAS G12C-Mutated Non-Small Cell Lung Cancer
Detailed Data to be Presented at an Upcoming Medical Congress
THOUSAND OAKS, Calif., Aug. 30, 2022 /PRNewswire/ -- Amgen (NASDAQ: AMGN) today announced that the global Phase 3 CodeBreaK 200 trial evaluating once daily oral LUMAKRAS® (sotorasib) met its primary endpoint of progression-free survival (PFS), demonstrating statistical significance and superiority over standard of care chemotherapy, intravenous docetaxel. The first randomized clinical trial for a KRASG12C inhibitor assessed the efficacy and safety of LUMAKRAS in 345 previously treated patients with KRAS G12C-mutated non-small cell lung cancer (NSCLC) who had received at minimum, prior platinum-based doublet chemotherapy and checkpoint inhibitor therapy.
"Further analyses of the data are ongoing, and we look forward to sharing detailed data at an upcoming medical meeting," said David M. Reese, M.D., executive vice president of Research and Development at Amgen. "We are grateful to all of the investigators and patients who participated in this first randomized, controlled clinical trial of a KRASG12C inhibitor."
About LUMAKRAS®/LUMYKRAS® (sotorasib)
Amgen took on one of the toughest challenges of the last 40 years in cancer research by developing LUMAKRAS/LUMYKRAS, a KRASG12C inhibitor.1 LUMAKRAS/LUMYKRAS has demonstrated a positive benefit-risk profile with rapid, deep, and durable anticancer activity in patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) harboring the KRAS G12C mutation with a once daily oral formulation.2
Amgen is progressing the largest and broadest global KRASG12C inhibitor development program with unparalleled speed and exploring more than 10 sotorasib combination regimens, including triplets, with clinical trial sites spanning five continents. To date, over 6,500 patients around the world have received LUMAKRAS/LUMYKRAS through the clinical development program, expanded access and commercial use.
In total, LUMAKRAS/LUMYKRAS is approved in over 44 markets around the world, including the United States, the European Union, Japan, United Arab Emirates, South Korea, Hong Kong, Switzerland, Taiwan and Qatar, and in Australia, Brazil, Canada, Great Britain, Israel and Singapore under the U.S. FDA's Project Orbis. Amgen has submitted MAAs in Argentina, Colombia, Kuwait, Malaysia, Mexico, Saudi Arabia, Thailand and Turkey.
LUMAKRAS/LUMYKRAS is also being studied in multiple other solid tumors.3
About CodeBreaK
The CodeBreaK clinical development program for Amgen's drug sotorasib is designed to study patients with an advanced solid tumor with the KRAS G12C mutation and address the longstanding unmet medical need for these cancers.
CodeBreaK 100, the Phase 1 and 2, first-in-human, open-label multicenter study, enrolled patients with KRAS G12C-mutant solid tumors.9 Eligible patients must have received a prior line of systemic anticancer therapy, consistent with their tumor type and stage of disease. The primary endpoint for the Phase 2 study was centrally assessed objective response rate. The Phase 2 trial in NSCLC enrolled 126 patients, 124 of whom had centrally evaluable lesions by RECIST at baseline.2 The Phase 2 trial in colorectal cancer (CRC) is fully enrolled and results have been published.10
CodeBreaK 200, the global Phase 3 randomized active-controlled study comparing sotorasib to docetaxel in KRAS G12C-mutated NSCLC completed enrollment of 345 patients. Eligible patients had previously treated, locally advanced and unresectable or metastatic KRAS G12C-mutated NSCLC. The primary endpoint is progression-free survival and key secondary endpoints include overall survival, objective response rate, and patient-reported outcomes.11
Amgen also has several Phase 1b studies investigating sotorasib monotherapy and sotorasib combination therapy across various advanced solid tumors (CodeBreaK 101) open for enrollment.12 A Phase 2 randomized study will evaluate sotorasib in patients with stage IV KRAS G12C-mutated NSCLC in need of first-line treatment (CodeBreaK 201).13
For information, please visit www.hcp.codebreaktrials.com.
LUMAKRAS® (sotorasib) U.S. Indication
LUMAKRAS is indicated for the treatment of adult patients with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.
This indication is approved under accelerated approval based on overall response rate (ORR) and duration of response (DOR). Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
LUMAKRAS® (sotorasib) Important U.S. Safety Information
Please see LUMAKRAS full Prescribing Information.
About Amgen Oncology
At Amgen Oncology, our mission to serve patients drives all that we do. That's why we're relentlessly focused on accelerating the delivery of medicines that have the potential to empower all angles of care and transform lives of people with cancer.
For the last four decades, we have been dedicated to discovering the firsts that matter in oncology and to finding ways to reduce the burden of cancer. Building on our heritage, Amgen continues to advance the largest pipeline in the Company's history, moving with great speed to advance those innovations for the patients who need them.
For more information, follow us on www.twitter.com/amgenoncology.
About Amgen
Amgen is committed to unlocking the potential of biology for patients suffering from serious illnesses by discovering, developing, manufacturing and delivering innovative human therapeutics. This approach begins by using tools like advanced human genetics to unravel the complexities of disease and understand the fundamentals of human biology.
Amgen focuses on areas of high unmet medical need and leverages its expertise to strive for solutions that improve health outcomes and dramatically improve people's lives. A biotechnology pioneer since 1980, Amgen has grown to be one of the world's leading independent biotechnology companies, has reached millions of patients around the world and is developing a pipeline of medicines with breakaway potential.
Amgen is one of the 30 companies that comprise the Dow Jones Industrial Average and is also part of the Nasdaq-100 index. In 2021, Amgen was named one of the 25 World's Best Workplaces™ by Fortune and Great Place to Work™ and one of the 100 most sustainable companies in the world by Barron's.
For more information, visit www.amgen.com and follow us on www.twitter.com/amgen.
View original content to download multimedia:https://www.prnewswire.com/news-releases/amgen-announces-topline-data-from-lumakras-sotorasib-phase-3-trial-in-non-small-cell-lung-cancer-301615013.html
SOURCE Amgen
Aug. 30, 2022 4:18 PM ET
By: Dulan Lokuwithana, SA News Editor
Amgen (NASDAQ:AMGN) announced Tuesday that the company's KRAS G12C inhibitor Lumakras met the primary endpoint over the standard of care chemotherapy with statistical significance in a late-stage trial for non-small cell lung cancer (NSCLC).
https://seekingalpha.com/symbol/AMGN
PUBLISHED5 September 2022 5 September 2022 07:00 BST
AstraZeneca’s Imfinzi (durvalumab) has been approved in the US for the treatment of adult patients with locally advanced or metastatic biliary tract cancer (BTC) in combination with chemotherapy (gemcitabine plus cisplatin).
The approval by the Food and Drug Administration (FDA) was based on the results from the TOPAZ-1 Phase III trial. In an interim analysis of TOPAZ-1, Imfinzi plus chemotherapy reduced the risk of death by 20% versus chemotherapy alone (hazard ratio [HR] 0.80; 95% confidence interval [CI] 0.66-0.97; p=0.021). An estimated one in four (25%) patients treated with Imfinzi plus chemotherapy were still alive at two years compared to one in 10 (10%) treated with chemotherapy alone. Results were consistent across all prespecified subgroups, regardless of PD-L1 expression or tumour location.
BTC is a group of rare and aggressive cancers that occur in the bile ducts and gallbladder.1,2 Approximately 23,000 people in the US are diagnosed with BTC each year.1 These patients have a poor prognosis, with approximately 5% to 15% of patients with BTC surviving five years.3
Aiwu Ruth He, MD, PhD, Associate Professor of Medicine, Leader of the GI Cancer Program, Georgetown Lombardi Comprehensive Cancer Center, Medstar Georgetown University Hospital, Washington DC, and a lead investigator in the TOPAZ-1 Phase III trial, said: “This approval represents a major step forward for patients with advanced biliary tract cancer, who urgently need new, well-tolerated and effective treatment options after more than a decade of limited innovation. The combination of durvalumab and chemotherapy should become a new standard of care in this setting, having demonstrated significantly improved survival for these patients who have historically faced a poor prognosis.”
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “For the first time, patients in the US with advanced biliary tract cancer have an immunotherapy-based treatment option that meaningfully extends survival and is well-tolerated. This approval for Imfinzi and chemotherapy advances our ambition to challenge treatment expectations and transform care for patients with gastrointestinal cancers with high unmet need.”
Stacie Lindsey, CEO, Cholangiocarcinoma Foundation, said: “Patients have been waiting a long time for a new, first-line treatment option for biliary tract cancer. The Foundation congratulates AstraZeneca for engaging in rare cancer research, which impacts patients and families nationwide. We are especially grateful to the patients who participated in this trial making it possible for the broader rare disease community to benefit from this treatment.”
The TOPAZ-1 Phase III trial results were presented at the 2022 American Society of Clinical Oncology Gastrointestinal Cancers (ASCO GI) Symposium and published in the New England Journal of Medicine Evidence. Imfinzi plus chemotherapy was generally well tolerated and did not increase the discontinuation rate due to adverse events compared to chemotherapy alone.
In July 2022, Imfinzi plus chemotherapy was added to the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) as a Category 1 preferred regimen as 1st-line therapy for locally advanced or metastatic BTC based on the data from TOPAZ-1.4
The US regulatory submission for TOPAZ-1 was reviewed under Project Orbis, which provides a framework for concurrent submission and review of oncology medicines among participating international partners. As part of Project Orbis, Imfinzi plus chemotherapy is also under regulatory review for the same indication by the Australian Therapeutic Goods Administration, the Brazilian Health Regulatory Agency (ANVISA), Health Canada, Israel’s Ministry of Health Pharmaceutical Administration, Singapore’s Health Sciences Authority, Switzerland’s Swissmedic and the UK’s Medicines and Healthcare products Regulatory Agency.
The approval was granted after securing Priority Review and Orphan Drug Designation for Imfinzi in the US in this setting. Regulatory applications are also currently under review in Europe, Japan and several other countries based on the TOPAZ-1 results.
TOPAZ-1
TOPAZ-1 is a randomised, double-blind, placebo controlled, multicentre, global Phase III trial of Imfinzi in combination with chemotherapy (gemcitabine plus cisplatin) versus placebo in combination with chemotherapy as a 1st-line treatment in 685 patients with unresectable advanced or metastatic BTC including intrahepatic and extrahepatic cholangiocarcinoma, and gallbladder cancer. Patients with ampullary carcinoma were excluded.
The primary endpoint is overall survival and key secondary endpoints included progression-free survival, objective response rate and safety. The trial was conducted in 105 centres across 17 countries including in the US, Europe, South America and several countries in Asia including South Korea, Thailand, Japan and China.
Imfinzi
Imfinzi (durvalumab) is a human monoclonal antibody that binds to the PD-L1 protein and blocks the interaction of PD-L1 with the PD-1 and CD80 proteins, countering the tumour’s immune-evading tactics and releasing the inhibition of immune responses.
In addition to the approval in BTC, Imfinzi is the only approved immunotherapy in the curative-intent setting of unresectable, Stage III non-small cell lung cancer (NSCLC) in patients whose disease has not progressed after chemoradiotherapy and is the global standard of care in this setting based on the PACIFIC Phase III trial.
Imfinzi is also approved in the US, EU, Japan, China and many other countries around the world for the treatment of extensive-stage small cell lung cancer (ES-SCLC) based on the CASPIAN Phase III trial. In 2021, updated results from the CASPIAN trial showed Imfinzi plus chemotherapy tripled patient survival at three years versus chemotherapy alone.
Imfinzi is also approved for previously treated patients with advanced bladder cancer in several countries.
Since the first approval in May 2017, more than 100,000 patients have been treated with Imfinzi.
As part of a broad development programme, Imfinzi is being tested as a single treatment and in combinations with other anti-cancer treatments for patients with SCLC, NSCLC, bladder cancer, several GI cancers, ovarian cancer, endometrial cancer, and other solid tumours.
Imfinzi combinations have demonstrated clinical benefit in multiple additional cancer settings with positive Phase III trials in unresectable advanced liver cancer (HIMALAYA) and metastatic NSCLC (POSEIDON).
AstraZeneca in GI cancers
AstraZeneca has a broad development programme for the treatment of GI cancers across several medicines and a variety of tumour types and stages of disease. In 2020, GI cancers collectively represented approximately 5.1 million new cancer cases leading to approximately 3.6 million deaths.6
Within this programme, the Company is committed to improving outcomes in gastric, liver, BTC, oesophageal, pancreatic, and colorectal cancers.
Imfinzi is being assessed in combinations in liver, BTC, oesophageal and gastric cancers in an extensive development programme spanning early to late-stage disease.
The Company aims to understand the potential of Enhertu (trastuzumab deruxtecan), a HER2-directed antibody drug conjugate, in the two most common GI cancers, colorectal and gastric cancers. Enhertu is jointly developed and commercialised by AstraZeneca and Daiichi Sankyo.
Lynparza (olaparib) is a first-in-class PARP inhibitor with a broad and advanced clinical trial programme across multiple GI tumour types including pancreatic and colorectal cancers. Lynparza is developed and commercialised in collaboration with MSD (Merck & Co., Inc. inside the US and Canada).
AstraZeneca in immunotherapy
Immunotherapy is a therapeutic approach designed to stimulate the body’s immune system to attack tumours. The Company’s Immuno-Oncology (IO) portfolio is anchored in immunotherapies that have been designed to overcome evasion of the anti-tumour immune response. AstraZeneca is invested in using IO approaches that deliver long-term survival for new groups of patients across tumour types.
The Company is pursuing a comprehensive clinical-trial programme that includes Imfinzi as a single treatment and in combination with tremelimumab and other novel antibodies in multiple tumour types, stages of disease, and lines of treatment, and where relevant using the PD-L1 biomarker as a decision-making tool to define the best potential treatment path for a patient.
In addition, the ability to combine the IO portfolio with radiation, chemotherapy, and targeted small molecules from across AstraZeneca’s oncology pipeline, and from research partners, may provide new treatment options across a broad range of tumours.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Sep. 05, 2022 5:37 AM ET
By: Ravikash, SA News Editor
BackPDF VersionSep 2, 2022
-With this approval, approximately 300 children with two copies of the F508del mutation will have a medicine to treat the underlying cause of their disease for the first time-
BOSTON--(BUSINESS WIRE)--Sep. 2, 2022-- Vertex Pharmaceuticals Incorporated (Nasdaq: VRTX) today announced the U.S. Food and Drug Administration (FDA) approved expanded use of ORKAMBI® (lumacaftor/ivacaftor) to include children with cystic fibrosis (CF) ages 12 to <24 months who are homozygous for the F508del mutation (F/F genotype) in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. ORKAMBI® was previously approved by the FDA for use in people with CF ages 2 years and older with two copies of the F508del mutation.
“Treating children with cystic fibrosis as early in life as possible is critically important, because early treatment has the potential to slow the progression of this devastating disease,” said Carmen Bozic, M.D., Executive Vice President, Global Medicines Development and Medical Affairs, and Chief Medical Officer, Vertex. “Today’s approval is another important step on our journey to reach people of all ages living with cystic fibrosis who may benefit from our medicines.”
ORKAMBI® was first approved in 2015 in the U.S. and is now available in more than 30 countries. For more information on ORKAMBI®, including prescribing information or patient assistance programs, visit Orkambi.com or VertexGPS.com.
About ORKAMBI® (lumacaftor/ivacaftor)
In people with two copies of the F508del mutation, the CFTR protein is not processed and trafficked normally within the cell, resulting in little to no CFTR protein at the cell surface.
ORKAMBI® (lumacaftor/ivacaftor) is an oral medicine that is a combination of lumacaftor and ivacaftor. Lumacaftor is designed to increase the amount of mature protein at the cell surface by targeting the processing and trafficking defect of the F508del-CFTR protein. Ivacaftor, which is known as a CFTR potentiator, is designed to facilitate the ability of CFTR proteins to transport salt and water across the cell membrane. The combined actions of lumacaftor and ivacaftor help hydrate and clear mucus from the airways.
The approval in children ages 12 to <24 months is based on a 24-week, Phase 3, open-label, multi-center study in 46 children ages 1 to less than 2 years with the F/F genotype. ORKAMBI® was generally well tolerated, and the safety profile and pharmacokinetics were similar to that observed in studies in patients ages 2 years and older. Additional study results, including reductions in sweat chloride concentration, suggest the potential for CF disease modification with the use of ORKAMBI®.
Results from this study were recently published in the American Journal of Respiratory and Critical Care Medicine (AJRCCM).
INDICATION AND USAGE
ORKAMBI® (lumacaftor/ivacaftor) is a prescription medicine used for the treatment of cystic fibrosis (CF) in patients aged 1 year and older who have two copies of the F508del mutation (F508del/F508del) in their CFTR gene.
ORKAMBI should not be used in patients other than those who have two copies of the F508del mutation in their CFTR gene.
It is not known if ORKAMBI is safe and effective in children under 1 year of age.
Please click here to see the full Prescribing Information for ORKAMBI (lumacaftor/ivacaftor).
View source version on businesswire.com: https://www.businesswire.com/news/home/20220902005252/en/
Source: Vertex Pharmaceuticals Incorporated
Sep. 02, 2022 2:09 PM ET
Vertex Pharmaceuticals Incorporated (VRTX)
By: Dulan Lokuwithana, SA News Editor
Aug 30, 2022 PDF Version
NEW YORK and PETAH TIKVA, Israel, Aug. 30, 2022 (GLOBE NEWSWIRE) -- Y-mAbs Therapeutics, Inc. (the “Company” or “Y-mAbs”) (Nasdaq: YMAB) a commercial-stage biopharmaceutical company focused on the development and commercialization of novel, antibody-based therapeutic products for the treatment of cancer and Takeda Israel, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited (TSE:4502/NSY:TAK) ("Takeda"), announced today that the Israeli Ministry of Health has approved DANYELZA in Israel for the treatment, in combination with granulocyte-macrophage colony-stimulating factor (“GM-CSF”), of pediatric patients 1 year of age and older and adult patients with relapsed or refractory high-risk neuroblastoma in the bone or bone marrow who have demonstrated a partial response, minor response, or stable disease to prior therapy.
In Israel, DANYELZA is expected to be commercialized by Takeda Israel, under the exclusive license and distribution agreement entered in 2020 between Takeda Israel and the Company.
“The regulatory approval of DANYELZA in Israel represents our first marketing authorization outside of the U.S. and is a milestone for our collaboration with Takeda and more importantly for the pediatric patients we are dedicated to serving,” said Thomas Gad, President, and Interim Chief Executive Officer. “The approval in Israel further demonstrates our commitment to expanding the reach of our commercial stage products internationally to patients with unmet medical needs.”
“We are extremely excited by the approval of DANYELZA in Israel,” said Arie Kramer, General Manager of Takeda Israel. “This registration, following the expedited reimbursement of the product last December by the Israeli Ministry of Health, allowing Takeda and Y-mAbs to offer a new innovative treatment for pediatric neuroblastoma patients in Israel, is in alignment with Takeda’s vision of Better Health and Brighter Future to every patient in the world.”
Researchers at Memorial Sloan Kettering Cancer Center (“MSK”) developed DANYELZA, which is exclusively licensed by MSK to Y-mAbs. As a result of this licensing arrangement, MSK has institutional financial interests in the compound.
About DANYELZA (naxitamab-gqgk)
DANYELZA (naxitamab-gqgk) is indicated, in combination with granulocyte-macrophage colony-stimulating factor (“GM-CSF”), for the treatment of pediatric patients 1 year of age and older and adult patients with relapsed or refractory high-risk neuroblastoma in the bone or bone marrow who have demonstrated a partial response, minor response, or stable disease to prior therapy. This indication was approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefits in a confirmatory trial. DANYELZA includes a Boxed Warning for serious infusion-related reactions, such as cardiac arrest and anaphylaxis, and neurotoxicity, such as severe neuropathic pain and transverse myelitis. See full Prescribing Information for complete Boxed Warning and other important safety information.
About Y-mAbs
Y-mAbs is a commercial-stage biopharmaceutical company focused on the development and commercialization of novel, antibody-based therapeutic cancer products. In addition to conventional antibodies, the Company’s technologies include bispecific antibodies generated using the Y-BiClone platform and the SADA platform. The Company’s broad and advanced product pipeline includes one FDA-approved product, DANYELZA® (naxitamab-gqgk), which targets tumors that express GD2, and one product candidate at the registration-stage, omburtamab, which targets tumors that express B7-3.
About Takeda Pharmaceutical Company Limited
Takeda Pharmaceutical Company Limited (TSE:4502/NYSE:TAK) is a global, values-based, R&D-driven biopharmaceutical leader headquartered in Japan, committed to bringing Better Health and a Brighter Future to patients by translating science into highly-innovative medicines. Takeda focuses its R&D efforts on four therapeutic areas: Oncology, Rare Diseases, Neuroscience, and Gastroenterology (“GI”). We also make targeted R&D investments in Plasma-Derived Therapies and Vaccines. We are focusing on developing highly innovative medicines that contribute to making a difference in people's lives by advancing the frontier of new treatment options and leveraging our enhanced collaborative R&D engine and capabilities to create a robust, modality-diverse pipeline. Our employees are committed to improving quality of life for patients and to working with our partners in health care in approximately 80 countries.
For more information, visit https://www.takeda.com.
DANYELZA®, OMBLASTYS® and Y-mAbs® are registered trademarks of Y-mAbs Therapeutics, Inc.
Source: Y-mAbs Therapeutics, Inc
Aug. 30, 2022 9:46 AM ET
Takeda Pharmaceutical Company Limited (TAK), YMAB
By: Ravikash, SA News Editor
PUBLISHED27 August 2022
Today, new results from a pre-specified, patient level, pooled analysis from the Phase III DAPA-HF and DELIVER trials demonstrated mortality benefit of FARXIGA® (dapagliflozin), compared to placebo, in patients with heart failure (HF). These results were presented at the European Society of Cardiology Congress 2022 in Barcelona, Spain and simultaneously published in Nature Medicine.1 The reduction in risk of cardiovascular (CV) death was consistent across pre-specified subgroups and is the first analysis to demonstrate a mortality benefit with an HF medication in patients with HF across the left ventricular ejection fraction (LVEF) range.
The analysis showed that FARXIGA reduced the risk of CV death by 14% (p=0.01, absolute risk reduction [ARR] 1.5%) over the median follow-up of 22 months, death from any cause by 10% (p=0.03, ARR 1.5%), total (first and repeat) hospitalization for HF (hHF) by 29% (p < 0.001, ARR 6%), and the composite of death from CV causes, myocardial infarction, or stroke by 10% (p=0.045, ARR 1.3%), in patients with HF irrespective of LVEF.1
Prof. John McMurray, Professor of Medical Cardiology and Deputy Director of the Institute of Cardiovascular and Medical Sciences at the University of Glasgow, UK, said: “In this patient-level meta-analysis including over 11,000 patients with heart failure across the full range of ejection fraction, dapagliflozin reduced the risk of both cardiovascular death and heart failure hospitalization. These results underpin the valuable role dapagliflozin can play in clinical practice, as we can initiate treatment right away while waiting for ejection fraction to be measured.”
Mene Pangalos, Executive Vice President, BioPharmaceuticals R&D, AstraZeneca, said:
“Heart failure remains one of the leading causes of death worldwide with high unmet need for some 64 million people. This analysis demonstrates FARXIGA’s ability to treat patients across the full left ventricular ejection fraction spectrum and reduce the risk of cardiovascular death.”
The DAPA-HF and DELIVER Phase III trials were randomized and double-blind, comparing FARXIGA to placebo. Each trial enrolled patients with a diagnosis of HF, functional limitation, and elevated natriuretic peptides. The principal difference between the two trials was that patients with an LVEF of 40% or less were randomized in DAPA-HF and those with a LVEF greater than 40% in DELIVER.2,3 The studies included 11,007 individuals with HF across 20 countries in each trial.
INDICATIONS AND LIMITATIONS OF USE for FARXIGA® (dapagliflozin)
FARXIGA is indicated:
FARXIGA is not recommended for patients with type 1 diabetes mellitus. It may increase the risk of diabetic ketoacidosis in these patients.
FARXIGA is not recommended for use to improve glycemic control in adults with type 2 diabetes mellitus with an eGFR less than 45 mL/min/1.73 m2. FARXIGA is likely to be ineffective in this setting based upon its mechanism of action.
FARXIGA is not recommended for the treatment of chronic kidney disease in patients with polycystic kidney disease or patients requiring or with a recent history of immunosuppressive therapy for kidney disease. FARXIGA is not expected to be effective in these populations.
DOSING
To improve glycemic control, the recommended starting dose is 5 mg orally once daily. Dose can be increased to 10 mg orally once daily for additional glycemic control.
For all other indications, the recommended dose is 10 mg orally once daily.
DAPA-HF
DAPA-HF (Dapagliflozin And Prevention of Adverse-outcomes in Heart Failure) was an international, multi-centre, parallel-group, randomized, double-blinded Phase III trial in 4,744 patients with HFrEF, with and without type 2 diabetes (T2D), designed to evaluate the effect of FARXIGA 10mg, compared with placebo, given once daily in addition to standard of care (SoC) consisting of an angiotensin-converting enzyme inhibitor (ACEi) or an angiotensin receptor blocker (ARB). The primary composite endpoint was time to the first occurrence of a worsening HF event (hospitalization or equivalent event, i.e., an urgent HF visit), or CV death. The median duration of follow-up was 18.2 months.3
The secondary endpoint included the total number of hHF (including repeat admissions) and CV deaths, change from baseline to 8 months in the total symptom score on the Kansas City Cardiomyopathy Questionnaire (KCCQ).3
DELIVER
DELIVER was an international, randomized, double-blind, parallel-group, placebo-controlled, event-driven Phase III trial designed to evaluate the efficacy of FARXIGA, compared with placebo, in the treatment of HF patients with LVEF greater than 40%, with or without T2D. FARXIGA was given once daily in addition to background therapy (regional SoC for all comorbidities, including diabetes and hypertension, with the exception of concomitant use of a sodium-glucose cotransporter 2 (SGLT2) inhibitor).2 DELIVER is the largest clinical trial to date in HF patients with LVEF above 40%, with 6,263 randomized patients.2
The primary endpoint was the time to first occurrence of CV death, hHF or an urgent HF visit. The secondary endpoint includes the total number of HF events (hHF or urgent HF visit) and CV death, change from baseline in the total symptom score of the KCCQ at eight months, time to the occurrence of CV death and time to the occurrence of death from any cause.2
AstraZeneca in CVRM
Cardiovascular, Renal and Metabolism (CVRM), part of BioPharmaceuticals, forms one of AstraZeneca’s main disease areas and is a key growth driver for the Company. By following the science to understand more clearly the underlying links between the heart, kidneys and pancreas, AstraZeneca is investing in a portfolio of medicines for organ protection and improving outcomes by slowing disease progression, reducing risks and tackling co-morbidities. The Company’s ambition is to modify or halt the natural course of CVRM diseases and potentially regenerate organs and restore function, by continuing to deliver transformative science that improves treatment practices and CV health for millions of patients worldwide.
About AstraZeneca
AstraZeneca is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialization of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. For more information, please visit www.astrazeneca-us.com and follow us on Twitter @AstraZenecaUS.
Aug. 29, 2022 5:16 AM ET AstraZeneca PLC (AZN)
By: Dulan Lokuwithana, SA News Editor
Disclosing data from a Phase 3 trial, AstraZeneca (NASDAQ:AZN) announced that its blockbuster diabetes therapy Farxiga (dapagliflozin) improved clinical outcomes in certain patients with heart failure (HF). The data were presented at the European Society of Cardiology Congress 2022 in Spain.
https://seekingalpha.com/symbol/AZN
Aug 29, 2022
Basel, August 29, 2022 — Novartis today announced that the European Commission (EC) has approved Scemblix® (asciminib) for the treatment of adult patients with Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase (Ph+ CML-CP), previously treated with two or more tyrosine kinase inhibitors (TKIs)1. Scemblix is the first CML treatment in Europe that works by specifically targeting the ABL myristoyl pocket (also known as a STAMP inhibitor in scientific literature), offering a reimagined treatment approach for patients who experience intolerance and/or resistance to currently available TKI therapies1,2.
“Until now, patients with CML in Europe had oral TKI therapies with the same mechanism of action to turn to, and those experiencing significant side effects or resistance to these treatment options would often cycle between these very similar therapies, with little success in controlling their disease or improving their quality of life,” said Dr. Andreas Hochhaus, Head of the Department of Hematology and Medical Oncology at Jena University Hospital in Germany. “The approval of Scemblix in Europe is a timely milestone that will help many patients find hope for the management of their CML.”
The EC approval for Scemblix follows a positive opinion by the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) in June, and the previous designation of Scemblix as an orphan drug; and it is applicable to all 27 European Union member states plus Iceland, Norway and Liechtenstein. The approval is based on results from the pivotal Phase III ASCEMBL trial, which showed a near doubling in MMR rate for patients treated with Scemblix vs. Bosulif®* (bosutinib) (25.5% vs. 13.2%, [P=.029]), with a more than three times lower discontinuation rate due to adverse reactions (5.8% vs. 21.1%), at the 24-week primary endpoint1,2. These results were confirmed in the 96-week longer-term follow-up where the MMR rate was more than double with Scemblix (37.6%, 95% CI: 29.99-45.65) compared with Bosulif (15.8%, 95% CI: 8.43-25.96) and the discontinuation rate due to adverse reactions was 7.7% for Scemblix and 26.3% for Bosulif. These data were shared as oral presentations during the annual meetings of the American Society for Clinical Oncology (ASCO) and the European Hematology Association (EHA) in June 20223,4. Based on all patients exposed to Scemblix in the ASCEMBL study and in the phase I study, the most common (incidence ≥ 20%) adverse reactions in patients receiving asciminib were musculoskeletal pain (37.1%), upper respiratory tract infections (28.1%), thrombocytopenia (27.5%), fatigue (27.2%), headache (24.2%), arthralgia (21.6%), increased pancreatic enzymes (21.3%), abdominal pain (21.3%), diarrhoea (20.5%) and nausea (20.2%)1.
“Approval of Scemblix from the European Commission is a critical milestone to help bring this novel treatment to patients living with CML in Europe,” said Haseeb Ahmad, President, Europe Innovative Medicines, Novartis. “Building on more than twenty years of innovation in CML, we are excited by the potential to once again transform the standard of care for more patients around the world.”
It is estimated that, every year, more than 6,300 people will be diagnosed with CML in Europe5. While many patients will benefit from available TKI therapies, a significant proportion may experience intolerance or resistance to these treatments6-13.
About Scemblix® (asciminib)
Scemblix is the first CML treatment that acts as a STAMP inhibitor, specifically targeting the ABL myristoyl pocket1,2. This novel mechanism of action may help address resistance in patients with CML previously treated with two or more TKIs and overcome mutations at the defective BCR::ABL1 gene, which is associated with the over-production of leukemic cells1,2,14-20.
Scemblix represents an important development for patients who experience resistance and/or intolerance to currently available TKI therapies, and it is being studied across multiple treatment lines for CML-CP, both as a monotherapy and in combination1,14-28. Specifically, the ASC4FIRST Phase III study (NCT04971226) evaluates Scemblix in newly diagnosed adult patients with Ph+ CML-CP vs. an investigator-selected TKI22.
Novartis has initiated regulatory filings for Scemblix in multiple countries and regions across the globe. In October 2021, the US FDA granted accelerated approval of Scemblix for adult patients with Ph+ CML-CP, previously treated with two or more TKIs based on MMR rate at 24 weeks, and full approval for adult patients with Ph+ CML-CP with the T315I mutation. In accordance with the Accelerated Approval Program, continued approval for the first indication may be contingent upon verification and description of clinical benefit from confirmatory evidence. The longer-term, 96-week efficacy and safety data have been shared with the FDA and are currently under evaluation through a priority review29.
Scemblix has received approval in several countries outside the US, including Japan, Switzerland, and the United Kingdom, for adult patients with Ph+ CML-CP with resistance or intolerance to at least two or more previous therapies.
About Novartis
Novartis is reimagining medicine to improve and extend people’s lives. As a leading global medicines company, we use innovative science and digital technologies to create transformative treatments in areas of great medical need. In our quest to find new medicines, we consistently rank among the world’s top companies investing in research and development. Novartis products reach nearly 800 million people globally and we are finding innovative ways to expand access to our latest treatments. About 108,000 people of more than 140 nationalities work at Novartis around the world. Find out more at https://www.novartis.com.
Aug. 29, 2022 6:06 AM ET
By: Dulan Lokuwithana, SA News Editor
Novartis (NYSE:NVS) (OTCPK:NVSEF) announced Monday that the European Commission greenlighted Scemblix (asciminib) for certain adult patients with chronic myeloid leukemia who had previously received tyrosine kinase inhibitors (TKI) such as Pfizer’s (PFE) blood cancer drug Bosulif.
https://seekingalpha.com/news/3877325-novartis-announces-european-approval-of-leukemia-therapy
https://seekingalpha.com/symbol/NVS
August 26, 2022 at 10:00 AM EDTPDF Version
– This marks the second indication for Pemazyre, which received accelerated FDA approval in 2020 for adults with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with a fibroblast growth factor receptor 2 (FGFR2) fusion or other rearrangement
– Pemazyre is the only FGFR inhibitor with multiple indications
WILMINGTON, Del.--(BUSINESS WIRE)--Aug. 26, 2022-- Incyte (Nasdaq:INCY) today announced that the U.S. Food and Drug Administration (FDA) has approved Pemazyre® (pemigatinib), a selective fibroblast growth factor receptor (FGFR) inhibitor, for the treatment of adults with relapsed or refractory myeloid/lymphoid neoplasms (MLNs) with FGFR1 rearrangement. MLNs with FGFR1 rearrangement are extremely rare and aggressive blood cancers that may impact less than 1 in 100,000 people in the United States1.
“The approval of Pemazyre represents an important treatment advancement for people living with MLNs with FGFR1 rearrangement who currently have limited treatment options,” said Hervé Hoppenot, Chief Executive Officer, Incyte. “These are complex hematologic malignancies with a range of presentations, and this approval highlights Incyte’s continued leadership and commitment to advancing care for patients with rare blood cancers.”
A patient with an MLN with FGFR1 rearrangement may present with bone marrow involvement with a chronic myeloid malignancy (such as myeloproliferative neoplasm [MPN], myelodysplastic syndrome/MPN) or a blast phase malignancy (such as B- or T-cell acute lymphoblastic leukemia/lymphoma, acute myeloid leukemia or mixed phenotype acute leukemia). Bone marrow involvement may or may not be accompanied by extramedullary disease (EMD); some patients may present with EMD only. MLNs with FGFR1 rearrangement are caused by chromosomal translocations involving the FGFR1 gene, with various partner genes resulting in constitutive activation of the FGFR1 receptor tyrosine kinase, impacting cell differentiation, proliferation and survival2. Patients often relapse because existing first-line therapies sometimes fail to induce durable clinical and cytogenetic responses.
The FDA approval was based on data from the Phase 2 FIGHT-203 study, a multicenter open-label, single-arm trial that evaluated the safety and efficacy of Pemazyre in 28 patients with relapsed or refractory MLNs with FGFR1 rearrangement. Patients could have relapsed after allogeneic hematopoietic stem cell transplantation (allo-HSCT) or after a disease modifying therapy or were not a candidate for allo-HSCT or other disease modifying therapies.
The most common (≥ 20%) adverse reactions were hyperphosphatemia (74%), nail toxicity (62%), alopecia (59%), stomatitis (53%), diarrhea (50%), dry eye (50%), fatigue (44%), rash (35%), abdominal pain (35%), anemia (35%), constipation (32%), dry mouth (32%), epistaxis (29%), retinal pigment epithelial detachment (26%), extremity pain (26%), decreased appetite (24%), dry skin (24%), dyspepsia (24%), back pain (24%), nausea (21%), blurred vision (21%), peripheral edema (21%) and dizziness (21%).
“In patients with relapsed or refractory MLNs with FGFR1 rearrangement treated with Pemazyre in FIGHT-203, the high rate of complete response and complete cytogenetic response in patients with chronic phase disease and the high rate of complete cytogenetic response in patients with blast phase disease is clinically meaningful, especially in light of the lack of these specific responses with existing first-line treatments,” said Dr. Srdan Verstovsek, M.D., Ph.D., Professor, Department of Leukemia, Division of Cancer Medicine, The University of Texas MD Anderson Cancer Center, Houston, TX, and principal investigator for the FIGHT-203 study.
The supplemental New Drug Application (sNDA) for Pemazyre for the treatment of adults with relapsed or refractory MLNs with FGFR1 rearrangement was reviewed by the FDA under Priority Review. The FDA grants Priority Review to medicines that may offer a major advance in treatment where none currently exists. The designation shortens the review period to six months compared to 10 months for Standard Review.
Incyte established its leadership in rare blood cancers more than 10 years ago with the development of the first JAK inhibitor approved by the FDA for the treatment of certain patients with myelofibrosis and polycythemia vera. Incyte continues to research additional pathways to address rare blood cancers through its LIMBER (Leadership In MPNs Beyond Ruxolitinib) clinical development program, designed to evaluate multiple therapies and investigational strategies to improve and expand treatments for patients living with MPNs and other related hematologic malignancies and conditions.
Incyte is committed to supporting patients and removing barriers to access medicines. Eligible patients in the U.S. who are prescribed Pemazyre have access to IncyteCARES (Connecting to Access, Reimbursement, Education and Support), a comprehensive program offering personalized patient support, including financial assistance and ongoing education and additional resources. More information about IncyteCARES is available by visiting www.incytecares.com or calling 1-855-452-5234.
About FIGHT-203
FIGHT-203 is a Phase 2, multicenter trial that enrolled patients 18 years and older with myeloid/lymphoid neoplasms (MLNs) with a fibroblast growth factor receptor 1 (FGFR1) rearrangement. Sponsored by Incyte, the study evaluated the safety and efficacy of pemigatinib for the treatment of adults with MLNs with FGFR1 rearrangement. Patients received pemigatinib 13.5 mg once daily in 21-day cycles, either on a continuous schedule (the approved recommended starting dosage for use in patients with MLNs with FGFR1 rearrangement) or as an intermittent schedule (14 days on, 7 days off, an unapproved dosage regimen in MLN with FGFR1 rearrangement). Pemigatinib was administered until disease progression or unacceptable toxicity or until patients were able to receive allo-HSCT. For more information about the study, please visit https://clinicaltrials.gov/ct2/show/NCT03011372.
About Pemazyre® (pemigatinib)
Pemazyre, a fibroblast growth factor receptor (FGFR) inhibitor, is the first targeted treatment approved for use in the United States for treatment of adults with relapsed or refractory myeloid/lymphoid neoplasms (MLNs) with FGFR1 rearrangement.
Pemazyre is also indicated for the treatment of adults with relapsed or refractory previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with a FGFR2 fusion or other rearrangement as detected by an FDA-approved test. This indication is approved under accelerated approval based on overall response rate and duration of response (DOR). Continued approval may be contingent on verification and description of clinical benefit in a confirmatory trial(s).
Please see the Full Prescribing Information, including Patient Information, for PEMAZYRE.
About Incyte
Incyte is a Wilmington, Delaware-based, global biopharmaceutical company focused on finding solutions for serious unmet medical needs through the discovery, development and commercialization of proprietary therapeutics. For additional information on Incyte, please visit Incyte.com and follow @Incyte.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220826005244/en/
Source: Incyte
Aug. 26, 2022 11:13 AM ET
By: Jonathan Block, SA News Editor
PUBLISHED25 August 2022
AstraZeneca and MSD’s Lynparza (olaparib) has been approved in Japan for the adjuvant treatment of patients with BRCA-mutated (BRCAm), HER2-negative early breast cancer at high risk of recurrence.
This approval by the Japanese Ministry of Health, Labour, and Welfare was based on results from the OlympiA Phase III trial published in The New England Journal of Medicine in June 2021.1 In the trial, Lynparza demonstrated a statistically significant and clinically meaningful improvement in invasive disease-free survival (iDFS), reducing the risk of invasive breast cancer recurrences, new cancers, or death by 42% versus placebo (based on a hazard ratio [HR] of 0.58; 99.5% confidence interval [CI] 0.41-0.82; p<0.0001).
Lynparza also demonstrated a statistically significant and clinically meaningful improvement in overall survival, reducing the risk of death by 32% versus placebo (based on an HR of 0.68; 98.5% CI 0.47-0.97; p=0.009). The safety and tolerability profile of Lynparza in this trial was in line with that observed in prior clinical trials.
Breast cancer is the most diagnosed cancer worldwide with an estimated 2.3 million patients diagnosed in 2020.2 In Japan, an estimated 95,000 people will be diagnosed with breast cancer in 2022, and over 15,000 will die from the disease.3,4 Out of the approximately 80% of patients with HER2-negative breast cancer, about 11% have BRCA mutations.5,6
Professor Andrew Tutt, Global Chair of the OlympiA Phase III trial and Professor of Oncology at The Institute of Cancer Research, London, and King’s College London, said: “Today’s approval marks a new era of care for patients in Japan. For patients with high-risk early-stage breast cancer, including those with germline BRCA mutations, recurrence rates remain unacceptably high, with more than one in four of these patients seeing their cancer return following surgery and systemic treatment. Today’s approval offers eligible patients in Japan an effective and targeted treatment that improves survival and helps to prevent cancer recurrence.”
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “Today’s approval marks a significant leap forward for breast cancer patients in Japan, where it is the most commonly diagnosed cancer among women. Patients with BRCA mutations have high rates of disease recurrence and lower survival, and Lynparza has been shown to significantly reduce both the risk of recurrence and death. We hope this approval sets a new, much-needed standard of care for these early breast cancer patients in Japan.”
Dr Eliav Barr, Senior Vice President, Head of Global Clinical Development and Chief Medical Officer, MSD Research Laboratories, said: “With this approval, Lynparza becomes the first and only PARP inhibitor available for patients with BRCA-mutated HER2-negative early breast cancer in Japan. This further reinforces the critical need to conduct BRCA testing at the point of diagnosis so that all eligible patients can be identified.”
In March 2022, Lynparza was approved in the US for the treatment of germline BRCAm (gBRCAm), HER2-negative high-risk early breast cancer, followed by approval in the EU in August 2022 based on the results of the OlympiA Phase III trial. Lynparza is also approved in the US, EU, Japan, and many other countries for the treatment of patients with gBRCAm, HER2-negative, metastatic breast cancer previously treated with chemotherapy based on results from the OlympiAD Phase III trial. In the EU, this indication also includes patients with locally advanced breast cancer.
OlympiA
OlympiA is a Phase III, double-blind, parallel group, placebo-controlled, multicentre trial testing the efficacy and safety of Lynparza tablets versus placebo as adjuvant treatment in patients with gBRCAm, high-risk HER2-negative early breast cancer, who have completed definitive local treatment and neoadjuvant or adjuvant chemotherapy.15
The primary endpoint of the trial was iDFS defined as time from randomisation to date of first locoregional or distant recurrence or new cancer or death from any cause.1
The OlympiA Phase III trial is led by the Breast International Group in partnership with the Frontier Science & Technology Research Foundation, NRG Oncology, the US National Cancer Institute, AstraZeneca and MSD. The trial is sponsored by NRG Oncology in the US and by AstraZeneca outside the US.
BRCA
BRCA1 and BRCA2 are human genes that produce proteins responsible for repairing damaged DNA and play an important role maintaining the genetic stability of cells.11 When either of these genes is mutated or altered such that its protein product either is not made or does not function correctly, DNA damage may not be repaired properly, and cells become unstable. As a result, cells are more likely to develop additional alterations that can lead to cancer. Cancers with BRCA mutations are more likely to be sensitive to PARP inhibitors including Lynparza.16-19
Lynparza
Lynparza (olaparib) is a first-in-class PARP inhibitor and the first targeted treatment to block DNA damage response (DDR) in cells/tumours harbouring a deficiency in homologous recombination repair (HRR), such as those with mutations in BRCA1 and/or BRCA2, or those where deficiency is induced by other agents (such as new hormonal agents – NHAs).
Inhibition of PARP proteins with Lynparza leads to the trapping of PARP bound to DNA single-strand breaks, stalling of replication forks, their collapse and the generation of DNA double-strand breaks and cancer cell death.
Lynparza is currently approved in a number of countries across PARP-dependent tumour types with defects and dependencies in the DDR pathway including maintenance treatment of platinum-sensitive relapsed ovarian cancer and as both monotherapy and in combination with bevacizumab for the 1st-line maintenance treatment of BRCA-mutated (BRCAm) and homologous recombination repair deficient (HRD)-positive advanced ovarian cancer, respectively; for gBRCAm, HER2-negative metastatic breast cancer (in the EU and Japan this includes locally advanced breast cancer); for gBRCAm, HER2-negative high-risk early breast cancer (in the EU and US); for gBRCAm metastatic pancreatic cancer; and HRR gene-mutated metastatic castration-resistant prostate cancer (BRCAm only in the EU and Japan).
Lynparza, which is being jointly developed and commercialised by AstraZeneca and MSD, is the foundation of AstraZeneca's industry-leading portfolio of potential new medicines targeting DDR mechanisms in cancer cells.
The AstraZeneca and MSD strategic oncology collaboration
In July 2017, AstraZeneca and Merck & Co., Inc., Kenilworth, NJ, US, known as MSD outside the US and Canada, announced a global strategic oncology collaboration to co-develop and co-commercialise Lynparza, the world’s first PARP inhibitor, and Koselugo (selumetinib), a mitogen-activated protein kinase (MEK) inhibitor, for multiple cancer types.
Working together, the companies will develop Lynparza and Koselugo in combination with other potential new medicines and as monotherapies. The companies will develop Lynparza and Koselugo in combination with their respective PD-L1 and PD-1 medicines independently.
AstraZeneca in breast cancer
Driven by a growing understanding of breast cancer biology, AstraZeneca is starting to challenge and redefine the current clinical paradigm for how breast cancer is classified and treated, to deliver even more effective treatments to patients in need – with the bold ambition to one day eliminate breast cancer as a cause of death.
AstraZeneca has a comprehensive portfolio of approved and promising compounds in development that leverage different mechanisms of action to address the biologically diverse breast cancer tumour environment.
AstraZeneca aims to continue to transform outcomes for HR-positive breast cancer with foundational medicines Faslodex and Zoladex and the next-generation oral selective oestrogen receptor degrader (SERD) and potential new medicine camizestrant.
The PARP inhibitor, Lynparza, is an approved targeted treatment option for early and metastatic breast cancer patients with an inherited BRCA mutation. AstraZeneca with MSD continue to research Lynparza in breast cancer patients with an inherited BRCA mutation.
Building on the initial approvals of Enhertu, a HER2-directed antibody drug conjugate (ADC), in previously treated HER2-positive metastatic breast cancer, AstraZeneca and Daiichi Sankyo are exploring its potential in earlier lines of treatment and in new breast cancer settings. Enhertu has received approval in the US in previously treated HER2-low metastatic breast cancer.
To bring much needed treatment options to patients with triple-negative breast cancer, an aggressive form of breast cancer, AstraZeneca is testing immunotherapy Imfinzi in combination with other oncology medicines, including Lynparza and Enhertu, evaluating the potential of AKT kinase inhibitor, capivasertib, in combination with chemotherapy, and collaborating with Daiichi Sankyo to explore the potential of TROP2-directed ADC, datopotamab deruxtecan.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by
Aug. 25, 2022 4:23 AM ET
By: Ravikash, SA News Editor
August 26, 2022 at 8:19 AM EDTPDF Version
-- Late-Breaking Data Show VASCEPA/VAZKEPA Significantly Reduced STEMI by 40% and NSTEMI by 27% in Pre-Specified, Post Hoc Analyses --
-- Data Presented During Late-Breaking Science Session at European Society of Cardiology (ESC) Congress 2022 in Barcelona --
DUBLIN, Ireland and BRIDGEWATER, N.J., Aug. 26, 2022 (GLOBE NEWSWIRE) -- Amarin Corporation plc (NASDAQ:AMRN) today announced that new REDUCE-IT data show that VASCEPA/VAZKEPA (icosapent ethyl) significantly reduced ST-segment elevation myocardial infarction (STEMI), non-ST segment elevated myocardial infarction (NSTEMI), and other MI subtypes in patients with established cardiovascular disease (CVD) or diabetes with risk factors.
The REDUCE-IT data presented today show STEMI was significantly reduced by 40% following treatment with icosapent ethyl (IPE) compared to placebo. IPE also significantly reduced NSTEMI by 27%. These data were presented today during a Late-Breaking Science Session at the European Society of Cardiology (ESC) Congress 2022 in Barcelona, Spain.
“This analysis of REDUCE-IT clearly shows that IPE 4 g/day as an adjunct to statin therapy in high-risk patients with residual hypertriglyceridemia provides a large and significant reduction in heart attacks,” said Deepak L. Bhatt, M.D., M.P.H., Executive Director of Interventional Cardiovascular Programs at Brigham and Women’s Hospital, Professor of Medicine at Harvard Medical School, and principal investigator of the REDUCE-IT trial. “Importantly, these new data show a significant reduction in the most important type of heart attack known as STEMI, as well as other MI subtypes.”
The REDUCE-IT study randomized 8,179 adult statin-treated patients with elevated triglycerides and either established CV disease or diabetes plus risk factors to IPE or placebo; median follow-up was 4.9 years. IPE treatment reduced the primary composite endpoint (CV death, nonfatal myocardial infarction [MI], nonfatal stroke, coronary revascularization, or hospitalization for unstable angina) and key secondary composite endpoint (CV death, nonfatal MI, or nonfatal stroke) endpoints 25% and 26%, respectively. Prespecified and post hoc analyses examined MI subtypes, which were independently adjudicated by a blinded Clinical Endpoint Committee.
In time to first event analyses, MI was significantly reduced with IPE treatment (HR=0.69; 95% CI 0.58, 0.81; P<0.0001) with a number needed to treat (NNT) of 39. IPE significantly reduced STEMI (HR=0.60; 95% CI 0.44, 0.81; P=0.0008) and NSTEMI (HR=0.73; 95% CI 0.60, 0.89; P=0.001). There were clinically important and statistically significant reductions in MI subtypes, including MI leading to cardiac arrest (HR=0.49; 95% CI 0.28, 0.87; P=0.01) and resuscitated MI (HR=0.34; 95% CI 0.15, 0.76; P=0.006). IPE also significantly reduced the overall burden of total (first and subsequent) STEMI (rate ratio [RR]=0.59; 95% CI 0.43, 0.80; P=0.0006) and total NSTEMI (RR=0.72; 95% CI 0.58, 0.88; P=0.002) versus placebo.
All analyses highlighted above were funded by Amarin. Dr. Bhatt received research funding paid to Brigham and Women’s Hospital from Amarin for his role as the Chair of REDUCE-IT.
About Amarin
Amarin is an innovative pharmaceutical company leading a new paradigm in cardiovascular disease management. From our scientific research foundation to our focus on clinical trials, and now our commercial expansion, we are evolving and growing rapidly. Amarin has offices in Bridgewater, New Jersey in the United States, Dublin in Ireland, and Zug in Switzerland as well as commercial partners and suppliers around the world. We are committed to rethinking cardiovascular risk through the advancement of scientific understanding of the impact on society of significant residual risk that exists beyond traditional therapies, such as statins for cholesterol management.
About REDUCE-IT®
REDUCE-IT was a global cardiovascular outcomes study designed to evaluate the effect of VASCEPA in adult patients with LDL-C controlled to between 41-100 mg/dL (median baseline 75 mg/dL) by statin therapy and various cardiovascular risk factors including persistent elevated triglycerides between 135-499 mg/dL (median baseline 216 mg/dL) and either established cardiovascular disease (secondary prevention cohort) or diabetes mellitus and at least one other cardiovascular risk factor (primary prevention cohort).
REDUCE-IT, conducted over seven years and completed in 2018, followed 8,179 patients at over 400 clinical sites in 11 countries with the largest number of sites located within the United States. REDUCE-IT was conducted based on a special protocol assessment agreement with FDA. The design of the REDUCE-IT study was published in March 2017 in Clinical Cardiology.6 The primary results of REDUCE-IT were published in The New England Journal of Medicine in November 2018.7 The total events results of REDUCE-IT were published in the Journal of the American College of Cardiology in March 2019.8 These and other publications can be found in the R&D section on the company’s website at www.amarincorp.com.
About VASCEPA®/VAZKEPA® (icosapent ethyl) Capsules
VASCEPA capsules are the first prescription treatment approved by the U.S. Food and Drug Administration (FDA) comprised solely of the active ingredient, icosapent ethyl, a unique form of eicosapentaenoic acid. VASCEPA was launched in the United States in January 2020 as the first and only drug approved by the U.S. FDA for treatment of the studied high-risk patients with persistent cardiovascular risk after statin therapy. VASCEPA was initially launched in the United States in 2013 based on the drug’s initial FDA approved indication for use as an adjunct therapy to diet to reduce triglyceride levels in adult patients with severe (≥500 mg/dL) hypertriglyceridemia. Since launch, VASCEPA has been prescribed over 18 million times. VASCEPA is covered by most major medical insurance plans. In addition to the United States, icosapent ethyl is approved and sold in Canada, Lebanon, Germany and the United Arab Emirates. In Europe, in March 2021 marketing authorization was granted to icosapent ethyl in the European Union for the reduction of risk of cardiovascular events in patients at high cardiovascular risk, under the brand name VAZKEPA. In April 2021 marketing authorization for VAZKEPA (icosapent ethyl) was granted in Great Britain. The Great Britain Marketing Authorization for VAZKEPA applies to England, Scotland and Wales.
United States
Indications and Limitation of Use
VASCEPA is indicated:
The effect of VASCEPA on the risk for pancreatitis in patients with severe hypertriglyceridemia has not been determined.
FULL U.S. FDA-APPROVED VASCEPA PRESCRIBING INFORMATION CAN BE FOUND AT WWW.VASCEPA.COM.
Europe
For further information about the Summary of Product Characteristics (SmPC) for VAZKEPA® in Europe, please click here.
Globally, prescribing information varies; refer to the individual country product label for complete information.
AMARIN, REDUCE-IT, VASCEPA and VAZKEPA are trademarks of Amarin Pharmaceuticals Ireland Limited. VAZKEPA is a registered trademark in Europe and other countries and regions and is pending registration in the United States.
Aug. 26, 2022 8:19 AM ET
By: Dulan Lokuwithana, SA News Editor
Announcing new data from its REDUCE-IT study, Ireland-based biotech Amarin Corporation (NASDAQ:AMRN) said Friday that its fish-oil-derived pill Vascepa caused a significant 40% reduction in a severe type of heart attack.
https://seekingalpha.com/news/3876986-amrn-stock-on-watch-after-new-data-for-fish-oil-drug
https://seekingalpha.com/symbol/AMRN
August 26, 2022 PDF Version
ANN ARBOR, Mich., Aug. 26, 2022 (GLOBE NEWSWIRE) -- Esperion (NASDAQ: ESPR) today announced that bempedoic acid (NEXLETOL®) is now recommended as an important oral non-statin therapy for LDL-cholesterol (LDL-C) lowering in the management of atherosclerotic cardiovascular disease (ASCVD) by the American College of Cardiology (ACC) task force on expert consensus decision pathways (ECDP). The recommendation, an update to the 2017 ECDP, was released on August 25, 2022 and published online in the Journal of the American College of Cardiology.
“The recommendation of bempedoic acid for LDL-C lowering in individuals at high-risk for ASCVD reflects an important management strategy for providers and the millions of patients who cannot reach their LDL-C goals with a statin alone,” said JoAnne Foody, MD, FACC, FAHA, Chief Medical Officer of Esperion. “LDL-C is causative in development of atherosclerotic CV disease and residual CV risk remains high if LDL-C remains elevated despite statins. While studies show reducing LDL-C levels with lipid-lowering agents lowers incidence of ASCVD events, most patients still do not reach their LDL-C goal. There is urgent need for the use of non-statin lipid-lowering therapies in appropriate patients to reduce LDL-C. The ACC ECDP recommendation underscores the importance of LDL-C lowering via multiple therapeutic options including oral therapies like bempedoic acid and ezetimibe and provides important guidance for the management of patients not served by current statin therapies.”
“These guidelines will be helpful for providers and payers to understand the value of non-statin therapies in helping patients achieve LDL-C goals,” said BJ Swartz, Chief Strategy Officer of Esperion. “They provide clear guidance on how new and existing non-statin adjunctive therapies will contribute to LDL-C goal attainment and have the potential to reduce cardiovascular risk in their patient populations.”
The 2022 ACC Expert Consensus statement seeks to address current gaps in care for LDL-C lowering to reduce ASCVD risk and provides recommendations for clinicians and patients regarding the use of non-statin therapies in high-risk patients who have a minimal response to statins or are statin intolerant. The recommendation of bempedoic acid, the first oral, once-daily, non-statin LDL-C lowering medicine approved since 2002, is based on robust Phase III clinical trial evidence including evidence from the CLEAR Tranquility (Evaluation of the Efficacy and Safety of Bempedoic Acid [ETC-1002] as Add-on to Ezetimibe Therapy in Patients With Elevated LDL-C) and CLEAR Serenity (Evaluation of the Efficacy and Safety of Bempedoic Acid in Patients With Hyperlipidemia and Statin Intolerant) trials, which demonstrated that monotherapy with bempedoic acid 180 mg daily in patients with statin-associated muscle symptoms on no statin therapy reduced LDL-C levels by approximately 24.5% compared with placebo.
In addition to the ACC ECDP, the American Association of Clinical Endocrinologists (AACE) and the American College of Endocrinology (ACE) recommend bempedoic acid for treating LDL-C to goal in their Dyslipidemia and Prevention of Cardiovascular Disease Algorithm. The potential ability of bempedoic acid to reduce cardiovascular risk in patients with statin-intolerance is currently being evaluated in a global cardiovascular outcomes trial (CVOT), known as CLEAR Outcomes. The Company anticipates top-line results from the trial in the first quarter of 2023.
CLEAR Cardiovascular Outcomes Trial
The effect of bempedoic acid on cardiovascular morbidity and mortality has not been determined. Esperion initiated a global cardiovascular outcomes trial (CVOT) to assess the effects of bempedoic acid on the occurrence of major cardiovascular events in patients with, or at high risk for, cardiovascular disease (CVD) who are only able to tolerate less than the lowest approved daily starting dose of a statin. The CVOT — known as CLEAR Cardiovascular Outcomes Trial — is an event-driven, global, randomized, double-blind, placebo-controlled study that completed enrollment in August 2019 of over 14,000 patients with hypercholesterolemia and high CVD risk at over 1,200 sites in 32 countries.
INDICATION
NEXLETOL is indicated as an adjunct to diet and maximally tolerated statin therapy for the treatment of adults with heterozygous familial hypercholesterolemia or established atherosclerotic cardiovascular disease who require additional lowering of LDL-C. Limitations of Use: The effect of NEXLETOL on cardiovascular morbidity and mortality has not been determined.
https://www.nexlizet.com/nexletol
Please see full Prescribing Information here.
Esperion Therapeutics
Esperion works hard to make our medicines easy to get, easy to take and easy to have. We discover, develop, and commercialize innovative medicines and combinations to lower cholesterol, especially for patients whose needs are not being met by the status quo. Our entrepreneurial team of industry leaders is inclusive, passionate and resourceful. We are singularly focused on managing cholesterol so you can improve your health easily. Esperion commercializes NEXLETOL® (bempedoic acid) and NEXLIZET® (bempedoic acid and ezetimibe) Tablets and is the leader in the development of convenient oral, once-daily non-statin LDL-cholesterol lowering drugs for patients with high levels of bad cholesterol. For more information, please visit www.esperion.com and follow us on Twitter at www.twitter.com/EsperionInc.
Aug. 26, 2022 9:09 AM ET
Esperion Therapeutics, Inc. (ESPR)
By: Dania Nadeem, SA News Editor
U.S. FDA Approves IMBRUVICA® (ibrutinib) as First and Only BTKi Treatment for Pediatric Patients with Chronic Graft-Versus-Host DiseaseIMBRUVICA® is now the only BTKi with 12 FDA approvals across seven indications, including five hematologic cancers and cGVHD
August 24, 2022 (HORSHAM, PA) – The Janssen Pharmaceutical Companies of Johnson & Johnson announced today that the U.S. Food and Drug Administration (FDA) has approved IMBRUVICA® (ibrutinib) for the treatment of pediatric patients one year and older with chronic graft-versus-host disease (cGVHD) after failure of one or more lines of systemic therapy. This milestone marks the first pediatric indication for IMBRUVICA® and the introduction of a new oral suspension formulation for patients ages one to less than 12. IMBRUVICA® is now the first FDA-approved therapy for these younger patients who previously had no approved treatment options for this life-threatening disease.
Chronic graft-versus-host disease is a life-threatening complication that can occur after a stem cell or bone marrow transplant when newly transplanted donor cells attack the transplant recipient's body.[1] Symptoms may include skin rash, mouth sores, dry eyes, liver inflammation, development of scar tissue in the skin and joints, and damage to the lungs.[1] Among children who undergo allogeneic transplants, 52-65 percent will develop cGVHD.[2]
“Imagine going through a transplant and then being told you have a moderate to severe chronic disease that can sometimes also be life-threatening,” said Dr. Paul A. Carpenter, attending physician at Seattle Children's Hospital and a study principal investigator.† “If these children were between one and 12 and didn’t respond to steroid treatment, we didn’t have any rigorously studied treatment options — until now. The iMAGINE trial showed encouraging safety results and sustained response rates in children, and the new IMBRUVICA oral suspension formulation helps address challenges children may have with swallowing capsules or tablets.”
“It’s heartbreaking for parents to watch their child struggle with the debilitating effects of cGVHD, especially since there are so few treatment options,” said Susan Stewart, Executive Director of BMT InfoNet,^ a non-profit organization dedicated to providing patients and their loved ones with emotional support and high quality, easy-to-understand information about blood stem cell transplants. “The FDA approval of IMBRUVICA puts another weapon in their arsenal and has the potential to truly make a difference for those who are faced with this challenging disease.”
The new indication is based on results from the Phase 1/2 iMAGINE study, which showed an overall response rate (ORR) through week 25 of 60 percent (Confidence Interval [CI] 95 percent; 44-74) in patients median age 13 years (range, one to 19 years) (n=47) with relapsed/refractory (R/R) moderate to severe cGVHD. Safety was consistent with the established profile for IMBRUVICA®, with observed adverse events (AEs) in pediatric patients being consistent with those observed in adult patients with moderate to severe cGVHD. IMBRUVICA® was approved to treat adults with cGVHD after failure of one or more lines of systemic therapy in 2017. Because of its unique kinase profile (e.g., inhibiting both BTK and interleukin-2-inducible T-cell kinase [ITK]), IMBRUVICA® has the potential to provide a clinical benefit for cGVHD.[3]
“The pediatric cGVHD community is a prime example of an underserved patient population with high unmet medical needs for whom Janssen is committed to developing life-saving therapies,” said Craig Tendler, M.D., Global Head of Late Development, Diagnostics & Medical Affairs, Hematology & Oncology Janssen Research & Development, LLC. “cGVHD has life-threatening implications for children, and we are deeply proud of the opportunity to make an impact for these young patients with IMBRUVICA and their families.”
About the iMAGINE Study
iMAGINE (PCYC-1146-IM) is an open-label, multi-center, single-arm trial of IMBRUVICA® for the treatment of pediatric and young adult patients aged one year to less than 22 years with moderate or severe cGVHD as defined by NIH Consensus Criteria. Primary endpoints included pharmacokinetics (PK) and safety, and secondary endpoints included ORR (complete response [CR]/partial response [PR]) per 2014 NIH criteria, overall survival, and duration of response (DOR). The study included 47 patients who required additional therapy after failure of one or more prior lines of systemic therapy. Patients aged 12 years and older were treated with IMBRUVICA® 420 mg orally once daily, and patients aged one year to less than 12 years were treated with IMBRUVICA® 240 mg/m2 orally once daily. The efficacy of IMBRUVICA® was established based on ORR through Week 25.
About IMBRUVICA®
IMBRUVICA® (ibrutinib) is a once-daily oral medication that is jointly developed and commercialized by Janssen Biotech, Inc. and Pharmacyclics LLC, an AbbVie company. IMBRUVICA® blocks the Bruton's tyrosine kinase (BTK) protein, which is needed by normal and abnormal B cells, including specific cancer cells, to multiply and spread. By blocking BTK, IMBRUVICA® may help move abnormal B cells out of their nourishing environments and inhibits their proliferation.[4],[5],[6]
IMBRUVICA® is approved in more than 100 countries and has been used to treat more than 250,000 patients worldwide. There are more than 50 company-sponsored clinical trials, including 18 Phase 3 studies, more than 11 years evaluating the efficacy and safety of IMBRUVICA®.
IMBRUVICA® was first approved by the U.S. Food and Drug Administration (FDA) in November 2013, and today is indicated for adult patients in six disease areas, including five hematologic cancers. These include indications to treat adults with chronic lymphocytic leukemia (CLL)/ small lymphocytic lymphoma (SLL) with or without 17p deletion (del17p), and adults with Waldenström’s macroglobulinemia (WM), and adult patients with previously treated mantle cell lymphoma (MCL)*, as well as to treat adult patients with previously treated marginal zone lymphoma (MZL) who require systemic therapy and have received at least one prior anti-CD20-based therapy*, and adult and pediatric patients aged one year and older with previously treated chronic graft-versus-host disease (cGVHD) after failure of one or more lines of systemic therapy.[7]
*Accelerated approval was granted for MCL and MZL based on overall response rate. Continued approval for MCL and MZL may be contingent upon verification and description of clinical benefit in confirmatory trials.
Since 2019, the National Comprehensive Cancer Network® (NCCN®), recommends ibrutinib (IMBRUVICA®) as a preferred regimen for the initial treatment of CLL/SLL and has Category 1 treatment status for treatment-naïve patients without deletion 17p/TP53 mutation and as a preferred treatment for treatment-naïve patients with deletion 17p/TP53 mutation. The NCCN Guidelines® also recommend IMBRUVICA®, with or without rituximab, as a preferred regimen for the treatment of relapsed/refractory MCL, as a Category 1 preferred regimen for both untreated and previously treated WM patients, and as a preferred regimen for relapsed/refractory MZL.[8]
For more information, visit www.IMBRUVICA.com.
Please see full Prescribing Information.
About the Janssen Pharmaceutical Companies of Johnson & Johnson
At Janssen, we’re creating a future where disease is a thing of the past. We’re the Pharmaceutical Companies of Johnson & Johnson, working tirelessly to make that future a reality for patients everywhere by fighting sickness with science, improving access with ingenuity, and healing hopelessness with heart. We focus on areas of medicine where we can make the biggest difference: Cardiovascular, Metabolism, & Retina; Immunology; Infectious Diseases & Vaccines; Neuroscience; Oncology; and Pulmonary Hypertension.
Learn more at www.janssen.com. Follow us at www.twitter.com/JanssenGlobal and www.twitter.com/JanssenUS. Janssen Research and Development, LLC and Janssen Biotech, Inc. are part of the Janssen Pharmaceutical Companies of Johnson & Johnson.
Aug. 25, 2022 5:38 AM ET
By: Ravikash, SA News Editor
PUBLISHED25 August 2022
Ultomiris (ravulizumab) has been approved in Japan for the treatment of adult patients with generalised myasthenia gravis (gMG) who are anti-acetylcholine receptor (AChR) antibody-positive and whose symptoms are difficult to control with high-dose intravenous immunoglobulin therapy (IVIg) or plasmaphaeresis. Japan’s Pharmaceuticals and Medical Devices Agency also indicated that Ultomiris can be considered for patients who cannot receive high-dose IVIg or plasmaphaeresis due to complications, adverse reactions or other limiting factors.
The approval of the first and only long-acting C5 complement inhibitor by the Japanese Ministry of Health, Labour and Welfare (MHLW) was based on positive results from the CHAMPION-MG Phase III trial, which showed Ultomiris was superior to placebo in the primary endpoint of change from baseline in the Myasthenia Gravis-Activities of Daily Living Profile (MG-ADL) total score at Week 26, a patient-reported scale that assesses patients’ abilities to perform daily activities.1 Additionally, in prolonged follow-up results from the open-label extension, clinical benefit of Ultomiris was observed through 60 weeks.1
gMG is a rare, debilitating, chronic, autoimmune neuromuscular disease that leads to a loss of muscle function and severe weakness.2 The diagnosed prevalence of gMG in Japan is estimated at approximately 22,000.3
Hiroyuki Murai, MD, PhD, Professor and Chairman, Department of Neurology, School of Medicine, International University of Health and Welfare, Narita, Japan, said: “C5 inhibition is a proven approach to manage gMG. The approval of Ultomiris is an important advance for the gMG community in Japan, offering patients and physicians a new long-acting C5 inhibitor that has demonstrated sustained improvement in activities of daily living through 60 weeks, with fewer infusions per year over current treatment.”
Marc Dunoyer, Chief Executive Officer, Alexion, said: “We are pleased that Ultomiris is now approved in Japan for adults with gMG, a disease that may impact their ability to work, meet family obligations and live their lives fully. The approval speaks to the strength and consistency of Ultomiris clinical data as demonstrated in the global CHAMPION-MG Phase III trial. We look forward to bringing this treatment option to people living with gMG in Japan as part of our broader strategy to expand global access to our medicines.”
In CHAMPION-MG, the safety profile of Ultomiris was comparable to placebo and consistent with that observed in Phase III trials of Ultomiris in paroxysmal nocturnal haemoglobinuria (PNH) and atypical haemolytic uraemic syndrome (aHUS). The most common adverse reactions in patients receiving Ultomiris were nausea, headache and diarrhoea.1
Results from the CHAMPION-MG trial were published online in NEJM Evidence and presented at the 2022 American Academy of Neurology Annual Meeting in April.
Ultomiris was approved in the US for adults with anti-AChR antibody-positive gMG in April, and regulatory reviews are ongoing in additional countries. It was recently recommended for marketing authorisation in the European Union as an add-on to standard therapy for the treatment of adult patients with gMG who are anti-AChR antibody-positive.
CHAMPION-MG
The global Phase III randomised, double-blind, placebo-controlled, multicentre 26-week trial evaluated the safety and efficacy of Ultomiris in adults with gMG. The trial enrolled 175 patients across North America, Europe, Asia-Pacific and Japan. Participants were required to have a confirmed myasthenia gravis diagnosis at least six months prior to the screening visit with a positive serologic test for anti-AChR antibodies, MG-ADL total score of at least 6 at trial entry and Myasthenia Gravis Foundation of America Clinical Classification Class II to IV at screening. Patients could stay on stable standard of care medicines, with a few exceptions, for the duration of the randomised control period.13
Patients were randomised 1:1 to receive Ultomiris or placebo for a total of 26 weeks. Patients received a single weight-based loading dose on Day 1, followed by regular weight-based maintenance dosing beginning on Day 15, every eight weeks. The primary endpoint of change from baseline in the MG-ADL total score at Week 26 was assessed along with multiple secondary endpoints evaluating improvement in disease-related and quality-of-life measures.
Patients who completed the randomised control period were eligible to continue into an open-label extension period evaluating the safety and efficacy of Ultomiris, which is ongoing.
Ultomiris
Ultomiris (ravulizumab), the first and only long-acting C5 complement inhibitor, offers immediate, complete and sustained complement inhibition. The medication works by inhibiting the C5 protein in the terminal complement cascade, a part of the body’s immune system. When activated in an uncontrolled manner, the complement cascade over-responds, leading the body to attack its own healthy cells. Ultomiris is administered intravenously every eight weeks in adult patients, following a loading dose.
Ultomiris is approved in the US and Japan for the treatment of certain adults with gMG.
Ultomiris is also approved in the US, EU and Japan for the treatment of certain adults with PNH and for certain children with PNH in the US and EU.
Additionally, Ultomiris is approved in the US, EU and Japan for certain adults and children with aHUS to inhibit complement-mediated thrombotic microangiopathy.
As part of a broad development programme, Ultomiris is being assessed for the treatment of additional haematology and neurology indications.
https://alexion.com/en/our-medicines/medicines/ultomiris
Alexion
Alexion, AstraZeneca Rare Disease, is the group within AstraZeneca focused on rare diseases, created following the 2021 acquisition of Alexion Pharmaceuticals, Inc. As a leader in rare diseases for nearly 30 years, Alexion is focused on serving patients and families affected by rare diseases and devastating conditions through the discovery, development and commercialisation of life-changing medicines. Alexion focuses its research efforts on novel molecules and targets in the complement cascade and its development efforts on haematology, nephrology, neurology, metabolic disorders, cardiology and ophthalmology. Headquartered in Boston, Massachusetts, Alexion has offices around the globe and serves patients in more than 50 countries.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Aug. 25, 2022 5:55 AM ET
By: Ravikash, SA News Editor2 Comments
PUBLISHED25 August 2022
Tagrisso is the only EGFR-targeted medicine approved in Japan for the
treatment of early-stage lung cancer after surgery
Approval based on results from the ADAURA Phase III trial
AstraZeneca’s Tagrisso (osimertinib) has been approved in Japan for the adjuvant treatment of patients with epidermal growth factor receptor-mutated (EGFRm) non-small cell lung cancer (NSCLC) after surgery. This approval by the Japanese Ministry of Health, Labour and Welfare was based on positive results from the global ADAURA Phase III trial.
While up to 30% of all patients with NSCLC may be diagnosed early enough to have surgery with curative intent, recurrence is still common in early-stage disease.1,2 Historically, over half of patients diagnosed in Stage II, and approximately three quarters of patients diagnosed in Stage III, have experienced recurrence within five years of resection.3,4 In Japan, lung cancer is the leading cause of cancer death and among patients with NSCLC, more than 35% have tumours with an EGFR mutation.5,6
Masahiro Tsuboi, MD, PhD, Chief and Director, Department of Thoracic Surgery & Oncology, National Cancer Center Hospital East, Japan, and principal investigator in the ADAURA trial, said: “Osimertinib was first approved in Japan over six years ago and it has since played a critical role in our treatment of patients with lung cancer, particularly given the high prevalence of EGFR mutations among Japanese patients. This approval of osimertinib for early-stage lung cancer means these patients will now have, for the first time, a targeted therapy option available earlier in their treatment journey, after surgery and chemotherapy as indicated.”
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “Patients diagnosed with lung cancer in Japan are more likely than patients anywhere else in the world to be alive five years after their diagnosis. Yet lung cancer remains the country’s leading cause of cancer death. With this approval of Tagrisso, early-stage lung cancer patients in Japan now have a targeted treatment option available after surgery that can dramatically change the course of their disease, potentially helping them live cancer-free even longer.”
In the ADAURA trial, Tagrisso demonstrated a statistically significant and clinically meaningful improvement in disease-free survival (DFS) in the primary analysis population of patients with Stage II and IIIA EGFRm NSCLC. The trial also showed a statistically significant and clinically meaningful improvement in DFS in the overall trial population of patients with Stage IB-IIIA disease, a key secondary endpoint. These results were published in The New England Journal of Medicine in October 2020.
The ADAURA trial is ongoing to assess overall survival (OS). Final DFS results will be presented at the upcoming European Society for Medical Oncology (ESMO) Congress 2022.
Tagrisso is approved to treat early-stage lung cancer in more than 85 countries, including in the US, EU and China, and additional global regulatory reviews are ongoing. In Japan, this is the third approved indication for Tagrisso following previous approvals for 2nd-line T790M and 1st-line EGFRm NSCLC in March 2016 and August 2018, respectively.
ADAURA
ADAURA is a randomised, double-blind, placebo-controlled, global Phase III trial in the adjuvant treatment of 682 patients with Stage IB, II, IIIA EGFRm NSCLC following complete tumour resection and, at physicians’ and patients’ discretion, adjuvant chemotherapy. Patients were treated with Tagrisso 80mg once-daily oral tablets or placebo for three years or until disease recurrence.
The trial enrolled in more than 200 centres across more than 20 countries, including the US, in Europe, South America, Asia and the Middle East. The primary endpoint was DFS in Stage II and IIIA patients and a key secondary endpoint was DFS in Stage IB, II and IIIA patients.
Though the primary data readout was originally anticipated in 2022, data from the trial were reported early following a recommendation from an Independent Data Monitoring Committee (IDMC) based on its determination of overwhelming efficacy. The trial is ongoing and will continue to assess the secondary endpoint of OS.
Tagrisso
Tagrisso (osimertinib) is a third-generation, irreversible EGFR-TKI with proven clinical activity in NSCLC, including against central nervous system metastases. Tagrisso (40mg and 80mg once-daily oral tablets) has been used to treat more than 600,000 patients across its indications worldwide and AstraZeneca continues to explore Tagrisso as a treatment for patients across multiple stages of EGFRm NSCLC.
In Phase III trials, Tagrisso is being tested in the neoadjuvant resectable setting (NeoADAURA), in the Stage IA2-IA3 adjuvant resectable setting (ADAURA2), in the Stage III locally advanced unresectable setting (LAURA), and in combination with chemotherapy (FLAURA2). AstraZeneca is also researching ways to address tumour mechanisms of resistance through the SAVANNAH and ORCHARD Phase II trials, and the SAFFRON Phase III trial, which test Tagrisso given concomitantly with savolitinib, an oral, potent and highly selective MET TKI, as well as other potential new medicines.
AstraZeneca in lung cancer
AstraZeneca is working to bring patients with lung cancer closer to cure through the detection and treatment of early-stage disease, while also pushing the boundaries of science to improve outcomes in the resistant and advanced settings. By defining new therapeutic targets and investigating innovative approaches, the Company aims to match medicines to the patients who can benefit most.
The Company’s comprehensive portfolio includes leading lung cancer medicines and the next wave of innovations, including Tagrisso (osimertinib) and Iressa (gefitinib); Imfinzi (durvalumab) and tremelimumab; Enhertu (trastuzumab deruxtecan) and datopotamab deruxtecan in collaboration with Daiichi Sankyo; Orpathys (savolitinib) in collaboration with HUTCHMED; as well as a pipeline of potential new medicines and combinations across diverse mechanisms of action.
AstraZeneca is a founding member of the Lung Ambition Alliance, a global coalition working to accelerate innovation and deliver meaningful improvements for people with lung cancer, including and beyond treatment.
AstraZeneca in onclogy
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company’s focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Aug. 25, 2022 4:35 AM ET AstraZeneca PLC (AZN)
By: Ravikash, SA News Editor
Aug. 23, 2022 8:30 AM ET
Bristol-Myers Squibb Company (BMY)
First and-only immunotherapy-based treatment for use before surgery for non-small cell lung cancer in Canada
Significant improvement in event-free survival and pathological complete response
MONTREAL, Aug. 23, 2022 /CNW/ - Today, Bristol Myers Squibb Canada (BMS) announced Health Canada's approval of OPDIVO® (nivolumab) 360 mg (injection for intravenous use) in combination with platinum-doublet chemotherapy every three weeks for three cycles for adult patients with resectable (tumors ≥4 cm or node positive) non-small cell lung cancer (NSCLC) in the neoadjuvant setting.i

"The approval of OPDIVO® plus platinum-doublet chemotherapy in the neoadjuvant setting represents a pivotal transformation in the way we can help patients with resectable non-small cell lung cancer," said Dr Jonathan Spicer, Associate Professor of Surgery, Department of Surgery (Thoracic and Upper GI Surgery), McGill University. "As thoracic surgeons, despite our best efforts to completely remove lung cancers, we see our patients experience recurrence of their disease all too often. My goal is to cure cancer and this new pre-operative treatment combination is now shown to prolong survival and increase the number of patients who have no signs of cancer in their tissues after surgery."
About Lung Cancer & Survival Rates
Lung cancer is the leading cause of cancer death for both males and females in Canada, accounting for over 25 percent of cancer deaths.iii Non–small cell lung cancer (NSCLC) is the most common type of lung cancer making up more than 85 percent of all cases. iii
"With the high rates of disease recurrence in patients with resectable NSCLC, this unique pre-operative treatment approach is very welcome news to our patient population," said Shem Singh, Executive Director, Lung Cancer Canada. "This is an important step towards improving the lives of thousands of Canadians living with lung cancer today and in the future.
Clinical Data for Approval
The Health Canada NOC was based on CheckMate-816, which is a Phase 3, randomized, open label trial evaluating OPDIVO® plus platinum-doublet chemotherapy compared to chemotherapy alone as neoadjuvant treatment in adult patients with resectable non-small cell lung cancer (NSCLC), regardless of PD-L1 expression. Published in the New England Journal of Medicine (NEJM), this study demonstrated that in patients with resectable NSCLC, treatment with neoadjuvant OPDIVO® plus chemotherapy resulted in significantly longer event-free survival (EFS) and a higher percentage of patients with a pathological complete response (pCR) than chemotherapy alone.iv
"At BMS, we are driven to understand the complexities of cancer and are leveraging the latest science and technology to develop immunotherapies and combination therapies that push beyond what is currently available. Today's approval is a demonstration to our multifaceted research approach," said Troy André, General Manager, BMS Canada. "We are committed to offering patients the possibility of a better life through treatment options that can make a difference earlier in the course of their disease."
About Bristol Myers Squibb Canada Co.
Bristol Myers Squibb Canada Co. is an indirect wholly-owned subsidiary of Bristol Myers Squibb Company, a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. Bristol Myers Squibb Canada Co. employs close to 300 people across the country. For more information, please visit https://www.bms.com/ca/en.
About Bristol Myers Squibb
Bristol Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information about Bristol Myers Squibb, visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
SOURCE Bristol Myers Squibb
https://seekingalpha.com/symbol/BMY
August 22, 2022
– European Commission Grants Marketing Authorization for Sunlenca, Helping to Address a Critical Unmet Clinical Need for People with Multi-Drug-Resistant HIV Who Have Very Limited Treatment Choices –
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the European Commission (EC) has granted Marketing Authorization for Sunlenca® (lenacapavir) injection and tablets for the treatment of HIV infection, in combination with other antiretroviral(s), in adults with multi-drug resistant HIV infection for whom it is otherwise not possible to construct a suppressive anti-viral regimen. Lenacapavir is a first-in-class capsid inhibitor with a multi-stage mechanism of action and has no known cross resistance to other existing drug classes, offering a new, every six-month treatment option for people with HIV whose virus no longer effectively responds to their current therapy.
“Lenacapavir helps to fill a critical unmet need for people with complex prior treatment histories and offers physicians a long-awaited twice-yearly option for these patients who are at greater risk of progressing to AIDS,” said Jean-Michel Molina, MD, Université Paris Cité, Professor of Infectious Diseases and Head of the Infectious Diseases Department at the Saint-Louis and Lariboisière Hospitals. “In the CAPELLA study, lenacapavir, in combination with other antiretroviral therapies, demonstrated sustained rates of virologic suppression and clinically meaningful CD4+ T-cell recovery in people with multi-drug resistant HIV. Lenacapavir provides an innovative long-acting HIV therapy option with the potential to transform the clinical landscape.”
The Marketing Authorization Application (MAA) for lenacapavir is supported by data from the Phase 2/3 CAPELLA study, which evaluated lenacapavir in combination with an optimized background regimen in people with multi-drug resistant HIV who are heavily treatment-experienced. In this patient population with significant unmet medical need, 83% (n=30/36) of participants receiving lenacapavir in addition to an optimized background regimen achieved an undetectable viral load (<50 copies/mL) at Week 52. Additionally, CAPELLA participants achieved a mean increase in CD4 count of 83 cells/µL.
“After more than three decades of driving advancements in HIV treatment and prevention, Gilead scientists have now delivered an innovative new option for long-acting care,” said Daniel O’Day, Chairman and Chief Executive Officer, Gilead Sciences. “Lenacapavir is a unique and potent medicine with the potential for flexible dosing options. Following today’s approval, it will now be the only twice-yearly treatment for people who struggle with multi-drug resistant HIV. Our goal is to deliver multiple long-acting options in the future, in the belief that this will make a fundamental difference in the journey to end the HIV epidemic.”
Despite the significant advances in ARV therapy, there remain numerous critical and pressing unmet needs for people with HIV. This is particularly true for people with HIV who are heavily treatment-experienced with limited therapy options and are unable to maintain virologic suppression due to resistance or challenges adhering to a complex regimen. This type of complexity further increases the chance of suboptimal adherence and treatment failure, underscoring the need for a new treatment option that is active against resistant variants of the virus with a novel mechanism of action.
Sunlenca is indicated in the European Union for the treatment of adults with multi-drug resistant HIV infection in combination with other antiretroviral(s), for whom it is otherwise not possible to construct a suppressive anti-viral regimen. Lenacapavir tablets are approved for oral loading prior to administration of long-acting lenacapavir injection. The marketing authorization applies to all 27 member states of the European Union, as well as Norway, Iceland and Liechtenstein.
The European Marketing Authorization is the latest milestone in the review of lenacapavir by a major regulatory authority. In July, the U.S. Food & Drug Administration (FDA) accepted for review the New Drug Application (NDA) resubmission for investigational lenacapavir. The FDA has assigned a Prescription Drug User Fee Act (PDUFA) action date of December 27, 2022. Additional regulatory filings and decisions by regulatory authorities are anticipated to continue throughout 2022.
For important safety information for Sunlenca, including dosing and method of administration, special warnings, drug interactions and adverse drug reactions, please see the Summary of Product Characteristics (SmPC) for Sunlenca, available from the European Medicines Agency website at www.ema.europa.eu.
There is currently no cure for HIV or AIDS.
About Sunlenca®
Sunlenca is a first-in-class, long-acting HIV capsid inhibitor approved in the European Union for the treatment of HIV infection, in combination with other antiretroviral(s), in people with multi-drug resistant HIV who are heavily-treatment experienced. Sunlenca’s multi-stage mechanism of action is distinguishable from other currently approved classes of antiviral agents and is designed to provide a new avenue for the development of a long-acting treatment option for individuals with multi-drug resistant HIV whose virus was no longer effectively responding to therapy. While most antivirals act on just one stage of viral replication, Sunlenca is designed to inhibit HIV at multiple stages of its lifecycle and has no known cross resistance to other existing drug classes. Sunlenca is the only HIV treatment option administered twice-yearly.
About CAPELLA (NCT04150068)
CAPELLA is a Phase 2/3, double-blinded, placebo-controlled global multicenter study designed to evaluate the antiviral activity of lenacapavir administered every six months as a subcutaneous injection in heavily treatment-experienced people with multi-drug resistant HIV-1 infection. CAPELLA includes men and women with HIV-1 and is being conducted at research centers in North America, Europe and Asia.
In CAPELLA, 36 participants with multi-class HIV-1 drug resistance and a detectable viral load while on a failing regimen were randomly allocated to receive oral lenacapavir or placebo in a 2:1 ratio for 14 days, in addition to continuing their failing regimen (functional monotherapy). An additional 36 participants were enrolled in a separate treatment cohort. Both cohorts are part of the ongoing maintenance period of the study evaluating the safety and efficacy of subcutaneous lenacapavir administered every six months in combination with an optimized background regimen. The primary endpoint was the proportion of participants randomly allocated to receive lenacapavir or placebo for 14 days, in addition to continuing their failing regimen, achieving ≥0.5 log10 copies/mL reduction from baseline in HIV-1 RNA at the end of the functional monotherapy period.
Following the 14-day functional monotherapy period, participants randomly allocated to receive lenacapavir or placebo, in addition to continuing their failing regimen, started open-label lenacapavir and an optimized background regimen, while those enrolled in a separate treatment cohort received open-label lenacapavir and an optimized background regimen on Day 1. This ongoing maintenance period is evaluating the additional study endpoints of safety and efficacy of subcutaneous lenacapavir administered every six months in combination with an optimized background regimen.
The New England Journal of Medicine published the primary outcome results of the CAPELLA study in its May 11, 2022 issue - Capsid Inhibition with Lenacapavir in Multidrug-Resistant HIV-1 Infection.
For further information, please see https://clinicaltrials.gov/ct2/show/NCT04150068.
About Gilead Sciences
Gilead Sciences, Inc. is a biopharmaceutical company that has pursued and achieved breakthroughs in medicine for more than three decades, with the goal of creating a healthier world for all people. The company is committed to advancing innovative medicines to prevent and treat life-threatening diseases, including HIV, viral hepatitis and cancer.
For 35 years, Gilead has been a leading innovator in the field of HIV, driving advances in treatment, prevention and cure research. Gilead researchers have developed 12 HIV medications, including the first single-tablet regimen to treat HIV, the first antiretroviral for pre-exposure prophylaxis (PrEP) to reduce the risk of acquiring HIV infection, and the first, long-acting injectable HIV treatment medication administered twice-yearly. Our advances in medical research have helped to transform HIV into a preventable, chronic condition for millions of people.
Gilead is committed to continued scientific innovation to provide solutions for the evolving needs of people affected by HIV around the world. Through partnerships and collaborations, the company also aims to improve education, expand access and address barriers to care, with the goal of ending the HIV epidemic for everyone, everywhere. Gilead was recognized as the number one philanthropic funder of HIV-related programs in a report released by Funders Concerned About AIDS.
Gilead operates in more than 35 countries worldwide, with headquarters in Foster City, California.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220819005367/en/
Source: Gilead Sciences, Inc.
Aug. 22, 2022 5:12 AM ET Gilead Sciences, Inc. (GILD)
By: Ravikash, SA News Editor
August 22, 2022 Vaccines
- Indonesia National Agency for Drug and Food Control, BPOM, Approved QDENGA (TAK-003) for Use in Individuals Six Years to 45 Years of Age1
- QDENGA is the Only Dengue Vaccine Approved in Indonesia for Use in Individuals Without Need for Pre-vaccination Testing
- Indonesia Approval Marks the First for QDENGA, Takeda’s First Marketed Vaccine Outside of Japan
OSAKA, Japan, and CAMBRIDGE, Massachusetts, August 22, 2022 – Takeda (TSE:4502/NYSE:TAK) today announced the company’s dengue vaccine, QDENGA® (Dengue Tetravalent Vaccine [Live, Attenuated]) (TAK-003), was approved by the Indonesia National Agency for Drug and Food Control, Badan Pengawas Obat dan Makanan (BPOM), for the prevention of dengue disease caused by any serotype in individuals six years to 45 years of age. The use of QDENGA should be in accordance with official recommendations. QDENGA is the only dengue vaccine approved in Indonesia for use in individuals regardless of previous dengue exposure and without the need for pre-vaccination testing.1
“Dengue can affect anyone living in or traveling to endemic areas - regardless of age, health and socio-economic circumstances,” said Gary Dubin, president of Takeda’s Vaccine Business Unit. “Developing this innovative dengue vaccine has been an exciting challenge, and its approval in Indonesia is an important achievement for Takeda and for public health. We’re proud to introduce QDENGA as a new dengue prevention tool to the people of Indonesia, and we will continue to work with additional regulatory agencies to make QDENGA available globally.”
Dengue is a mosquito-borne viral disease that poses a significant global public health threat, with prevalence in over 125 countries.2 In recent years, Indonesia has experienced almost half of the dengue disease burden within Southeast Asia and continues to suffer from one of the highest burdens of dengue in the world.2,3 In the first half of 2022 alone, Indonesia reported over 63,000 dengue cases and nearly 600 deaths spread across 455 cities in 34 provinces.5
“As a doctor, I have seen firsthand the burden dengue disease places on the patients and communities I serve in Indonesia. There is an ongoing fear of an outbreak and contracting the disease, experiencing the physical setbacks dengue can cause as well as the potential financial impacts,” said Dr. Anggraini Alam, Sp.A(K), pediatric infectious disease consultant. “Vaccination will offer health care providers in Indonesia a welcomed advancement in dengue prevention, along with vector control, allowing us to reduce the burden of dengue and protect the broader population.”
The approval of QDENGA is based on results through three years after vaccination from the ongoing Phase 3
Tetravalent Immunization against Dengue Efficacy Study (TIDES) trial that enrolled over 20,000 healthy children and adolescents ages four to 16 years living in dengue-endemic areas in Asia and Latin America. QDENGA demonstrated continued overall protection against dengue illness and hospitalization three years after vaccination, regardless of an individual’s previous dengue exposure.1 QDENGA has been generally well tolerated, with no important safety risks identified in the TIDES trial, to date.6 Takeda recently presented long-term safety and efficacy results from the TIDES trial through 54 months of follow-up, which further validated the vaccine’s efficacy and safety profile.
Takeda is proud to make QDENGA available to health care providers and their eligible patients in Indonesia and to work with BPOM and local health experts to make the vaccine accessible in the coming months. QDENGA is currently undergoing regulatory review for the prevention of dengue in children and adults in the European Union (EU) and in dengue-endemic countries outside the EU through the EU-M4all (previously Article 58) procedure. It is not approved for use in other countries.
QDENGA® (TAK-003) is a dengue vaccine that is based on a live-attenuated dengue serotype 2 virus, which provides the genetic “backbone” for all four dengue virus serotypes and is designed to protect against any of these serotypes.7
In Indonesia, QDENGA is indicated for the prevention of dengue disease caused by any dengue virus serotype in individuals six years to 45 years of age and should be administered subcutaneously as a 0.5 mL dose at a two-dose (0 and 3 months) schedule pursuant to approved dosing regimen.1 The use of QDENGA should be in accordance with official recommendations.
QDENGA was assessed across a robust clinical development program that included various Phase 1, Phase 2 and Phase 3 trials, and more than 28,000 participants, including Takeda’s pivotal Tetravalent Immunization against Dengue Efficacy Study (TIDES) trial. The TIDES trial met its primary endpoint of overall vaccine efficacy (VE) against virologically-confirmed dengue (VCD) with 80.2% efficacy at 12-months follow-up. The trial also met all secondary endpoints for which there were a sufficient number of dengue cases at 18-months follow-up.8,9 The VE result in preventing hospitalization due to VCD fever was 90.4%. Up to three years after the second dose, VE in preventing VCD was shown for all four serotypes in baseline dengue seropositive subjects. In baseline seronegative subjects, VE was shown for DENV-1 and DENV-2, but not shown for DENV-3 and could not be shown for DENV-4 due to lower incidence of cases.1 Through four and a half years (54 months after the second dose), QDENGA demonstrated continued overall protection, with sustained overall VE of 61.2% and 84.1% VE against hospitalized dengue.6
QDENGA has been generally well tolerated, with no evidence of disease enhancement in vaccine recipients, and no important safety risks have been identified in the TIDES trial, to date.6
Please consult the Summary of Product Characteristics (SmPC) before prescribing.
Guidance for use: QDENGA should be administered by subcutaneous injection preferably in the upper arm in the region of deltoid. QDENGA must not be injected intravascularly, intradermally or intramuscularly. Vaccination should be postponed in subjects suffering from an acute severe febrile illness. The presence of a minor infection, such as a cold, should not result in a deferral of vaccination. Vaccination should be preceded by a review of the individual’s medical history (especially with regards to previous vaccination and possible hypersensitivity reactions which occurred after vaccination). Appropriate medical treatment and supervision must always be readily available in the event of a rare anaphylactic reaction following administration of the vaccine. Anxiety-related reactions, including vasovagal reactions (syncope), hyperventilation or stress‐related reactions may occur in association with vaccination as a psychogenic response to the needle injection. It is important that precautions are in place to avoid injury from fainting. A protective immune response with QDENGA may not be elicited in all vaccinees against all serotypes of dengue virus and may decline over time. It is currently unknown whether a lack of protection could result in an increased severity of dengue. It is recommended to continue personal protection measures against mosquito bites after vaccination. Individuals should seek medical care if they develop dengue symptoms or dengue warning signs.
Contraindications: Hypersensitivity to the active substances or excipients listed, or to previous QDENGA dose. Individuals with congenital or acquired immune deficiency, including immunosuppressive therapies such as chemotherapy or high doses of systemic corticosteroids (eg, 20 mg/day or 2 mg/kg body weight/day of prednisone for 2 weeks or more) within 4 weeks prior to vaccination. Individuals with symptomatic HIV infection or asymptomatic HIV infection with impaired immune function. Pregnant and breast-feeding women.
The double-blind, randomized, placebo-controlled Phase 3 Tetravalent Immunization against Dengue Efficacy Study (TIDES) trial is evaluating the safety and efficacy of two doses of TAK-003 in the prevention of laboratory-confirmed symptomatic dengue fever of any severity and due to any of the four dengue virus serotypes in children and adolescents.8 Study participants were randomized 2:1 to receive two doses of TAK-003 0.5 mL or placebo on Months 0 and 3, administered subcutaneously.8 The study is comprised of five parts. Part 1 and the primary endpoint analysis evaluated vaccine efficacy (VE) and safety through 12 months after the second dose.8 Part 2 continued for an additional six months to complete the assessment of the secondary endpoints of VE by serotype, baseline serostatus and disease severity, including VE against hospitalized dengue.8 Part 3 evaluated VE and long-term safety by following participants for an additional two and a half to three years, as per World Health Organization (WHO) recommendations.11 Part 4 will evaluate efficacy and safety for 13 months following booster vaccination, and Part 5 will evaluate long-term efficacy and safety for one year after completion of Part 4.11
The trial is taking place at sites in dengue-endemic areas in Latin America (Brazil, Colombia, Panama, the Dominican Republic and Nicaragua) and Asia (Philippines, Thailand and Sri Lanka) where there are unmet needs in dengue prevention and where severe dengue is a leading cause of serious illness and death among children.11 Baseline blood samples were collected from all individuals participating in the trial to allow for evaluation of safety and efficacy based on serostatus. Takeda and an independent Data Monitoring Committee of experts are actively monitoring safety on an ongoing basis.
Takeda is a global, values-based, R&D-driven biopharmaceutical leader headquartered in Japan, committed to discover and deliver life-transforming treatments, guided by our commitment to patients, our people and the planet. Takeda focuses its R&D efforts on four therapeutic areas: Oncology, Rare Genetics and Hematology, Neuroscience, and Gastroenterology (GI). We also make targeted R&D investments in Plasma-Derived Therapies and Vaccines. We are focusing on developing highly innovative medicines that contribute to making a difference in people’s lives by advancing the frontier of new treatment options and leveraging our enhanced collaborative R&D engine and capabilities to create a robust, modality-diverse pipeline. Our employees are committed to improving quality of life for patients and to working with our partners in health care in approximately 80 countries and regions. For more information, visit https://www.takeda.com.
Aug. 23, 2022 5:57 AM ET
Takeda Pharmaceutical Company Limited (TAK)
By: Ravikash, SA News Editor1 Comment
PUBLISHED16 August 2022
First PARP inhibitor to demonstrate clinical benefit in combination with a new hormonal
agent irrespective of homologous recombination repair (HRR) gene mutations
AstraZeneca’s supplemental New Drug Application (sNDA) for LYNPARZA® (olaparib) in combination with abiraterone and prednisone or prednisolone has been accepted and granted Priority Review in the US for the treatment of adult patients with metastatic castration-resistant prostate cancer (mCRPC).
LYNPARZA is being jointly developed and commercialized by AstraZeneca and Merck & Co., Inc., known as MSD outside the US and Canada.
The Food and Drug Administration (FDA) grants Priority Review to applications for medicines that offer significant advantages over available options by demonstrating safety or efficacy improvements, preventing serious conditions, or enhancing patient compliance.1 The Prescription Drug User Fee Act date, the FDA action date for their regulatory decision, is anticipated during the fourth quarter of 2022.
In the US, prostate cancer is the second most common cancer in male patients and is projected to cause approximately 35,000 deaths in 2022.2 Overall survival for patients with mCRPC is approximately three years in clinical trial settings, and even shorter in the real world.3-6 Approximately half of patients with mCRPC may receive only one line of active treatment, with diminishing benefit of subsequent therapies.6-11
Susan Galbraith, Executive Vice President, Oncology R&D, AstraZeneca, said: “There remains a critical unmet need among patients diagnosed with metastatic castration-resistant prostate cancer, where the prognosis remains poor and treatment options are limited. Today’s news is another step towards bringing forward a new, much-needed treatment option in this setting. If approved, LYNPARZA with abiraterone will become the first combination of a PARP inhibitor and a new hormonal agent for patients with this disease.”
Dr. Eliav Barr, Senior Vice President, Head of Global Clinical Development and Chief Medical Officer, Merck Research Laboratories, said: “Merck is committed to developing new treatment options for patients with metastatic castration-resistant prostate cancer, a complex disease that urgently needs more therapies. We look forward to working with the FDA towards the goal of bringing a new option to patients with mCRPC with or without HRR gene mutations.”
The sNDA was based on results from the PROpel Phase III trial presented at the 2022 American Society of Clinical Oncology (ASCO) Genitourinary Cancers Symposium and later published in NEJM Evidence.
These results showed LYNPARZA in combination with abiraterone reduced the risk of disease progression or death by 34% versus abiraterone alone (based on a hazard ratio [HR] of 0.66; 95% confidence interval [CI] 0.54-0.81; p<0.0001). Median radiographic progression-free survival (rPFS) was 24.8 months for LYNPARZA plus abiraterone versus 16.6 for abiraterone alone. The safety and tolerability of LYNPARZA in combination with abiraterone was in line with that observed in prior clinical trials and the known profiles of the individual medicines.12
LYNPARZA is approved in the US for patients with HRR gene-mutated mCRPC (BRCA-mutated and other HRR gene mutations) who have progressed following prior treatment with enzalutamide or abiraterone; and in the EU, Japan and China for patients with BRCA-mutated mCRPC who have progressed following prior therapy that included a new hormonal agent. These approvals were based on the data from the PROfound Phase III trial.
IMPORTANT SAFETY INFORMATION
CONTRAINDICATIONS
There are no contraindications for LYNPARZA.
WARNINGS AND PRECAUTIONS
Myelodysplastic Syndrome/Acute Myeloid Leukemia (MDS/AML): Occurred in approximately 1.5% of patients exposed to LYNPARZA monotherapy, and the majority of events had a fatal outcome. The median duration of therapy in patients who developed MDS/AML was 2 years (range: <6 months to >10 years). All of these patients had previous chemotherapy with platinum agents and/or other DNA-damaging agents, including radiotherapy.
Do not start LYNPARZA until patients have recovered from hematological toxicity caused by previous chemotherapy (≤Grade 1). Monitor complete blood count for cytopenia at baseline and monthly thereafter for clinically significant changes during treatment. For prolonged hematological toxicities, interrupt LYNPARZA and monitor blood count weekly until recovery.
If the levels have not recovered to Grade 1 or less after 4 weeks, refer the patient to a hematologist for further investigations, including bone marrow analysis and blood sample for cytogenetics. Discontinue LYNPARZA if MDS/AML is confirmed.
INDICATIONS
LYNPARZA is a poly (ADP-ribose) polymerase (PARP) inhibitor indicated:
First-Line Maintenance BRCAm Advanced Ovarian Cancer
For the maintenance treatment of adult patients with deleterious or suspected deleterious germline or somatic BRCA-mutated (gBRCAm or sBRCAm) advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in complete or partial response to first-line platinum-based chemotherapy. Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
First-Line Maintenance HRD-Positive Advanced Ovarian Cancer in Combination with Bevacizumab
In combination with bevacizumab for the maintenance treatment of adult patients with advanced epithelial ovarian, fallopian tube or primary peritoneal cancer who are in complete or partial response to first-line platinum-based chemotherapy and whose cancer is associated with homologous recombination deficiency (HRD) positive status defined by either:
Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
Maintenance Recurrent Ovarian Cancer
For the maintenance treatment of adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer, who are in complete or partial response to platinum-based chemotherapy.
Advanced gBRCAm Ovarian Cancer
For the treatment of adult patients with deleterious or suspected deleterious germline BRCA-mutated (gBRCAm) advanced ovarian cancer who have been treated with 3 or more prior lines of chemotherapy. Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
Adjuvant Treatment of gBRCAm, HER2-Negative, High-Risk Early Breast Cancer
For the adjuvant treatment of adult patients with deleterious or suspected deleterious gBRCAm, human epidermal growth factor receptor 2 (HER2)-negative high-risk early breast cancer who have been treated with neoadjuvant or adjuvant chemotherapy. Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
gBRCAm, HER2-Negative Metastatic Breast Cancer
For the treatment of adult patients with deleterious or suspected deleterious gBRCAm, human epidermal growth factor receptor 2 (HER2)-negative metastatic breast cancer who have been treated with chemotherapy in the neoadjuvant, adjuvant, or metastatic setting. Patients with hormone receptor (HR)-positive breast cancer should have been treated with a prior endocrine therapy or be considered inappropriate for endocrine therapy. Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
First-Line Maintenance gBRCAm Metastatic Pancreatic Cancer
For the maintenance treatment of adult patients with deleterious or suspected deleterious gBRCAm metastatic pancreatic adenocarcinoma whose disease has not progressed on at least 16 weeks of a first-line platinum-based chemotherapy regimen. Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
HRR Gene-mutated Metastatic Castration-Resistant Prostate Cancer
For the treatment of adult patients with deleterious or suspected deleterious germline or somatic homologous recombination repair (HRR) gene-mutated metastatic castration-resistant prostate cancer (mCRPC) who have progressed following prior treatment with enzalutamide or abiraterone. Select patients for therapy based on an FDA-approved companion diagnostic for LYNPARZA.
Please see complete Prescribing Information, including Medication Guide.
Notes
Metastatic castration-resistant prostate cancer
Metastatic prostate cancer is associated with a significant mortality rate.13 Development of prostate cancer is often driven by male sex hormones called androgens, including testosterone.14
In patients with mCRPC, their prostate cancer grows and spreads to other parts of the body despite the use of androgen-deprivation therapy to block the action of male sex hormones.7 Approximately 10-20% of men with advanced prostate cancer will develop castration-resistant prostate cancer (CRPC) within five years, and at least 84% of these men will have metastases at the time of CRPC diagnosis.7 Of patients with no metastases at CRPC diagnosis, 33% are likely to develop metastases within two years.6
Despite the advances in mCRPC treatment in the past decade with taxane and new hormonal agent (NHA) treatment, there is high unmet need in this population.7,9,10,15
PROpel
PROpel is a randomized, double-blind, multi-center Phase III trial testing the efficacy, safety, and tolerability of LYNPARZA versus placebo when given in addition to abiraterone in men with mCRPC who had not received prior chemotherapy or NHAs in the mCRPC setting.
Men in both treatment groups will also receive either prednisone or prednisolone twice daily. The primary endpoint is rPFS and secondary endpoints include overall survival, time to secondary progression or death, and time to first subsequent therapy.
For more information about the trial please visit ClinicalTrials.gov.
LYNPARZA
LYNPARZA (olaparib) is a first-in-class PARP inhibitor and the first targeted treatment to block DNA damage response (DDR) in cells/tumors harboring a deficiency in HRR, such as those with mutations in BRCA1 and/or BRCA2, or those where deficiency is induced by other agents (such as NHAs).
Inhibition of PARP with LYNPARZA leads to the trapping of PARP bound to DNA single-strand breaks, stalling of replication forks, their collapse and the generation of DNA double-strand breaks and cancer cell death. In the PROpel Phase III trial, LYNPARZA is combined with abiraterone, an NHA which targets the androgen receptor (AR) pathway.
Androgen receptor signaling engages a transcriptional program that is critical for tumor cell growth & survival in prostate cancer.16,17 Preclinical models have identified interactions between PARP signaling and the AR pathway which support the observation of a combined anti-tumor effect of LYNPARZA and NHAs, like abiraterone, in both HRR deficient and HRR proficient prostate cancer.18,19,20
The PARP1 protein has been reported to be required for the transcriptional activity of androgen receptors; therefore inhibiting PARP with LYNPARZA may impair the expression of androgen receptor target genes and enhance the activity of NHAs.16,19,21 Additionally, it is thought that abiraterone may alter/inhibit the transcription of some HRR genes which may induce HRR deficiency and increase sensitivity to PARP inhibition.18,20,22,23
LYNPARZA is currently approved in a number of countries across PARP-dependent tumor types with defects and dependencies in the DDR pathway including maintenance treatment of platinum-sensitive relapsed ovarian cancer and as both monotherapy and in combination with bevacizumab for the 1st-line maintenance treatment of BRCA-mutated (BRCAm) and homologous recombination repair deficient (HRD)-positive advanced ovarian cancer, respectively; for germline BRCAm (gBRCAm) HER2-negative metastatic breast cancer (in the EU and Japan this includes locally advanced breast cancer); for gBRCAm, HER2-negative high-risk early breast cancer; for gBRCAm metastatic pancreatic cancer; and HRR gene-mutated metastatic castration-resistant prostate cancer (BRCAm only in the EU and Japan).
LYNPARZA, which is being jointly developed and commercialized by AstraZeneca and Merck, is the foundation of AstraZeneca's industry-leading portfolio of potential new medicines.
The AstraZeneca and Merck strategic oncology collaboration
In July 2017, AstraZeneca and Merck & Co., Inc., Kenilworth, NJ, US, known as MSD outside the US and Canada, announced a global strategic oncology collaboration to co-develop and co-commercialize LYNPARZA, the world’s first PARP inhibitor, and selumetinib, a mitogen-activated protein kinase (MEK) inhibitor, for multiple cancer types.
Working together, the companies will develop LYNPARZA and selumetinib and other potential new medicines as monotherapies. The companies will also develop LYNPARZA and selumetinib in combination with their respective PD-L1 and PD-1 medicines independently.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyze changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
About AstraZeneca
AstraZeneca is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialization of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit www.astrazeneca-us.com and follow the Company on Twitter @AstraZenecaUS.
Aug. 16, 2022 4:20 AM ET
AstraZeneca PLC (AZN), JNJ, MRK
By: Ravikash, SA News Editor
Tuesday, August 23, 2022 - 06:45am
NEW YORK & MAINZ, Germany--(BUSINESS WIRE)-- Pfizer Inc. (NYSE: PFE) and BioNTech SE (Nasdaq: BNTX) today announced updated efficacy results from a Phase 2/3 trial evaluating a three 3-µg dose series of the Pfizer-BioNTech COVID-19 Vaccine in children 6 months through 4 years of age, reinforcing previously reported interim vaccine efficacy data collected in March and April 2022. Emergency Use Authorization (EUA) of this vaccine was granted by the U.S. Food and Drug Administration (FDA) for this age group on June 17 and an application for conditional Marketing Authorization in this age group is under review by the European Medicines Agency (EMA).
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20220823005341/en/
Participants in the study received either the Pfizer-BioNTech COVID-19 Vaccine (3-μg) as a three-dose series or placebo (2:1 randomization). Vaccine efficacy, a secondary endpoint in the trial, was 73.2% (2-sided 95% CI: 43.8%, 87.6%) among children 6 months through 4 years of age without evidence of prior COVID-19 infection. This analysis was based on 13 cases in the Pfizer-BioNTech COVID-19 Vaccine group (n=794) and 21 cases in the placebo group (n=351), diagnosed from March to June 2022. The study protocol specified that this formal efficacy analysis should be performed when at least 21 total symptomatic COVID-19 cases were identified, each at least seven days after the third dose. Consistent with the time period when the cases occurred, sequencing of viral RNA from illness visit nasal swabs indicated that observed cases were primarily caused by Omicron BA.2. Omicron BA.4 and BA.5 strains were emerging during the period of the study, with only a few cases accrued and efficacy results against these strains were inconclusive. Consistent with our approach in adults, we are working with the FDA to prepare an EUA application for an Omicron BA.4/BA.5-adapted bivalent vaccine in children 6 months through 11 years of age.
“Building on the strong safety and immunogenicity data that led to FDA authorization of our COVID-19 vaccine for children 6 months through 4 years, we are pleased to share confirmatory evidence that a full course of vaccination helps protect against symptomatic disease, particularly during a time when the Omicron BA.2 strain was predominant,” said Albert Bourla, Chairman and Chief Executive Officer, Pfizer.
“While these results confirm that three 3-µg doses of our COVID-19 vaccine provide young children with a high level of protection at a time when the Omicron BA.2 strain was highly prevalent with a favorable safety profile, we are also developing an Omicron BA.4/BA.5-adapted bivalent vaccine in this age group to address these sublineages,” said Prof. Ugur Sahin, M.D., CEO and Co-Founder of BioNTech.
Among children ages 6 through 23 months, the vaccine was 75.8% (2-sided 95% CI: 9.7%, 94.7%) effective at preventing COVID-19, based on 4 cases in the vaccine group (n=296) and 8 cases in the placebo group (n=147), after a median of 1.9 months (range: 0.0, 4.9) follow-up after the third dose. For children ages 2 through 4 years of age, the vaccine was 71.8% (2-sided 95% CI: 28.6%, 89.4%) effective at preventing COVID-19, based on 9 cases in the vaccine group (n=498) and 13 cases in the placebo group (n=204), after a median of 2.4 months (range: 0.0, 4.9) follow-up after the third dose.
Three 3-µg doses of the Pfizer-BioNTech COVID-19 Vaccine continue to be well-tolerated in this age group. The majority of adverse events observed in this age group have been mild or moderate, with a safety profile similar to placebo.
On July 8, Pfizer and BioNTech submitted safety and immunogenicity data to the European Medicines Agency (EMA) requesting an update to the Conditional Marketing Authorization (CMA) in the European Union (EU) to include children ages 6 months through 4 years. The companies plan to submit the updated efficacy data to the FDA, EMA and other regulatory agencies around the world in the coming weeks.
The Pfizer-BioNTech COVID-19 Vaccine, which is based on BioNTech’s proprietary mRNA technology, was developed by both BioNTech and Pfizer. BioNTech is the Marketing Authorization Holder for BNT162b2 (COMIRNATY®) in the United States, the European Union, the United Kingdom, Canada and other countries, and the holder of emergency use authorizations or equivalents in the United States (jointly with Pfizer) and other countries. Submissions to pursue regulatory approvals in those countries where emergency use authorizations or equivalent were initially granted are planned.
About the Phase 1/2/3 Trial in Children
The Phase 1/2/3 trial has enrolled more than 10,000 children ages 6 months to under 12 years of age in the United States, Finland, Poland, Brazil and Spain from more than 90 clinical trial sites. The trial evaluated the safety, tolerability, and immunogenicity of three doses of the Pfizer-BioNTech COVID-19 Vaccine in three age groups: ages 5 to under 12 years; ages 2 to under 5 years; and ages 6 months to under 2 years. Based on the Phase 1 dose-escalation portion of the trial, children ages 5 to under 12 years received a two-dose schedule of 10 µg each while children under age 5 received a lower 3 µg dose for each injection in the Phase 2/3 study. The trial enrolled children with or without prior evidence of SARS-CoV-2 infection.
U.S. Indication & Authorized Use
Pfizer-BioNTech COVID-19 Vaccine is FDA authorized under Emergency Use Authorization (EUA) for active immunization to prevent coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in individuals 6 months of age and older. Pfizer-BioNTech COVID-19 Vaccine is FDA authorized to provide:
Primary Series
Booster Series
COMIRNATY®INDICATION
COMIRNATY® (COVID-19 Vaccine, mRNA) is a vaccine approved for active immunization to prevent coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in individuals 12 years of age and older.
HOW IS COMIRNATY ® GIVEN?
COMIRNATY® is administered as an injection into the muscle as a 2-dose primary series, 3 weeks apart.
COMIRNATY® AUTHORIZED USES
COMIRNATY® (COVID-19 Vaccine, mRNA) is FDA authorized under Emergency Use Authorization (EUA) to provide:
Primary Series
Booster Dose
Emergency Use Authorization
Emergency uses of the vaccine have not been approved or licensed by FDA, but have been authorized by FDA, under an Emergency Use Authorization (EUA) to prevent Coronavirus Disease 2019 (COVID 19) in individuals 6 months of age and older. The emergency uses are only authorized for the duration of the declaration that circumstances exist justifying the authorization of emergency use of the medical product under Section 564(b)(1) of the FD&C Act unless the declaration is terminated or authorization revoked sooner.
About Pfizer: Breakthroughs That Change Patients’ Lives
At Pfizer, we apply science and our global resources to bring therapies to people that extend and significantly improve their lives. We strive to set the standard for quality, safety and value in the discovery, development and manufacture of health care products, including innovative medicines and vaccines. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments and cures that challenge the most feared diseases of our time. Consistent with our responsibility as one of the world’s premier innovative biopharmaceutical companies, we collaborate with health care providers, governments and local communities to support and expand access to reliable, affordable health care around the world. For more than 170 years, we have worked to make a difference for all who rely on us. We routinely post information that may be important to investors on our website at www.Pfizer.com. In addition, to learn more, please visit us on www.Pfizer.com and follow us on Twitter at @Pfizer and @Pfizer News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
About BioNTech
Biopharmaceutical New Technologies is a next generation immunotherapy company pioneering novel therapies for cancer and other serious diseases. The Company exploits a wide array of computational discovery and therapeutic drug platforms for the rapid development of novel biopharmaceuticals. Its broad portfolio of oncology product candidates includes individualized and off-the-shelf mRNA-based therapies, innovative chimeric antigen receptor T cells, bispecific immune checkpoint modulators, targeted cancer antibodies and small molecules. Based on its deep expertise in mRNA vaccine development and in-house manufacturing capabilities, BioNTech and its collaborators are developing multiple mRNA vaccine candidates for a range of infectious diseases alongside its diverse oncology pipeline. BioNTech has established a broad set of relationships with multiple global pharmaceutical collaborators, including Genmab, Sanofi, Genentech, a member of the Roche Group, Regeneron, Genevant, Fosun Pharma, and Pfizer. For more information, please visit www.BioNTech.com.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220823005341/en/
Source: Pfizer Inc.
Aug. 23, 2022 7:13 AM ET
By: Ravikash, SA News Editor1 Comment
Pfizer (NYSE:PFE) and BioNTech's (NASDAQ:BNTX) COVID-19 vaccine showed 73.2% efficacy in children ages six months through four years of age in a phase 2/3 trial.
https://seekingalpha.com/symbol/PFE
https://seekingalpha.com/symbol/BNTX
Aug 22, 2022
Omnipod 5 is the first tubeless AID System cleared in the U.S. for preschool children
ACTON, Mass.--(BUSINESS WIRE)--Aug. 22, 2022-- Insulet Corporation (NASDAQ: PODD) (Insulet or the Company), the global leader in tubeless insulin pump technology with its Omnipod® brand of products, today announced it has received clearance from the U.S. Food and Drug Administration (FDA) for its Omnipod® 5 Automated Insulin Delivery System (Omnipod 5) for individuals aged two years and older with type 1 diabetes (T1D). Omnipod 5, the first tubeless automated insulin delivery (AID) system in the U.S., was originally cleared for use in individuals aged six and older in January 2022.
“We received tremendous first-hand reports of how Omnipod 5 made diabetes management easier for our pivotal trial participants, and the clinical data demonstrated impressive glycemic improvements as well,” said Dr. Trang Ly MBBS, FRACP, PhD, Insulet Senior Vice President and Medical Director. “This expanded indication for younger children gives us great pride, knowing we can further ease the burden of glucose management for these children and their caregivers with our simple to use, elegant, automated insulin delivery system.”
Omnipod 5 is the first tubeless AID system in the U.S. that integrates with the Dexcom G6 CGM system and a compatible smartphone1 to automatically adjust insulin and help protect against high and low glucose levels 2. The system3 consists of the tubeless Pod enhanced with SmartAdjustTM technology, the Omnipod 5 mobile app with its integrated SmartBolus Calculator, and the Dexcom G6 CGM.
“Omnipod 5 has allowed our family to think less about diabetes,” said Kara Hornbuckle, an Omnipod 5 user with two children also using the system. “Omnipod 5 has provided more control and greater peace of mind for all three of us, especially during the nighttime when we are all most vulnerable. The impact that this device has had on my family cannot be understated. We are just so grateful!”
In a recent publication by Sherr et al. in Diabetes Care4, Omnipod 5 significantly improved time in range, and reduced HbA1c and time in hypoglycemia (<70 mg/dL) in very young children (aged 2 – 5.9 years) with type 1 diabetes. In addition, parents and caregivers of study participants reported improved sleep quality as assessed by the Pittsburgh Sleep Quality Index (PSQI)5, a questionnaire considered to be the gold standard in measuring subjective sleep quality.
Healthcare professionals can now prescribe Omnipod 5 to their patients with insurance coverage and patients can access their prescription through the pharmacy channel, which means there is no contract and no commitment. The Omnipod brand is the only insulin pump in the U.S. available through the pharmacy.
About Insulet Corporation:
Insulet Corporation (NASDAQ: PODD), headquartered in Massachusetts, is an innovative medical device company dedicated to simplifying life for people with diabetes and other conditions through its Omnipod product platform. The Omnipod Insulin Management System provides a unique alternative to traditional insulin delivery methods. With its simple, wearable design, the disposable Pod provides up to three days of non-stop insulin delivery, without the need to see or handle a needle. Insulet’s latest innovation, the Omnipod 5 Automated Insulin Delivery System, is a tubeless automated insulin delivery system, integrated with a continuous glucose monitor to manage blood sugar with no multiple daily injections, zero fingersticks6, and is fully controlled by a compatible personal smartphone. Insulet also leverages the unique design of its Pod by tailoring its Omnipod technology platform for the delivery of non-insulin subcutaneous drugs across other therapeutic areas. For more information, please visit: insulet.com and omnipod.com.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220822005158/en/
Source: Insulet Corporation
Aug. 22, 2022 6:35 AM ET
Insulet Corporation (PODD)DXCM
By: Dulan Lokuwithana, SA News Editor
August 15, 2022
–Trodelvy is the First TROP-2 Directed ADC to Show a Significant Improvement in Overall Survival in HR+/HER2- Breast Cancer –
– Planned SecondInterim Analysis Demonstrated a Statistically Significant and Clinically Meaningful Benefit for Overall Survival –
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced statistically significant and clinically meaningful results from the second interim analysis of the key secondary endpoint of overall survival (OS) in the Phase 3 TROPiCS-02 study evaluating Trodelvy® (sacituzumab govitecan-hziy) in patients with HR+/HER2- metastatic breast cancer who received prior endocrine therapy, CDK4/6 inhibitors and two to four lines of chemotherapy. Detailed OS results will be presented at an upcoming medical conference. The safety profile for Trodelvy was consistent with prior studies, and no new safety signals emerged in this patient population.
“These survival results from the TROPiCS-02 study are important for the breast cancer community and we are encouraged by the potential this may have in helping patients who otherwise have limited alternatives,” said Merdad Parsey, MD, PhD, Chief Medical Officer, Gilead Sciences. “We look forward to discussing these results with global health authorities, as pre-treated HR+/HER2- metastatic disease patients currently have limited treatment options and poor quality of life.”
Gilead has submitted a supplemental Biologics License Application (sBLA) to the U.S. Food & Drug Administration (FDA). These data will also be shared with health authorities outside the U.S.
In June 2022, based on the positive primary results from the TROPiCS-02 study presented at the 2022 American Society of Clinical Oncology (ASCO) Annual Meeting, the National Comprehensive Cancer Network® (NCCN®) Clinical Practice Guidelines in Oncology (NCCN Guidelines®)i included a category 2A preferred recommendation for sacituzumab govitecan-hziy for the investigational use in patients with HR+/HER2- advanced breast cancer after prior treatment including endocrine therapy, a CDK4/6 inhibitor and at least two lines of chemotherapy. The TROPiCS-02 study enrolled HR+/HER2- metastatic breast cancer patients, which included patients with HER2-low and IHC 0 status.
TROPiCS-02 is an event driven study with planned analyses based on a pre-specified number of events. This interim analysis was executed based on the pre-specified criteria.
Trodelvy has not been approved by any regulatory agency for the treatment of HR+/HER2- metastatic breast cancer. Its safety and efficacy have not been established for this indication. Trodelvy has a Boxed Warning for severe or life-threatening neutropenia and severe diarrhea; please see below for additional Important Safety Information.
About the TROPiCS-02 Study
The TROPiCS-02 study is a global, multicenter, open-label, Phase 3 study, randomized 1:1 to evaluate Trodelvy versus physicians’ choice of chemotherapy (eribulin, capecitabine, gemcitabine, or vinorelbine) in 543 patients with HR+/HER2- metastatic breast cancer who were previously treated with endocrine therapy, CDK4/6 inhibitors and two to four lines of chemotherapy for metastatic disease. The primary endpoint is progression-free survival per Response Evaluation Criteria in Solid Tumors (RECIST 1.1) as assessed by blinded independent central review (BICR) for participants treated with Trodelvy compared to those treated with chemotherapy. Secondary endpoints include overall survival, overall response rate, clinical benefit rate and duration of response, as well as assessment of safety and tolerability and quality of life measures. In the study, HER2 negativity was defined per American Society of Clinical Oncology (ASCO) and the College of American Pathologists (CAP) criteria as immunohistochemistry (IHC) score of 0, IHC 1+ or IHC 2+ with a negative in-situ hybridization (ISH) test.
More information about TROPiCS-02 is available at https://clinicaltrials.gov/ct2/show/NCT03901339.
About Trodelvy
Trodelvy® (sacituzumab govitecan-hziy) is a first-in-class Trop-2 directed antibody-drug conjugate. Trop-2 is a cell surface antigen highly expressed in multiple tumor types, including in more than 90% of breast and bladder cancers. Trodelvy is intentionally designed with a proprietary hydrolyzable linker attached to SN-38, a topoisomerase I inhibitor payload. This unique combination delivers potent activity to both Trop-2 expressing cells and the microenvironment.
Trodelvy is approved in more than 35 countries, with multiple additional regulatory reviews underway worldwide, for the treatment of adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (TNBC) who have received two or more prior systemic therapies, at least one of them for metastatic disease. Trodelvy is also approved in the U.S. under the accelerated approval pathway for the treatment of adult patients with locally advanced or metastatic urothelial cancer (UC) who have previously received a platinum-containing chemotherapy and either programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor.
Trodelvy is also being developed for potential investigational use in other TNBC and metastatic UC populations, as well as a range of tumor types where Trop-2 is highly expressed, including hormone receptor-positive/human epidermal growth factor receptor 2-negative (HR+/HER2-) metastatic breast cancer, metastatic non-small cell lung cancer (NSCLC), metastatic small cell lung cancer (SCLC), head and neck cancer, and endometrial cancer.
U.S. Indications for Trodelvy
In the United States, Trodelvy is indicated for the treatment of:
View source version on businesswire.com: https://www.businesswire.com/news/home/20220814005018/en/
Source: Gilead Sciences, Inc.
Aug. 15, 2022 8:53 AM ET
By: Dulan Lokuwithana, SA News Editor7 Comments
FDA accepts supplemental Biologics License Application for Roche’s Polivy combination for people with previously untreated diffuse large B-cell lymphoma
August 16, 2022
Basel, 16 August 2022 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the U.S. Food and Drug Administration (FDA) has accepted the company’s supplemental Biologics License Application (sBLA) for Polivy® (polatuzumab vedotin-piiq) in combination with Rituxan® (rituximab) plus cyclophosphamide, doxorubicin and prednisone (R-CHP) for the treatment of people with previously untreated diffuse large B-cell lymphoma (DLBCL). The FDA is expected to make a decision on approval by 2 April 2023.
“The POLARIX study results suggest that Polivy plus R-CHP could transform the treatment of this aggressive malignancy, and we are working with the FDA to bring this combination to newly diagnosed DLBCL patients as soon as possible,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “We hope it will become the new standard of care for the first-line treatment of DLBCL, potentially reducing the need for subsequent treatments and limiting patient burden.”
DLBCL is an aggressive blood cancer. Although DLBCL often responds to initial treatment, it is not cured with the current standard of care in four out of 10 people. Most relapses occur within two years of starting treatment and the majority of those who require subsequent lines of therapy have poor outcomes.
The sBLA is based on results from the pivotal phase III POLARIX trial, which is the first in two decades to show a clinically meaningful improvement in progression-free survival (PFS) compared to the current standard of care Rituxan plus cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP). The risk of disease progression, relapse or death was reduced by 27% with Polivy plus R-CHP compared with R-CHOP after a median follow-up of 28.2 months (hazard ratio [HR] 0.73; 95% confidence interval [CI]: 0.57–0.95; p<0.02). Safety outcomes were consistent with those seen in previous clinical trials, and the safety profile was comparable for Polivy plus R-CHP versus R-CHOP, including rates of Grade 3-4 adverse events (AEs; 57.7% versus 57.5%), serious AEs (34.0% versus 30.6%), Grade 5 AEs (3.0% versus 2.3%), and AEs leading to dose reduction (9.2% versus 13.0%).
Based on pivotal data from the POLARIX study, the European Commission approved Polivy in combination with R-CHP in May 2022 for the treatment of adult patients with previously untreated DLBCL. Polivy is currently approved as a readily available, fixed-duration treatment option for relapsed or refractory (R/R) DLBCL in combination with bendamustine and Mabthera/Rituxan in more than 70 countries worldwide, including in the EU and in the United States.
Roche continues to explore areas of unmet need where Polivy has the potential to deliver additional benefit, including in ongoing studies investigating combinations of Polivy with the company’s CD20xCD3 T-cell engaging bispecific antibodies Lunsumio® (mosunetuzumab) and glofitamab, and with Rituxan in combination with gemcitabine and oxaliplatin in the phase III POLARGO study.
About the POLARIX study
POLARIX [NCT03274492] is an international phase III, randomised, double-blind, placebo- controlled study evaluating the efficacy, safety and pharmacokinetics of Polivy® (polatuzumab vedotin-piiq) plus Rituxan® (rituximab), cyclophosphamide, doxorubicin, and prednisone (R-CHP) versus rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP) in people with previously untreated diffuse large B-cell lymphoma. Eight-hundred and seventy-nine patients were randomised 1:1 to receive either Polivy plus R- CHP plus a vincristine placebo for six cycles, followed by rituximab for two cycles; or R-CHOP plus a Polivy placebo for six cycles, followed by two cycles of rituximab. The primary outcome measure is progression-free survival (PFS) as assessed by the investigator using the Lugano Response Criteria for malignant lymphoma. POLARIX is being conducted in collaboration with The Lymphoma Study Association (LYSA) and The Lymphoma Academic Research Organisation (LYSARC). Other clinical investigators from around the world also participated in the trial.
About Polivy® (polatuzumab vedotin-piiq)
Polivy is a first-in-class anti-CD79b antibody-drug conjugate (ADC). The CD79b protein is expressed specifically in the majority of B-cells, an immune cell impacted in some types of non-Hodgkin lymphoma (NHL), making it a promising target for the development of new therapies. Polivy binds to cancer cells such as CD79b and destroys these B-cells through the delivery of an anti-cancer agent, which is thought to minimise the effects on normal cells. Polivy is being developed by Roche using Seagen ADC technology and is currently being investigated for the treatment of several types of NHL. Polivy is currently marketed in the EU for the treatment of relapsed or refractory diffuse large B-cell lymphoma.
About Roche in haematology
Roche has been developing medicines for people with malignant and non-malignant blood diseases for more than 20 years; our experience and knowledge in this therapeutic area runs deep. Today, we are investing more than ever in our effort to bring innovative treatment options to patients across a wide range of hematological diseases. Our approved medicines include MabThera/Rituxan® (rituximab), Gazyvaro/Gazyva® (obinutuzumab), Polivy® (polatuzumab vedotin-piiq), Venclexta/Venclyxto® (venetoclax) in collaboration with AbbVie, and Hemlibra® (emicizumab). Our pipeline of investigational haematology medicines includes T-cell engaging bispecific antibodies, glofitamab and Lunsumio®( mosunetuzumab), targeting both CD20 and CD3, and cevostamab, targeting both FcRH5 and CD3; Tecentriq® (atezolizumab), a monoclonal antibody designed to bind with PD-L1 and crovalimab, an anti-C5 antibody engineered to optimise complement inhibition. Our scientific expertise, combined with the breadth of our portfolio and pipeline, also provides a unique opportunity to develop combination regimens that aim to improve the lives of patients even further.
About Roche
Founded in 1896 in Basel, Switzerland, as one of the first industrial manufacturers of branded medicines, Roche has grown into the world’s largest biotechnology company and the global leader in in-vitro diagnostics. The company pursues scientific excellence to discover and develop medicines and diagnostics for improving and saving the lives of people around the world. We are a pioneer in personalised healthcare and want to further transform how healthcare is delivered to have an even greater impact. To provide the best care for each person we partner with many stakeholders and combine our strengths in Diagnostics and Pharma with data insights from the clinical practice.
In recognising our endeavor to pursue a long-term perspective in all we do, Roche has been named one of the most sustainable companies in the pharmaceuticals industry by the Dow Jones Sustainability Indices for the thirteenth consecutive year. This distinction also reflects our efforts to improve access to healthcare together with local partners in every country we work.
Genentech, in the United States, is a wholly owned member of the Roche Group. Roche is the majority shareholder in Chugai Pharmaceutical, Japan.
For more information, please visit www.roche.com.
All trademarks used or mentioned in this release are protected by law.
Aug. 16, 2022 4:41 AM ET
Roche Holding AG (RHHBY), RHHBF
By: Ravikash, SA News Editor1 Comment
Friday, August 12, 2022 - 06:45am
NEW YORK--(BUSINESS WIRE)-- Pfizer Inc. (NYSE:PFE) today announced positive top-line results from its pivotal U.S. Phase 3 study (NCT04382326) in infants evaluating its 20-valent pneumococcal conjugate vaccine candidate (20vPnC) for the prevention of invasive pneumococcal disease (IPD) caused by the 20 Streptococcus pneumoniae (pneumococcus) serotypes contained in the vaccine for the pediatric population.
The study had two co-primary objectives, associated with immunogenicity responses one month after the third and fourth doses of the four-dose vaccination series, respectively: non-inferiority (NI) of the percentage of participants with predefined serotype-specific immunoglobin G (IgG) concentrations after Dose 3 and NI of IgG geometric mean concentrations (GMCs) after Dose 4. All 20 serotypes met the co-primary objective of NI of IgG GMCs after Dose 4. Fourteen of the 20 serotypes met the co-primary objective of NI of the percentage of participants with predefined IgG levels after Dose 3 (two serotypes missed by a wider margin while four narrowly missed), and all serotypes met noninferiority for the key secondary objective of IgG GMCs after Dose 3. All 20 serotypes elicited robust functional responses (OPA) and increases in antibody responses after Dose 4, with the totality of data supporting the potential benefit of all serotypes in this 20-valent vaccine candidate.
“We are encouraged by today’s data which show that if approved for a pediatric indication, 20vPnC would have the potential to cover more of the clinically significant remaining burden of infant pneumococcal disease than any other available pneumococcal conjugate vaccine,” said Annaliesa Anderson, Ph.D., Senior Vice President and Chief Scientific Officer, Vaccine Research and Development, Pfizer. “We are grateful to everyone who made this study possible, including the study investigators and in particular the trial participants and their parents/guardians for their contribution to this important research.”
Overall, the safety profile of the 20vPnC candidate was consistent with Prevnar 13 given in the same schedule. A similar percentage of infants receiving either vaccine experienced local reactions (pain at the injection site, redness, and swelling), fever, and other systemic events (decreased appetite, drowsiness, and irritability). The study also met non-inferiority objectives for responses to co-administered routinely used pediatric vaccines.
Based on the totality of positive safety and immunogenicity data, Pfizer plans to submit a supplemental Biologics License Application (sBLA) by the end of this year, subject to discussions with the U.S. Food and Drug Administration. Pfizer will seek to present and publish outcomes from this clinical trial at a future date once safety and immunogenicity data have been fully analyzed. Additional top-line results from other pediatric 20vPnC clinical trials are expected to read out in the second half of 2022, with discussions with other regulatory bodies planned once those pivotal data become available.
About the 20vPnC Phase 3 Pediatric Program
In 2020, Pfizer initiated the Phase 3 clinical trial program for the pediatric indication for 20vPnC. Four core Phase 3 pediatric studies will help expand the data on the safety, tolerability, and immunogenicity of 20vPnC. These studies collectively enrolled approximately 4,700 infants and 800 toddlers and children of all ages including:
About 20vPnC
Pfizer’s 20vPnC pediatric vaccine candidate includes 13 serotypes already included in Prevnar 13® – 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, and 23F. The seven new serotypes included in 20vPnC are global causes of invasive pneumococcal disease (IPD), and are associated with high case-fatality rates, antibiotic resistance, and/or meningitis. Together, the 20 serotypes included in 20vPnC are responsible for the majority of currently circulating pneumococcal disease in the U.S. and globally.
On August 14, 2020, Pfizer’s 20vPnC received the FDA’s Breakthrough Therapy Designation for the prevention of disease caused by Streptococcus pneumoniae serotypes in the vaccine in infants, children, and adolescents.
The FDA previously granted Fast Track Designation for 20vPnC in May 2017 for the pediatric indication.
On June 8, 2021, the FDA approved PREVNAR 20® for the prevention of invasive disease and pneumonia caused by the 20 Streptococcus pneumoniae (pneumococcus) serotypes in the vaccine in adults ages 18 years and older.
INDICATIONS FOR PREVNAR 13®
INDICATIONS FOR PREVNAR 20®
View source version on businesswire.com: https://www.businesswire.com/news/home/20220812005073/en/
Source: Pfizer Inc.
Aug. 12, 2022 7:12 AM ET
By: Dulan Lokuwithana, SA News Editor
Pfizer (NYSE:PFE) announced Friday that the company’s 20-valent pneumococcal conjugate vaccine candidate named 20vPnC met the co-primary endpoints in a Phase 3 study involving children.
https://seekingalpha.com/symbol/PFE
https://prevnar20.pfizerpro.com/
PUBLISHED12 August 2022
AstraZeneca and Daiichi Sankyo’s Enhertu (trastuzumab deruxtecan) has been approved in the US for the treatment of adult patients with unresectable or metastatic non-small cell lung cancer (NSCLC) whose tumours have activating HER2 (ERBB2) mutations, as detected by a Food and Drug Administration (FDA)-approved test, and who have received a prior systemic therapy.
This indication is approved under accelerated approval based on objective response rate (ORR) and duration of response (DoR). Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
Enhertu is a specifically engineered HER2-directed antibody drug conjugate (ADC) being jointly developed and commercialised by AstraZeneca and Daiichi Sankyo.
The accelerated approval by the FDA was based on the results from the DESTINY-Lung02 Phase II trial. An interim efficacy analysis in a pre-specified patient cohort showed Enhertu (5.4mg/kg) demonstrated a confirmed ORR of 57.7% (n=52; 95% confidence interval [CI] 43.2-71.3), as assessed by blinded independent central review (BICR), in patients with previously treated unresectable or metastatic non-squamous HER2-mutant (HER2m) NSCLC. Complete responses (CR) were seen in 1.9% of patients and partial responses (PR) in 55.8% of patients with a median DoR of 8.7 months (95% CI 7.1-NE). Results from the DESTINY-Lung02 trial will be presented at an upcoming medical meeting.
Bob T. Li, MD, PhD, MPH, Medical Oncologist and Physician-Scientist, Memorial Sloan Kettering Cancer Center, US, said: “The approval of trastuzumab deruxtecan in non-small cell lung cancer is an important milestone for patients and the oncology community. After two decades of research into the role of targeting HER2 in lung cancer, the approval of the first HER2-directed treatment option validates HER2 as an actionable target in lung cancer and marks an important step forward for treating this patient population with unmet medical needs.”
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “HER2-mutant non-small cell lung cancer is an aggressive form of disease which commonly affects young patients who have faced limited treatment options and a poor prognosis to date. Today’s news provides these patients with the opportunity to benefit from a targeted therapy and highlights the importance of testing for predictive markers, including HER2 in lung cancer, at the time of diagnosis to ensure patients receive the most appropriate treatment for their specific disease.”
Ken Keller, Global Head of Oncology Business, and President and CEO, Daiichi Sankyo, Inc, said: “We are excited that the FDA has granted accelerated approval for Enhertu for patients with HER2-mutant metastatic non-small cell lung cancer. Enhertu has now been approved in three different tumour types, underscoring its significant potential across several HER2-targetable tumours. We are continuing to evaluate the efficacy and safety of Enhertu versus standard chemotherapy in our DESTINY clinical trials in lung cancer.”
The safety of Enhertu was evaluated in an analysis of 101 patients with unresectable or metastatic HER2m NSCLC who received at least one dose of Enhertu (5.4mg/kg) in the DESTINY-Lung02 trial. In the analysis, the safety profile of Enhertu was consistent with previous clinical trials with no new safety concerns identified.
Concurrently with this approval, the FDA also approved companion diagnostic tests to detect HER2 mutations in lung tumour tissue and plasma. This is the third tumour type approved by the FDA for Enhertu in three years, following approval in breast and gastric cancers. The approval follows the recently received Priority Review in the US as well as the Breakthrough Therapy Designation granted in 2020 by the FDA for this specific type of lung cancer based on the results of the DESTINY-Lung01 trial.
HER2m NSCLC
Lung cancer is the second most common form of cancer globally, with more than two million patients diagnosed in 2020.1 In the US, lung cancer is the second most commonly diagnosed cancer, with more than 236,000 patients expected to be diagnosed in 2022.2 For patients with metastatic NSCLC, prognosis is particularly poor, as only approximately 8% will live beyond five years after diagnosis.3
HER2 is a tyrosine kinase receptor growth-promoting protein expressed on the surface of many types of tumours, including lung, breast, gastric and colorectal cancers. Certain HER2 gene alterations (called HER2 mutations) have been identified in patients with non-squamous NSCLC as a distinct molecular target, and occur in approximately 2-4% of patients with this type of lung cancer.4,5 Next-generation sequencing is used to identify HER2 (ERBB2) mutations.6
While HER2 gene mutations can occur in a range of patients, they are more commonly found in patients with NSCLC who are younger, female and have never smoked.7 HER2 gene mutations have been independently associated with cancer cell growth and poor prognosis, with an increased incidence of brain metastases.8 Although the role of anti-HER2 treatment is well established in breast and gastric cancers, there were no approved HER2-directed therapies in NSCLC prior to the accelerated approval of Enhertu in unresectable or metastatic NSCLC.8,9
DESTINY-Lung02
DESTINY-Lung02 is a global Phase II trial evaluating the safety and efficacy of two doses (5.4mg/kg or 6.4mg/kg) of Enhertu in patients with HER2m metastatic NSCLC with disease recurrence or progression during or after at least one regimen of prior anticancer therapy that must have contained a platinum-based chemotherapy.
The primary endpoint of the trial is ORR as assessed by BICR. Secondary endpoints include disease control rate (DCR), DoR, progression-free survival (PFS), investigator-assessed ORR, overall survival (OS) and safety.
DESTINY-Lung02 enrolled 152 patients at multiple sites, including Asia, Europe and North America. For more information about the trial, visit ClinicalTrials.gov.
DESTINY-Lung01
DESTINY-Lung01 is a global Phase II, open-label, two-cohort trial evaluating the safety and efficacy of Enhertu in patients with HER2m (6.4mg/kg) or HER2-overexpressing (defined as IHC 3+ or IHC 2+) [6.4mg/kg and 5.4mg/kg] unresectable and/or metastatic non-squamous NSCLC who had progressed after one or more systemic therapies. The primary endpoint is confirmed ORR by independent central review (ICR). Key secondary endpoints include DoR, DCR, PFS, OS and safety. DESTINY-Lung01 enrolled 181 patients at multiple sites, including Asia, Europe and North America.
Data from the DESTINY-Lung01 Phase II trial was published in The New England Journal of Medicine.10 Primary results from previously-treated patients with HER2-mutations (cohort 2) of DESTINY-Lung01 demonstrated an ORR of 54.9% (95% CI 44.2-65.4) in patients treated with Enhertu (6.4mg/kg) as assessed by ICR. One (1.1%) CR and 49 (53.8%) PRs were observed. A confirmed DCR of 92.3% was seen with a reduction in tumour size observed in most patients. After a median follow-up of 13.1 months, the median DoR for Enhertu was 9.3 months. The median PFS was 8.2 months and the median OS was 17.8 months.
The safety profile of the most common adverse events with Enhertu in DESTINY-Lung01 was consistent with previous clinical trials with no new safety concerns identified. For more information about the trial, visit ClinicalTrials.gov.
Enhertu
Enhertu is a HER2-directed ADC. Designed using Daiichi Sankyo’s proprietary DXd ADC technology, Enhertu is the lead ADC in the oncology portfolio of Daiichi Sankyo and the most advanced programme in AstraZeneca’s ADC scientific platform.
Enhertu consists of a HER2 monoclonal antibody attached to a topoisomerase I inhibitor payload, an exatecan derivative, via a stable tetrapeptide-based cleavable linker.
Enhertu (5.4mg/kg) is approved in more than 30 countries for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received a (or one or more) prior anti-HER2-based regimen either in the metastatic setting, or in the neoadjuvant or adjuvant setting and have developed disease recurrence during or within six months of completing therapy based on the results from the DESTINY-Breast03 trial.
Enhertu (5.4mg/kg) is approved in several countries for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received two or more prior anti-HER2-based regimens based on the results from the DESTINY-Breast01 trial.
Enhertu (5.4mg/kg) is approved in the US for the treatment of adult patients with unresectable or metastatic HER2-low (IHC 1+ or IHC 2+/ISH-) breast cancer who have received a prior chemotherapy in the metastatic setting or developed disease recurrence during or within six months of completing adjuvant chemotherapy based on the results from the DESTINY-Breast04 trial.
Enhertu (5.4mg/kg) is approved in the US for the treatment of adult patients with unresectable or metastatic NSCLC whose tumours have activating HER2 (ERBB2) mutations, as detected by an FDA-approved test, and who have received a prior systemic therapy based on the results from the DESTINY-Lung02 trial.
Enhertu (6.4mg/kg) is approved in several countries for the treatment of adult patients with locally advanced or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma who have received a prior trastuzumab-based regimen based on the results from the DESTINY-Gastric01 trial.
Enhertu development programme
A comprehensive development programme is underway globally, evaluating the efficacy and safety of Enhertu monotherapy across multiple HER2-targetable cancers, including breast, gastric, lung and colorectal cancers. Trials in combination with other anticancer treatments, such as immunotherapy, are also underway.
Regulatory applications for Enhertu in breast and gastric cancer are currently under review in several countries based on the DESTINY-Breast01, DESTINY-Breast03, DESTINY-Breast04, DESTINY-Gastric01 and DESTINY-Gastric02 trials, respectively.
Daiichi Sankyo collaboration
Daiichi Sankyo Company, Limited (TSE: 4568) [referred to as Daiichi Sankyo] and AstraZeneca entered into a global collaboration to jointly develop and commercialise Enhertu (a HER2-directed ADC) in March 2019, and datopotamab deruxtecan (a TROP2-directed ADC) in July 2020, except in Japan where Daiichi Sankyo maintains exclusive rights. Daiichi Sankyo is responsible for the manufacturing and supply of Enhertu and datopotamab deruxtecan.
AstraZeneca in lung cancer
AstraZeneca is working to bring patients with lung cancer closer to cure through the detection and treatment of early-stage disease, while also pushing the boundaries of science to improve outcomes in the resistant and advanced settings. By defining new therapeutic targets and investigating innovative approaches, the Company aims to match medicines to the patients who can benefit most.
The Company’s comprehensive portfolio includes leading lung cancer medicines and the next wave of innovations, including Tagrisso (osimertinib) and Iressa (gefitinib); Imfinzi (durvalumab) and tremelimumab; Enhertu (trastuzumab deruxtecan) and datopotamab deruxtecan in collaboration with Daiichi Sankyo; Orpathys (savolitinib) in collaboration with HUTCHMED; as well as a pipeline of potential new medicines and combinations across diverse mechanisms of action.
AstraZeneca is a founding member of the Lung Ambition Alliance, a global coalition working to accelerate innovation and deliver meaningful improvements for people with lung cancer, including and beyond treatment.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Aug. 12, 2022 7:17 AM ET
AstraZeneca PLC (AZN), DSKYF, DSNKY
By: Ravikash, SA News Editor
PUBLISHED15 August 2022 15 August 2022 07:00 BST
https://seekingalpha.com/symbol/AZN
Thursday, Aug 11, 2022
South San Francisco, CA -- August 11, 2022 --
Genentech, a member of the Roche Group (SIX: RO, ROG; OTCQX: RHHBY), today announced that the U.S. Food and Drug Administration (FDA) has approved a supplemental New Drug Application (sNDA) for Xofluza® (baloxavir marboxil) for the treatment of acute uncomplicated influenza in otherwise healthy children aged five to less than 12 years of age who have been symptomatic for no more than 48 hours. This marks the first single-dose oral influenza medicine approved for children in this age group. Additionally, the FDA approved Xofluza for the prevention (post-exposure prophylaxis) of influenza in children aged five to less than 12 years of age following contact with someone with influenza.
“Despite the ongoing COVID-19 pandemic, influenza continues to be a threat to public health, and effective influenza antivirals remain critical to alleviating the burden on healthcare systems,” said Levi Garraway, M.D., Ph.D., chief medical officer and head of Global Product Development. “Xofluza has proven to be an important tool in fighting and preventing influenza in adults as well as adolescents, and we are pleased to now offer households and younger children our single-dose oral treatment.”
According to the Centers for Disease Control and Prevention, influenza can be a serious illness for young children. During the ongoing COVID-19 pandemic, there have been significantly fewer influenza cases likely due in large part to social distancing and mask wearing. However, in the U.S. 2018-2019 influenza season, there were more than 6 million illnesses, thousands of hospitalizations and more than 100 deaths for children aged five to 17 caused by influenza.
“Historically, school-aged children have played a significant role in the community transmission of influenza. The annual influenza vaccine continues to be the most important first step to prevent illness in children, though there can still be breakthrough cases where antiviral treatment is needed,” said Dr. Pedro Piedra, miniSTONE-2 study investigator and professor of molecular virology and microbiology, pediatrics at Baylor College of Medicine. “Today’s FDA approval provides children with a single-dose antiviral option, Xofluza, to treat influenza."
The FDA approval is based on results from two Phase III studies, miniSTONE-2 and BLOCKSTONE. miniSTONE-2 evaluated Xofluza compared with oseltamivir in otherwise healthy children and included patients aged five to less than 12 years with an influenza infection and displaying influenza symptoms for no more than 48 hours. BLOCKSTONE evaluated Xofluza compared with placebo as a preventive treatment for household members (adults and children) who were living with someone with influenza. The results from these studies were published in The Pediatric Infectious Disease Journal and The New England Journal of Medicine respectively.
Adverse events reported in at least 5% of pediatric patients (ages five to 11 years) treated with Xofluza included vomiting (5%) and diarrhea (5%).
Xofluza is already FDA-approved to treat influenza in people 12 years of age and older who have had influenza symptoms for no more than 48 hours and who are otherwise healthy or at high risk of developing influenza-related complications. Xofluza is also approved to prevent influenza in people 12 years of age and older following contact with someone with influenza (known as post-exposure prophylaxis). Xofluza is available as a one-dose, single tablet.
About Xofluza® (baloxavir marboxil)
Xofluza is a first-in-class, single-dose oral medicine with an innovative proposed mechanism of action that has demonstrated efficacy in a wide range of influenza viruses, including in vitro activity against oseltamivir-resistant strains and avian strains (H7N9, H5N1) in non-clinical studies. Xofluza is the first in a class of antivirals designed to inhibit the cap-dependent endonuclease protein, which is essential for viral replication.
In October 2018, Xofluza was first approved by the FDA for the treatment of acute, uncomplicated influenza in otherwise healthy people 12 years of age and older who have been symptomatic for no more than 48 hours, representing the first new antiviral to treat influenza in the U.S. in over 20 years.
Xofluza is being further studied in a Phase III development program, including children under the age of one (NCT03653364) as well as to assess the potential to reduce direct transmission of influenza from otherwise healthy patients to household contacts (NCT03969212).
Xofluza was discovered by Shionogi & Co., Ltd. and is being further developed and commercialized globally in collaboration with the Roche Group (which includes Genentech in the U.S.) and Shionogi & Co., Ltd. Under the terms of this agreement, Roche holds worldwide rights to Xofluza excluding Japan and Taiwan, which will be retained exclusively by Shionogi & Co., Ltd.
Xofluza U.S. Indication
XOFLUZA is a prescription medicine used to:
XOFLUZA does not treat or prevent illness that is caused by infections other than the influenza virus.
XOFLUZA does not prevent bacterial infections that may happen with the flu.
It is not known if XOFLUZA is safe and effective for the treatment and prevention of the flu in children less than 5 years of age. XOFLUZA is not for use in children less than 5 years of age.
Please see full Prescribing Information, including Patient Product Information.
About Genentech
Founded more than 40 years ago, Genentech is a leading biotechnology company that discovers, develops, manufactures and commercializes medicines to treat patients with serious and life-threatening medical conditions. The company, a member of the Roche Group, has headquarters in South San Francisco, California. For additional information about the company, please visit http://www.gene.com.
Aug. 11, 2022 4:22 PM ET
Roche Holding AG (RHHBY), RHHBF
By: Anuron Mitra, SA News Editor
08/10/2022 CATEGORY:
Abecma is the first BCMA-directed CAR T cell therapy to demonstrate superiority versus standard regimens in relapsed and refractory multiple myeloma
PRINCETON, N.J., & CAMBRIDGE, Mass.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) and 2seventy bio, Inc. (Nasdaq: TSVT) today announced positive topline results from KarMMa-3, a Phase 3, global, randomized, multicenter, open-label study evaluating Abecma (idecabtagene vicleucel) compared to standard combination regimens in adults with multiple myeloma that is relapsed and refractory after two to four prior lines of therapy and refractory to the last regimen. KarMMa-3 is the first randomized clinical trial to evaluate a CAR T cell therapy in multiple myeloma. Results of a pre-specified interim analysis conducted through an independent review committee showed that KarMMa-3 met its primary endpoint of demonstrating a statistically significant improvement in progression-free survival. Treatment with Abecma also showed an improvement in the key secondary endpoint of overall response rate compared to standard regimens. Follow-up for overall survival, a key secondary endpoint, remains ongoing.
Safety results in the trial were consistent with the well-established and predictable safety profile of Abecma previously demonstrated in the pivotal KarMMa trial. No new safety signals were reported in this study.
“Results from the KarMMa-3 study clearly demonstrate the clinical benefit of using a CAR T cell therapy earlier in the multiple myeloma treatment paradigm,” said Anne Kerber, senior vice president, head of Cell Therapy Development, Bristol Myers Squibb. “These data reinforce our commitment to unlocking the full potential of cell therapy as we strive to build on the company’s heritage of innovation in blood cancers and transform patients’ lives through science.”
“We are extremely pleased to have met the KarMMa-3 primary endpoint at an interim analysis. These results help to advance our efforts to make Abecma available for earlier lines of treatment for patients and we look forward to discussing these results with regulatory authorities,” said Steve Bernstein, M.D., chief medical officer, 2seventy bio. “Today’s results are another important proof point for the transformative potential of autologous cell therapy and underscore the importance of continuing to study Abecma in earlier treatment settings for multiple myeloma.”
Bristol Myers Squibb and 2seventy bio will complete a full evaluation of the KarMMa-3 data and work with investigators to present detailed results at an upcoming medical meeting, as well as discuss these results with health authorities. The companies thank the patients and investigators who are participating in the KarMMa-3 clinical trial.
Abecma was approved by the U.S. Food and Drug Administration (FDA) in March 2021 for the treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody. Please see the Important Safety Information section below, including Boxed WARNINGS for Abecma regarding cytokine release syndrome, neurologic toxicities, Hemophagocytic Lymphohistiocytosis/Macrophage Activation Syndrome and Prolonged Cytopenia. Abecma is also approved in the European Union, Switzerland, Japan, Canada and the United Kingdom for adult patients with relapsed or refractory multiple myeloma who have received at least three prior therapies.
About KarMMa-3
KarMMa-3 (NCT03651128) is a pivotal, Phase 3, global, randomized, multicenter trial evaluating Abecma compared to standard regimens in patients with multiple myeloma that is relapsed and refractory after two to four prior lines of treatment and refractory to the last treatment regimen. Patients were randomized to receive Abecma or standard regimens that consisted of combinations that included daratumumab, pomalidomide, dexamethasone, bortezomib, ixazomib, lenalidomide, carfilzomib or elotuzumab. The primary endpoint evaluated in this study is progression-free survival, defined as time from randomization to the first documentation of progressive disease or death due to any cause, whichever occurs first. Key secondary endpoints include overall response rate and overall survival.
About Abecma
Abecma is the first-in-class B-cell maturation antigen (BCMA)-directed chimeric antigen receptor (CAR) T cell immunotherapy approved in the U.S. for the treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody. Abecma recognizes and binds to BCMA on the surface of multiple myeloma cells leading to CAR T cell proliferation, cytokine secretion, and subsequent cytolytic killing of BCMA-expressing cells. Abecma is being jointly developed and commercialized in the U.S. as part of a Co-Development, Co-Promotion, and Profit Share Agreement between Bristol Myers Squibb and 2seventy bio.
The companies’ broad clinical development program for Abecma includes clinical studies (KarMMa-2, KarMMa-3, KarMMa-7) in earlier lines of treatment for patients with multiple myeloma. For more information visit clinicaltrials.gov.
Please see full Prescribing Information, including Boxed WARNINGS and Medication Guide.
About Bristol Myers Squibb
Bristol Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information about Bristol Myers Squibb, visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
About 2seventy bio
Our name, 2seventy bio, reflects why we do what we do - TIME. Cancer rips time away, and our goal is to work at the maximum speed of translating human thought into action – 270 miles per hour – to give the people we serve more time. We are building the leading immuno-oncology cell therapy company, focused on discovering and developing new therapies that truly disrupt the cancer treatment landscape With a deep understanding of the human body’s immune response to tumor cells and how to translate cell therapies into practice, we’re applying this knowledge to deliver next generation cellular therapies that focus on a broad range of hematologic malignancies, including the first FDA-approved CAR T cell therapy for multiple myeloma, as well as solid tumors. Our research and development is focused on delivering therapies that are designed with the goal to “think” smarter and faster than the disease. Importantly, we remain focused on accomplishing these goals by staying genuine and authentic to our “why” and keeping our people and culture top of mind every day.
For more information, visit www.2seventybio.com.
Follow 2seventy bio on social media: Twitter and LinkedIn.
2seventy bio is a trademark of 2seventy bio, Inc.
Source: Bristol Myers Squibb
Aug. 10, 2022 7:16 AM ET
Bristol-Myers Squibb Company (BMY) TSVT
By: Dulan Lokuwithana, SA News Editor1 Comment
Bristol Myers Squibb (NYSE:BMY) and its partner 2seventy bio (TSVT) announced on Wednesday that a Phase 3 trial for Abecma, a CAR T cell therapy they jointly develop, reached the main goal in certain adult patients with multiple myeloma.
2seventy bio (TSVT) added ~18% after the news, while Bristol Myers (BMY) traded flat.
https://seekingalpha.com/symbol/TSVT
https://seekingalpha.com/symbol/BMY
August 10, 2022 at 6:49 AM EDT PDF Version
Abecma is the first BCMA-directed CAR T cell therapy to demonstrate superiority versus standard regimens in relapsed and refractory multiple myeloma
(PRINCETON, N.J., & CAMBRIDGE, Mass., AUGUST 10, 2022) --
Bristol Myers Squibb (NYSE: BMY) and 2seventy bio, Inc. (Nasdaq: TSVT)
Aug. 11, 2022 8:00 AM ET AstraZeneca PLC (AZN)
First and only approved medicine targeting germline BRCA mutations in high-risk early breast cancer
MISSISSAUGA, ON, Aug. 11, 2022 /CNW/ - Health Canada has granted a Notice of Compliance with Conditions (NOC/c) for Lynparza® (olaparib) for the adjuvant treatment of adult patients with deleterious or suspected deleterious germline BRCA-mutated (gBRCAm), human epidermal growth factor receptor 2 (HER2)-negative high-risk early breast cancer who have been treated with neoadjuvant or adjuvant chemotherapy.1 Patients must have confirmation of a germline BRCA mutation before Lynparza treatment is initiated.1

This approval was granted under Health Canada's accelerated review pathway and based on results from the OlympiA Phase III trial,1 presented during the 2021 American Society of Clinical Oncology Annual Meeting and published in The New England Journal of Medicine. The OlympiA trial demonstrated a reduction in the risk of invasive breast cancer recurrences, second cancers or death by 42 per cent versus placebo (based on a hazard ratio [HR] of 0.58; 95% confidence interval [CI] 0.41-0.82; p<0.0001).1
"Although most HER2-negative early breast cancers now have excellent outcomes, the risk of disease recurrence for persons with high-risk breast cancers and BRCA mutations remains unacceptably high, with a significant potential to advance to advanced disease, where a cure is no longer an option," said Dr. Karen Gelmon, Professor of Medicine, University of British Columbia and Medical Oncologist, BC Cancer. "Today's approval of olaparib is an important step forward, offering these patients a much-needed option that can improve survival for persons with high-risk early breast cancer. It also stresses the importance of BRCA testing as soon as possible after diagnosis to identify eligible patients."
In Canada, it is estimated that 94 per cent of all breast cancers are detected at an early stage.2 Despite advances in the treatment of early breast cancer, up to 30 per cent of patients with high-risk clinical and/or pathologic features recur within the first few years.3 The risk is particularly high for patients with germline BRCA mutations, who are more likely to be diagnosed at a younger age than those without these mutations.2
"This approval is fantastic news for the many women with high-risk early breast cancer who live with the ongoing fear of recurrence and what that means for their future," said Cathy Ammendolea, Chair of the Board, Canadian Breast Cancer Network. "New treatment options can provide hope for patients and their families, and we look forward to Canadian women having access to this treatment."
"Hereditary breast cancer is often diagnosed at a younger age and Rethink has supported and advocated for young people with breast cancer for more than 20 years. Facing their mortality at a young age, they want what all cancer patients want – care that will lead to the best possible outcome, including reducing their future cancer risk," said MJ DeCoteau, Founder and Executive Director of Rethink Breast Cancer. "They want to survive, and eventually thrive, and now patients with a BRCA-mutated breast cancer will have a treatment that is shown to help them do just that."
About the OlympiA Phase III Trial
OlympiA is a Phase III, double-blind, placebo-controlled, multicentre trial testing the efficacy and safety of Lynparza as adjuvant treatment in patients with gBRCAm HER2-negative high-risk early breast cancer, who have completed definitive local treatment and neoadjuvant or adjuvant chemotherapy.1 The primary endpoint was invasive disease-free survival (iDFS), and key secondary outcome measures include distant disease-free survival (DDFS) and overall survival (OS).1
In the key secondary endpoint, results showed an improvement of distant disease-free survival (DDFS) in the intention to treat (ITT) population, where Lynparza reduced the risk of distant disease recurrence or death by 43 per cent (based on a HR of 0.57 (99.5% CI:0.39-0.83; P < 0.001).1 Interim overall survival (8% maturity, DCO 27 March 2020) did not meet statistical significance.1
The safety and tolerability profile of Lynparza in this trial was in line with that observed in prior clinical trials.1
About Lynparza (olaparib)
Lynparza was the first oral (PARP) inhibitor approved in Canada. Lynparza exploits tumour DNA damage response (DDR) in cells/tumours harbouring a deficiency in homologous recombination repair (HRR), such as mutations in BRCA1 and/or BRCA2, to selectively kill cancer cells.1 Lynparza is the only PARP inhibitor currently approved in multiple tumour types in Canada including breast, ovarian, pancreatic and prostate cancers.
Lynparza has been issued conditional marketing authorization for the adjuvant treatment of adult patients with deleterious or suspected deleterious gBRCAm, HER2-negative high-risk early breast cancer who have been treated with neoadjuvant or adjuvant chemotherapy pending the results of studies to verify its clinical benefit. Patients should be advised of this conditional marketing authorization.
About AstraZeneca (AZN)
AstraZeneca is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialization of prescription medicines in Oncology, Rare Diseases, and Biopharmaceuticals, including Cardiovascular, Renal & Metabolism (CVRM), and Respiratory & Immunology. The company employs more than 1,200 people across Canada, including 700 employees at our head office and clinical research hub in Mississauga, Ontario. For more information, please visit the company's website at www.astrazeneca.ca.
SOURCE AstraZeneca Canada Inc.
https://seekingalpha.com/symbol/AZN
PUBLISHED9 August 2022
Initial results from the TROPION-Lung02 Phase Ib trial showed that datopotamab deruxtecan (Dato-DXd) in combination with pembrolizumab with or without platinum chemotherapy demonstrated promising clinical activity and a tolerable safety profile in patients with previously untreated or pretreated, advanced or metastatic non-small cell lung cancer (NSCLC) without actionable genomic alterations. Results were presented during a late-breaking presentation (#MA13.07) today at the International Association for the Study of Lung Cancer 2022 World Conference on Lung Cancer (WCLC).
Datopotamab deruxtecan is a specifically designed TROP2-directed DXd antibody drug conjugate (ADC) being jointly developed by AstraZeneca and Daiichi Sankyo.
NSCLC is diagnosed at an advanced stage in nearly 50% of patients and often has a poor prognosis with worsening outcomes after each line of subsequent therapy.1-3 While 1st-line treatment consisting of immunotherapy with or without chemotherapy has improved outcomes for patients with NSCLC without actionable genomic alterations, disease progression still occurs in majority of patients and additional treatment strategies in this setting are needed.4,5
Benjamin Philip Levy, MD, Clinical Director of Medical Oncology, Johns Hopkins Sidney Kimmel Cancer Center at Sibley Memorial Hospital, Associate Professor of Oncology at Johns Hopkins University School of Medicine and investigator in the TROPION-Lung02 trial, said: “Many patients with advanced non-small cell lung cancer still experience disease progression following initial treatment, underscoring the need for new therapeutic approaches. The initial results from the TROPION-Lung02 trial show encouraging efficacy and safety results when combining datopotamab deruxtecan and pembrolizumab with or without platinum chemotherapy and warrant further study in the 1st-line metastatic setting.”
Cristian Massacesi, Chief Medical Officer and Oncology Chief Development Officer, AstraZeneca, said: “Building on preliminary findings of datopotamab deruxtecan combination therapy in triple-negative breast cancer shared earlier this year, these initial results from TROPION-Lung02 reflect the broader promise of combining existing treatments with antibody drug conjugates. We look forward to continuing this important research with the goal of providing a new, effective treatment option for patients with advanced non-small cell lung cancer.”
Gilles Gallant, Senior Vice President, Global Head, Oncology Development, Oncology R&D, Daiichi Sankyo, said: “These early findings from TROPION-Lung02 are promising and represent the first lung cancer trial to report results combining a TROP2-directed ADC with an immune checkpoint inhibitor with or without platinum chemotherapy in patients with advanced or metastatic non-small cell lung cancer. These data support the initiation of the TROPION-Lung08 Phase III trial to further evaluate datopotamab deruxtecan in combination with pembrolizumab as a 1st-line combination treatment in patients with advanced non-small cell lung cancer without actionable genomic alterations.”
An interim analysis of the ongoing TROPION-Lung02 trial in patients with previously untreated or pretreated, advanced or metastatic NSCLC without actionable genomic alterations demonstrated a promising overall response rate (ORR) in the overall population of 37% (median follow-up of 6.5 months) in patients treated with datopotamab deruxtecan and pembrolizumab (doublet therapy) and an ORR of 41% (median follow-up of 4.4 months) in patients receiving datopotamab deruxtecan, pembrolizumab and platinum chemotherapy (triplet therapy). A disease control rate (DCR) of 84% was seen with both the doublet and triplet combination therapy in the overall population that comprised both 1st-line and 2nd-line settings.
In previously untreated patients, ORRs of 62% (eight of the 13 patients receiving doublet therapy) and 50% (10 of 20 patients receiving triplet therapy) were observed. Eight partial responses (PRs) were seen in patients receiving doublet therapy and 10 PRs (three pending confirmation) were seen in patients receiving triplet therapy. A DCR of 100% was observed with doublet therapy and a DCR of 90% was observed with triplet therapy.
Combinations with datopotamab deruxtecan demonstrated a tolerable safety profile, which supports further evaluation in ongoing studies. Grade 3 or greater treatment-emergent adverse events (TEAEs) occurred in 40% and 60% of patients in the doublet and triplet cohorts, respectively. The most frequent TEAEs of any Grade in the doublet and triplet cohorts respectively were stomatitis (56% and 29%), nausea (41% and 48%), decreased appetite (28% and 38%), fatigue (25% and 36%) and anaemia (16% and 36%). There were four interstitial lung disease (ILD) events determined as drug-related by an independent adjudication committee across both cohorts; two were adjudicated as Grade 1/2 events and two were adjudicated as Grade 3 events. No Grade 4 or Grade 5 ILD events were adjudicated as drug-related. At the time of the data cut-off, there were three potential ILD events pending adjudication. Three deaths occurred (two within the doublet cohort, one in the triplet cohort), none of which were determined as drug-related. Treatment discontinuations due to adverse events occurred in less than 22% of patients and datopotamab deruxtecan dose discontinuation occurred in 13% of patients.
Patients in TROPION-Lung02 receiving doublet therapy were previously treated with one median line of therapy, including platinum chemotherapy (60%) and immunotherapy (30%). In the triplet cohort, patients previously received platinum chemotherapy (35%) and immunotherapy (38%). Datopotamab deruxtecan-based combination as a 1st-line of therapy accounted for 33% and 63% of patients in doublet and triplet cohorts, respectively. As of the 2 May 2022 data cut-off, 53% and 77% of patients remained on the doublet and triplet therapy, respectively.
TROPION-Lung02
TROPION-Lung02 is an ongoing global, open-label, Phase Ib trial evaluating the safety and efficacy of datopotamab deruxtecan at two dose levels (4mg/kg and 6mg/kg) in combination with pembrolizumab (200mg) with or without platinum chemotherapy (carboplatin or cisplatin), in both previously untreated and pretreated patients with advanced or metastatic NSCLC without actionable genomic alterations (e.g., EGFR, ALK, ROS1, NTRK, BRAF, RET, MET or other known actionable alterations).
The primary endpoints of TROPION-Lung02 are dose-limiting toxicities and treatment-emergent adverse events. Secondary endpoints include ORR, duration of response, progression-free survival, overall survival, pharmacokinetics and anti-drug antibodies for datopotamab deruxtecan and pembrolizumab.
Datopotamab deruxtecan (Dato-DXd)
Datopotamab deruxtecan (Dato-DXd) is an investigational TROP2-directed ADC. Designed using Daiichi Sankyo’s proprietary DXd ADC technology, datopotamab deruxtecan is one of the most advanced programmes in AstraZeneca’s ADC scientific platform, and one of the three leading ADCs in the oncology pipeline of Daiichi Sankyo. Datopotamab deruxtecan is comprised of a humanised anti-TROP2 IgG1 monoclonal antibody, developed in collaboration with Sapporo Medical University, attached to a number of topoisomerase I inhibitor payloads, an exatecan derivative, via tetrapeptide-based cleavable linkers.
A comprehensive development programme called TROPION is underway globally with trials evaluating the efficacy and safety of datopotamab deruxtecan across multiple solid tumours, including triple negative breast cancer, HR-positive/HER2-negative breast cancer, NSCLC, small cell lung cancer, urothelial, gastric and oesophageal cancer. Trials in combination with other anticancer treatments, such as immunotherapy, also are underway.
Daiichi Sankyo collaboration
Daiichi Sankyo Limited (TSE: 4568) [referred to as Daiichi Sankyo] and AstraZeneca entered into a global collaboration to jointly develop and commercialise datopotamab deruxtecan in July 2020, except in Japan where Daiichi Sankyo maintains exclusive rights. Daiichi Sankyo is responsible for the manufacturing and supply of datopotamab deruxtecan.
AstraZeneca in lung cancer
AstraZeneca is working to bring patients with lung cancer closer to cure through the detection and treatment of early-stage disease, while also pushing the boundaries of science to improve outcomes in the resistant and advanced settings. By defining new therapeutic targets and investigating innovative approaches, the Company aims to match medicines to the patients who can benefit most.
The Company’s comprehensive portfolio includes leading lung cancer medicines and the next wave of innovations, including Tagrisso (osimertinib) and Iressa (gefitinib); Imfinzi (durvalumab) and tremelimumab; Enhertu (trastuzumab deruxtecan) and datopotamab deruxtecan in collaboration with Daiichi Sankyo; Orpathys (savolitinib) in collaboration with HUTCHMED; as well as a pipeline of potential new medicines and combinations across diverse mechanisms of action.
AstraZeneca is a founding member of the Lung Ambition Alliance, a global coalition working to accelerate innovation and deliver meaningful improvements for people with lung cancer, including and beyond treatment.
AstraZeneca in oncology
AstraZeneca is leading a revolution in oncology with the ambition to provide cures for cancer in every form, following the science to understand cancer and all its complexities to discover, develop and deliver life-changing medicines to patients.
The Company's focus is on some of the most challenging cancers. It is through persistent innovation that AstraZeneca has built one of the most diverse portfolios and pipelines in the industry, with the potential to catalyse changes in the practice of medicine and transform the patient experience.
AstraZeneca has the vision to redefine cancer care and, one day, eliminate cancer as a cause of death.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Aug. 09, 2022 7:10 AM ET
AstraZeneca PLC (AZN), DSNKY, DSKYFMRK
By: Ravikash, SA News Editor
AstraZeneca (NASDAQ:AZN) and Daiichi Sankyo (OTCPK:DSKYF) (OTCPK:DSNKY) said their medicine datopotamab deruxtecan (Dato-DXd) in combination with Merck's (MRK) Keytruda (pembrolizumab), with or without platinum chemotherapy, showed promising clinical activity and a tolerable safety profile in certain patients with a type of lung cancer.
https://seekingalpha.com/symbol/DSKYF
https://seekingalpha.com/symbol/DSNKY
https://seekingalpha.com/symbol/AZN
August 09, 2022
https://www.daiichisankyo.com/
PUBLISHED8 August 2022
Preliminary results from the SAVANNAH Phase II trial showed that Tagrisso (osimertinib) plus savolitinib demonstrated an objective response rate (ORR) of 49% (95% confidence interval [CI], 39-59%) in patients with epidermal growth factor receptor-mutated (EGFRm) non-small cell lung cancer (NSCLC) with high levels of MET overexpression and/or amplification, defined as IHC90+ and/or FISH10+, whose disease progressed on treatment with Tagrisso.
The highest ORR was observed in patients with high levels of MET who were not treated with prior chemotherapy (52% [95% CI, 41-63%]). In patients whose tumours did not show high levels of MET, the ORR was 9% (95% CI, 4-18%). These results are being shared at the International Association for the Study of Lung Cancer 2022 World Conference on Lung Cancer, taking place 6-9 August in Vienna, Austria.
Savolitinib, marketed in China under the brand name Orpathys, is an oral, potent, and highly selective MET tyrosine kinase inhibitor (TKI) being jointly developed and commercialised by AstraZeneca and HUTCHMED.
While EGFR-targeted therapy can provide a durable survival benefit to patients with EGFRm NSCLC, most will eventually develop resistance to their treatment, with MET being the most common resistance biomarker.1 Among patients screened for enrolment in SAVANNAH, all of whom experienced disease progression on Tagrisso, 62% had tumours with MET overexpression and/or amplification, and more than one-third (34%) met the defined high MET level cut-off.
Myung-Ju Ahn, MD, PhD, Professor of Hemato-Oncology at the Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, South Korea, and Principal Investigator in the SAVANNAH Phase II trial, said: “Acquired resistance to targeted therapy and disease progression are difficult realities for most patients with EGFR-mutated non-small cell lung cancer. These preliminary SAVANNAH results potentially support a novel approach for identifying patients with MET overexpression and/or amplification who are most likely to benefit from a MET-directed therapy, like savolitinib. They also suggest that with the right biomarker testing strategy, MET may be a more prevalent target among resistant patients than previously understood, supporting further investigation of the osimertinib plus savolitinib regimen.”
Cristian Massacesi, Chief Medical Officer and Oncology Chief Development Officer, AstraZeneca, said: “The current standard of care for patients with EGFR-mutated lung cancer who progress on targeted treatment is chemotherapy. The results from SAVANNAH suggest savolitinib added to Tagrisso at the time of disease progression could possibly provide these biomarker-selected patients with a potentially less toxic, more effective treatment option. We look forward to better understanding the potential of the Tagrisso plus savolitinib regimen in this trial and in the SAFFRON Phase III trial.”
Weiguo Su, Chief Executive Officer and Chief Scientific Officer, HUTCHMED, said: “It is encouraging to see the savolitinib and Tagrisso combination regimen progress into a global Phase III study, SAFFRON, with a well-supported patient selection strategy that could benefit more patients than previously recognized. The preliminary results of the SAVANNAH study also affirm the role of molecular testing prior to initiating subsequent treatment for non-small cell lung cancer patients who experience disease progression on an EGFR-targeted therapy. We are aligned in pursuing a selective, patient-centric approach in development efforts for savolitinib in this setting.”
In this analysis, patients’ MET overexpression and/or amplification levels were determined by two tests: immunohistochemistry (IHC), which detects if cancer cells have a particular protein or marker on their surface, and fluorescence in situ hybridisation (FISH), which detects a specific DNA sequence from cancer cells. All patients in this analysis (n=193) had at least IHC50+ and/or FISH5+, and were treated with savolitinib 300mg once daily added to Tagrisso 80mg once daily following disease progression on Tagrisso alone.
SAVANNAH
SAVANNAH is an ongoing global, randomised, single-arm Phase II trial studying the efficacy of savolitinib added to Tagrisso in patients with EGFRm, locally advanced or metastatic NSCLC with MET overexpression and/or amplification who progressed following treatment with Tagrisso. Patients were treated with savolitinib 300 or 600 mg once-daily (QD) or 300 mg twice-daily, in combination with oral osimertinib 80 mg QD.
The trial has enrolled 294 patients to date in more than 80 centres globally, including centres in the US, Canada, Europe, South America and Asia. The primary endpoint is ORR. Key secondary endpoints include PFS, DoR and safety.
Tagrisso
Tagrisso (osimertinib) is a third-generation, irreversible EGFR-TKI with proven clinical activity in NSCLC, including against central nervous system metastases. Tagrisso (40mg and 80mg once-daily oral tablets) has been used to treat approximately 575,000 patients across indications worldwide and AstraZeneca continues to explore Tagrisso as a treatment for patients across multiple stages of EGFRm NSCLC.
In Phase III trials, Tagrisso is being investigated in the neoadjuvant resectable setting (NeoADAURA), in the Stage IA2-IA3 adjuvant resectable setting (ADAURA2), in the Stage III locally advanced unresectable setting following chemoradiation therapy (LAURA), and in combination with chemotherapy in the advanced setting (FLAURA2). AstraZeneca is also researching ways to address tumour mechanisms of resistance through the SAVANNAH and ORCHARD Phase II trials, and the SAFFRON Phase III trial, which test Tagrisso given concomitantly with savolitinib, an oral, potent and highly selective MET TKI, as well as other potential new medicines.
Savolitinib
Savolitinib is an oral, potent, and highly selective MET TKI that has demonstrated clinical activity in advanced solid tumours. It blocks atypical activation of the MET receptor tyrosine kinase pathway that occurs because of mutations (such as exon 14 skipping alterations or other point mutations), gene amplification or protein overexpression.
Savolitinib is marketed in China under the brand name Orpathys for the treatment of patients with NSCLC with MET exon 14 skipping alterations who have progressed following prior systemic therapy or are unable to receive chemotherapy. It is currently under clinical development for multiple tumour types, including lung, kidney, and gastric cancers, as a single treatment and in combination with other medicines.
AstraZeneca and HUTCHMED collaboration
In 2011, AstraZeneca and HUTCHMED entered a global licensing and collaboration agreement to jointly develop and commercialise savolitinib. Joint development of savolitinib in China is led by HUTCHMED, while AstraZeneca leads development outside of China. HUTCHMED is responsible for the marketing authorisation, manufacturing and supply of savolitinib in China. AstraZeneca is responsible for the commercialisation of savolitinib in China and worldwide. Sales of savolitinib are recognised by AstraZeneca.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development, and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Aug. 08, 2022 8:11 AM ET AstraZeneca PLC (AZN), HCM
By: Ravikash, SA News Editor
AstraZeneca (NASDAQ:AZN) said its drug Tagrisso and savolitinib, developed with Hutchmed (China) (NASDAQ:HCM), showed response in certain patients with non-small cell lung cancer (NSCLC) in a phase 2 trial called SAVANNAH.
https://seekingalpha.com/symbol/HCM
https://seekingalpha.com/symbol/AZN
8 Aug 2022
— MET is the most common biomarker in patients with EGFR-mutated lung cancer who develop resistance to targeted therapy —
— Global SAFFRON Phase III trial evaluating this combination is underway —
Hong Kong, Shanghai & Florham Park, NJ — Monday, August 8, 2022: HUTCHMED (China) Limited (“HUTCHMED”) (Nasdaq/AIM: HCM, HKEX: 13) and AstraZeneca PLC (“AstraZeneca”) (LON/STO/Nasdaq: AZN)
https://www.hutch-med.com/savannah-study-wclc2022/
https://www.hutch-med.com/pipeline-and-products/our-pipeline/
PUBLISHED5 August 2022
AstraZeneca’s new tablet formulation of Calquence (acalabrutinib) has been approved in the US for all current indications, including adult patients with chronic lymphocytic leukaemia (CLL), small lymphocytic lymphoma (SLL) and for patients with relapsed or refractory mantle cell lymphoma (MCL), which is approved under accelerated approval based on overall response rate.
The approval by the US Food and Drug Administration (FDA) was based on results from the ELEVATE-PLUS trials presented during the 63rd American Society of Hematology (ASH) Annual Meeting & Exposition in December 2021.1
In the trials, results showed the Calquence capsule and tablet formulations are bioequivalent, indicating the same efficacy and safety profile can be expected with the same dosing strength and schedule.1 The tablet can be taken with gastric acid-reducing agents, including proton pump inhibitors (PPIs), antacids and H2-receptor antagonists (H2RAs).1,2 The majority of observed adverse events (AEs) in these studies were mild with no new safety concerns identified.1
John C. Byrd, MD, Chair of the Department of Internal Medicine at the University of Cincinnati, said: “Patients with blood cancers like chronic lymphocytic leukaemia and mantle cell lymphoma are often older and may face multiple medical conditions that may need intervention, including acid reflux or peptic ulcer disease. The US approval of acalabrutinib in a tablet form enables co-administration of the acalabrutinib tablet alongside a proton pump inhibitor. This provides another option for some patients with chronic lymphocytic leukaemia and relapsed or refractory mantle cell lymphoma, enabling more patients to potentially benefit from this treatment.”
Dave Fredrickson, Executive Vice President, Oncology Business Unit, AstraZeneca, said: “Today’s approval of the new Calquence tablet formulation will offer physicians and patients increased flexibility when devising treatment plans for chronic lymphocytic leukaemia and mantle cell lymphoma. This new option is a result of our focus on understanding the wants and needs of this community and providing patient-focused solutions for their treatment.”
Calquence is also approved as a capsule formulation for the same indications as the tablet in the US and in many other countries worldwide.3 Indications may vary by market.
ELEVATE-PLUS
ELEVATE-PLUS is comprised of three Phase I, open-label, single-dose, cross-over studies conducted in 116 healthy subjects. The trials established bioequivalence between acalabrutinib tablets (100mg) and acalabrutinib (100mg) capsules, evaluated the PPI effect of acalabrutinib tablets administered in the presence versus absence of PPI rabeprazole and investigated the effect of food by comparing acalabrutinib tablets administered with a high-fat diet versus fasted.1
Calquence
Calquence (acalabrutinib) is a next-generation, selective inhibitor of Bruton’s tyrosine kinase (BTK). Calquence binds covalently to BTK, thereby inhibiting its activity.3,10 In B cells, BTK signalling results in activation of pathways necessary for B-cell proliferation, trafficking, chemotaxis and adhesion.3
Calquence is available for prescribing in capsule and tablet formulations in the US. Calquence tablets and capsules are approved in the US for the treatment of CLL and SLL, and for the treatment of adult patients with MCL who have received at least one prior therapy. Capsules have restrictions in relation to use with gastric acid reducing agents. The tablets are not licensed in the European Union.
Calquence capsules are approved for CLL in the EU and many other countries worldwide and approved in Japan for relapsed or refractory CLL and SLL. A Phase I trial is currently underway in Japan for the treatment of front-line CLL.
In the US and several other countries, Calquence capsules are also approved for the treatment of adult patients with MCL who have received at least one prior therapy. The US MCL indication is approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials. Calquence is not currently approved for the treatment of MCL in Europe or Japan.
As part of an extensive clinical development programme, AstraZeneca and Acerta Pharma are currently evaluating Calquence in more than 20 company-sponsored clinical trials. Calquence is being evaluated for the treatment of multiple B-cell blood cancers, including CLL, MCL, diffuse large B-cell lymphoma, Waldenström’s macroglobulinaemia, follicular lymphoma and marginal zone lymphoma.
AstraZeneca
AstraZeneca (LSE/STO/Nasdaq: AZN) is a global, science-led biopharmaceutical company that focuses on the discovery, development and commercialisation of prescription medicines in Oncology, Rare Diseases, and BioPharmaceuticals, including Cardiovascular, Renal & Metabolism, and Respiratory & Immunology. Based in Cambridge, UK, AstraZeneca operates in over 100 countries and its innovative medicines are used by millions of patients worldwide. Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca
Aug. 08, 2022 5:13 AM ET
By: Ravikash, SA News Editor1 Comment
August 8, 2022Download PDF
Since the medicine's first approval in 2016, nearly 130,000 people in the U.S. have been treated with Taltz
INDIANAPOLIS, Aug. 8, 2022 /PRNewswire/ -- Eli Lilly and Company (NYSE: LLY) announced today the availability of a new, citrate-free formulation of Taltz® (ixekizumab) injection 80 mg/mL. The new formulation, which was recently approved by The U.S. Food and Drug Administration in May 2022, includes the same active ingredient as the original formulation. The new Taltz formulation significantly reduced injection site pain experienced by some people immediately following injection as shown by an 86% decrease in a visual analog scale (VAS) of pain versus the original formulation. Taltz is approved to treat adults and children six years and older with moderate to severe plaque psoriasis who are candidates for systemic therapy or phototherapy, adults with active psoriatic arthritis, active ankylosing spondylitis (AS) and active non-radiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation.
"Taltz has long delivered effective treatment with a well-established safety profile that addresses symptoms for people living with plaque psoriasis, psoriatic arthritis, AS and nr-axSpA," said Ashley Diaz-Granados, vice president, U.S. Immunology at Lilly. "We're proud of our investment in research that keeps the patient experience at the center. This new formulation provides yet another reason to choose Taltz, and we look forward to introducing it to patients who have not yet tried Taltz and providing a seamless transition for those already enjoying the medicine's benefits."
Taltz citrate-free demonstrated a safety profile consistent with the original formulation. The safety information for Taltz can be found below.
Existing Taltz patients will not need a new prescription, nor should they experience a gap in their therapy. The new formulation is currently being shipped across the U.S. with anticipated broad availability for both new and existing Taltz patients by the end of the month. In the interim, the original formulation of Taltz continues to be available until it is replaced by the citrate-free formulation. The citrate-free formulation of Taltz was approved by the European Medicines Agency in December 2021 with several markets launching now and many more anticipated in the coming months.
"Today is an exciting milestone for the nearly 30 million people around the world who live with the challenging symptoms of these autoimmune diseases that affect the skin and joints," said April Armstrong, M.D., MPH, professor of dermatology and associate dean of clinical research, Keck School of Medicine at the University of Southern California. "In my six years of prescribing Taltz, I've seen firsthand the significant impact Taltz has had for patients across multiple indications. The availability of Taltz as a citrate-free formulation represents an important advance in patient care that will allow more patients to experience less injection-site pain."
Lilly is committed to improving experiences for people treated with Taltz, providing the same active ingredient in a new citrate-free formulation. Lilly's investment into patient-centric research is evident as Taltz has been studied in more than 10,000 people in clinical trials globally and has been available in most markets for more than five years.1 In the U.S., more people living with psoriasis are treated with Taltz compared to any other IL-17A antagonist, adding to the nearly 130,000 people in the U.S. who have been treated with the medicine.2
To learn more about real success stories with Taltz, please visit Taltz.com.
IMPORTANT SAFETY INFORMATION FOR TALTZ
CONTRAINDICATIONS
Taltz is contraindicated in patients with a previous serious hypersensitivity reaction, such as anaphylaxis, to ixekizumab or to any of the excipients.
Please see full Prescribing Information and Medication Guide for Taltz. See Instructions for Use included with the device.
About Taltz®
Taltz® (ixekizumab) is a monoclonal antibody that selectively binds with interleukin 17A (IL-17A) cytokine and inhibits its interaction with the IL-17 receptor. IL-17A is a naturally occurring cytokine that is involved in normal inflammatory and immune responses. Taltz inhibits the release of pro-inflammatory cytokines and chemokines.
About the Citrate-Free Injection Pain Study
The citrate-free injection pain study (N=70) was a subject-blind, randomized, crossover study in healthy participants ages 18-75 years to determine injection site pain differences between Taltz citrate-free formulation compared to the original formulation of Taltz. The primary endpoint, pain intensity on injection, was measured by the Visual Analog Scale (VAS) of pain 0-100mm.3
About Lilly
Lilly unites caring with discovery to create medicines that make life better for people around the world. We've been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world's most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer's disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we're motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn.
View original content to download multimedia:https://www.prnewswire.com/news-releases/lillys-taltz-ixekizumab-now-available-in-new-citrate-free-formulation-to-reduce-injection-site-pain-for-improved-patient-experience-301601067.html
SOURCE Eli Lilly and Company
Aug. 08, 2022 7:31 AM ET
By: Ravikash, SA News Editor
Eli Lilly (NYSE:LLY) said that a new, citrate-free formulation of its arthritis/psoriasis drug Taltz (ixekizumab) injection 80 mg/mL was now available.
https://seekingalpha.com/symbol/LLY
This press-release (German version) is protected by “doc-check”
SPECIALIZED PRESS
Monday - August 8, 2022
Not intended for U.S. and UK Media
Darolutamide now has indications in both non-metastatic castration-resistant prostate cancer (nmCRPC) for men at high risk of developing metastatic disease and metastatic hormone-sensitive prostate cancer (mHSPC) / Today’s approval under the FDA’s Real-Time Oncology Review (RTOR) pilot program was based on the pivotal Phase III ARASENS trial, which showed a significant overall survival (OS) benefit with darolutamide plus androgen deprivation therapy (ADT) and docetaxel compared to ADT plus docetaxel / Darolutamide is the only androgen receptor inhibitor (ARi) approved in combination with docetaxel, demonstrating a 32.5% reduction in the risk of death compared to docetaxel alone and with a proven tolerability profile
Berlin, August 8, 2022 – Bayer announced today the U.S. Food and Drug Administration (FDA) has approved a supplemental New Drug Application (sNDA) for the oral androgen receptor inhibitor (ARi) darolutamide in combination with docetaxel for the treatment of patients with metastatic hormone-sensitive prostate cancer (mHSPC).
The approval is based on positive results of the Phase III ARASENS trial that demonstrated darolutamide plus androgen deprivation therapy (ADT) and docetaxel significantly reduced the risk of death by 32.5% compared to ADT plus docetaxel. These results were recently published in The New England Journal of Medicine. Darolutamide is approved in more than 70 markets around the world, including the U.S., the European Union (EU), Japan and China, under the brand name Nubeqa™, for the treatment of patients with non-metastatic castration-resistant prostate cancer (nmCRPC), who are at high risk of developing metastatic disease. The compound is also being investigated in further studies across various stages of prostate cancer.
“Darolutamide plus ADT and docetaxel has shown significant benefit in overall survival and a favorable safety profile for patients with metastatic hormone-sensitive prostate cancer,” said Matthew Smith, M.D., Ph.D., Director of the Genitourinary Malignancies Program, Massachusetts General Hospital Cancer Center. “This new indication for darolutamide is particularly meaningful, as it highlights its proven tolerability and provides a new option for patients.”
Prostate cancer remains the second leading cancer-related cause of death among men in the U.S., with up to one-third of patients developing metastatic disease.1,2 The incidence of mHSPC has increased by 72% in the U.S. over the past 10 years.3 Approximately one in three patients who are diagnosed with mHSPC survive the disease five years or longer, with most eventually experiencing progression to castration-resistant prostate cancer (CRPC).2,3
“With compelling data from the Phase III ARASENS and ARAMIS trials, darolutamide has now demonstrated significant efficacy and tolerability in mHSPC, in addition to high-risk nmCRPC, and extends benefit to a much broader population,” said Christine Roth, Member of the Executive Committee of Bayer’s Pharmaceuticals Division and Head of the Oncology SBU at Bayer. “This approval reaffirms Bayer’s commitment to patients across different stages of prostate cancer, providing these eligible patients with a reliable treatment option.”
“Prostate cancer is the most common cancer among men in the U.S., with chances of survival decreasing dramatically for those diagnosed with mHSPC compared to localized prostate cancer,” said Charles J. Ryan, M.D., President and Chief Executive Officer, Prostate Cancer Foundation (PCF). “This approval adds a different treatment approach for mHSPC patients and their physicians to choose from.”
The application received Priority Review designation granted by the FDA and was submitted under the FDA’s Real-Time Oncology Review (RTOR) pilot program, which aims to provide a more efficient review process of applications to ensure that safe and effective cancer treatments are available to patients as early as possible. Ongoing reviews are also being conducted under the FDA Oncology Center of Excellence's (OCE) Project Orbis initiative, which provides a framework for concurrent submission and review of cancer treatments among participating international health authorities.
Darolutamide is developed jointly by Bayer and Orion Corporation, a globally operating Finnish pharmaceutical company.
About the ARASENS Trial
The ARASENS trial is the only randomized, Phase III, multi-center, double-blind, trial which was prospectively designed to compare the use of a second-generation oral androgen receptor inhibitor (ARi) darolutamide plus ADT and chemotherapy docetaxel to ADT plus docetaxel (a guideline recommended standard-of-care) in metastatic hormone-sensitive prostate cancer (mHSPC). A total of 1,306 newly diagnosed patients were randomized in a 1:1 ratio to receive 600 mg of darolutamide twice a day or matching placebo, plus ADT and docetaxel.
The primary endpoint of this trial was overall survival (OS). Secondary endpoints included time to castration-resistant prostate cancer (CRPC), time to pain progression, time to first symptomatic skeletal event (SSE), time to initiation of subsequent anticancer therapy, all evaluated at 12‐week intervals, as well as adverse events (AEs) as a measure of safety and tolerability. Results from this trial were published in the New England Journal of Medicine4. The ARASENS trial demonstrated that darolutamide plus androgen deprivation therapy (ADT) and docetaxel significantly reduced the risk of death by 32.5% compared to ADT plus docetaxel4. Improvements in the secondary endpoints supported the benefit observed in the primary endpoint, overall survival4.
About Nubeqa™ (darolutamide)
Darolutamide is an oral androgen receptor inhibitor (ARi) with a distinct chemical structure that binds to the receptor with high affinity and exhibits strong antagonistic activity, thereby inhibiting the receptor function and the growth of prostate cancer cells. The low potential for blood-brain barrier penetration for darolutamide is supported by preclinical models and neuroimaging data in healthy humans. This is supported by the overall low incidence of central nervous system (CNS)-related adverse events (AEs) compared to placebo as seen in the ARAMIS Phase III trial and the improved verbal learning and memory observed in the darolutamide arm of the Phase II ODENZA trial.
The product is approved under the brand name Nubeqa™ in more than 70 markets around the world, including the U.S., EU, Japan, China, for the treatment of patients with non-metastatic castration-resistant prostate cancer (nmCRPC), who are at high risk of developing metastatic disease. The compound is also being investigated in further studies across various stages of prostate cancer, including in the ARANOTE Phase III trial evaluating darolutamide plus androgen deprivation therapy (ADT) versus ADT alone for metastatic hormone-sensitive prostate cancer (mHSPC), as well as the Australian and New Zealand Urogenital and Prostate Cancer Trials Group (ANZUP) led international Phase III co-operative group DASL-HiCaP (ANZUP1801) trial evaluating darolutamide as an adjuvant treatment for localized prostate cancer with very high risk of recurrence. Information about these trials can be found at www.clinicaltrials.gov.
About Bayer
Bayer is a global enterprise with core competencies in the life science fields of health care and nutrition. Its products and services are designed to help people and the planet thrive by supporting efforts to master the major challenges presented by a growing and aging global population. Bayer is committed to driving sustainable development and generating a positive impact with its businesses. At the same time, the Group aims to increase its earning power and create value through innovation and growth. The Bayer brand stands for trust, reliability and quality throughout the world. In fiscal 2021, the Group employed around 100,000 people and had sales of 44.1 billion euros. R&D expenses before special items amounted to 5.3 billion euros. For more information, go to www.bayer.com.
Aug. 08, 2022 4:38 AM ET
Bayer Aktiengesellschaft (BAYRY), BAYZF
By: Ravikash, SA News Editor

