Moorestown, New Jersey, United States


February 9, 2022 5:00 pm ET
KENILWORTH, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, today announced the publication of results from the Phase 3 KEYNOTE-522 trial in the Feb. 10, 2022 edition of the New England Journal of Medicine. Results showed that neoadjuvant KEYTRUDA, Merck’s anti-PD-1 therapy, in combination with chemotherapy followed by adjuvant KEYTRUDA as monotherapy (the KEYTRUDA regimen), significantly prolonged event-free survival (EFS) compared with neoadjuvant chemotherapy followed by adjuvant placebo (the chemotherapy-placebo regimen) in patients with high-risk early-stage triple-negative breast cancer (TNBC).
As previously reported, after a median follow-up of 39 months, the KEYTRUDA regimen reduced the risk of events or death by 37% (HR=0.63 [95% CI, 0.48-0.82]; p<0.001) versus the chemotherapy-placebo regimen. A total of 15.7% (n= 123/784) of patients who received the KEYTRUDA regimen had an EFS event compared to 23.8% (n= 93/390) of patients who received the chemotherapy-placebo regimen. The estimated three-year EFS rates were 84.5% (95% CI, 81.7-86.9) in the KEYTRUDA group compared with 76.8% (95% CI, 72.2-80.7) in the chemotherapy-placebo group. Event-free survival was defined as the time from randomization to the first occurrence of either disease progression that precluded definitive surgery, a local/distant recurrence, a second primary malignancy, or death from any cause. The safety profile of the KEYTRUDA regimen was consistent with the known profiles of each regimen, and no new safety concerns were identified. Additional detailed efficacy and safety data are also featured in the publication.
KEYTRUDA in combination with chemotherapy as neoadjuvant treatment and then continued as a single agent as adjuvant treatment after surgery is approved in the U.S. for the treatment of patients with high-risk early-stage TNBC. Additional global regulatory submissions are ongoing.
“These data, which supported the FDA approval and updates to the NCCN guidelines, establish that KEYTRUDA plus chemotherapy as neoadjuvant therapy followed by adjuvant KEYTRUDA could change clinical practice for the treatment of patients with high-risk early-stage TNBC,” said Dr. Peter Schmid, lead, Centre for Experimental Cancer Medicine, Barts Cancer Institute in London, England. “KEYNOTE-522 is the first prospective randomized Phase 3 trial to show an improvement in event-free survival among patients with stage II and stage III TNBC.”
“The study of KEYTRUDA in earlier disease states has long been a critical aspect of our clinical program,” Dr. Roy Baynes, senior vice president and head of global clinical development, chief medical officer, Merck Research Laboratories. “We are incredibly proud that the New England Journal of Medicine has chosen to publish data from this pivotal neoadjuvant/adjuvant trial of KEYTRUDA in high-risk early-stage TNBC for the second time. Results from KEYNOTE-522, in which the KEYTRUDA regimen showed a significant 37% reduction in the risk of EFS events compared to neoadjuvant chemotherapy, represent an important advance and support earlier intervention with KEYTRUDA in these patients.”
Merck is rapidly advancing a broad portfolio in gynecologic and breast cancers with an extensive clinical development program for KEYTRUDA and several other investigational and approved medicines across these areas.
Study Design and Additional Data From KEYNOTE-522
KEYNOTE-522 is a Phase 3, randomized, double-blind trial (ClinicalTrials.gov, NCT03036488). The dual primary endpoints are pathological complete response (pCR), defined as pathological stage ypT0/Tis ypN0 at the time of definitive surgery, and EFS, defined as the time from randomization to the first occurrence of either disease progression that precluded definitive surgery, a local/distant recurrence, a second primary cancer, or death from any cause in all patients randomized. Secondary endpoints include pCR rate using alternative definitions, overall survival (OS) in all patients randomized, pCR rate according to all definitions, EFS and OS in patients whose tumors express PD-L1 (Combined Positive Score [CPS] ≥1), safety and health-related quality of life assessments. The study enrolled 1,174 patients who were randomized 2:1 to receive either:
As previously announced, KEYNOTE-522 met the success criterion for the dual primary endpoint of pCR at the first interim analysis; pCR was observed in 64.8% of patients who received KEYTRUDA plus chemotherapy (n=260/401), an increase of 13.6% (p=0.00055) from 51.2% in patients who received placebo plus chemotherapy (n=103/201). At the fourth interim analysis, KEYNOTE-522 met the success criterion for the dual primary endpoint of EFS. The trial also evaluated OS, a key secondary endpoint. Although the OS data did not cross the boundary for statistical significance at the fourth interim analysis, there was a 28% reduction in the risk of death with the KEYTRUDA regimen versus the chemotherapy-placebo regimen (HR=0.72 [95% CI, 0.51-1.02]). The study is continuing to allow for additional follow-up of OS.
For the combined neoadjuvant and adjuvant phases, treatment-related adverse events Grade 3 or higher occurred in 77.1% of patients receiving the KEYTRUDA regimen (n=783) and in 73.3% of patients receiving the chemotherapy-placebo regimen (n=389). Treatment-related adverse events led to death in 0.5% of patients receiving the KEYTRUDA regimen (n=4) and 0.3% of patients receiving the chemotherapy-placebo regimen (n=1). No new safety concerns were identified.
Immune-mediated adverse events (AEs) of any grade occurred in 33.5% of patients receiving the KEYTRUDA regimen and 11.3% of patients receiving the chemotherapy-placebo regimen. A higher incidence of endocrine disorders was observed with the KEYTRUDA regimen as compared to the chemotherapy-placebo regimen. Immune-mediated AEs led to death in 0.3% of patients receiving the KEYTRUDA regimen (n=2) and no patients receiving the chemotherapy-placebo regimen. Most immune-mediated AEs occurred during the neoadjuvant phase rather than the adjuvant phase.
About KEYTRUDA® (pembrolizumab) Injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,700 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of adult and pediatric (12 years and older) patients with stage IIB, IIC, or III melanoma following complete resection.
Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [tumor proportion score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [combined positive score (CPS) ≥1] as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy.
KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Carcinoma
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
Non-muscle Invasive Bladder Cancer
KEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin (BCG)-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with MSI-H central nervous system cancers have not been established.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC).
Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or gastroesophageal (GEJ) (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
KEYTRUDA, in combination with chemotherapy, with or without bevacizumab, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
KEYTRUDA is indicated for the adjuvant treatment of patients with RCC at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions.
Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf .
Source: Merck & Co., Inc.
https://seekingalpha.com/symbol/MRK

February 11, 2022
-- Data Supports the Continued Use of Veklury for Treatment of COVID-19 for Current SARS-CoV-2 Variants --
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today released data demonstrating the in vitro activity of Veklury® (remdesivir) against ten SARS-CoV-2 variants, including Omicron. Results of Gilead studies are consistent with other in vitro studies independently conducted by researchers from institutions in other countries, including Belgium, Czech Republic, Germany, Poland, and the United States, which confirmed Veklury’s antiviral activity against multiple previously identified variants of SARS-CoV-2, including Alpha, Beta, Gamma, Delta and Omicron.
The study analyzed in vitro antiviral activity by two methods to understand the susceptibility of ten major SARS-CoV-2 variants to Veklury. The study results showed similar activity of Veklury against the variants and an early ancestral A lineage isolate detected in Seattle, WA (WA1 strain). Specifically, Delta and Omicron variants both remained fully susceptible to Veklury, and these laboratory results demonstrate that Veklury has remained active against all major variants isolated over the past two years.
Veklury directly inhibits viral replication inside host cells by targeting the SARS-CoV-2 RNA-dependent RNA polymerase. On entering the body, Veklury is transformed into the active triphosphate metabolite, which is then incorporated into the viral RNA and stops replication of the virus within the infected cells. The study analyzed nearly 6 million publicly available variant isolate sequences and confirmed that the nsp12 protein, the RNA polymerase target of Veklury, is highly conserved across all variants. Further characterization confirmed that none of the few identified nsp12 mutations prevalent in some of the SARS-CoV-2 variants affects the virus susceptibility to Veklury.
“These results provide evidence of the consistent and durable antiviral activity of remdesivir across known variants that have emerged throughout the pandemic, including Omicron and support its continued use for the treatment of COVID-19 for current SARS-CoV-2 variants,” said Tomas Cihlar, Senior Vice President of Virology Research, Gilead Sciences. “Now with a new version of Omicron (BA.2 subvariant) increasing in circulation around the world, these latest data also suggest that remdesivir will retain antiviral activity against this new subvariant because the viral RNA polymerase that remdesivir targets does not contain any additional unique mutations. Gilead continuously evaluates the activity of Veklury against viral variants.”
The results of this study have been submitted for publication in a peer-reviewed journal and have been uploaded as preprint at BioRxiv available here.
About Veklury
Veklury (remdesivir) is a nucleotide analog invented by Gilead, building on more than a decade of the company’s antiviral research. Veklury is the antiviral standard of care for the treatment of hospitalized patients with COVID-19 and is a recommended treatment for reducing disease progression in non-hospitalized patients at high risk of disease progression. At this time, more than half of patients hospitalized with COVID-19 in the United States are treated with Veklury. It can help reduce disease progression across a spectrum of disease severity and enable patients to recover faster, freeing up limited hospital resources and saving healthcare systems money.
Veklury was approved by the U.S. Food and Drug Administration (FDA) on October 22, 2020 for adults and pediatric patients 12 years of age and older and weighing at least 40 kg for the treatment of COVID-19 requiring hospitalization. On January 21, 2022, the FDA approved a supplemental new drug application (sNDA) for Veklury to expand the indication to the treatment of non-hospitalized adult and adolescent patients who are at high risk of progression to severe COVID-19, including hospitalization or death. The expanded indication allows for Veklury to be administered in qualified outpatient settings that can administer daily intravenous (IV) infusions over three consecutive days. Veklury is contraindicated in patients who are allergic to Veklury or any of its components; please see below for additional Important Safety Information for Veklury.
Veklury is approved or authorized for temporary use in approximately 50 countries worldwide. To date, Veklury and generic remdesivir have been made available to more than 10 million patients around the world, including nearly 7 million people in 127 middle- and low-income countries through Gilead’s voluntary licensing program. These licenses currently remain royalty-free, reflecting Gilead’s existing commitment to enabling broad patient access to remdesivir.
U.S. Indication for Veklury
Veklury® (remdesivir 100 mg for injection) is indicated for the treatment of COVID-19 in adults and pediatric patients (12 years of age and older and weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, who are:
Veklury should only be administered in settings in which health care providers have immediate access to medications to treat a severe infusion or hypersensitivity reaction, such as anaphylaxis, and the ability to activate the emergency medical system (EMS), as necessary. Veklury must be administered by intravenous infusion. Veklury is contraindicated in patients who are allergic to Veklury or any of its components. For more information, please see the U.S. full Prescribing Information available at www.gilead.com.
U.S. full Prescribing Information for Veklury is available at www.gilead.com.
Veklury, Gilead and the Gilead logo are registered trademarks of Gilead Sciences, Inc., or its related companies.
For more information about Gilead, please visit the company’s website at www.gilead.com , follow Gilead on Twitter (@Gilead Sciences) or call Gilead Public Affairs at 1-800-GILEAD-5 or 1-650-574-3000.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220211005143/en/
Source: Gilead Sciences, Inc.
Feb. 11, 2022 8:46 AM ET
By: Jonathan Block, SA News Editor


Basel, 11 February 2022 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that new two-year data from its phase III studies of Vabysmo™ (faricimab) and Susvimo™ (previously called Port Delivery System with ranibizumab) will be presented at Angiogenesis, Exudation and Degeneration 2022 on 12 February. These longer-term results from the Vabysmo YOSEMITE and RHINE studies in diabetic macular edema (DME) and the Susvimo Archway study in neovascular or “wet” age-related macular degeneration (nAMD) further reinforce the potential to allow for longer time between treatments and fewer eye injections for people with these conditions, while still achieving and maintaining vision gains seen with previous standard-of-care injections. Neovascular AMD and DME are two leading causes of vision loss, together affecting around 40 million people worldwide, which require treatment with eye injections as often as once a month.1-4
“Results from these three studies reinforce the potential of Vabysmo and Susvimo to redefine standards of care and reduce treatment burden for people living with diabetic macular edema and neovascular AMD,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “These two first-of-their-kind treatments are the culmination of over a decade of pioneering research, aiming to better address the needs of people with retinal conditions.”
In the YOSEMITE and RHINE studies, at least 60% of people eligible for extended dosing with Vabysmo could be treated every four months at two years – a 10 percentage point increase since the primary analyses at one year – while achieving non-inferior vision gains versus aflibercept given every two months. Furthermore, nearly 80% of people eligible for extended dosing with Vabysmo could be treated every three months or longer. In the Archway study, Susvimo allowed 95% of people to go six months between treatments at two years – the fourth complete refill-exchange interval – while maintaining vision outcomes that were non-inferior to monthly ranibizumab injections. Across all three studies, with longer follow-up, Vabysmo and Susvimo continued to be generally well-tolerated, with favourable benefit-risk profiles. Safety will continue to be monitored closely in the post-market setting.
VabysmoTM (faricimab-svoa) is the first bispecific antibody for the eye approved by the U.S. Food and Drug Administration (FDA), and the only injectable eye medicine approved for treatments from one to four months apart in the first year following four initial monthly doses, based on evaluation of the patient’s anatomy and vision outcomes.5 Vabysmo is designed to block two disease pathways linked to a number of vision-threatening retinal conditions by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A). Ang-2 and VEGF-A are thought to contribute to vision loss by destabilising blood vessels, which may cause new leaky blood vessels to form and increase inflammation. While additional research continues, inhibition of both pathways has been shown in preclinical studies to have potentially complementary benefits, stabilising vessels and thereby reducing vessel leakage and inflammation more than inhibition of VEGF-A alone.4
Susvimo is the first nAMD treatment in 15 years to provide an alternative to standard-of-care eye injections. By continuously delivering medicine into the eye through a refillable implant, SusvimoTM (ranibizumab injection) 100 mg/mL for intravitreal use via ocular implant is the only FDA-approved treatment that may help people with nAMD maintain their vision with as few as two treatments per year.6
Vabysmo: YOSEMITE and RHINE Two-Year Results
In the YOSEMITE and RHINE studies, DME patients received Vabysmo, given either every two months or up to every four months using a treat and extend approach, or aflibercept given every two months. Two-year results showed Vabysmo patients maintained the vision improvements achieved in the first year and vision gains continued to be non-inferior to those achieved by aflibercept patients. In YOSEMITE, the average vision gains from baseline at two years were +10.7 eye chart letters in both the Vabysmo treat and extend and two-month arms, and +11.4 letters in the aflibercept arm. In RHINE, the average vision gains from baseline at two years were +10.1 and +10.9 letters in the Vabysmo treat and extend and two-month arms, respectively, and +9.4 letters in the aflibercept arm.
Importantly, 60% (n=162/270) of Vabysmo treat and extend patients in YOSEMITE and 64.5% (n=185/287) in RHINE achieved four-month dosing at two years. This is an increase over one-year results, which showed 52.8% (n=151/286) of Vabysmo treat and extend patients in YOSEMITE and 51% (n=157/308) in RHINE achieved four-month dosing. An additional 18.1% (n=49/270) of Vabysmo treat and extend patients in YOSEMITE and 13.6% (n=39/287) in RHINE achieved three-month dosing. Combined, almost 80% of Vabysmo treat and extend patients were able to go three months or longer between treatments at the end of the second year. Across study arms, Vabysmo showed consistent two-step or better improvement in diabetic retinopathy according to the Early Treatment Diabetic Retinopathy Study – Diabetic Retinopathy Severity Score (ETDRS-DRSS). At two years, 42.8% of Vabysmo treat and extend patients in YOSEMITE and 44.3% in RHINE achieved a two-step or better improvement from baseline. In the two-month Vabysmo arms, 51.4% and 53.5% of patients in YOSEMITE and RHINE, respectively, achieved a two-step or better improvement in diabetic retinopathy severity. Vabysmo given at intervals of up to four months continued to demonstrate greater reductions in central subfield thickness (CST) compared to aflibercept given every two months in both studies. Safety results were consistent across study arms, with no reported cases of retinal vasculitis or retinal occlusive events.
One-year results from the YOSEMITE and RHINE studies and the TENAYA and LUCERNE studies in nAMD were recently published in The Lancet.4,7
Susvimo: Archway Two-Year Results
Neovascular AMD patients in Archway received either Susvimo refilled every six months or monthly ranibizumab 0.5 mg eye injections. Two-year results showed vision was maintained by Susvimo patients and continued to be non-inferior to that achieved with monthly ranibizumab injections. Susvimo patients averaged -1.1 eye chart letters in visual acuity from baseline at two years, while monthly ranibizumab patients averaged -0.5 letters from baseline. In addition, 95% of Susvimo patients were able to go six months without needing additional treatment in the second, third and fourth refill-exchange intervals. In Archway, Susvimo was generally well-tolerated, with a favourable benefit-risk profile. The most common adverse events of special interest (≥5%) were cataract, conjunctival bleb and vitreous haemorrhage. The safety profile of Susvimo in the clinical trial setting is well understood and will continue to be monitored closely.
In addition to Archway results, two-year interim data from the ongoing phase III Portal study will be presented at the Angiogenesis meeting. Portal is an extension study evaluating the long-term safety and efficacy of Susvimo in nAMD.
Vabysmo is approved by the FDA for the treatment of nAMD and DME.5 Susvimo is approved by the FDA for the treatment of people with nAMD who have previously responded to at least two anti-VEGF injections.6 Vabysmo is currently under review by the European Medicines Agency for the treatment of nAMD and DME and Susvimo is under review for the treatment of nAMD. Submissions to other regulatory authorities around the world are ongoing.
Roche has a robust phase III clinical development programme for Vabysmo and Susvimo. For Vabysmo, the programme includes AVONELLE-X, an extension study of TENAYA and LUCERNE evaluating the long-term safety and tolerability of Vabysmo in nAMD, and RHONE-X, an extension study of YOSEMITE and RHINE evaluating the long-term safety and tolerability of Vabysmo in DME.8,9 Additionally, the COMINO and BALATON trials are also underway, evaluating the efficacy and safety of Vabysmo in people with macular edema following retinal vein occlusion.10,11
For Susvimo, the clinical development programme includes the Portal, Pagoda, Pavilion and Velodrome studies.12-15 Portal is an extension study evaluating the long-term safety and efficacy of Susvimo in nAMD.12 Pagoda is evaluating Susvimo for the treatment of DME, while Pavilion is a study of Susvimo in diabetic retinopathy without DME.13,14 Velodrome is evaluating Susvimo refilled every nine months in nAMD.15
About the YOSEMITE and RHINE studies7
YOSEMITE (NCT03622580) and RHINE (NCT03622593) are two identical, randomised, multicentre, double-masked, global phase III studies evaluating the efficacy and safety of Vabysmo™ (faricimab) compared to aflibercept in 1,891 people with diabetic macular edema (940 in YOSEMITE and 951 in RHINE). The studies each have three treatment arms: Vabysmo 6.0 mg administered up to every four months after four initial monthly doses using a treat and extend approach; Vabysmo 6.0 mg administered at two-month intervals after six initial monthly doses; and aflibercept administered at fixed two-month intervals after five initial monthly doses. Dosing schedule for patients within the treat-and-extend arm was determined by central subfield thickness (CST) and visual acuity. In all three arms, sham injections were administered at study visits when treatment injections were not scheduled to maintain the masking of investigators and participants.
The primary endpoint of the studies is the average change in best-corrected visual acuity (BCVA) score (the best distance vision a person can achieve – including with correction such as glasses – when reading letters on an eye chart) from baseline at one year, averaged over weeks 48, 52 and 56. Secondary endpoints include: safety; the percentage of participants in the treat and extend arm receiving Vabysmo every one, two, three and four months, at week 52; the percentage of participants achieving a two-step or greater improvement from baseline in diabetic retinopathy severity at week 52; the percentage of participants achieving a gain, and the percentage avoiding a loss, of 15 letters or more in BCVA from baseline over time; change in CST from baseline over time; and percentage of patients with absence of intraretinal fluid over time.
About the Archway study16
Archway (NCT03677934) was a randomised, multicentre, open-label phase III study evaluating the efficacy and safety of Susvimo™ (previously called Port Delivery System with ranibizumab), refilled every six months at fixed intervals, compared to monthly intravitreal injections of ranibizumab 0.5 mg in 415 people living with neovascular or “wet” age-related macular degeneration. Patients enrolled in Archway were responders to prior treatment with anti-vascular endothelial growth factor (VEGF) therapy. In both study arms, patients were treated with at least three anti-VEGF injections within the six months prior to their Archway screening visit. The primary endpoint of the study was the change in best-corrected visual acuity (BCVA) score from baseline at the average of Week 36 and Week 40. Secondary endpoints include safety, overall change in vision (BCVA) from baseline and change from baseline in centre point thickness over time.
About Vabysmo™ (faricimab)
Vabysmo™ (faricimab) is the first bispecific antibody designed for the eye. It targets and inhibits two disease pathways linked to a number of vision-threatening retinal conditions by neutralising angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A). Ang-2 and VEGF-A contribute to vision loss by destabilising blood vessels, causing new leaky blood vessels to form and increasing inflammation. By blocking both pathways involving Ang-2 and VEGF-A, Vabysmo is designed to stabilise blood vessels.4
About Susvimo™ (previously called Port Delivery System with ranibizumab)
Susvimo™ (previously called Port Delivery System with ranibizumab) is a refillable eye implant surgically inserted into the eye during a one-time, outpatient procedure. Susvimo continuously delivers a customised formulation of ranibizumab over time. Susvimo is indicated for intravitreal use via the Susvimo eye implant only. Ranibizumab is a vascular endothelial growth factor (VEGF) inhibitor designed to bind to and inhibit VEGF-A, a protein that has been shown to play a critical role in the formation of new blood vessels and the leakiness of the vessels.6
Susvimo is different from the ranibizumab intravitreal injection, a medicine marketed as Lucentis®* (ranibizumab injection), which is approved to treat nAMD and other retinal diseases. Lucentis* was first approved for nAMD by the FDA in 2006.24
For more information, please visit www.roche.com.
Feb. 11, 2022 6:43 AM ET
Roche Holding AG (RHHBY), RHHBF
By: Ravikash, SA News Editor
Roche's (OTCQX:RHHBY) (OTCQX:RHHBF) unit Genentech reported positive two-year data from its phase 3 studies of Vabysmo (faricimab-svoa) and Susvimo (ranibizumab injection).
https://seekingalpha.com/symbol/RHHBY
https://seekingalpha.com/symbol/RHHBF
https://www.gene.com/patients/medicines/vabysmo
https://www.gene.com/patients/medicines/susvimo

Feb. 10, 2022 7:00 AM ET AbbVie Inc. (ABBV)
MONTREAL, Feb. 10, 2022 /CNW/ - AbbVie (ABBV), a research-based global biopharmaceutical company, announced today that an agreement was reached with the pan-Canadian Pharmaceutical Alliance (pCPA) for VENCLEXTA® (venetoclax) in combination with azacitidine for the treatment of patients with newly diagnosed acute myeloid leukemia (AML) who are 75 years or older, or who have comorbidities that preclude use of intensive induction chemotherapy.i Effective February 2nd, on Québec's Liste des medicaments-établissements, effective February 1st on Saskatchewan Cancer Agency drug formulary, and effective February 24th on Manitoba's Drug Benefits and Interchangeability Formulary, VENCLEXTA is listed in combination with azacitidine, for first line treatment of patients with newly diagnosed AML who are 75 years or older, or who have comorbidities that preclude use of intensive induction chemotherapy. For full criteria, consult the list of medications in effect.ii, iii, iv"Acute myeloid leukemia is a blood cancer with a survival rate that still needs to be improved. Intensive chemotherapy treatments lead to toxicity, which can limit their use. However, our understanding of this disease has improved considerably over the past few years and, thanks to new treatment options such as the combination of venetoclax and azacitidine, we are now able to effectively treat a greater proportion of patients," explains Dr. Julie Bergeron, MD, FRCPC, associate clinical professor, head of the Optilab CHUM cluster of hematology laboratories, and hematologist at the CEMTL Maisonneuve-Rosemont facility.In Canada, the five-year net survival rate is approximately 21% for people diagnosed with AML in the general population. v"Every day, we aim to transform the standard of care in Oncology. Having effective and proven treatment options is vital for patients and their families impacted by AML. It is great news that VENCLEXTA plus azacitidine is now reimbursed in Quebec, Saskatchewan and Manitoba for people living with AML," says Tracey Ramsay, Vice President and General Manager, AbbVie Canada.VENCLEXTA in combination with azacitidine was approved by Health Canada in December 2020. Health Canada's approval was granted under Project Orbis, an FDA initiative which provides a framework for concurrent submission and accelerated review of oncology products among international partners.VENCLEXTA is jointly commercialized by AbbVie and Genentech, a member of the Roche Group, in the U.S. and by AbbVie outside of the U.S.
For more information about AbbVie, please visit us at www.abbvie.ca. Follow @abbviecanada on Twitter or find us on LinkedIn. SOURCE AbbVie Canada
https://seekingalpha.com/symbol/ABBV
(ay-za-SYE-ti-deen)
Trade Name(s): Vidaza®, Onureg®
Other Name(s): 5-azacitadine
https://chemocare.com/chemotherapy/drug-info/Vidaza.aspx
VENCLEXTA is a prescription medicine used:
It is not known if VENCLEXTA is safe and effective in children.

02/10/22 at 1:24 AM ESTPDF Version
– Pseudovirus results being shared with government and regulatory authorities;
publication in bioRxiv anticipated in the coming week; additional live virus testing underway –
SAN FRANCISCO, Feb. 09, 2022 (GLOBE NEWSWIRE) -- Vir Biotechnology, Inc. (Nasdaq: VIR) today announced that preclinical data suggest that sotrovimab, an investigational monoclonal antibody authorized for emergency use in the United States and developed in conjunction with GlaxoSmithKline, retains neutralizing activity against the BA.2 subvariant of Omicron. These pseudovirus results are being shared with government and regulatory authorities around the world and the Company expects to publish data on bioRxiv in the coming week, with live virus data to follow.
“Our view of the data is that they support the ongoing role of sotrovimab as a critical treatment in the fight against the continuously evolving SARS-CoV-2 virus,” said George Scangos, Vir’s chief executive officer. “We note recent conclusions from another lab, which state that no approved or authorized monoclonal antibodies for treatment retain activity against all subvariants of Omicron. We are therefore pleased to share that, based on our pseudovirus and extensive pharmacokinetic data, we believe that the 500 mg dose of sotrovimab is sufficient to retain activity against the BA.2 variant, just as it has against all other variants of concern and interest.”
About sotrovimab
Sotrovimab is an investigational SARS-CoV-2 neutralizing monoclonal antibody. The antibody binds to an epitope on SARS-CoV-2 shared with SARS-CoV-1 (the virus that causes SARS), indicating that the epitope is highly conserved, which may make it more difficult for resistance to develop. Sotrovimab, which incorporates Xencor, Inc.’s Xtend™ technology, has also been designed to achieve high concentration in the lungs to ensure optimal penetration into airway tissues affected by SARS-CoV-2 and to have an extended half-life.
About global access to sotrovimab
Sotrovimab is authorized for emergency use in the US and has been granted a marketing authorization in the European Union (EU), conditional marketing authorization in Great Britain, provisional marketing authorization in Australia, and conditional marketing authorization in Saudi Arabia. It has also been approved via Japan’s Special Approval for Emergency Pathway. Temporary authorizations for sotrovimab have also been granted in 12 other countries.
Sotrovimab is supplied in several countries worldwide, including through national agreements in the US, UK, Japan, Australia, Canada, Singapore, Switzerland, and the United Arab Emirates. Vir and GlaxoSmithKline are also supplying sotrovimab to participating Member States of the EU through a Joint Procurement Agreement with the European Commission. Additional agreements are yet to be disclosed due to confidentiality or regulatory requirements.
Sotrovimab in the United States
The following is a summary of information for sotrovimab. Healthcare providers in the US should review the Fact Sheets for information about the authorized use of sotrovimab and mandatory requirements of the Emergency Use Authorization (EUA). Please see the Food and Drug Administration (FDA) Letter of Authorization, full Fact Sheet for Healthcare Providers and full Fact Sheet for Patients, Parents, and Caregivers.
Sotrovimab has been authorized by the US FDA for the emergency use described below. Sotrovimab is not FDA-approved for this use.
Sotrovimab is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of sotrovimab under section 564(b)(1) of the Food, Drug, and Cosmetic Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.
Authorized use
The FDA has issued an EUA to permit the emergency use of the unapproved product sotrovimab for the treatment of mild-to-moderate coronavirus disease 2019 (COVID-19) in adults and pediatric patients (12 years of age and older weighing at least 40kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death.
Limitations of authorized use
Sotrovimab is not authorized for use in patients:
Benefit of treatment with sotrovimab has not been observed in patients hospitalized due to COVID-19. SARS-CoV-2 monoclonal antibodies may be associated with worse clinical outcomes when administered to hospitalized patients with COVID19 requiring high flow oxygen or mechanical ventilation.
Important Safety Information
CONTRAINDICATIONS
Sotrovimab is contraindicated in patients who have a history of anaphylaxis to sotrovimab or to any of the excipients in the formulation.
We routinely post information that may be important to investors on our website at www.vir.bio.
Source: Vir Biotechnology, Inc.
Feb. 10, 2022 5:28 AM ET Vir Biotechnology, Inc. (VIR), GSK By: Ravikash, SA News Editor

February 8, 2022
JUVÉDERM® VOLBELLA® XC, PART OF THE NUMBER ONE CHOSEN COLLECTION OF FILLERS, IS THE FIRST AND ONLY DERMAL FILLER TO RECEIVE FDA APPROVAL FOR THE IMPROVEMENT OF INFRAORBITAL HOLLOWS[1,2]
IRVINE, Calif., Feb. 8, 2022 /PRNewswire/ -- Allergan Aesthetics, an AbbVie company (NYSE: ABBV), announces the FDA approval of JUVÉDERM® VOLBELLA® XC for improvement of infraorbital hollows in adults over the age of 21.2 According to clinical trial data, 90% of subjects reported satisfaction through one year after treatment.2 With this approval, Allergan Aesthetics continues the expansion of its treatment portfolio to better address unmet patient needs. Per FDA requirement for this new indication, Allergan Aesthetics is providing a product training program for all interested providers, which includes facial anatomy and considerations for safe injection in this area, as well as identification and management of potential complications. Successful completion of this training is necessary prior to administration of JUVÉDERM® VOLBELLA® XC for this new indication.
"This additional indication for JUVÉDERM® VOLBELLA® XC demonstrates Allergan Aesthetics' continued commitment to innovation. The eye area, including the undereye hollow, is a top concern among patients," says Carrie Strom, President, Global Allergan Aesthetics and Senior Vice President, AbbVie. "Allergan Aesthetics offers the broadest portfolio of treatment options designed to address the delicate eye area from topical skin care with SkinMedica®, to crow's feet lines with BOTOX® Cosmetic (onabotulinumtoxinA) and now, with this approval, the infraorbital hollows, commonly referred to as tear troughs, with JUVÉDERM® VOLBELLA® XC."
Patient safety and consumer satisfaction are a top priority at Allergan Aesthetics. As the JUVÉDERM® Collection of Fillers continues to be at the forefront of innovation, Allergan Aesthetics is committed to providing best-in-class training to our providers through the Allergan Medical Institute (AMI). During the required infraorbital hollows training, providers will be educated on how to assess facial anatomy holistically where JUVÉDERM® VOLUMA® XC may be added as part of a treatment plan to address volume loss in the midface. The safety and efficacy of combined use of JUVÉDERM® VOLUMA® XC and JUVÉDERM® VOLBELLA® XC has not been studied. The required training can be accessed and completed at VolbellaTraining.com.
"The undereye area is one of the most frequently requested treatment sites among patients, regardless of race and ethnicity, but it is undertreated.3 This is in part because it is a sensitive area to inject as it takes great skill and precision," says AMI trainer, Board Certified Oculofacial Plastic Surgeon and Ophthalmologist, Dr. Julie Woodward. "The approval of JUVÉDERM® VOLBELLA® XC is a milestone in offering providers, like myself, a safe and effective treatment option to address the undereye area for my patients. The characteristics of JUVÉDERM® VOLBELLA® XC with lower amounts of hyaluronic acid molecules and low water affinity provides a soft, smooth formulation appropriate for treating undereye hollows and I am excited to work with Allergan Aesthetics on a robust injector and patient education plan to ensure safe and effective outcomes in this challenging to treat area. The results of the clinical trial demonstrate significant improvements in the appearance of undereye hollows and overall appearance. In addition, 80% of subjects reported they were a little or not at all bothered by how tired and old the under-eye area looked at 3 months compared to 15% and 30% before treatment, respectively.2"
According to the clinical studies, the primary effectiveness criteria were met in the treatment group's responder rate of 83.1% and was statistically significantly greater (p<0.0001) than the responder rate for the no–treatment control group (15.6%) based on the mITT population with multiple imputation. The mean improvement was clinically significant (≥ 1 point), with the majority of subjects demonstrating improvement through one year.5 In addition, 90.1% of patients were willing to recommend the treatment to a friend.5
Consumers and new patients who receive aesthetic treatment from the JUVÉDERM® Collection of Fillers, can also enroll in Allē, Allergan Aesthetics loyalty rewards program to unlock access to curated content, exclusive offers, and personalized rewards that can be used for savings on the Allergan Aesthetics portfolio of products and redeemed at a participating provider's office, subject to program terms and conditions that apply. Allē is the first and only loyalty program in the aesthetics market to also offer consumers the ability to earn points on over 40 non-Allergan Aesthetics treatments and brands.
The majority of subjects in the clinical study experienced a side effect, such as tenderness to touch, bruising, swelling, lumps/bumps, redness, pain after injection, firmness, discoloration (not redness or swelling), or itching as reported in their 30-day daily diaries. A majority of these side effects were mild (easily tolerated) in severity, although a few subjects experienced mild swelling more than 30 days after treatment. The swelling was treated with antibiotics for 1 subject; the other subjects did not require treatment. All of these events resolved within 45 days.4
JUVÉDERM® VOLBELLA® XC was first FDA–approved in 2016 for use in the lips and perioral rhytids.2 As the category leader, the JUVÉDERM® Collection of Fillers offers the broadest portfolio of specifically tailored treatment options, and this latest approval marks the sixth approved indication in the U.S.
For more information on the JUVÉDERM® Collection of Fillers, visit Juvéderm.com and follow @JUVÉDERM on Instagram.
For more information about AbbVie, please visit us at www.abbvie.com.
INDICATIONS
JUVÉDERM® VOLUMA® XC injectable gel is indicated for deep (subcutaneous and/or supraperiosteal) injection for cheek augmentation to correct age-related volume deficit in the mid-face and for augmentation of the chin region to improve the chin profile in adults over the age of 21.
JUVÉDERM® VOLLURE® XC injectable gel is indicated for injection into the mid-to-deep dermis for correction of moderate to severe facial wrinkles and folds (such as nasolabial folds) in adults over the age of 21.
JUVÉDERM® VOLBELLA® XC injectable gel is indicated for injection into the lips for lip augmentation and correction of perioral rhytids, and for the improvement of infraorbital hollowing in adults over the age of 21.
JUVÉDERM® Ultra Plus XC and JUVÉDERM® Ultra XC injectable gels are indicated for injection into the mid-to-deep dermis for correction of moderate to severe facial wrinkles and folds (such as nasolabial folds).
JUVÉDERM® Ultra XC injectable gel is also indicated for injection into the lips and perioral area for lip augmentation in adults over the age of 21.
Feb. 08, 2022 8:32 AM ET AbbVie Inc. (ABBV)
By: Ravikash, SA News Editor3 Comments

02/03/2022
– Full Data from Phase 3 ECHELON-1 Clinical Trial to be Submitted for Presentation at Upcoming Medical Meeting –
BOTHELL, Wash.--(BUSINESS WIRE)-- Seagen Inc. (Nasdaq:SGEN) today announced that the phase 3 ECHELON-1 clinical trial demonstrated a statistically significant improvement in overall survival (OS) (p=0.009) in patients with advanced classical Hodgkin lymphoma (cHL) following treatment with ADCETRIS (brentuximab vedotin) in combination with chemotherapy. With approximately six years median follow up, patients receiving ADCETRIS plus doxorubicin, vinblastine, and dacarbazine (A+AVD) in the frontline setting had a 41 percent reduction in the risk of death (HR 0.59; [95% CI: 0.396 to 0.879]) compared with patients receiving doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD). The safety profile of ADCETRIS was consistent with previous studies and no new safety signals were observed.
“These groundbreaking results are important for patients with advanced classical Hodgkin lymphoma given that an improvement in overall survival has rarely been shown in frontline treatment of this disease,” said Roger Dansey, M.D., Chief Medical Officer at Seagen. “We look forward to presentation of the results at an upcoming medical meeting.”
ECHELON-1 is an open-label, international, randomized, phase 3 trial evaluating the safety and efficacy of frontline ADCETRIS plus AVD versus ABVD in 1,334 adult patients with stage III or IV cHL. Patients were randomly assigned to receive A+AVD or ABVD intravenously on days 1 and 15 of each 28-day cycle for up to six cycles. OS is the key secondary endpoint of the trial. The primary endpoint, modified progression free survival, served as the basis for global regulatory approvals.
Please see Important Safety Information, including BOXED WARNING, for ADCETRIS below.
ADCETRIS is approved for certain types of relapsed or refractory Hodgkin lymphoma (HL) including previously untreated Stage III/IV cHL and previously untreated peripheral T-cell lymphoma (PTCL). It has received marketing authorization in more than 75 countries and is being evaluated globally in more than 70 corporate- and investigator-sponsored clinical trials across multiple settings in lymphoma and other diseases.
About ADCETRIS
ADCETRIS is an antibody-drug conjugate (ADC) comprising an anti-CD30 monoclonal antibody attached by a protease-cleavable linker to a microtubule disrupting agent, monomethyl auristatin E (MMAE), utilizing Seagen’s proprietary technology. The ADC employs a linker system that is designed to be stable in the bloodstream but to release MMAE upon internalization into CD30-expressing cells.
ADCETRIS is indicated for the treatment of adult patients with:
Seagen and Takeda are jointly developing ADCETRIS. Under the terms of the collaboration agreement, Seagen has U.S. and Canadian commercialization rights and Takeda has rights to commercialize ADCETRIS in the rest of the world. Seagen and Takeda are funding joint development costs for ADCETRIS on a 50:50 basis, except in Japan where Takeda is solely responsible for development costs.
For more information on the company’s marketed products and robust pipeline, visit www.seagen.com and follow @SeagenGlobal on Twitter.
1 https://www.cancer.org/cancer/hodgkin-lymphoma.html
View source version on businesswire.com: https://www.businesswire.com/news/home/20220203005330/en/
Source: Seagen Inc.
Feb. 03, 2022 12:50 PM ET Seagen Inc. (SGEN) By: Ravikash, SA News Editor

The Dexcom G6 CGM System is now covered by Alberta Health for people with diabetes who are under 18 years old and require ongoing use of insulin or insulin pump therapy (Photo: Business Wire)
February 03, 2022 05:19 PM Eastern Standard Time
BURNABY, British Columbia--(BUSINESS WIRE)--Dexcom, Inc. (NASDAQ: DXCM), the global leader in real-time continuous glucose monitoring (CGM), announced today that people with diabetes who are under 18 years old and require ongoing use of insulin or insulin pump therapy are eligible for public coverage of the Dexcom G6 CGM System through Alberta Health.
“Today’s announcement means more pediatric patients will have access to this life-changing technology to manage their diabetes.”
Tweet this
Alberta joins five other jurisdictions in providing public coverage of real-time CGM systems under provincial health plans. The Non-Insured Health Benefits Program also recently announced coverage for First Nations and Inuit children. With expanded public coverage for CGM, more children and youth can access this standard of care technology, helping them manage their diabetes.
For more information about Dexcom CGM, visit www.dexcom.com.
Feb. 03, 2022 5:27 PM ET DexCom, Inc. (DXCM)
By: Ravikash, SA News Editor

JANUARY, 31, 2022 DOWNLOAD(OPENS IN NEW WINDOW)
Approval based on a comprehensive submission package including efficacy and safety data approximately six months after second dose
SPIKEVAX has received approval by regulators in more than 70 countries, including Canada, Japan, the European Union, the UK, Israel
807 million doses of Moderna's COVID-19 vaccine shipped globally in 2021; approximately 25% of those doses shipped to low- and middle-income countries
CAMBRIDGE, MA / ACCESSWIRE / January 31, 2022 / Moderna, Inc. (Nasdaq:MRNA), a biotechnology company pioneering messenger RNA (mRNA) therapeutics and vaccines, today announced the U.S. Food and Drug Administration (FDA) has approved the Biologics License Application (BLA) for SPIKEVAX (COVID-19 Vaccine, mRNA) to prevent COVID-19 in individuals 18 years of age and older.
"Our COVID-19 vaccine has been administered to hundreds of millions of people around the world, protecting people from COVID-19 infection, hospitalization and death. The totality of real-world data and the full BLA for Spikevax in the United States reaffirms the importance of vaccination against this virus. This is a momentous milestone in Moderna's history as it is our first product to achieve licensure in the U.S.," said Stéphane Bancel, Chief Executive Officer of Moderna. "The full licensure of Spikevax in the U.S. now joins that in Canada, Japan, the European Union, the UK, Israel, and other countries, where the adolescent indication is also approved. We are grateful to the U.S. FDA for their thorough review of our application. We are humbled by the role that Spikevax is playing to help end this pandemic."
The FDA based its decision on the totality of scientific evidence shared by the Company in its submission package, which included follow-up data from the Phase 3 COVE study showing high efficacy and favorable safety approximately six months after the second dose. Moderna also submitted manufacturing and facilities data required by the FDA for licensure. SPIKEVAX has received approval by regulators in more than 70 countries.
Moderna's COVID-19 vaccine was available under Emergency Use Authorization (EUA) in the U.S. from December 18, 2020. Under an EUA, the FDA has the authority to allow medical products to be used in an emergency to diagnose, treat, or prevent serious or life-threatening diseases or conditions during a declared public health emergency when there are no adequate, approved, and available alternatives. A booster dose of the Moderna COVID-19 vaccine at the 50 µg dose level is authorized for emergency use in the U.S. under EUA for adults 18 years and older. A third dose of the Moderna COVID-19 vaccine at the 100 µg dose level is authorized for emergency use in immunocompromised individuals 18 years of age or older in the United States who have undergone solid organ transplantation, or who are diagnosed with conditions that are considered to have an equivalent level of immunocompromise.
INDICATION (U.S.)
SPIKEVAX (COVID-19 Vaccine, mRNA) is a vaccine indicated for active immunization to prevent coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in individuals 18 years of age and older.
To learn more, visit www.modernatx.com.
SOURCE: Moderna, Inc.
View source version on accesswire.com:
https://www.accesswire.com/686397/Moderna-Receives-Full-US-FDA-Approval-for-COVID-19-Vaccine-Spikevax
Jan. 31, 2022 11:21 AM ET Moderna, Inc. (MRNA)PFE, BNTX
By: Dulan Lokuwithana, SA News Editor4 Comments
JANUARY 31, 2022 DOWNLOAD(OPENS IN NEW WINDOW)
Approval based on a comprehensive submission package including efficacy and safety data approximately six months after second doseSPIKEVAX has received approval by regulators in more than 70 countries, including Canada, Japan, the European Union, the UK, Israel807 million doses of Moderna's COVID-19 vaccine shipped globally in 2021; approximately 25% of those doses shipped to low- and middle-income countriesCAMBRIDGE, MA / ACCESSWIRE / January 31, 2022 / Moderna, Inc. (Nasdaq:MRNA), a biotechnology company pioneering messenger RNA (mRNA) therapeutics and vaccines, today announced the U.S. Food and Drug Administration (FDA) has approved the Biologics License Application (BLA) for SPIKEVAX (COVID-19 Vaccine, mRNA) to prevent COVID-19 in individuals 18 years of age and older.https://investors.modernatx.com/news/news-details/2022/Moderna-Receives-Full-U.S.-FDA-Approval-for-COVID-19-Vaccine-Spikevax/default.aspxhttps://seekingalpha.com/symbol/MRNA

FEB 2, 2022
Robotic prostatectomy performed by Doctor Alexandre Mottrie at Onze-Lieve-Vrouw Ziekenhuis (OLV) in Aalst, Belgium
DUBLIN and AALST, Belgium, Feb. 2, 2022 /PRNewswire(opens new window)/ -- Medtronic plc (NYSE:MDT), a global leader in healthcare technology, and OLV Hospital Aalst today announced that the first clinical procedure in Europe was performed with the Hugo™ robotic-assisted surgery (RAS) system. The robotic prostatectomy was performed by Prof. Alexandre Mottrie, M.D., head of urology at OLV Hospital in Aalst, Belgium, and chief executive officer and founder of the Belgium-based ORSI Academy, a multidisciplinary center for training, research and development, and data analysis to improve minimally invasive surgery best practices.
 View File Download FileMedtronic-Surgical-Robotics
View File Download FileMedtronic-Surgical-Robotics"Performing Europe's very first procedure with the Hugo RAS system is a career highlight for me," said Dr. Mottrie. "With more than two decades and 4,000 robotic-assisted surgery procedures under my belt, I am intimately aware of the barriers that have kept the benefits of surgical robotics from physicians, hospitals, and patients. Now, I believe we are entering a new era filled with greater access and flexibility."
A form of minimally invasive surgery, robotic-assisted surgery offers fewer complications, shorter hospital stays, faster return to normal activities, and smaller scars than open surgery.1–3,†
"This is an exciting and important moment for healthcare in Europe and we're proud to share it with Dr. Mottrie and the team at OLV," said Megan Rosengarten, president of the Surgical Robotics business, which is part of the Medical Surgical Portfolio at Medtronic. "Dr. Mottrie has left a meaningful mark on our program over the many years we've worked together, and now, through our partnership with OLV, Medtronic's journey to bring the benefits of robotic-assisted surgery to more patients in Europe is well underway."
The Hugo RAS system — Medtronic's solution to historic cost and utilization barriers that have kept surgical robotics out of reach for many hospitals — is a modular, multi-quadrant platform designed for a broad range of soft-tissue procedures. It combines wristed instruments, 3D visualization, and Touch Surgery™ Enterprise, a cloud-based surgical video capture and management solution, with dedicated support teams specializing in robotics program optimization, service, and training.
In 2021, Medtronic announced the first urologic and gynecologic procedures with the Hugo system in Latin America and Asia-Pacific. Those procedures and cases in Europe will become part of the Hugo RAS system patient registry, which is collecting clinical data to support regulatory submissions around the world.
"The Hugo RAS system introduces the long-awaited power of choice in the category and will redefine all that robotic-assisted surgery can make possible," said Henk Westendorp, senior country director Benelux at Medtronic. "Medtronic thoughtfully designed the Hugo RAS system with surgeons in mind and patients at heart to tackle today's barriers to adoption in a future-proofed way. We know that by innovating real solutions for the way surgeons want to work — alongside partners like OLV Hospital Aalst who share our passion for advancing patient care — we can make a substantial impact."
"We're incredibly proud to have left our stamp on medical history as the very first center in the region to embrace surgical robotics in 1999," said Peter Verhulst, chief executive officer, OLV Hospital Aalst. "Decades later, we are delighted to be recognized as a robotic surgery center of excellence, leaving another indelible mark as the first hospital in all of Europe to offer the Hugo RAS system and the first in the world to have Medtronic's two RAS platforms — the Hugo system for soft tissue and the Mazor™ system for spinal surgery. The OLV Hospital closely monitors innovation in the medical world and often plays a pioneering role in the introduction of new minimally invasive techniques. The worldwide reputation of our OLV doctors in the field of robotic surgery and other minimally invasive procedures is a result of this. With the Hugo RAS system, we are again at the forefront, with the latest medical innovation that is designed with the patient at heart."
The Hugo RAS system is commercially available in certain geographies. Regulatory requirements of individual countries and regions will determine approval, clearance, or market availability. In the EU, the Hugo RAS system is CE marked. In Canada, the Hugo RAS system has a medical device licence. The Hugo RAS system is approved in Australia. In the U.S., the Hugo RAS system is an investigational device not for sale. Touch Surgery Enterprise is not intended to direct surgery, or aid in diagnosis or treatment of a disease or condition.
For more information, visit medtronic.com/hugo(opens new window).
About OLV Hospital in Aalst, Belgium
The OLV Hospital offers a wide range of medical consultations, examinations, interventions, treatments and aftercare - both in the outpatient center and inpatient setting - at the campus in Aalst, the branch in Asse and the medical centre in Ninove. OLV Hospital always strives to limit the impact of an intervention on the patient as much as possible so that a quicker recovery is possible. As a result, OLV Hospital is also a pioneer in the field of minimally invasive techniques and precision surgery with the support of the operating robot. The physicians and staff at OLV Hospital do everything possible to provide Top in Care. Besides the physical health of patients, they also want to look after their well-being in all its aspects and in every stage of life.
With 2,554 employees and more than 300 doctors and independent care professionals, OLV Hospital is the largest employer in the region, and with 959 beds (114 of which in outpatient care) and more than 90,000 admissions (hospitalisation and outpatient care) per year, OLV Hospital is one of the largest non-university hospitals in Flanders.
For more information on Medtronic (NYSE:MDT), visit www.Medtronic.com(opens new window) and follow @Medtronic(opens new window) on Twitter and LinkedIn(opens new window).
Feb. 02, 2022 8:18 AM ET Medtronic plc (MDT) By: Ravikash, SA News Editor

February 02, 2022Download PDFPhase 2/3 trial data, the basis for submission, demonstrates Rylaze maintains a clinically meaningful level of nadir serum asparaginase activity throughout the entire duration of treatment for adult and pediatric patients with acute lymphoblastic leukemia and lymphoblastic lymphoma
Submission will be reviewed under FDA's Real-Time Oncology Review Program
DUBLIN, Feb. 2, 2022 /PRNewswire/ -- Jazz Pharmaceuticals plc (Nasdaq: JAZZ) today announced that the Company has completed the submission of a Supplemental Biologics License Application (sBLA) to the U.S. Food and Drug Administration (FDA) seeking approval for a Monday/Wednesday/Friday (M/W/F) intramuscular (IM) dosing schedule for Rylaze™ (asparaginase erwinia chrysanthemi (recombinant)-rywn), for use as a component of a multi-agent chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia (ALL) and lymphoblastic lymphoma (LBL) in adult and pediatric patients one month and older who have developed hypersensitivity to E. coli-derived asparaginase. The submission will be reviewed under the Real-Time Oncology Review (RTOR) program, an initiative of FDA's Oncology Center of Excellence designed for efficient review of safe and effective cancer treatments, and follows Rylaze's initial approval under the RTOR program in June 2021.
"We were pleased Rylaze, a much-needed therapeutic option, was approved under the RTOR program while the clinical trial was ongoing. Our science-led and patient-focused development program has enabled us to deliver a clinically significant advancement for patients," said Rob Iannone, M.D., M.S.C.E., executive vice president, global head of research and development of Jazz Pharmaceuticals. "With a dosing schedule of Rylaze administered 25/25/50 mg/m2 on Monday/Wednesday/Friday, patients maintain a clinically meaningful level of nadir serum asparaginase activity through the entire duration of treatment. We look forward to submitting two additional regulatory applications this year to ensure as many patients as possible can have access to a reliable and high-quality supply of this important therapy, including another regulatory application to FDA to support the intravenous route of administration and an additional application in Europe later this year."
The sBLA submitted by Jazz is supported by data from the three-cohort intramuscular administration part of the Phase 2/3 trial of Rylaze in adult and pediatric patients with ALL and LBL who have developed hypersensitivity to an E. coli-derived asparaginase. The trial studied three dosing regimens of Rylaze, with cohort 1a receiving 25 mg/m2 administered M/W/F, cohort 1b receiving 37.5 mg/m2 administered M/W/F and cohort 1c receiving 25 mg/m2 administered Monday and Wednesday and 50 mg/m2 administered on Friday. Initial results showed that in cohort 1c, a dosing regimen of Rylaze administered 25 mg/m2 on Monday and Wednesday and 50 mg/m2 on Friday demonstrated a positive benefit-to-risk profile, showing that Rylaze maintains a clinically meaningful level of nadir serum asparaginase activity (NSAA) ≥0.1 IU/mL at both 48 and 72 hours (from Friday to Monday). In addition, the safety profile of Rylaze was consistent with the reported safety information for patients with ALL/LBL receiving asparaginase with combination chemotherapy. Initial results from the trial were presented at the 63rd American Society of Hematology (ASH) Annual Meeting in December 2021.1
The sBLA follows FDA approval of Rylaze in June 2021 under the RTOR program.2 Rylaze was also granted orphan drug designation for the treatment of ALL/LBL in June 2021 and was added to the National Comprehensive Cancer Network® Clinical Practice Guidelines in Oncology (NCCN Guidelines®) in July 2021.3
About Rylaze™ (asparaginase erwinia chrysanthemi (recombinant)-rywn)
Rylaze, also known as JZP458, is approved in the U.S. for use as a component of a multi-agent chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia (ALL) and lymphoblastic lymphoma (LBL) in adult and pediatric patients one month or older who have developed hypersensitivity to E. coli-derived asparaginase. Rylaze has orphan drug designation for the treatment of ALL/LBL in the United States. Rylaze is a recombinant erwinia asparaginase that uses a novel Pseudomonas fluorescens expression platform. JZP458 was granted Fast Track designation by the U.S. Food and Drug Administration (FDA) in October 2019 for the treatment of this patient population. Rylaze was approved as part of the Real-Time Oncology Review program, an initiative of the FDA's Oncology Center of Excellence designed for efficient delivery of safe and effective cancer treatments to patients.
The full U.S. Prescribing Information for Rylaze is available at: https://pp.jazzpharma.com/pi/rylaze.en.USPI.pdf
For more information, please visit www.jazzpharmaceuticals.com and follow @JazzPharma on Twitter.
View original content to download multimedia:https://www.prnewswire.com/news-releases/jazz-pharmaceuticals-completes-us-fda-supplemental-biologics-license-application-for-rylaze-asparaginase-erwinia-chrysanthemi-recombinant-rywn-mondaywednesdayfriday-dosing-schedule-301473413.html
SOURCE Jazz Pharmaceuticals plc
Feb. 02, 2022 9:36 AM ET
Jazz Pharmaceuticals plc (JAZZ)
By: Ravikash, SA News Editor
Jazz Pharmaceuticals (JAZZ -0.5%) completed the submission of a supplemental biologics license application to the U.S. Food and Drug Administration seeking approval for a Monday/Wednesday/Friday intramuscular dosing schedule for Rylaze for use as a component of a multi-agent chemotherapeutic regimen to treat acute lymphoblastic leukemia and lymphoblastic lymphoma in adult and pediatric patients one month and older who have developed hypersensitivity to E. coli-derived asparaginase.
https://seekingalpha.com/symbol/JAZZ

Cabenuva is now approved for administration as few as six times a year for virologically suppressed adults living with HIV without prior treatment failure or resistance to cabotegravir or rilpivirine
London, 1 February 2022 – ViiV Healthcare, the global specialist HIV company majority-owned by GlaxoSmithKline plc (GSK), with Pfizer Inc. (Pfizer) and Shionogi Limited (Shionogi) as shareholders, today announced that the US Food and Drug Administration (FDA) approved Cabenuva (cabotegravir, rilpivirine) for every-two-month dosing for the treatment of HIV-1 in virologically suppressed adults (HIV-1 RNA less than 50 copies per millilitre [c/ml]) on a stable regimen, with no history of treatment failure, and with no known or suspected resistance to either cabotegravir or rilpivirine.
Cabenuva is the first and only complete long-acting HIV treatment regimen and was first approved by the US FDA in January 2021 as a once-monthly treatment for HIV-1 in virologically suppressed adults.1 It contains ViiV Healthcare’s cabotegravir extended-release injectable suspension in a single-dose vial and rilpivirine extended-release injectable suspension in a single-dose vial, a product of Janssen Sciences Ireland Unlimited Company, one of the Janssen Pharmaceutical Companies of Johnson & Johnson. The US FDA approval allows Cabenuva to be dosed monthly or every two months.
About ATLAS-2M (NCT03299049)
The ATLAS-2M phase IIIb trial is an ongoing, randomised, open-label, active-controlled, multicentre, parallel-group trial designed to assess the non-inferior antiviral activity and safety of long-acting cabotegravir and rilpivirine administered every eight weeks (every two months, 3ml dose of each medicine) compared to every four weeks (once monthly, 2ml dose of each medicine) over a 48-week treatment period in 1,045 adults living with HIV-1.2 Subjects were required to be virologically suppressed for six months or greater, on a first or second antiretroviral regimen, with no prior virologic failure. The primary outcome measure for the trial was the proportion of participants with HIV-1 RNA ≥ 50 c/ml at Week 48 using the US FDA Snapshot algorithm (intent-to-treat exposed population). ATLAS-2M is part of ViiV Healthcare’s extensive and innovative clinical trial programme. It is being conducted at research centres in Australia, Argentina, Canada, France, Germany, Italy, Mexico, Russia, South Africa, South Korea, Spain, Sweden and the United States.
For further information, please see https://clinicaltrials.gov/ct2/show/NCT03299049.
About Cabenuva (cabotegravir, rilpivirine)
Cabenuva is indicated as a complete regimen for the treatment of HIV-1 infection in adults to replace the current antiretroviral regimen in those who are virologically suppressed (HIV-1 RNA <50 c/ml) on a stable antiretroviral regimen with no history of treatment failure and with no known or suspected resistance to either cabotegravir or rilpivirine.
The complete regimen combines the integrase strand transfer inhibitor (INSTI) cabotegravir, developed by ViiV Healthcare, with rilpivirine, a non-nucleoside reverse transcriptase inhibitor (NNRTI) developed by Janssen. Rilpivirine is approved in the US as a 25mg tablet taken once a day to treat HIV-1 in combination with other antiretroviral agents in antiretroviral treatment-naïve patients 12 years of age and older and weighing at least 35kg with a viral load ≤100,000 HIV RNA c/ml.
INSTIs inhibit HIV replication by preventing the viral DNA from integrating into the genetic material of human immune cells (T-cells). This step is essential in the HIV replication cycle and is also responsible for establishing chronic disease. Rilpivirine is an NNRTI that works by interfering with an enzyme called reverse transcriptase, which stops the virus from multiplying.
Long-acting cabotegravir and rilpivirine are approved for use every two months in Canada under the name Cabenuva and in the EU as Vocabria and Rekambys.
Trademarks are owned by or licensed to the ViiV Healthcare group of companies.
Important Safety Information for Cabenuva (cabotegravir; rilpivirine) extended-release injectable suspensions
Cabenuva is indicated as a complete regimen for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults to replace the current antiretroviral regimen in those who are virologically suppressed (HIV-1 RNA less than 50 copies per ml) on a stable antiretroviral regimen with no history of treatment failure and with no known or suspected resistance to either cabotegravir or rilpivirine.
CONTRAINDICATIONS
Please see full Prescribing Information.
About ViiV Healthcare
ViiV Healthcare is a global specialist HIV company established in November 2009 by GlaxoSmithKline (LSE/NYSE: GSK) and Pfizer (NYSE: PFE) dedicated to delivering advances in treatment and care for people living with HIV and for people who are at risk of becoming infected with HIV. Shionogi joined in October 2012. The company’s aims are to take a deeper and broader interest in HIV and AIDS than any company has done before and take a new approach to deliver effective and innovative medicines for HIV treatment and prevention, as well as support communities affected by HIV.
For more information on the company, its management, portfolio, pipeline, and commitment, please visit www.viivhealthcare.com.
GSK is a science-led global healthcare company. For further information please visit www.gsk.com/about-us.
Feb. 01, 2022 9:23 AM ET GlaxoSmithKline plc (GSK)SGIOY, JNJ, PFE, SGIOF By: Dulan Lokuwithana, SA News Editor

January 31, 2022
– Updated Toxicity Management Strategy Can Improve Certain Adverse Events Without Compromising the Activity of Yescarta –
SANTA MONICA, Calif.--(BUSINESS WIRE)-- Kite, a Gilead Company (Nasdaq: GILD), today announced the U.S. Food and Drug Administration (FDA) has approved an update to the prescribing information for Yescarta® (axicabtagene ciloleucel) to include use of prophylactic corticosteroids across all approved indications. Yescarta is now the first and only chimeric antigen receptor (CAR) T-cell therapy with information in the label to help physicians manage, and potentially prevent, treatment side effects.
The label update is based on the results of a new safety management cohort (Cohort 6) of the pivotal ZUMA-1 study, which was designed to assess the impact of prophylactic use of corticosteroids and earlier treatment with corticosteroids and/or tocilizumab and prophylactic levetiracetam on the incidence and severity of cytokine release syndrome (CRS) and neurologic events. In the cohort, no Grade ≥3 CRS events occurred [0% (0/39) of patients in Cohort 6 compared to 13% (14/108) in the pivotal Cohorts 1/2]. Grade ≥3 neurologic events occurred in 13% of patients at the time of data cut-off, and one patient experienced a late onset Grade 5 event following the data cut-off [13% (5/39) of patients in Cohort 6 compared to 31% (33/108) in the pivotal Cohorts 1/2]. Cohort 6 shows CRS median time to onset of five days with a range from 1-15 days, and neurotoxicity median time to onset of six days with a range from 1-274 days in patients that experienced these complications.
Additional data recently published shows 68% of patients had no CRS or neurologic events within 72 hours of Yescarta infusion (27/40).1 Helping to address the potential concern that steroid use might impact efficacy, the Cohort 6 one-year update presented at the American Society of Hematology (ASH) in December 2021 suggest that Cohort 6 toxicity management strategy can improve certain adverse events without compromising the activity of Yescarta.2 Patients in Cohort 4 and Cohort 6 were found to have received median cumulative steroid doses that were lower than those used in matched Cohorts 1/2 who received steroids to manage CRS or neurologic events when they occurred.3 [1Oluwole OO, et al. Br J Haematol. 2021;194:690-700 and 2Oluwole OO, et al. 2021 ASH Annual Meeting and Exposition; December 11-14, 2021; Atlanta, GA. Abstract 2832 and 3Topp et al. Br J Haematol. 2021;195(3):388-398.]
“These new data will enable doctors to more easily and confidently manage treatment for patients,” said Frank Neumann, MD, PhD, Kite’s Global Head of Clinical Development. “Since the first approval of Yescarta, Kite has worked closely with physicians to optimize all aspects of CAR T-cell therapy to enable as many patients as possible to have the chance to benefit from this treatment. Our responsibility includes research to expand into new diseases and earlier lines of treatment, but also continuously improving the efficacy and safety of our existing CAR T therapies.”
The ZUMA-1 study also included Cohort 4 which was added to the FDA label in May 2021 and evaluated the earlier treatment with corticosteroids and/or tocilizumab and prophylactic levetiracetam. Cohort 6 builds on this growing knowledge regarding how to manage and minimize side effects gained over many years of developing Yescarta. Cohort 6 evaluated 39 patients with relapsed or refractory LBCL. Patients received dexamethasone 10 mg orally once daily for three days, starting prior to Yescarta infusion. In Cohort 6, corticosteroids and tocilizumab were started earlier, at lower grades of CRS and neurologic events, than in the ZUMA-1 pivotal cohorts (Cohorts 1 and 2). All 39 patients received three prophylactic doses of corticosteroids.
Patients can currently access Kite’s CAR T-cell therapies through 111 authorized treatment centers across the U.S. that are experienced in administering and managing patients being treated with CAR T-cell therapy. The Yescarta prescribing information does not specify a treatment setting, whether in the hospital or in near-by outpatient offices with close monitoring. It is up to a patient’s physician to determine the most appropriate treatment setting for their care based on individual circumstances. The Yescarta label update may help physicians manage potential complications and determine appropriate setting for care of their patients.
Yescarta was the first CAR T-cell therapy to be approved by the U.S. FDA for the treatment of adult patients with relapsed or refractory large B-cell lymphoma (LBCL) after two or more lines of systemic therapy. Yescarta is also currently under review in the U.S. and European Union for use as an earlier treatment of adult patients with second-line relapsed or refractory LBCL.
About Yescarta
Please see full Prescribing Information, including BOXED WARNING and Medication Guide.
Yescarta is a CD19-directed genetically modified autologous T cell immunotherapy currently indicated for the treatment of:
For more information on Kite, please visit www.kitepharma.com.
U.S. Prescribing Information for Yescarta including BOXED WARNING, is available at www.kitepharma.com and www.gilead.com.
Kite, the Kite logo, Yescarta, Tecartus, XLP and GILEAD are trademarks of Gilead Sciences, Inc. or its related companies.
For more information on Kite, please visit the company’s website at www.kitepharma.com. Follow Kite on social media on Twitter (@KitePharma) and LinkedIn.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220131005477/en/
Source: Gilead Sciences, Inc.
Jan. 31, 2022 9:45 AM ET Gilead Sciences, Inc. (GILD) By: Dulan Lokuwithana, SA News Editor

January 31, 2022
CAMBRIDGE, Massachusetts, January 31, 2022 – Takeda Pharmaceutical Company Limited (TSE:4502/NYSE:TAK) (“Takeda”) today announced that the U.S. Food & Drug Administration (FDA) approved VONVENDI® [von Willebrand factor (Recombinant)] for routine prophylaxis to reduce the frequency of bleeding episodes in patients with severe Type 3 von Willebrand disease (VWD) receiving on-demand therapy.
VONVENDI is the only recombinant von Willebrand factor (VWF) replacement therapy, and the first and only treatment to reduce the frequency of bleeding episodes for severe Type 3 VWD approved by the FDA for routine prophylactic use. VONVENDI is now indicated for routine prophylaxis in adults with severe Type 3 VWD receiving on-demand therapy, as well as on-demand and perioperative bleed management in adults with VWD.
“This approval is a major advancement for those living with severe Type 3 VWD and is a testament to Takeda's commitment to improve VWD care,” said Heather Dean, Vice President, U.S. Hematology Franchise Head, Takeda. “With routine prophylactic treatment, there is now a proactive strategy available for management of bleeding episodes and may offer people living with severe Type 3 VWD hope that bleed reduction is possible.”
VWD is an inherited disorder that affects women and men equally.1 It is caused by a deficiency or defective function of VWF, one of several types of proteins in the blood that are needed for proper blood clotting.1 Due to this defective function or deficiency, blood is not able to clot effectively in people with VWD. VONVENDI is an infused product that is specifically designed to replace the body's missing or dysfunctional VWF.1
“VWD is a complex disease where both patients and providers may experience stress and uncertainty due to the unpredictable disease course and limited treatment options,” said Miguel A. Escobar, M.D., professor in the Department of Pediatrics and Internal Medicine at the McGovern Medical School at The University of Texas Health Science Center at Houston and an investigator in the VONVENDI prophylaxis study*. “A prophylactic treatment option may allow for greater disease control and the potential to enhance the standard of care.”
The approval is based on data from a prospective, open-label, international multicenter study to evaluate efficacy and safety of prophylactic treatment in reducing the frequency of bleeding episodes in 10 adult patients diagnosed with severe Type 3 VWD who were previously treated on-demand. Based on descriptive statistics, the median annualized bleeding rates (ABR) for all bleeds (treated and untreated spontaneous and traumatic bleeding events) was reduced from historical median ABR 5.0 (range: 3.0, 159.0) to an on-study median ABR of 2.3 (range: 0, 157.9), which is a 54.7% reduction. The most common adverse reactions (≥2% of subjects) observed in adult patients treated with VONVENDI in clinical trials were headache, vomiting, nausea, dizziness, arthralgia, joint injury, vertigo, ALT increased and generalized pruritus.
VONVENDI is used in adults (age 18 years and older) diagnosed with von Willebrand disease to:
For more information, visit https://www.takeda.com.
Jan. 31, 2022 10:40 AM ET
Takeda Pharmaceutical Company Limited (TAK)
By: Dulan Lokuwithana, SA News Editor

Jan 28, 2022
ACTON, Mass.--(BUSINESS WIRE)--Jan. 28, 2022-- Insulet Corporation (NASDAQ: PODD) (Insulet or the Company), the global leader in tubeless insulin pump technology with its Omnipod® brand of products, today announced it has received clearance from the U.S. Food and Drug Administration (FDA) for its Omnipod® 5 Automated Insulin Delivery System (Omnipod 5) for individuals aged six years and older with type 1 diabetes. Omnipod 5 is the first tubeless automated insulin delivery (AID) system that integrates with the Dexcom G6 Continuous Glucose Monitoring (CGM) System and a compatible smartphone to automatically adjust insulin and help protect against highs and lows.
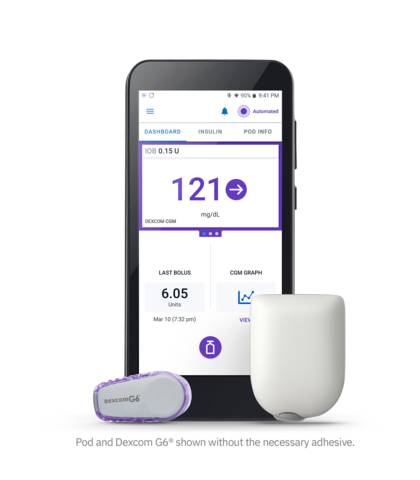
Omnipod® 5 Automated Insulin Delivery System (Photo: Business Wire)
Omnipod 5 is designed to make it easier than ever to manage glucose with no multiple daily injections, no tubes, and zero fingersticks1 to help simplify life with diabetes.
“Omnipod 5 is a life-changing technology that we believe will revolutionize the market and the lives of people with diabetes,” said Shacey Petrovic, President and Chief Executive Officer of Insulet. “We are incredibly proud of this simple-to-use, elegant system, designed to deliver unmatched freedom and to greatly simplify insulin management and improve glucose control for our users.”
The Omnipod 5 System2 consists of the tubeless Pod enhanced with SmartAdjust™ technology, the Dexcom G6 CGM, and the Omnipod 5 mobile app with its integrated SmartBolus Calculator. The user has the option to download this app onto a compatible personal smartphone or to use the Omnipod 5 Controller, which is provided free with the first prescription.
Every five minutes, SmartAdjust receives a Dexcom CGM value and trend, and predicts where glucose will be 60 minutes into the future. The system then increases, decreases, or pauses insulin delivery using the user’s desired and customized glucose target, helping to protect against highs and lows.
“As the pioneer of integrated CGM, we are excited to see our years of collaborative work with Insulet culminate into the first and only FDA-cleared tubeless automated insulin delivery system,” said Kevin Sayer, Chairman, President and CEO of Dexcom. “Omnipod 5 combines the accuracy and unmatched user experience of the Dexcom G6 CGM with the simplicity of tubeless insulin delivery to offer people with diabetes a revolutionary new way to optimize time in range.”
Omnipod 5 will launch through the pharmacy channel, providing customer benefits of no contract, no commitment, and no obligation. In the coming days, Insulet will offer Omnipod 5 in a limited market release so that the Company can incorporate learnings to deliver a best-in-class product experience. Omnipod 5 is expected to be broadly available shortly after the limited market release.
Omnipod 5 was recently selected as a CES® 2022 Innovation Award honoree in two categories: Health and Wellness and Wearable Technologies. The CES Innovation Awards is an annual competition honoring outstanding design and engineering in consumer technology products.
People with diabetes can enjoy the benefits of Pod Therapy today through OmnipodPromise™, which allows new and existing users to start on Omnipod DASH® and upgrade to Omnipod 5 at no additional cost when coverage is available.
To learn more, visit the Omnipod website.
Insulet will provide additional details about today’s announcement on the Company’s fourth quarter 2021 earnings conference call on February 23, 2022 at 4:30 p.m. (Eastern Time). The link to the live call will be available here and will be archived for future replay. To participate in the live call via phone, please pre-register online here to receive a telephone number, passcode, and a unique registrant ID required to enter the call.
For more information, please visit insulet.com and omnipod.com.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220127006047/en/
Source: Insulet Corporation
Jan. 28, 2022 6:32 AM ET Insulet Corporation (PODD) By: Mamta Mayani, SA News Editor
January 31, 2022 at 12:59 AM EST Back
TARRYTOWN, N.Y. and PARIS, Jan. 31, 2022 /PRNewswire/ --
Recommendation based on pivotal trial that showed Dupixent significantly reduced severe asthma attacks and improved lung function in children aged 6 to 11 years
Dupixent is the only biologic to show improved lung function in a randomized Phase 3 trial for children
Data further reinforce well-established safety profile of Dupixent
Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) and Sanofi today announced that the European Medicines Agency's Committee for Medicinal Products for Human Use (CHMP) has adopted a positive opinion, recommending to extend the approval of Dupixent® (dupilumab) in the European Union (EU) to include add-on maintenance treatment for children aged 6 to 11 years with severe asthma with type 2 inflammation characterized by raised blood eosinophils and/or raised fractional exhaled nitric oxide (FeNO) who are inadequately controlled on two maintenance therapies. The European Commission is expected to announce a final decision on the Dupixent application in the coming months.
The CHMP positive opinion is supported by Phase 3 data recently published in the New England Journal of Medicine showing that Dupixent added to standard of care significantly reduced the rate of severe asthma attacks and rapidly improved lung function within two weeks, with sustained improvement up to 52 weeks, in children with uncontrolled moderate-to-severe asthma. The safety results from the trial were generally consistent with the known safety profile of Dupixent in patients aged 12 years and older with uncontrolled moderate-to-severe asthma. Adverse events more commonly observed with Dupixent compared to placebo included injection site reactions, viral upper respiratory tract infections and eosinophilia. Helminth infections were also more commonly observed with Dupixent compared to placebo in patients aged 6 to 11 years.
Asthma is one of the most common chronic diseases in children. Up to 85% of children with asthma may have type 2 inflammation and are more likely to have higher disease burden. Despite treatment with current standard-of-care inhaled corticosteroids (ICS) and bronchodilators, these children may continue to experience serious symptoms such as coughing, wheezing and difficulty breathing. Severe asthma may impact children's developing airways and cause potentially life-threatening exacerbations. Children with severe asthma also may require the use of multiple courses of systemic corticosteroids that carry significant risks. Uncontrolled severe asthma can interfere with day-to-day activities like sleeping, attending school and playing sports.
On October 20, 2021, Dupixent was approved by the FDA as an add-on maintenance treatment for patients aged 6 to 11 years with moderate-to-severe asthma characterized by an eosinophilic phenotype or with oral corticosteroid-dependent asthma. The use of Dupixent in children aged 6 to 11 years with uncontrolled severe asthma is investigational in the EU and is not yet approved.
About Dupixent
Dupixent, which was invented using Regeneron's proprietary VelocImmune® technology, is a fully human monoclonal antibody that inhibits the signaling of the interleukin-4 (IL-4) and interleukin-13 (IL-13) pathways and is not an immunosuppressant. IL-4 and IL-13 are key and central drivers of the type 2 inflammation that plays a major role in atopic dermatitis, asthma and chronic rhinosinusitis with nasal polyposis (CRSwNP).
Dupixent is currently approved in Europe for adults and adolescents 12 years and older as an add-on maintenance treatment for severe asthma with type 2 inflammation characterized by raised blood eosinophils and/or raised FeNO, who are inadequately controlled with high-dose ICS plus another medicinal product for maintenance treatment. It is also approved in the U.S., Japan and other countries around the world for use in certain patients with asthma, specific patients with moderate-to-severe atopic dermatitis as well as CRSwNP in different age populations. Dupixent is also approved in one or more of these indications in more than 60 countries around the world, and more than 350,000 patients have been treated globally.
Dupilumab Development Program
Dupilumab is being jointly developed by Regeneron and Sanofi under a global collaboration agreement. To date, dupilumab has been studied across 60 clinical trials involving more than 10,000 patients with various chronic diseases driven in part by type 2 inflammation.
In addition to the currently approved indications, Regeneron and Sanofi are studying dupilumab in a broad range of diseases driven by type 2 inflammation or other allergic processes including chronic obstructive pulmonary disease with evidence of type 2 inflammation (Phase 3), pediatric atopic dermatitis (6 months to 5 years of age, Phase 3), eosinophilic esophagitis (Phase 3), bullous pemphigoid (Phase 3), prurigo nodularis (Phase 3), chronic spontaneous urticaria (Phase 3), chronic inducible urticaria-cold (Phase 3), chronic rhinosinusitis without nasal polyposis (Phase 3), allergic fungal rhinosinusitis (Phase 3), allergic bronchopulmonary aspergillosis (Phase 3) and peanut allergy (Phase 2). These potential uses of dupilumab are currently under clinical investigation, and the safety and efficacy in these conditions have not been fully evaluated by any regulatory authority.
U.S. Indications
DUPIXENT is a prescription medicine used:
Please see accompanying full Prescribing Information including Patient Information.
For additional information about the company, please visit www.regeneron.com or follow @Regeneron on Twitter.
View original content:https://www.prnewswire.com/news-releases/chmp-recommends-approval-of-dupixent-dupilumab-for-children-aged-6-to-11-years-with-severe-asthma-with-type-2-inflammation-301470938.html
SOURCE Regeneron Pharmaceuticals, Inc.
Jan. 31, 2022 5:50 AM ET Regeneron Pharmaceuticals, Inc. (REGN), SNY By: Mamta Mayani, SA News Editor
February 1, 2022 at 4:29 PM EST
TARRYTOWN, N.Y. and PARIS, Feb. 1, 2022 /PRNewswire/ --
Late-breaking pivotal data show significant disease improvements in eosinophilic esophagitis and also in chronic spontaneous urticaria
18 abstracts add to body of evidence that IL-4 and IL-13 pathways inhibited by Dupixent are key drivers of the type 2 inflammation underlying certain diseases, accounting for the impact of Dupixent in its approved and investigational indications
Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) and Sanofi today announced positive Dupixent® (dupilumab) data across five diseases with underlying type 2 inflammation will be presented at the American Academy of Allergy, Asthma and Immunology (AAAAI) Annual Meeting from February 25 to 28.

Friday, Jan 28, 2022
South San Francisco, CA -- January 28, 2022 --
Genentech, a member of the Roche Group (SIX: RO, ROG; OTCQX: RHHBY), today announced that the U.S. Food and Drug Administration (FDA) has approved Vabysmo ™ (faricimab-svoa) for the treatment of wet, or neovascular, age-related macular degeneration (AMD) and diabetic macular edema (DME). Wet AMD and DME are two leading causes of vision loss among U.S. adults. Vabysmo targets and inhibits two disease pathways linked to a number of vision-threatening retinal conditions by neutralizing angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A). Vabysmo is the first and only FDA-approved injectable eye medicine for wet AMD and DME that improves and maintains vision with treatments from one to four months apart in the first year following four initial monthly doses, based on evaluation of the patient’s anatomy and vision outcomes. Standard of care for wet AMD and DME typically requires eye injections every one to two months.
“Vabysmo represents an important step forward for ophthalmology. It is the first bispecific antibody approved for the eye and a major advance in treating retinal conditions such as wet AMD and diabetic macular edema,” said Charles Wykoff, M.D., Ph.D., Director of Research at Retina Consultants of Texas in Houston and a Vabysmo Phase III investigator. “With Vabysmo, we now have the opportunity to offer patients a medicine that could improve their vision, potentially lowering treatment burden with fewer injections over time.”
The approval is based on positive results across four Phase III studies in wet AMD and DME. The studies consistently showed that patients treated with Vabysmo given at intervals of up to four months achieved non-inferior vision gains versus aflibercept given every two months in the first year. Vabysmo was generally well tolerated in all four studies, with a favorable benefit-risk profile. The most common adverse reaction (≥5%) reported in patients receiving Vabysmo was conjunctival hemorrhage (7%).
Two scientific papers and an editorial on these one-year results were recently published in The Lancet.
Vabysmo is designed to block pathways involving Ang-2 and VEGF-A. Ang-2 and VEGF-A are thought to contribute to vision loss by destabilizing blood vessels, which may cause new leaky blood vessels to form and increase inflammation. While additional research continues, inhibition of both pathways has been shown in preclinical studies to have potentially complementary benefits, stabilizing vessels and thereby reducing vessel leakage and inflammation.
“Vabysmo provides a new approach to treating vision-threatening retinal conditions through a mechanism of action that targets two pathways simultaneously,” said Levi Garraway, M.D., Ph.D., chief medical officer and head of Global Product Development. “This is our second FDA approval in ophthalmology in recent months, underscoring our commitment to people living with retinal conditions.”
With Vabysmo, people with wet AMD initially receive four monthly treatments. Based on anatomical and vision outcomes, they may receive subsequent treatments every two, three or four months. People with DME are initially given four monthly treatments. Subsequently their treatment may be extended or reduced based on anatomical and vision outcomes, with a range of one to four months between doses. A second approved treatment regimen for DME involves six monthly loading doses, followed by treatment every two months. Some people with wet AMD and DME may be treated monthly if needed, although additional efficacy was not demonstrated in most patients given Vabysmo every month.
Genentech has ongoing long-term extension studies for Vabysmo in people with wet AMD and DME. These include AVONELLE-X, an extension study of TENAYA and LUCERNE evaluating the long-term safety and tolerability of Vabysmo in wet AMD, and RHONE-X, an extension study of YOSEMITE and RHINE evaluating the long-term safety and tolerability of Vabysmo in DME. Additionally, the COMINO and BALATON trials are also underway, evaluating the efficacy and safety of Vabysmo in people with macular edema following retinal vein occlusion.
Vabysmo will be available in the United States in the coming weeks. The European Medicines Agency is also currently evaluating the Vabysmo Marketing Authorization Application for the treatment of wet AMD and DME.
Genentech is committed to helping people access the medicines they are prescribed and will be offering comprehensive services for people prescribed Vabysmo to help minimize barriers to access and reimbursement. Patients can call 833-EYE-GENE for more information. For people who qualify, Genentech plans to offer patient assistance programs through Genentech Access Solutions. More information is also available at 866-4ACCESS/866-422-2377 or http://www.Genentech-Access.com.
About the TENAYA and LUCERNE Studies
TENAYA (NCT03823287) and LUCERNE (NCT03823300) are two identical, randomized, multicenter, double-masked, global Phase III studies evaluating the efficacy and safety of Vabysmo compared to aflibercept in 1,329 people living with wet age-related macular degeneration (671 in TENAYA and 658 in LUCERNE). The studies each have two treatment arms: Vabysmo 6.0 mg administered at intervals of two, three, or four months, following four initial monthly doses, selected based on objective assessment of disease activity as measured by optical coherence tomography and visual acuity evaluations at weeks 20 and 24; and aflibercept 2.0 mg administered at fixed two-month intervals after three initial monthly doses. In both arms, sham injections were administered at study visits when treatment injections were not scheduled, to maintain the masking of investigators and participants.
The primary endpoint of the studies is the average change in best-corrected visual acuity (BCVA) score (the best distance vision a person can achieve – including with correction such as glasses – when reading letters on an eye chart) from baseline, averaged over weeks 40, 44 and 48. Secondary endpoints include: safety; the percentage of participants in the Vabysmo arm receiving treatment every two, three and four months; the percentage of participants achieving a gain, and the percentage avoiding a loss, of 15 letters or more in BCVA from baseline over time; change in central subfield thickness (CST) from baseline over time; and change in total area of choroidal neovascularization (CNV) lesion and leakage from baseline over time.
Both studies met their primary endpoint, with Vabysmo given at intervals of up to every four months consistently shown to offer visual acuity gains that were non-inferior to aflibercept given every two months. In TENAYA and LUCERNE, the average vision gains from baseline at one year in the Vabysmo arms were +5.8 and +6.6 letters, respectively, compared to +5.1 and +6.6 letters in the aflibercept arms.
The studies also measured the proportion of people in the Vabysmo arm that were treated on dosing schedules of every three or four months during the first year. In both studies, comparable reductions in central subfield thickness (CST) and CNV size and area of leakage were observed with Vabysmo given at intervals of up to four months versus aflibercept given every two months in the first year. Vabysmo was generally well-tolerated in both studies, with a favorable benefit-risk profile. In TENAYA and LUCERNE, the most common adverse reactions (≥3% of patients) included conjunctival hemorrhage, vitreous floaters, retinal pigment epithelial (RPE) tears, increase of intraocular pressure and eye pain. Safety results were consistent across study arms.
About the YOSEMITE and RHINE Studies
YOSEMITE (NCT03622580) and RHINE (NCT03622593) are two identical, randomized, multicenter, double-masked, global Phase III studies evaluating the efficacy and safety of Vabysmo compared to aflibercept in 1,891 people with diabetic macular edema (940 in YOSEMITE and 951 in RHINE). The studies each have three treatment arms: Vabysmo 6.0 mg administered up to every four months after four initial monthly doses using a treat-and-extend approach; Vabysmo 6.0 mg administered at two-month intervals after six initial monthly doses; and aflibercept administered at fixed two-month intervals after five initial monthly doses. Dosing schedule for patients within the treat-and-extend arm was determined by CST and visual acuity. In all three arms, sham injections were administered at study visits when treatment injections were not scheduled, to maintain the masking of investigators and participants.
The primary endpoint of the studies is the average change in BCVA score from baseline at one year, averaged over weeks 48, 52 and 56. Secondary endpoints included: safety; the percentage of participants in the treat-and-extend arm receiving Vabysmo every one, two, three and four months, at week 52; the percentage of participants achieving a two-step or greater improvement from baseline in diabetic retinopathy severity at week 52; the percentage of participants achieving a gain, and the percentage avoiding a loss, of 15 letters or more in BCVA from baseline over time; change in CST from baseline over time; and percentage of patients with absence of intraretinal fluid over time.
Both studies met their primary endpoint, with Vabysmo at intervals of up to every four months consistently shown to offer visual acuity gains that were non-inferior to aflibercept given every two months. In YOSEMITE, the average vision gains from baseline at one year were +11.6 and +10.7 eye chart letters in the Vabysmo treat-and-extend and two-month arms, respectively, and +10.9 letters in the aflibercept arm. In RHINE, the average vision gains from baseline at one year were +10.8 and +11.8 letters in the Vabysmo treat-and-extend and two-month arms, respectively, and +10.3 letters in the aflibercept arm.
A secondary endpoint in both studies measured the proportion of people in the Vabysmo treat-and-extend arm that achieved dosing schedules of every three or four months at the end of the first year. In both studies, greater reductions in CST and intraretinal fluid were observed with Vabysmo given at intervals of up to four months versus aflibercept given every two months in the first year. Vabysmo was generally well-tolerated in both studies, with a favorable benefit-risk profile. In YOSEMITE and RHINE, the most common adverse reactions (≥3% of patients) included conjunctival hemorrhage, vitreous floaters and increase of intraocular pressure. Safety results were consistent across study arms.
About Vabysmo™ (faricimab-svoa)
Vabysmo (faricimab-svoa) is the first bispecific antibody approved for the eye. It targets and inhibits two disease pathways linked to a number of vision-threatening retinal conditions by neutralizing angiopoietin-2 (Ang-2) and vascular endothelial growth factor-A (VEGF-A). Ang-2 and VEGF-A contribute to vision loss by destabilizing blood vessels, causing new leaky blood vessels to form and increasing inflammation. By blocking pathways involving Ang-2 and VEGF-A, Vabysmo is designed to stabilize blood vessels.
Vabysmo U.S. Indications
Vabysmo (faricimab-svoa) is a prescription medicine given by injection into the eye, used to treat adults with neovascular (wet) age‑related macular degeneration (AMD) and diabetic macular edema (DME).
Please see additional Important Safety Information in the full Vabysmo Prescribing Information.
For additional information about the company, please visit http://www.gene.com.
Jan. 30, 2022 11:50 PM ET Roche Holding AG (RHHBY)
By: Mamta Mayani, SA News Editor

Friday, January 28, 2022 - 09:46am
Approval based on results from Phase 3 CROWN trial, showing LORVIQUA reduced risk of disease progression or death by 72% in newly diagnosed individuals compared to XALKORI® (crizotinib)
NEW YORK--(BUSINESS WIRE)-- Pfizer Inc. (NYSE: PFE) announced today that the European Commission (EC) granted marketing authorization for LORVIQUA® (lorlatinib, available in the U.S. under the brand name LORBRENA®) as monotherapy for the treatment of adult patients with anaplastic lymphoma kinase (ALK)- positive advanced non-small cell lung cancer (NSCLC) previously not treated with an ALK inhibitor.
“For more than a decade, Pfizer has worked tirelessly in its pursuit to help transform the trajectory for people living with advanced, biomarker-driven lung cancers,” said Andy Schmeltz, Global President & General Manager, Pfizer Oncology. “The European Commission’s approval of LORVIQUA as a first-line therapy is a significant milestone that we hope will help bring a needed and meaningful difference to those impacted by this deadly disease in Europe.”
The approval for the first-line use of LORVIQUA was based on the results of the pivotal Phase 3 CROWN trial, in which LORVIQUA reduced the risk of disease progression or death by 72% compared to XALKORI® (crizotinib). As a secondary endpoint, the confirmed objective response rate (ORR) was 76% (95% CI, 68 to 83) with LORVIQUA and 58% (95% CI, 49 to 66) with XALKORI. In patients with measurable brain metastases, 82% of patients in the LORVIQUA arm experienced an intracranial response (71% had an intracranial complete response), compared to 23% of XALKORI patients. The CROWN trial is a randomized, open-label, parallel 2-arm trial in which 296 people with previously untreated advanced ALK-positive NSCLC were randomized 1:1 to receive LORVIQUA monotherapy (n=149) or XALKORI monotherapy (n=147).
“The expanded approval for LORVIQUA in Europe is a considerable advancement – especially for the close to 40 percent of patients with ALK-positive metastatic NSCLC who are faced with brain metastases at diagnosis,” said Professor Benjamin Solomon, MBBS, PhD., Department of Medical Oncology at the Peter MacCallum Cancer Centre in Melbourne, Australia. “It is exciting to see the significant data generated from the CROWN trial continuing to support expanded use around the world and providing physicians in Europe with a highly effective option from the onset of their patients’ treatment journey.”
The EC approval of LORVIQUA follows a positive opinion from the Committee for Medicinal Products for Human Use (CHMP) in December 2021. LORVIQUA is approved in the U.S. by the Food and Drug Administration (FDA) under the brand name LORBRENA for the treatment of adults with metastatic NSCLC whose tumors are ALK-positive as detected by an FDA-approved test. In 2019, the EC granted conditional marketing authorization for LORVIQUA as a monotherapy for the treatment of adult patients with ALK-positive advanced NSCLC whose disease has progressed after alectinib or ceritinib as the first ALK tyrosine kinase inhibitor (TKI) therapy, or crizotinib and at least one other ALK TKI.
About the CROWN Trial of LORVIQUA
In the CROWN trial, patients were required to have an ECOG performance status of 0-2 and ALK-positive NSCLC as identified by the VENTANA ALK (D5F3) CDx assay. The primary endpoint of the CROWN trial was progression-free survival (PFS) based on blinded independent central review (BICR). Secondary endpoints included overall survival (OS) and tumor assessment related data by BICR, including ORR, and duration of response (DOR). In patients with measurable central nervous system (CNS) metastases at baseline, additional outcome measures were intracranial (IC)-ORR and IC-DOR by BICR. The trial is continuing in order to further evaluate the secondary endpoint of OS, which was not mature at the time of analysis.
Overall, the safety profile of LORVIQUA was similar to that reported in previous studies. The most frequent adverse events (AEs) in ≥20% of 149 patients treated with LORVIQUA were edema, weight gain, peripheral neuropathy, cognitive effects, diarrhea, dyspnea and hypertriglyceridemia. Serious AEs occurred in 34% of people treated with LORVIQUA; the most frequently reported serious AEs were pneumonia, dyspnea, respiratory failure, cognitive effects and pyrexia. Fatal AEs occurred in 3.4% of people treated with LORVIQUA. Permanent discontinuation of LORVIQUA due to AEs occurred in 6.7% of people. Detailed results from the CROWN study were published in the November 2020 issue of the New England Journal of Medicine.
About LORVIQUA® (lorlatinib)
LORVIQUA is a TKI that has been shown to be highly active in preclinical lung cancer models harboring chromosomal rearrangements of ALK. LORVIQUA was specifically developed to inhibit tumor mutations that drive resistance to other ALK inhibitors and to penetrate the blood brain barrier.
The full U.S. prescribing information for LORBRENA can be found here.
IMPORTANT LORBRENA® (lorlatinib) SAFETY INFORMATION FROM THE U.S. PRESCRIBING INFORMATION
Contraindications: LORBRENA is contraindicated in patients taking strong CYP3A inducers, due to the potential for serious hepatotoxicity.
In addition, to learn more, please visit us on www.Pfizer.com and follow us on Twitter at @Pfizer and @Pfizer News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
Jan. 28, 2022 10:25 AM ET
By: Dulan Lokuwithana, SA News Editor
Jan. 27, 2022 8:30 AM ET Eli Lilly and Company (LLY)
The Verzenio Phase 3 monarchE trial was a randomized (1:1), open-label, multicentre study in adult women and men with HR+ HER2-, node-positive, resected, EBC with clinical and pathological features consistent with a high risk of disease recurrence. In the trial, patients were randomized to receive two years of Verzenio 150 mg twice daily plus physician's choice of standard endocrine therapy (ET), or standard ET alone. Patients in both treatment arms were instructed to continue to receive adjuvant endocrine therapy for up to 5-10 years as recommended by their clinician. At the second interim efficacy analysis, with a median follow-up of 15.5 months, a statistically significant improvement in invasive disease–free survival (IDFS) was observed in patients who received Verzenio plus endocrine therapy versus those who received endocrine therapy alone. Patients treated with Verzenio plus endocrine therapy had a 25.3% reduction in risk of IDFS recurrence or death compared to patients treated with endocrine therapy alone. Consistent benefit was observed across patient stratification subgroups of geographic region, prior chemotherapy, and menopausal status.
At the final IDFS analysis, efficacy results were updated for the intent to treat (ITT) population, with a median follow-up of 19.1 months. Patients treated with Verzenio plus endocrine therapy had a 28.7% reduction in risk of recurrence or death compared to patients treated with ET alone (IDFS HR=0.713, 95% CI: 0.583, 0.871) and the 2-year IDFS rates were 92.3% and 89.3%, respectively. Also, patients treated with Verzenio plus ET had a 31.3% reduction in risk of distant recurrence or death compared to patients treated with ET alone [distant relapse free survival (DRFS) HR=0.687, 95% CI: 0.551, 0.858] and the 2 year DRFS rates were 93.8% and 90.8%, respectively. The most common sites of distance recurrence were bone, liver, and lung. In this efficacy analysis, overall survival data were immature and a total of 106 patients had died (55 deaths (2.0%) in patients treated with Verzenio plus ET and 51 deaths (1.8%) with ET alone).2
Having achieved the study's primary endpoint in the entire enrolled population, a pre-specified analysis of IDFS was also conducted in patients with high-risk clinical and pathological factors and a Ki-67 score ≥20%. This subgroup analysis (N=2,003) included patients with ≥4 positive axillary lymph nodes (ALN), or 1-3 positive ALN with either Grade 3 disease and/or tumour size ≥5 cm, and whose tumours had a Ki-67 score of ≥20%. There was also a statistically significant improvement in IDFS for this pre-specified subgroup of patients receiving Verzenio plus ET compared to those who received ET alone (HR=0.643, 95% CI: 0.475, 0.872, p=0.0042).2
About the monarchE Study
The efficacy of Verzenio plus physician's choice of standard endocrine therapy was evaluated in monarchE, a randomized (1:1), open-label, multicenter study in adult women and men with HR-positive, HER2-negative, node-positive, resected, early breast cancer (EBC) with clinical and pathological features consistent with a high risk of disease recurrence. To be enrolled, all patients had to have HR positive, HER2-negative EBC with tumor involvement in at least 1 axillary lymph node (pALN). Two cohorts of patients were enrolled. To be enrolled in cohort 1, patients needed to have either ≥4 pALN, or pALN 1-3 and tumor grade 3 and/or tumor size ≥ 50 mm. To be enrolled in cohort 2, patients were required to have pALN 1-3 and Ki-67 score of ≥20% as measured in untreated breast tumor tissue, using a clinical trial assay at a central laboratory. For patients in cohort 1 with available untreated breast tissue samples, retrospective central testing of tissue was conducted to establish if the Ki-67 score was ≥20% or < 20% (specified in the protocol as "Ki-67 high" or "Ki-67 low", respectively). The intent to treat (ITT) population included patients from both cohort 1 (n=5120) and cohort 2 (n=517). Randomization to treatment was stratified by prior treatment (neoadjuvant chemotherapy versus adjuvant chemotherapy versus no chemotherapy); menopausal status (premenopausal versus postmenopausal); and region (North America/Europe versus Asia versus other). Men were stratified as postmenopausal.1,2
About Verzenio® (abemaciclib)
Verzenio® (abemaciclib) is a targeted treatment known as a CDK4/6 inhibitor. Verzenio is a non-chemotherapy oral tablet.
Verzenio works inside the cell to block CDK4/6 activity and help stop the growth of cancer cells, so they may eventually die (based on preclinical studies). Cyclin-dependent kinases (CDK)4/6 are activated by binding to D-cyclins. In estrogen receptor-positive (ER+) breast cancer cell lines, cyclin D1 and CDK4/6 promote phosphorylation of the retinoblastoma protein (Rb), cell cycle progression, and cell proliferation.
In vitro, continuous exposure to Verzenio inhibited Rb phosphorylation and blocked progression from G1 to S phase of the cell cycle, resulting in senescence and apoptosis (cell death). Preclinically, Verzenio dosed daily without interruption resulted in reduction of tumour size. Inhibiting CDK4/6 in healthy cells can result in side effects, some of which may be serious. Clinical evidence also suggests that Verzenio crosses the blood-brain barrier. In patients with advanced cancer, including breast cancer, concentrations of Verzenio and its active metabolites (M2 and M20) in cerebrospinal fluid are comparable to unbound plasma concentrations.
Verzenio is Lilly's first solid oral dosage form to be made using a faster, more efficient process known as continuous manufacturing. Continuous manufacturing is a new and advanced type of manufacturing within the pharmaceutical industry, and Lilly is one of the first companies to use this technology.
INDICATIONS FOR VERZENIO
Early Breast Cancer
Verzenio® (abemaciclib) (tablets) is indicated in combination with endocrine therapy for the adjuvant treatment of adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, node-positive, early breast cancer at high risk of disease recurrence based on clinicopathological features and a Ki-67 score ≥20 per cent.
Advanced or Metastatic Breast Cancer
Verzenio (abemaciclib) (tablets) is indicated for the treatment of hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer.

Jan 28, 2022 5:00 AM
BRUKINSA was granted its first breakthrough therapy designation in China with this application
The application is supported by SEQUOIA trial results, which demonstrated BRUKINSA’s superiority in efficacy over chemoimmunotherapy
BRUKINSA received the China NMPA conditional approval for the treatment of patients with relapsed or refractory chronic lymphocytic leukemia in June 2020
CAMBRIDGE, Mass. & BEIJING--(BUSINESS WIRE)-- BeiGene (NASDAQ: BGNE; HKEX: 06160; SSE: 688235), a global, science-driven biotechnology company focused on developing innovative and affordable medicines, today announced that the Center for Drug Evaluation (CDE) of the China National Medical Products Administration (NMPA) has accepted a supplemental new drug application (sNDA) for BeiGene’s BTK inhibitor BRUKINSA (zanubrutinib) as a treatment for adult patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) and granted BRUKINSA breakthrough therapy designation (BTD).
“This is BRUKINSA’s first filing in treatment-naïve CLL supported by the positive global Phase 3 SEQUOIA trial, a remarkable step forward in its global registration program. As presented at ASH, BRUKINSA significantly prolonged progression-free survival and was generally well-tolerated in these patients, with demonstrated superiority over chemoimmunotherapy in the SEQUOIA trial,” commented Jane Huang, M.D., Chief Medical Officer of Hematology at BeiGene. “Together with the filing in Waldenström’s macroglobulinemia, we are hoping to expand the clinical use of this potential best-in-class BTK inhibitor from relapsed or refractory setting to frontline care for the blood cancer community in China.”
The sNDA is supported by clinical results from the randomized, multicenter, global Phase 3 SEQUOIA trial (NCT03336333) comparing BRUKINSA to bendamustine in combination with rituximab (B+R) in patients with treatment-naïve CLL.
As assessed by an independent review committee (IRC), BRUKINSA demonstrated superiority in progression-free survival (PFS) over B+R. With a median follow-up of 26.15 months, the 24-month PFS was 85.5% (95% CI: 80.1, 89.6) with BRUKINSA, compared to 69.5% (95% CI: 62.4, 75.5) with B+R, and the hazard ratio (HR) was 0.42 (95% CI: 0.27, 0.63), p<0.0001. BRUKINSA was generally well tolerated with a safety profile consistent with its broad clinical program, including a low rate of atrial fibrillation.
About Chronic Lymphocytic Leukemia
Chronic lymphocytic leukemia (CLL) is the most common form of leukemia in adults, with a global incidence of approximately 114,000 new cases in 2017. CLL affects white blood cells or lymphocytes in the bone marrow, Proliferation of cancer cells (leukemia) in the marrow result in reduced ability to fight infection and spread into the blood, which affects other parts of the body including the lymph nodes, liver and spleen.1,2,3 The BTK pathway is a known route that signals malignant B cells and contributes to the onset of CLL.4 Small lymphocytic lymphoma (SLL) is a non-Hodgkin’s lymphoma affecting the B-lymphocytes of the immune system, which shares many similarities to CLL but with cancer cells found mostly in lymph nodes.5
About BRUKINSA
BRUKINSA is a small molecule inhibitor of Bruton’s tyrosine kinase (BTK) discovered by BeiGene scientists that is currently being evaluated globally in a broad clinical program as a monotherapy and in combination with other therapies to treat various B-cell malignancies. Because new BTK is continuously synthesized, BRUKINSA was specifically designed to deliver complete and sustained inhibition of the BTK protein by optimizing bioavailability, half-life, and selectivity. With differentiated pharmacokinetics compared to other approved BTK inhibitors, BRUKINSA has been demonstrated to inhibit the proliferation of malignant B cells within a number of disease relevant tissues.
BRUKINSA has received 20 approvals covering 43 countries and regions:
To date, more than 20 marketing authorization applications have been submitted for BRUKINSA for various indications.
* This indication was approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
** This indication was approved under conditional approval. Complete approval for this indication may be contingent upon results from ongoing randomized, controlled confirmatory clinical trials.
Please see full U.S. Prescribing Information at www.beigene.com/PDF/BRUKINSAUSPI.pdf and Patient Information at www.beigene.com/PDF/BRUKINSAUSPPI.pdf.
To learn more about BeiGene, please visit www.beigene.ca and follow us on Twitter at @BeiGeneGlobal.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220128005107/en/
Source: BeiGene
Jan. 28, 2022 5:55 AM ET BeiGene, Ltd. (BGNE)
By: Mamta Mayani, SA News Editor
01/28/2022CATEGORY:
Recommendation for approval based on results from TRANSCEND NHL 001, the largest pivotal trial of patients with large B-cell lymphoma after at least two prior therapies, and TRANSCEND WORLD
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) today announced the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) has recommended approval of Breyanzi (lisocabtagene maraleucel; liso-cel), a CD19-directed chimeric antigen receptor (CAR) T cell therapy for the treatment of adult patients with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL), primary mediastinal large B-cell lymphoma (PMBCL), and follicular lymphoma grade 3B (FL3B) after two or more lines of systemic therapy. The CHMP recommendation will now be reviewed by the European Commission (EC), which has the authority to approve medicines for the European Union (EU).
“This positive CHMP opinion is an important milestone that recognizes Breyanzi as a differentiated cell therapy with the potential to address unmet needs for patients in the European Union with aggressive lymphomas who have limited treatment options,” said Anne Kerber, senior vice president, Cellular Therapy Development, Bristol Myers Squibb. “We look forward to the European Commission’s decision as we continue to accelerate our research in cell therapy to develop new treatment options for patients living with difficult-to-treat blood cancers.”
The CHMP adopted a positive opinion based on results from TRANSCEND NHL 001, the largest pivotal trial of patients with R/R LBCL after at least two prior therapies, and additional data from the TRANSCEND WORLD study. The studies evaluated patients with R/R DLBCL, PMBCL and FL3B, including those with a broad range of histologies and high-risk disease, and patients who received Breyanzi in the inpatient and outpatient setting.
The EC is expected to deliver its final decision within 67 days of receipt of the CHMP opinion. The decision will be applicable to all European Union member states and Iceland, Norway and Liechtenstein.*
*Centralized Marketing Authorization does not include approval in Great Britain (England, Scotland and Wales).
About TRANSCEND NHL 001
TRANSCEND NHL 001 is an open-label, multicenter, pivotal Phase 1 study conducted in the United States (U.S.) to determine the safety, antitumor activity and pharmacokinetics of Breyanzi in patients with R/R LBCL, including DLBCL, high-grade B-cell lymphoma (HGL), PMBCL and FL3B. The primary outcome measures included treatment-related adverse events, dose-limiting toxicities and objective response rate. Key secondary outcome measures included complete response rate, duration of response, progression-free survival and overall survival. The TRANSCEND program is a broad clinical program evaluating Breyanzi in multiple hematologic indications and treatment lines.
About TRANSCEND WORLD
TRANSCEND WORLD is a single-arm, multi-cohort, multicenter, Phase 2 study to determine the efficacy and safety of Breyanzi in patients with aggressive B-cell non-Hodgkin lymphoma (NHL). The primary outcome measure was overall response rate. Secondary outcome measures included safety, complete response rate, event-free survival, progression-free survival, overall survival, duration of response, pharmacokinetics and health-related quality of life. The study was conducted in Europe and Japan.
About Breyanzi
Breyanzi is a CD19-directed chimeric antigen receptor (CAR) T cell therapy with a defined composition and 4-1BB costimulatory domain. Breyanzi is administered as a defined composition to reduce variability of the CD8 and CD4 component dose. The 4-1BB signaling domain enhances the expansion and persistence of the CAR T cells.
Breyanzi is approved by the U.S. Food and Drug Administration for the treatment of adult patients with relapsed or refractory (R/R) large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B cell lymphoma (DLBCL) not otherwise specified (including DLBCL arising from indolent lymphoma), high-grade B-cell lymphoma, primary mediastinal large B-cell lymphoma, and follicular lymphoma grade 3B. Breyanzi is also approved in Japan for the treatment of patients with third-line plus R/R LBCL and follicular lymphoma.
Please see full Prescribing Information, including Boxed WARNINGS and Medication Guide.
For more information about Bristol Myers Squibb, visit us at BMS.com
View source version on businesswire.com: https://www.businesswire.com/news/home/20220113005630/en/
Source: Bristol Myers Squibb
Jan. 28, 2022 7:27 AM ET Bristol-Myers Squibb Company (BMY)
By: Dulan Lokuwithana, SA News Editor
Bristol Myers Squibb (NYSE:BMY) announced that an expert panel of the European Medicines Agency (EMA) recommended its chimeric antigen receptor (CAR) T cell therapy, Breyanzi, as a treatment for three forms of lymphoma.
JANUARY 27, 2022 • INVESTOR RELATIONS
CAMBRIDGE, Mass., Jan. 27, 2022 (GLOBE NEWSWIRE) -- Biogen Inc. (Nasdaq: BIIB) and Eisai Co., Ltd. (Tokyo, Japan) today announced additional details about the Phase 4 post-marketing confirmatory study, ENVISION, of ADUHELM® (aducanumab-avwa) 100 mg/mL injection for intravenous use in early Alzheimer’s disease, including details of the study’s goal for diverse enrollment and primary endpoint.
Biogen aims to enroll 18 percent of U.S. participants in ENVISION from Black/African American and Latinx populations. This goal is reflective of Biogen’s ongoing commitment to increase diversity in clinical trials.
“Historically, patients from diverse backgrounds have been poorly represented in Alzheimer’s disease clinical trials, and we are committed to changing this,” said Priya Singhal M.D., M.P.H., Head of Global Safety & Regulatory Sciences and interim Head of Research & Development at Biogen. “This goal matches the diversity among Americans diagnosed with early Alzheimer’s disease, while at the same time, the trial will generate substantial data to verify the effectiveness of ADUHELM.”
Biogen will implement multiple strategies to help overcome barriers to diverse patient enrollment in Alzheimer’s disease trials, such as, lack of access to medical centers, familiarity with benefit/risk profile of treatment, and financial or logistical burdens.
“It’s important to see this ambitious focus on diversity being prioritized in enrollment and integrated as a key part of the ENVISION clinical trial, so that we can have data from patients who more closely represent what we see in the clinic,” said Dylan Wint, M.D., Cleveland Clinic Lou Ruvo Center for Brain Health, Nevada.
The companies also announced today that the primary endpoint for the global, placebo-controlled ENVISION trial will be measured by the Clinical Dementia Rating–Sum of Boxes (CDR-SB) at 18 months after treatment initiation with ADUHELM. The CDR-SB endpoint is a validated measure of both cognition and function that is widely used in clinical trials of patients with early symptomatic Alzheimer’s disease, is consistent with ADUHELM’s Phase 3 EMERGE and ENGAGE studies, and capable of generating robust outcomes. The update also includes an increase in the previously announced enrollment, from 1,300 to 1,500 people with early Alzheimer’s disease (Mild Cognitive Impairment due to Alzheimer’s disease and mild Alzheimer’s disease), with confirmation of amyloid beta pathology, to further strengthen the data provided by the study.
Although ENVISION and other ADUHELM clinical trials are already planned or underway, the Centers for Medicare and Medicaid Services (CMS) recently released a draft National Coverage Determination (NCD), which would restrict Medicare coverage of ADUHELM and other amyloid-targeting therapies to patients enrolled in an additional clinical trial. Biogen is committed to engaging with CMS to avoid unnecessary duplication of clinical trials and work towards finding a path to offer immediate access to patients to the first FDA approved treatment for Alzheimer’s disease since 2003.
In addition to the primary endpoint, CDR-SB, secondary endpoints include Alzheimer's Disease Assessment Scale-Cognitive Subscale (ADAS-Cog 13), Alzheimer's Disease Cooperative Study - Activities of Daily Living Inventory - Mild Cognitive Impairment Version (ADCS-ADL-MCI), Integrated Alzheimer's Disease Rating Scale (iADRS), Mini-Mental State Examination (MMSE) and Neuropsychiatric Inventory (NPI-10).
The initiation of patient screening for ENVISION is planned for May 2022. Based on enrollment rates from the previous Phase 3 trials with ADUHELM, the primary completion date is expected to be approximately four years after the study begins. The companies are grateful to the healthcare professionals, medical centers, patients and families who will participate in this trial.
Previously, in July 2021, the companies set another substantial diversity goal in the observational Phase 4 ICARE AD trial, which aims to enroll a total of approximately 6,000 patients.
About ADUHELM® (aducanumab-avwa) 100 mg/mL injection for intravenous use
ADUHELM is indicated for the treatment of Alzheimer’s disease. Treatment with ADUHELM should be initiated in patients with mild cognitive impairment or mild dementia stage of disease, the population in which treatment was initiated in clinical trials. There are no safety or effectiveness data on initiating treatment at earlier or later stages of the disease than were studied. This indication is approved under accelerated approval based on reduction in amyloid beta plaques observed in patients treated with ADUHELM. Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trial(s).
ADUHELM is a monoclonal antibody directed against amyloid beta. The accumulation of amyloid beta plaques in the brain is a defining pathophysiological feature of Alzheimer’s disease. The accelerated approval of ADUHELM has been granted based on data from clinical trials showing the effect of ADUHELM on reducing amyloid beta plaques, a surrogate biomarker that is reasonably likely to predict clinical benefit, in this case a reduction in clinical decline.
ADUHELM can cause serious side effects including: Amyloid Related Imaging Abnormalities or “ARIA”. ARIA is a common side effect that does not usually cause any symptoms but can be serious. Although most people do not have symptoms, some people may have symptoms such as: headache, confusion, dizziness, vision changes and nausea. The patient’s healthcare provider will do magnetic resonance imaging (MRI) scans before and during treatment with ADUHELM to check for ARIA. ADUHELM can also cause serious allergic reactions. The most common side effects of ADUHELM include: swelling in areas of the brain, with or without small spots of bleeding in the brain or on the surface of the brain (ARIA); headache; and fall. Patients should call their healthcare provider for medical advice about side effects.
As of October 2017, Biogen and Eisai Co., Ltd. are collaborating on the global co-development and co-promotion of aducanumab.
Please click here for full Prescribing Information, including Medication Guide, for ADUHELM.
To learn more, please visit www.biogen.com
For more information about Eisai Co., Ltd., please visit https://www.eisai.com.
Jan. 27, 2022 8:06 AM ET Biogen Inc. (BIIB), ESALFESALYBy: Mamta Mayani, SA News Editor1 Comment
Monday, Jan 24, 2022
South San Francisco, CA -- January 24, 2022 --
Genentech, a member of the Roche Group (SIX: RO, ROG; OTCQX: RHHBY), today announced that the U.S. Food and Drug Administration (FDA) has granted priority review of a supplemental new drug application (sNDA) for the use of Evrysdi® (risdiplam) to treat pre-symptomatic babies under two months of age with spinal muscular atrophy (SMA). The sNDA submission incorporates interim data from the RAINBOWFISH study, which shows the majority of pre-symptomatic babies treated with Evrysdi achieved key milestones such as sitting, standing, walking and maintained the ability to swallow following 12 months of treatment.
“Treating very young babies with Evrysdi before SMA symptoms arise may help them to achieve milestones such as standing and walking within timeframes typical of healthy infants,” said Levi Garraway, M.D., Ph.D., Genentech’s chief medical officer and head of Global Product Development. “Extending treatment access for the youngest members of the SMA community is crucial and we look forward to working with the FDA on this application.”
Evrysdi is designed to treat SMA by increasing and sustaining production of the survival motor neuron (SMN) protein in the central nervous system (CNS) and peripheral tissues. SMN protein is found throughout the body and is critical for maintaining healthy motor neurons and movement. Evrysdi’s existing FDA label is for the treatment of SMA in adults, children and babies two months and older. If approved, Evrysdi would be the first medicine administered at home for pre-symptomatic babies with SMA.
Initial interim data from the RAINBOWFISH study, presented at the World Muscle Society (WMS) Virtual Congress 2021, showed that of the babies included in the interim efficacy analysis, all (5/5), maintained the ability to swallow and were able to feed exclusively orally after 12 months of treatment. Eighty percent (4/5) treated with Evrysdi for at least 12 months achieved milestones such as standing and walking independently within World Health Organization windows for healthy children. All participants (n=5) met Hammersmith Infant Neurological Examination, Section 2 (HINE-2) motor milestones of head control, sitting upright, rolling and crawling after 12 months of treatment with Evrysdi.
No treatment-related serious adverse events were reported in any of the babies treated with Evrysdi through the interim safety analysis period (n=12). Four treatment-emergent adverse events were reported, and all were resolved or were resolving with ongoing treatment with Evrysdi. The most common adverse events (AEs) were nasal congestion (33%), cough (25%), teething (25%), vomiting (25%), eczema (17%), abdominal pain (17%), diarrhea (17%), gastroenteritis (17%), papule (17%) and pyrexia (17%). The AEs were reflective of the age of the babies rather than the underlying SMA. The RAINBOWFISH study is currently recruiting.
The latest results from the RAINBOWFISH study will be presented at the Muscular Dystrophy Association (MDA) Clinical and Scientific Conference in March 2022.
Genentech leads the clinical development of Evrysdi as part of a collaboration with the SMA Foundation and PTC Therapeutics.
About Evrysdi® (risdiplam)
Evrysdi is a survival motor neuron 2 (SMN2) splicing modifier designed to treat SMA caused by mutations in chromosome 5q that lead to survival motor neuron (SMN) protein deficiency. Evrysdi is administered daily at home in liquid form by mouth or by feeding tube.
Evrysdi is designed to treat SMA by increasing and sustaining the production of SMN protein levels in the central nervous system (CNS) and peripheral tissues as demonstrated in animal models. SMN protein is found throughout the body and is critical for maintaining healthy motor neurons and movement.
Evrysdi was granted PRIME designation by the European Medicines Agency (EMA) in 2018 and Orphan Drug Designation by the U.S Food and Drug Administration in 2017. In 2021, Evrysdi was awarded Drug Discovery of the Year by the British Pharmacological Society as well as the Society for Medicines Research Award for Drug Discovery. Evrysdi is currently approved in 70 countries and the dossier is under review in a further 31 countries.
Evrysdi is currently being evaluated in five multicenter trials in people with SMA:
What is Evrysdi?
Evrysdi is a prescription medicine used to treat spinal muscular atrophy (SMA) in adults and children 2 months of age and older.
It is not known if Evrysdi is safe and effective in children under 2 months of age.
Please see the full Prescribing Information for additional Important Safety Information.
For additional information about the company, please visit http://www.gene.com.
Jan. 25, 2022 1:48 AM ET Roche Holding AG (RHHBY), PTCT By: Mamta Mayani, SA News Editor
JAN 24, 2022
Studies show significant reduction in pain and improved quality of life for patients treated with spinal cord stimulation compared to medication management alone
DUBLIN, Jan. 24, 2022 /PRNewswire(opens new window)/ -- Medtronic plc (NYSE:MDT), a global leader in healthcare technology, today announced it has received U.S. Food and Drug Administration approval of its Intellis™ rechargeable neurostimulator and Vanta™ recharge-free neurostimulator for the treatment of chronic pain associated with diabetic peripheral neuropathy (DPN).
DPN is a debilitating and progressive neurological disorder that affects approximately 30% of people with diabetes, significantly impacting both quality of life and functional ability, including mood, social relationships, and sleep.1 DPN occurs when high blood sugar (glucose) damages nerves in the body, most often in the legs and feet, leading to numbness and burning or stabbing pain. In some patients, the pain can become progressively worse and excruciating. Patients may be treated with medications, but they are often only partially effective and can result in serious side effects.
This new indication offers patients with DPN access to Medtronic's industry-leading spinal cord stimulation (SCS) portfolio of rechargeable and recharge-free platforms, which include multiple programming options to personalize patient therapy, unrestricted MRI access2, unrivaled battery chemistry and performance, and the Medtronic TYRX™ Neuro Absorbable Antibacterial Envelope. The surgical management of diabetic patients can be a challenge for health care providers as they often have additional risk factors and are at greater risk for infection.3 The TYRX™ antibacterial surgical mesh envelope has been shown to stabilize device placement and help reduce infection by over 60%.4-10 Clinicians may also advise patients to take advantage of CareGuidePro™, which serves as a virtual guide for patients throughout their SCS journey.
Independent studies show patients with DPN achieve significant pain relief when treated with SCS compared to conventional treatments alone.11,12 Overall, 70% of patients receiving treatment with SCS experienced relief of their pain symptoms compared to 6% of patients receiving only conventional treatments. Those treated with SCS experienced a 53% average reduction in pain, compared to 0% among patients receiving only conventional treatments. A recent meta-analysis showed a significant improvement in health-related quality of life in patients treated with SCS compared to those receiving only conventional treatments.13 A long-term analysis of patients treated in one of the studies using Medtronic SCS technology showed 80% of patients treated with SCS continued to use their devices at five years to treat their pain.14
"DPN is a significant challenge for patients with diabetes, leading to disability and a diminished quality of life," said Charlie Covert, vice president and general manager, Pain Therapies within the Neuromodulation business, which is part of the Neuroscience Portfolio at Medtronic. "This new indication enables us to apply Medtronic's more than 40 years of proven SCS experience, as well as the company's deep diabetes expertise, to deliver better care to even greater numbers of diabetes patients."
Medtronic estimates that up to 800,000 US patients suffer from moderate to severe DPN symptoms that are not resolved through conventional medical management approaches, like drugs. The company views these patients as potential candidates for SCS, representing an approximate $1.8 billion annual market opportunity. Today, Medtronic estimates that the US market revenue for SCS treatment of chronic pain associated with DPN, sometimes also referred to as Painful Diabetic Neuropathy (PDN), is approximately $70 million, and the company expects market revenue to grow to $300 million by fiscal year 2026.
For more information on Medtronic (NYSE:MDT), visit www.Medtronic.com(opens new window), and follow @Medtronic(opens new window) on Twitter and LinkedIn(opens new window).
Jan. 24, 2022 12:54 PM ET Medtronic plc (MDT) By: Dulan Lokuwithana, SA News Editor
Chronic Pain

January 21, 2022
-- Approval Based on Phase 3 Data Showing Veklury Significantly Reduced Risk of Hospitalization By 87% Compared with Placebo --
-- NIH Guidelines Recommend Veklury for the Treatment of Non-Hospitalized Patients at High Risk --
-- FDA Expands Pediatric Emergency Use Authorization (EUA) to Include Treatment of Non-Hospitalized Pediatric Patients at High Risk --
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the U.S. Food and Drug Administration (FDA) has granted expedited approval of a supplemental new drug application (sNDA) for Veklury® (remdesivir) for the treatment of non-hospitalized adult and adolescent patients who are at high risk of progression to severe COVID-19, including hospitalization or death. This approval expands the role of Veklury, which is the antiviral standard of care for the treatment of patients hospitalized with COVID-19. The expanded indication allows for Veklury to be administered in qualified outpatient settings that can administer daily intravenous (IV) infusions over three consecutive days. The FDA has also expanded the pediatric Emergency Use Authorization (EUA) of Veklury to include non-hospitalized pediatric patients younger than 12 years of age who are at high risk of disease progression.
These actions by the FDA come amidst a surge in COVID-19 cases and the reduced susceptibility to several anti-SARS-CoV-2 monoclonal antibodies (mAbs) due to the Omicron variant. In contrast, Veklury targets the highly conserved viral RNA polymerase, thereby retaining activity against existing SARS-CoV-2 variants of concern. In vitro laboratory testing shows that Veklury retains activity against the Omicron variant. To date, no major genetic changes have been identified in any of the known variants of concern that would significantly alter the viral RNA polymerase targeted by Veklury.
The FDA sNDA approval, pediatric EUA expansion and recently updated National Institutes of Health (NIH) Treatment Guidelines for COVID-19 that additionally recommend Veklury for treatment in non-hospitalized settings, are based on results from the PINETREE Phase 3 randomized, double-blind, placebo-controlled trial. The trial evaluated the efficacy and safety of a three-day course of Veklury for intravenous (IV) use for the treatment of COVID-19 in non-hospitalized patients at high risk for disease progression. An analysis of 562 participants randomly assigned in a 1:1 ratio to receive Veklury or placebo, demonstrated that treatment with Veklury resulted in a statistically significant 87% reduction in risk for the composite primary endpoint of COVID-19 related hospitalization or all-cause death by Day 28 (0.7% [2/279]) compared with placebo (5.3% [15/283]) p=0.008. In the study, no deaths were observed in either arm by Day 28. The safety profile was similar between Veklury and placebo across the variety of outpatient settings in this trial, with the most common treatment emergent adverse events (≥5%) in patients taking Veklury being nausea and headache.
“Remdesivir has now helped to treat more than 10 million people around the world with COVID-19 and continues to play a key role in helping to reduce the burden of the pandemic. Based on the most recent data, we now understand that remdesivir is also effective in the early stages of COVID-19 infection, in addition to helping patients who are hospitalized with the disease,” said Daniel O’Day, Chairman and Chief Executive Officer, Gilead Sciences. “While we continue to advance remdesivir to benefit more patients in multiple settings, we are also advancing our investigational oral compounds. These are based on the same antiviral mechanism of action as remdesivir and a Phase 1 trial for our oral COVID-19 antiviral, GS-5245 is now underway.”
In the United States, Veklury is indicated for the treatment of COVID-19 in adults and pediatric patients (12 years of age and older and weighing at least 40 kg) who are either hospitalized or not hospitalized and are at high risk for progression to severe COVID-19, including hospitalization or death. Veklury is additionally authorized for these uses under the EUA for pediatric patients weighing 3.5 kg to less than 40 kg or pediatric patients less than 12 years of age weighing at least 3.5 kg. Veklury is contraindicated in patients who are allergic to Veklury or any of its components; please see below for additional Important Safety Information for Veklury.
Under the expanded indication for Veklury, non-hospitalized adult and pediatric patients (12 years of age and older and weighing at least 40 kg) with confirmed SARS-CoV-2 infection who are at high risk for COVID-19 disease progression can be treated with a recommended treatment duration of three days to help prevent hospitalization. For hospitalized patients not on mechanical ventilation and/or ECMO, a 5-day course of treatment is recommended, with the option to extend to a total of 10-days as needed. Critically ill patients who require mechanical ventilation and/or ECMO should receive a 10-day course of treatment.
About Veklury
Veklury (remdesivir) is a nucleotide analog invented by Gilead, building on more than a decade of the company’s antiviral research. Veklury is the antiviral standard of care for the treatment of hospitalized patients with COVID-19. At this time, more than half of patients hospitalized with COVID-19 in the United States are treated with Veklury. It can help reduce disease progression across a spectrum of disease severity and enable hospitalized patients to recover faster, freeing up limited hospital resources and saving healthcare systems money.
Veklury is approved or authorized for temporary use in approximately 50 countries worldwide. To date, Veklury and generic remdesivir have been made available to more than 10 million patients around the world, including 7 million people in 127 middle- and low-income countries through Gilead’s voluntary licensing program. These licenses currently remain royalty-free, reflecting Gilead’s existing commitment to enabling broad patient access to remdesivir.
U.S. Indication for Veklury
Veklury® (remdesivir 100 mg for injection) is indicated for the treatment of COVID-19 in adults and pediatric patients (12 years of age and older and weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, who are:
Veklury should only be administered in settings in which health care providers have immediate access to medications to treat a severe infusion or hypersensitivity reaction, such as anaphylaxis, and the ability to activate the emergency medical system (EMS), as necessary. Veklury must be administered by intravenous infusion. Veklury is contraindicated in patients who are allergic to Veklury or any of its components. For more information, please see the U.S. full Prescribing Information available at www.gilead.com.
U.S. full Prescribing Information for Veklury is available at www.gilead.com.
Veklury, Gilead and the Gilead logo are registered trademarks of Gilead Sciences, Inc., or its related companies.
For more information about Gilead, please visit the company’s website at www.gilead.com , follow Gilead on Twitter (@Gilead Sciences) or call Gilead Public Affairs at 1-800-GILEAD-5 or 1-650-574-3000.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220121005359/en/
Source: Gilead Sciences, Inc.
Jan. 21, 2022 10:35 PM ETGilead Sciences, Inc. (GILD)By: Jonathan Block, SA News Editor8 Comments
Innovent and Eli Lilly and Company Announced Final Clinical Results and Biomarker Analysis of Phase Ib Study of TYVYT® (Sintilimab Injection) Plus Bevacizumab Biosimilar Injection for Advanced Hepatocellular Carcinoma at ASCO GI Annual Meeting 2022Published on: Jan 22nd, 2022Back
SAN FRANCISCO, U.S. and SUZHOU, China, Jan 21, 2022 — Innovent Biologics, Inc. (“Innovent”, HKEX: 01801), a world-class biopharmaceutical company that develops, manufactures and commercializes high-quality medicines for the treatment of cancer, metabolic, autoimmune and other major diseases, today jointly announced with Eli Lilly and Company (“Lilly”, NYSE: LLY) the final clinical outcome and biomarker analysis of the open label, phase Ib study (NCT04072679)of sintilimab plus bevacizumab biosimilar injection for advanced hepatocellular carcinoma at the 2022 ASCO Gastrointestinal Cancers Symposium (Abstract# 455).
The study, which was conducted in China, evaluated the treatment of sintilimab plus bevacizumab biosimilar injection for patients with local advanced or metastatic HCC with or without previous systemic treatment or systemic treatment failure. The Phase Ib study included dose escalation and dose expansion stages. In the dose escalation stage, patients were randomly assigned to receive sintilimab 200mg plus bevacizumab biosimilar 7.5 mg/kg (low-dose group) or 15 mg/kg (high-dose group). In the dose expansion stage safety and efficacy were assessed for each dose group. A total of fifty patients were enrolled in final analysis, with 29 patients administered bevacizumab biosimilar 7.5 mg/kg and 21 patients 15 mg/kg. The safety profile was consistent with that observed in previously reported studies of sintilimab and bevacizumab biosimilar, without new or unexpected safety signals. The most common treatment-related adverse events (TRAE) were as hypertension (32%), proteinuria (26%) and fever (26%). The incidence of grade ≥ 3 adverse events in the high-dose group was 28.6%, while 13.8% in the low-dose group.
The overall response rate (ORR) was 34% (17/50) and ORRs for the low-dose and high-dose groups were 31% and 38%, respectively. The disease control rate (DCR) was 78% (39/50). Median progression-free survival (PFS) was 10.5 months (95%CI, 8.4-12.7) and median overall survival (OS) was 20.2 months (95%CI, 16.1 -24.3). Further biomarker analysis showed that patients with a high level of CD137 ≥ 31.8 pg/mL have longer PFS (mPFS:14.2 vs. 4.1months, p=0.001) and OS (mOS: NR vs.15.6months, p=0.023). In addition, the Tumor immune Microenvironment(TiME)analysis demonstrated that the high density of M1 macrophages (CD68+, CD163-) in stroma was related to higher efficacy (p=0.033), longer PFS (p=0. 024) and OS (p =0.046).
Professor Zhou Aiping from the Cancer Hospital of the Chinese Academy of Medical Sciences stated:"This study not only showed the safety and efficacy data for sintilimab in combination with different doses of bevacizumab, but also identified biomarkers that could predict the efficacy of the combined treatment regimen for hepatocellular carcinoma, which lays the groundwork for more individualized treatment.”
About NCT04072679 study
NCT04072679 study is a Phase 1b study assessing the safety, tolerability and anti-tumor activity of sintilimab in combination with bevacizumab biosimilar in subjects with local advanced or metastatic HCC with or without previous systemic treatment or systemic treatment failure in China. The study has two stages, the first is a dose escalation stage and the second is a dose expansion stage. In the dose escalation stage, subjects will receive either 7.5 mg/kg (low-dose group) or 15 mg/kg (high-dose group) of bevacizumab biosimilar (IBI305) in combination with a fixed dose of 200 mg of sintilimab, respectively, and then entered into the dose expansion stage.
About Sintilimab
Sintilimab, marketed as TYVYT® (sintilimab injection) in China, is an innovative PD-1 inhibitor with global quality standards jointly developed by Innovent and Eli Lilly and Company. Sintilimab is an immunoglobulin G4 monoclonal antibody, which binds to PD-1 molecules on the surface of T-cells, blocks the PD-1 / PD-Ligand 1 (PD-L1) pathway, and reactivates T-cells to kill cancer cells. Innovent is currently conducting more than 20 clinical studies of sintilimab worldwide, to evaluate its safety and efficacy in a wide variety of cancer indications, including more than 10 registrational or pivotal clinical trials.
In China, sintilimab has been approved and included in the National Reimbursement Drug List (NRDL) for four indications, including:
Additionally, sintilimab currently has three regulatory submissions accepted for review in NMPA, including:
Additionally, two clinical studies of sintilimab have met their primary endpoints:
In May 2021, the U.S. FDA accepted for review the Biologics License Application (BLA) for sintilimab in combination with pemetrexed and platinum chemotherapy for the first-line treatment of non-squamous non-small cell lung cancer.
About BYVASDA® (bevacizumab biosimilar injection)
BYVASDA®, also known as IBI305, is a bevacizumab biosimilar and a recombinant humanized anti-VEGF monoclonal antibody drug. Vascular endothelial growth factor (VEGF) is an important factor in angiogenesis that is highly expressed by the endothelial cells in most human tumors. An anti-VEGF antibody binds VEGF-A selectively with high affinity and blocks its binding to VEGF-2 receptors on the surface of vascular endothelial cells, thereby inhibiting signaling pathways such as PI3K-Akt/PKB and Ras-Raf-MEK-ERK. BYVASDA® produces anti-tumor effects by inhibiting the growth, proliferation and migration of vascular endothelial cells, blocking angiogenesis, reducing vascular permeability, blocking blood supply to tumor tissues, inhibiting the proliferation and metastasis of tumor cells and inducing apoptosis in tumor cells. Since its launch, bevacizumab has been approved for the treatment of patients with multiple malignant tumors globally, including non-small cell lung cancer, metastatic colorectal cancer, glioblastoma, renal cell carcinoma, cervical cancer, and epithelial ovarian, fallopian tube, or primary peritoneal cancer. The efficacy and safety of bevacizumab in these tumor types have been well recognized worldwide.
In China, BYVASDA® (bevacizumab biosimilar injection) is approved for indications including advanced non-small cell lung cancer, metastatic colorectal cancer, adult recurrent glioblastoma, and advanced or unresectable hepatocellular carcinoma.
For more information, please visit: www.innoventbio.com.
To learn more about Lilly, please visit us at www.lilly.com
and http://newsroom.lilly.com/social-channels.
About Eli Lilly and Company’s strategic cooperation with Innovent Biologics
Lilly entered into a strategy collaboration with Innovent focused on biological medicine in March 2015 – a groundbreaking partnership between a Chinese pharmaceutical company and a multinational pharmaceutical company. Under the agreement, Lilly and Innovent will co-develop and commercialize oncology medicines, including Tyvyt® (sintilimab injection) in China. In October 2015, the two companies announced the extension of their existing collaboration to include co-development of three additional oncology antibodies targeting oncology indications. In August 2019, Innovent further entered a licensing agreement with Lilly to develop and commercialize a potentially global best-in-class diabetes medicine in China. Its collaboration with Lilly indicates that Innovent has established a comprehensive level of cooperation between China’s innovative pharmaceuticals sector and the international pharmaceuticals sector in fields such as R&D, CMC, clinical development and commercialization.
TYVYT® (sintilimab injection) is not an approved product in the United States.
BYVASDA® (bevacizumab biosimilar injection), SULINNO®, and HALPRYZA® (rituximab biosimilar injection) are not approved products in the United States.
TYVYT® (sintilimab injection, Innovent)
BYVASDA® (bevacizumab biosimilar injection, Innovent)
HALPRYZA® (rituximab biosimilar injection, Innovent)
SULINNO® (adalimumab biosimilar injection, Innovent)
Pemazyre® (pemigatinib oral inhibitor, Incyte Corporation). Pemazyre® was discovered by Incyte Corporation and licensed to Innovent for development and commercialization in Mainland China, Hong Kong, Macau and Taiwan.
Jan. 24, 2022 2:13 AM ET
Innovent Biologics, Inc. (IVBIY)
SAN FRANCISCO and SUZHOU, China, Jan. 24, 2022 /PRNewswire/ -- Innovent Biologics, Inc. (IVBIY) (Innovent) (HKEX: 01801), a world-class biopharmaceutical company that develops, manufactures and commercializes high-quality medicines for the treatment of cancer, metabolic, autoimmune and other major diseases announced that the Hong Kong Department of Health (DH) has approved Pemazyre® (pemigatinib) for the treatment of adults with locally advanced or metastatic cholangiocarcinoma with a fibroblast growth factor receptor 2 (FGFR2) fusion or rearrangement that have progressed after at least one prior line of systemic therapy.
Pemazyre, discovered by Incyte and licensed to Innovent for development and commercialization in Mainland China, Hong Kong, Macau and Taiwan, is the first tyrosine kinase inhibitor approved for the treatment of cholangiocarcinoma, a type of biliary tract cancer, in Hong Kong market, marking a new milestone following the successful approval of Pemazyre in Taiwan in June 2021 and the New Drug Applicant(NDA) acceptance by China's NMPA in July 2021.
The approval was based on the FIGHT-202 study, which is a Phase 2, multi-center, open-label, single-arm study (NCT02924376) evaluating the safety and efficacy of Pemazyre – a selective fibroblast growth factor receptor (FGFR) inhibitor – in adult (age ≥18 years) patients with previously treated, locally advanced or metastatic cholangiocarcinoma with documented FGFR2 fusion or rearrangement. The major efficacy outcome measure was overall response rate (ORR) determined by an independent review committee Per RECIST V1.1. As data cut of (April 4th,2020), a total of 108 subjects with FGFR 2 fusion/rearrangement was enrolled in the study and received 13.5mg pemigatinib per day, the ORR was 37.0% (95% CI: 27.94%, 46.86%), including 4 complete responses. The median duration of response (DOR) was 8.08 months with responses lasting ≥ 6 months in 26 of the 40 (66.0%) responding patients and ≥ 12 months in 15 (37.5%) patients. The safety analysis, including 147 patients, demonstrated that pemigatinib was generally well tolerated. Hyperphosphatemia was the most common (58.5%) treatment-emergent adverse event (TEAE). TEAEs grade 3 or higher were reported in 68.7% of patients; the most frequent of which were hypophosphatemia (14.3%), arthralgia (6.1%), stomatitis 6.1%), hyponatremia (5.4%), abdominal pain (5.4%) and fatigue (5.4%).
For more information FIGHT-202, please visit https://clinicaltrials.gov/ct2/show/NCT02924376 or https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(20)30109-1/fulltext.
Dr. Yongjun Liu, President of Innovent, stated: "The approval of Pemazyre in Hong Kong, following the approval in Taiwan earlier this year, further broadens Innovent's product portfolio and market coverage. Currently Innovent has developed a robust pipeline of 26 clinical assets and more than 80 projects in preclinical stage, along the way we have established strategic collaborations with a dozen of global and regional partners. We look forward to launching more innovative drugs to help fulfill unmet needs, overcome current therapeutic limitation and improve the life quality of patients."
"Cholangiocarcinoma is the second most common malignancy originated in the liver with a high incidence in Asia. This cancer is usually not diagnosed until it has already developed into an advanced unresectable and/or metastatic stage. There are limitations on current treatment options, which call for innovative drugs that could greatly enhance disease control as well as provide better life quality," stated Dr. Hui Zhou, Senior Vice President of Innovent. "Data from previous clinical trials of Pemazyre in participants with advanced cholangiocarcinoma with FGFR2 fusion that have progressed after at least one prior line of systemic therapy has not only shown satisfactory safety results but also revealed compelling efficacy signals. In addition, Pemazyre is an orally administered medication that could improve convenience and patient compliance. The approval is a new milestone for Innovent; we look forward to hearing good news from NMPA as well and to bringing the therapeutic benefit of Pemazyre in the treatment of eligible patients with cholangiocarcinoma in the Greater China market."
About Pemazyre® (pemigatinib)
In April 2020, the U.S. Food and Drug Administration (FDA) approved Incyte's Pemazyre® (pemigatinib), a selective, oral inhibitor of FGFR isoforms 1, 2 and 3, for the treatment of adults with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with a fibroblast growth factor receptor 2 (FGFR2) fusion or rearrangement as detected by an FDA-approved test. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
In Japan, Pemazyre® is approved for the treatment of patients with unresectable biliary tract cancer with a FGFR2 fusion gene, worsening after cancer chemotherapy. In Europe, Pemazyre® is approved for the treatment of adults with locally advanced or metastatic cholangiocarcinoma with a FGFR2 fusion or rearrangement that have progressed after at least one prior line of systemic therapy.
Pemazyre® is marketed by Incyte in the United States, Europe and Japan.
In December 2018, Innovent and Incyte entered into a strategic collaboration for three clinical-stage product candidates discovered and developed by Incyte, including pemigatinib (FGFR1/2/3 inhibitor). Under the terms of the agreement, Innovent has received the rights to develop and commercialize the three assets in Mainland China, Hong Kong, Macau and Taiwan. In March 2020, Innovent announced that the first patient was dosed in the pivotal registrational trial evaluating pemigatinib in patients with advanced cholangiocarcinoma in China.
In June 2021, Taiwan Food and Drug Administration (TFDA) approved Pemazyre® (pemigatinib) for the treatment of adults with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with a FGFR2 fusion or rearrangement.
For more information, please visit: www.innoventbio.com. and www.linkedin.com/company/innovent-biologics/.
SOURCE Innovent Biologics
(IVBIY)
https://seekingalpha.com/symbol/IVBIY
https://www.pemazyre.com/?gtm_debug=x
Jan 24, 2022 5:00 AM
In the RATIONALE 305 trial, tislelizumab combined with chemotherapy prolonged overall survival for patients with PD-L1 expression
Additional follow-up is needed to assess overall survival benefits in intention-to-treat patient population
Safety findings of tislelizumab were consistent with that observed in previous trials
RATIONALE 305 is the sixth positive Phase 3 trial in tislelizumab’s broad clinical program
CAMBRIDGE, Mass. & BEIJING--(BUSINESS WIRE)-- BeiGene (NASDAQ: BGNE; HKEX: 06160; SSE: 688235), a global, science-driven biotechnology company focused on developing innovative and affordable medicines to improve treatment outcomes and access for patients worldwide, today announced positive findings from the global Phase 3 RATIONALE 305 trial of tislelizumab versus placebo in combination with chemotherapy as a first-line treatment for patients with locally advanced, unresectable or metastatic gastric or gastroesophageal junction (G/GEJ) adenocarcinoma. At the interim analysis, tislelizumab in combination with chemotherapy met the primary endpoint of overall survival (OS) in patients with PD-L1 expression, with additional follow-up needed to assess OS benefits in the intention-to-treat (ITT) population. The safety profile of tislelizumab was consistent with that observed in previous trials, with no new safety signals identified with the addition of chemotherapy.
“The addition of tislelizumab to chemotherapy significantly extended the overall survival for previously untreated patients with advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumor expressed PD-L1. We will continue to follow up to determine OS benefits across the patient population in the trial,” commented Yong (Ben) Ben, M.D., Chief Medical Officer, Solid Tumors at BeiGene.
RATIONALE 305: Tislelizumab vs. Placebo in Combination with Chemotherapy in First-Line G/GEJ Adenocarcinoma
RATIONALE 305 is a randomized, double-blind, placebo-controlled global Phase 3 trial (NCT03777657) comparing the efficacy and safety of tislelizumab combined with platinum and fluoropyrimidine chemotherapy and placebo combined with platinum and fluoropyrimidine chemotherapy as a first-line treatment for patients with locally advanced, unresectable or metastatic G/GEJ adenocarcinoma. The primary endpoint of the trial is OS. Secondary endpoints include progression-free survival (PFS), overall response rate (ORR), duration of response (DoR), and safety. A total of 997 patients from 13 countries and regions globally, including close to 50 percent from outside of China, were enrolled and randomized 1:1 to receive either tislelizumab and chemotherapy or placebo and chemotherapy.
To learn more about BeiGene, please visit www.beigene.com
About Tislelizumab
Tislelizumab (BGB-A317) is a humanized IgG4 anti-PD-1 monoclonal antibody specifically designed to minimize binding to FcγR on macrophages. In pre-clinical studies, binding to FcγR on macrophages has been shown to compromise the anti-tumor activity of PD-1 antibodies through activation of antibody-dependent macrophage-mediated killing of T effector cells. Tislelizumab is the first drug from BeiGene’s immuno-oncology biologics program and is being developed internationally as a monotherapy and in combination with other therapies for the treatment of a broad array of both solid tumor and hematologic cancers.
The China National Medical Products Administration (NMPA) has approved tislelizumab in six indications, including full approval for first-line treatment of patients with advanced squamous non-small cell lung cancer (NSCLC) in combination with chemotherapy, for first-line treatment of patients with advanced non-squamous NSCLC in combination with chemotherapy, and for second- or third-line treatment of patients with locally advanced or metastatic NSCLC who progressed on prior platinum-based chemotherapy. NMPA also granted conditional approval for the treatment of patients with classical Hodgkin’s lymphoma (cHL) who received at least two prior therapies, for the treatment of patients with locally advanced or metastatic urothelial carcinoma (UC) with PD-L1 high expression whose disease progressed during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy, and for the treatment of patients with hepatocellular carcinoma (HCC) who have received at least one systemic therapy. Full approval for these indications is contingent upon results from ongoing randomized, controlled confirmatory clinical trials.
In addition, three supplemental Biologics License Applications for tislelizumab are under review by the Center for Drug Evaluation (CDE) of the NMPA, including for patients with previously treated, locally advanced unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair-deficient (dMMR) solid tumors, for the treatment of patients with locally advanced or metastatic esophageal squamous cell carcinoma (ESCC) who have disease progression following or are intolerant to first-line standard chemotherapy, and for first-line treatment of patients with recurrent or metastatic nasopharyngeal cancer (NPC).
In the U.S., a Biologics License Application for tislelizumab as a treatment for patients with unresectable recurrent locally advanced or metastatic ESCC after prior systemic therapy is currently under review by the U.S. Food and Drug Administration with a PDUFA target action date of July 12, 2022.
BeiGene has initiated or completed 17 potentially registration-enabling clinical trials in China and globally, including 13 Phase 3 trials and four pivotal Phase 2 trials.
In January 2021, BeiGene and Novartis entered into a collaboration and license agreement granting Novartis rights to develop, manufacture, and commercialize tislelizumab in North America, Europe, and Japan.
Tislelizumab is not approved for use outside of China.
About the Tislelizumab Clinical Program
Clinical trials of tislelizumab include:
View source version on businesswire.com: https://www.businesswire.com/news/home/20220124005223/en/
Source: BeiGene
Jan. 24, 2022 6:10 AM ETBeiGene, Ltd. (BGNE)By: Mamta Mayani, SA News Editor
Karyopharm Receives Orphan Drug Designation from FDA for Eltanexor for the Treatment of Myelodysplastic Syndromes
NEWTON, Mass., Jan. 24, 2022 /PRNewswire/ -- Karyopharm Therapeutics Inc. (Nasdaq: KPTI), a commercial-stage pharmaceutical company pioneering novel cancer therapies, today announced that the U.S. Food and Drug Administration (FDA) has granted orphan drug designation for eltanexor, a novel oral, Selective Inhibitor of Nuclear Export (SINE) compound, for the treatment of myelodysplastic syndromes (MDS). MDS are a group of diseases characterized by ineffective production of the components of the blood due to poor bone marrow function with a risk of progression to acute myeloid leukemia.
Karyopharm is currently investigating eltanexor in an ongoing open-label Phase 1/2 study as a single-agent or in combination with approved and investigational agents in patients with several types of hematologic and solid tumor cancers (KCP-8602-801; NCT02649790). Previously, Karyopharm reported positive data from an investigator-sponsored Phase 1 study evaluating single-agent eltanexor in patients with hypomethylating agent (HMA)-refractory MDS, where eltanexor demonstrated a 53% overall response rate and median overall survival of 9.9 months. This compares favorably to historical survival of four to six months for HMA-refractory MDS patients.
Approximately 15,000 people are diagnosed with intermediate-to-high risk MDS each year in the U.S.1 HMAs are the current standard of care for newly diagnosed, higher-risk MDS patients. However, only 40-60% of patients respond, with these responses typically lasting less than two years.2 The prognosis in HMA-refractory disease is poor, with a median overall survival of four to six months.3,4 There are currently no approved therapies for HMA- refractory MDS.
"We are pleased to receive the FDA's orphan drug designation for eltanexor in MDS and believe it reinforces eltanexor's potential to improve clinical outcomes for patients with HMA-refractory MDS," said Richard Paulson, President and Chief Executive Officer of Karyopharm. "We are focused on advancing our ongoing clinical trials and remain steadfast in our commitment to bringing this new treatment option to patients and their families."
Orphan drug designation by the FDA is granted to promote the development of drugs that target conditions affecting 200,000 or fewer U.S. patients annually and are expected to provide a significant therapeutic advantage over existing treatments. Orphan designation qualifies a company for certain incentives that apply across all stages of drug development, including the potential for seven years of market exclusivity following marketing approval, tax credits on qualified U.S. clinical trials, eligibility for orphan drug grants, and exemption from certain administrative fees.
About Eltanexor
Eltanexor (KPT-8602) is an investigational novel SINE compound that, like selinexor, functions by binding with, and inhibiting, the nuclear export protein, XPO1, leading to the accumulation of tumor suppressor proteins in the cell nucleus. This reinitiates and amplifies their tumor suppressor function and is believed to lead to the selective induction of apoptosis in cancer cells, while largely sparing normal cells.
In preclinical models, eltanexor has a broad therapeutic window with minimal penetration of the blood brain barrier and, therefore, has the potential to serve as another SINE compound for cancer indications. Following oral administration, animals treated with eltanexor show lower percentage of body weight loss and improved food consumption than animals similarly treated with selinexor. This allows more frequent dosing of eltanexor, enabling a longer period of exposure than is possible with selinexor.
Eltanexor is an investigational medicine and has not been approved by the United States Food and Drug Administration or any other regulatory agency.
For more information about our people, science and pipeline, please visit www.karyopharm.com, and follow us on Twitter at @Karyopharm and LinkedIn.
XPOVIO® and NEXPOVIO® are registered trademarks of Karyopharm Therapeutics Inc. Any other trademarks referred to in this release are the property of their respective owners.
Jan. 24, 2022 7:16 AM ET
Karyopharm Therapeutics Inc. (KPTI)
By: Dulan Lokuwithana, SA News Editor1 Comment
January 21, 2022
- SKYRIZI® (risankizumab-rzaa) met the primary endpoint of ACR20 at week 24 in two pivotal studies, demonstrating significant improvement in joint symptoms, including swollen, tender and painful joints, compared to placebo[1,2,3]
- SKYRIZI also showed improvement in dactylitis and enthesitis - inflammation of fingers, toes and sites at which tendons or ligaments attach to bone[1,2,3]
- SKYRIZI is now the only IL-23 inhibitor approved for adults with moderate to severe plaque psoriasis and active psoriatic arthritis that can be administered with a single injection four times a year (after two starter doses at weeks 0 and 4)[1]
NORTH CHICAGO, Ill., Jan. 21, 2022 /PRNewswire/ -- AbbVie (NYSE: ABBV) today announced the U.S. Food and Drug Administration (FDA) has approved SKYRIZI® (risankizumab-rzaa) for the treatment of adults with active psoriatic arthritis (PsA), a systemic inflammatory disease that affects the skin and joints and impacts approximately 30 percent of patients with psoriasis.1,4-7
The FDA approval is supported by data from two pivotal studies, KEEPsAKE-1 and KEEPsAKE-2, which evaluated the efficacy and safety of SKYRIZI in adults with active PsA, including those who had responded inadequately or were intolerant to biologic therapy and/or non-biologic disease-modifying antirheumatic drugs (DMARDs).2,3 Across the two Phase 3 studies, SKYRIZI met the primary endpoint of ACR20 response at week 24 compared to placebo and demonstrated significant improvements across several other manifestations of PsA, including swollen, tender and painful joints.2,3
"Patients often do not suspect a connection between their psoriasis skin symptoms and the joint pain, swelling and stiffness they may be experiencing, potentially leading to a delay in diagnosis and treatment of psoriatic arthritis," said Thomas Hudson, M.D., senior vice president, research and development, chief scientific officer, AbbVie. "We're proud to expand the use of SKYRIZI to patients with psoriatic arthritis who are living with the debilitating combination of skin and joint symptoms."
SKYRIZI maintains a dosing regimen for PsA that is consistent with the existing regimen for moderate to severe plaque psoriasis patients – a single 150 mg subcutaneous injection four times a year (after two starter doses at weeks 0 and 4) – and can be administered alone or in combination with DMARDs.1
"In the pivotal KEEPsAKE trials, SKYRIZI demonstrated improvements across a number of psoriatic arthritis symptoms, including joint pain, enthesitis and dactylitis," said Alan J. Kivitz, M.D., CPI, founder and medical director of the Altoona Center for Clinical Research and Altoona Arthritis and Osteoporosis Center in Duncansville, Pa. and KEEPsAKE clinical trial investigator. "This approval provides both dermatologists and rheumatologists with an option that helps improve skin and joint symptoms in patients with active psoriatic arthritis, alongside a quarterly dosing schedule that may fit their patients' lifestyle."
SKYRIZI is part of a collaboration between Boehringer Ingelheim and AbbVie, with AbbVie leading development and commercialization of SKYRIZI globally.
Highlights from the Pivotal Phase 3 Program1,2,3
About KEEPsAKE-1 and KEEPsAKE-22,3
KEEPsAKE-1 and KEEPsAKE-2 are both Phase 3, multicenter, randomized, double-blind, placebo-controlled studies designed to evaluate the safety and efficacy of SKYRIZI in adult patients with active psoriatic arthritis. KEEPsAKE-1 evaluated SKYRIZI in patients who had an inadequate response or intolerance to at least one DMARD. KEEPsAKE-2 evaluated SKYRIZI in patients who had an inadequate response or intolerance to biologic therapy and/or DMARDs. Patients were randomized to SKYRIZI 150 mg or placebo followed by SKYRIZI 150 mg at week 24 in the open-label extension. Patients randomized to SKYRIZI received four maintenance doses a year, following two initiation doses.
The primary endpoint for both studies was the achievement of ACR20 response at week 24. Some of the ranked secondary endpoints included change from baseline in Health Assessment Questionnaire-Disability Index and the achievement of PASI 90 at week 24. The studies are ongoing, and patients in the long-term extension remain blinded to the original randomized allocation for the duration of the study, which will evaluate the long-term safety, tolerability and efficacy of SKYRIZI in patients who have completed the placebo-controlled period.
More information on these trials can be found on www.clinicaltrials.gov (KEEPsAKE-1: NCT03675308; KEEPsAKE-2: NCT03671148).
About SKYRIZI® (risankizumab-rzaa)
SKYRIZI is an interleukin-23 (IL-23) inhibitor that selectively blocks IL-23 by binding to its p19 subunit.8 IL-23, a cytokine involved in inflammatory processes, is thought to be linked to a number of chronic immune-mediated diseases, including psoriasis.8 SKYRIZI is approved in the U.S. to treat moderate to severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy, as well as to treat active psoriatic arthritis in adults. The approved dose for SKYRIZI is 150 mg (one 150 mg pre-filled pen or pre-filled syringe) administered by subcutaneous injection at weeks 0 and 4, and every 12 weeks thereafter. Phase 3 trials of SKYRIZI in psoriasis, Crohn's disease, ulcerative colitis and psoriatic arthritis are ongoing.9-17
For more information about AbbVie, please visit us at www.abbvie.com.
Jan. 21, 2022 5:06 PM ET AbbVie Inc. (ABBV)
By: Dulan Lokuwithana, SA News Editor13 Comments

PUBLISHED18 January 2022
A single priming dose of tremelimumab plus IMFINZI every four
weeks reduced risk of death by 22% in HIMALAYA Phase III trial
Combination also showed no increase in severe liver toxicity and fewer
discontinuations due to treatment-related adverse events vs. sorafenib
Positive results from the HIMALAYA Phase III trial showed a single priming dose of tremelimumab added to IMFINZI® (durvalumab) demonstrated a statistically significant and clinically meaningful improvement in overall survival (OS) versus sorafenib as a 1st-line treatment for patients with unresectable hepatocellular carcinoma (HCC) who had not received prior systemic therapy and were not eligible for localized treatment.
This novel dose and schedule of IMFINZI and tremelimumab, an anti-CTLA4 antibody, is called the STRIDE regimen (Single Tremelimumab Regular Interval Durvalumab). Results from the trial will be presented on January 21 at the 2022 American Society of Clinical Oncology (ASCO) Gastrointestinal Cancers Symposium.
Liver cancer, of which HCC is the most common type, is the third-leading cause of cancer death and the sixth most commonly diagnosed cancer worldwide.1,2 Approximately 80,000 people in the US, Europe and Japan and 260,000 people in China present with advanced, unresectable HCC each year.3 Only 7% of patients with advanced disease survive five years.4
Ghassan Abou-Alfa, MD, MBA, Attending Physician at Memorial Sloan Kettering Cancer Center and principal investigator in the HIMALAYA Phase III trial, said: “Patients with unresectable liver cancer face a dismal prognosis, and new treatment options are critical to improving long-term survival. The three-year overall survival rate and favorable safety profile seen with the STRIDE regimen set a new benchmark in this setting and underscore the potential of this innovative treatment approach.”
Susan Galbraith, Executive Vice President, Oncology R&D, AstraZeneca, said: “The HIMALAYA trial reinforces our scientific approach for tremelimumab, tapping into the potential of CTLA-4 inhibition and a unique dosing regimen to prime the immune system to help patients live longer and with minimal side effects. We look forward to bringing potential new treatment options to patients with unresectable liver cancer, an area of high unmet need, as quickly as possible.”
Patients treated with the STRIDE regimen experienced a 22% reduction in the risk of death versus sorafenib (based on a hazard ratio [HR] of 0.78, 96.02% confidence interval [CI] 0.65-0.93; p=0.0035). Median OS was 16.4 months versus 13.8 for sorafenib. An estimated 31% of patients were still alive at three years versus 20% for sorafenib.
Results also showed an increase in objective response rate (ORR) with the STRIDE regimen versus sorafenib (20.1% vs. 5.1%). Median duration of response (DoR) was 22.3 months with the STRIDE regimen versus 18.4 with sorafenib. The addition of tremelimumab to IMFINZI did not increase severe liver toxicity, and no bleeding risk was observed.
HIMALAYA also tested IMFINZI monotherapy, which demonstrated non-inferior OS to sorafenib (HR 0.86; 95.67% CI 0.73-1.03; non-inferiority margin 1.08) with a median OS of 16.6 months versus 13.8, and an improved tolerability profile versus sorafenib.
Indications:
IMFINZI is indicated for the treatment of adult patients with unresectable Stage III non-small cell lung cancer (NSCLC) whose disease has not progressed following concurrent platinum-based chemotherapy and radiation therapy.
IMFINZI, in combination with etoposide and either carboplatin or cisplatin, is indicated for the first-line treatment of adult patients with extensive-stage small cell lung cancer (ES-SCLC).
Please see complete Prescribing Information, including Medication Guide.
About IMFINZI® (durvalumab)
IMFINZI is a human monoclonal antibody that binds to the PD-L1 protein and blocks the interaction of PD-L1 with PD-1 and CD80, countering the tumor’s immune-evading tactics and releasing the inhibition of immune responses.
IMFINZI is the only approved immunotherapy in the curative-intent setting of unresectable, Stage III non-small cell lung cancer (NSCLC) in patients whose disease has not progressed after chemoradiation therapy and is the global standard of care in this setting based on the PACIFIC Phase III trial.
IMFINZI is also approved in the US, EU, Japan, China and many other countries around the world for the treatment of extensive-stage small cell lung cancer (ES-SCLC) based on the CASPIAN Phase III trial.
IMFINZI is also approved for previously treated patients with advanced bladder cancer in several countries. Since the first approval in May 2017, more than 100,000 patients have been treated with IMFINZI.
As part of a broad development program, IMFINZI is being tested as a single treatment and in combinations with other anti-cancer treatments for patients with NSCLC, SCLC, bladder cancer, HCC, biliary tract cancer (BTC), esophageal cancer, gastric and gastroesophageal cancer, cervical cancer, ovarian cancer, endometrial cancer, and other solid tumors.
About tremelimumab
Tremelimumab is a human monoclonal antibody and potential new medicine that targets the activity of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). Tremelimumab blocks the activity of CTLA-4, contributing to T-cell activation, priming the immune response to cancer and fostering cancer cell death.
Tremelimumab is being tested in a clinical trial program in combination with IMFINZI in NSCLC, SCLC, bladder cancer and liver cancer.
For more information, please visit www.astrazeneca-us.com and follow us on Twitter @AstraZenecaUS.
Jan. 18, 2022 6:29 PM ET AstraZeneca PLC (AZN) By: Jonathan Block, SA News Editor
Download as PDFJanuary 20, 2022
TOKYO and MIAMI, Jan. 20, 2022 (GLOBE NEWSWIRE) -- Pfizer Japan Inc. and OPKO Health, Inc. (NASDAQ: OPK) announced today that the next generation long-acting growth hormone injection, NGENLA® (somatrogon) Inj. 24 mg Pens and 60 mg Pens, has been approved by the Ministry of Health, Labour and Welfare (MHLW) in Japan. NGENLA® is a once-weekly long-acting recombinant human growth hormone, for the indication of short statue due to growth hormone deficiency without closed epiphyses. NGENLA® provides patients with pediatric growth hormone deficiency (GHD) with a new option that reduces treatment frequency from daily injections to once-weekly injections.
This approval is based on the results of a Phase 3 study conducted in Japanese subjects and a global Phase 3 clinical study, both of which were conducted in subjects with pediatric GHD, and both of which compared the efficacy and safety of once-weekly NGENLA® with GENOTROPIN® (somatropin), a recombinant human growth hormone for injection administered once-daily. In both studies, NGENLA® showed comparable efficacy to GENOTROPIN in the primary endpoint of annual height velocity at 12 months. NGENLA® was generally well tolerated in both studies, with comparable safety to that of GENOTROPIN administered once-daily with respect to the types, numbers and severity of the adverse events observed between the treatment arms.
“We are pleased to receive approval for once-weekly NGENLA®, which offers a new treatment option for pediatric GHD patients that can help reduce the burden associated with daily growth hormone administration. We wish to express our gratitude to the patients and their families who participated in the clinical studies and to all the sites conducting these trials,” said Taro Ishibashi, President of Pfizer R&D Japan G.K.
In 2014, Pfizer and OPKO Health entered into a worldwide agreement for the development and commercialization of somatrogon for the treatment of GHD. Under the agreement, OPKO is responsible for conducting the clinical program and Pfizer is responsible for registering and commercializing somatrogon for GHD.
【About NGENLA】
Product nameNGENLA® Inj.24mg Pens
NGENLA® Inj.60mg Pens General nameSomatrogon (recombination)INDICATIONSShort stature due to growth hormone deficiency without closed epiphysesDOSAGE AND ADMINISTRATION
Generally, Somatrogon (recombination) 0.66 mg per kilogram body weight is administered once-weekly by subcutaneous injection.
Marketing
Authorization Holder
Pfizer Japan Inc.
About the Japan Phase 3 Study
The Phase 3 study of NGENLA® in 44 treatment-naïve Japanese pre-pubertal children with pediatric GHD was a 12-month, open-label, randomized, active-controlled, parallel-group study of the efficacy and safety of weekly NGENLA® compared to recombinant human growth hormone (r-hGH), GENOTROPIN (somatropin) for injection treatment administered once-daily. Eligible patients were randomized in a 1:1 ratio to receive either once-weekly NGENLA® or GENOTROPIN administered once-daily (reference therapy, 0.025 mg/kg/day which is equivalent to 0.175 mg/kg/week). To obtain pharmacokinetic information of three different weekly doses in Japanese pediatric GHD patients, NGENLA® treated patients received 0.25 mg/kg/week for 2 weeks, followed by 0.48 mg/kg/week for 2 weeks followed by 0.66 mg/kg/week for the remaining 46 weeks.
About the Global Phase 3 Study
The Global Phase 3 study of NGENLA® in 224 treatment-naïve children with pediatric GHD in over 20 countries was a 12-month randomized, open-label, active-controlled study evaluating the safety and efficacy of weekly NGENLA® (somatrogon) injection compared to GENOTROPIN (somatropin) administered once-weekly. Eligible patients were randomized 1:1 into two arms: somatrogon administered at a dose of 0.66 mg/kg body weight once-weekly vs GENOTROPIN® (somatropin) administered at a dose of 0.034 mg/kg body weight once-daily.
About NGENLA® (somatrogon) injection
NGENLA® is a biologic product that is glycosylated and comprises the amino acid sequence of human growth hormone and one copy of the C-terminal peptide (CTP) from the beta chain of human chorionic gonadotropin (hCG) at the N-terminus and two copies of CTP (in tandem) at the C-terminus. The glycosylation and CTP domains account for the half-life of the molecule. NGENLA® was approved in Canada in October 2021 and in Australia in November 2021.
For more information, visit www.opko.com.
In addition, to learn more, please visit us on www.pfizer.com
Jan. 20, 2022 11:07 AM ET Pfizer Inc. (PFE), OPK
By: Ravikash, SA News Editor5 Comments
Approval Based on Pivotal CodeBreaK 100 Data Demonstrating Durable Responses and a Favorable Benefit-Risk Profile With LUMAKRAS
LUMAKRAS is the Only KRAS G12C Inhibitor Approved Anywhere in the World
THOUSAND OAKS, Calif., Jan. 20, 2022 /PRNewswire/ -- Amgen (NASDAQ: AMGN) today announced that LUMAKRAS® (sotorasib) has been approved in Japan for the treatment of KRAS G12C-mutated positive, unresectable, advanced and/or recurrent non-small cell lung cancer (NSCLC) that has progressed after systemic anticancer therapy.
"Today's approval of LUMAKRAS as the first and only KRASG12C inhibitor marks a paradigm shift in the treatment of patients with non-small cell lung cancer in Japan," said David M. Reese, M.D., executive vice president of Research and Development at Amgen. "In just over three years since the first patient was dosed in the pivotal CodeBreaK 100 trial, LUMAKRAS is now approved in nearly 40 countries, illustrating our commitment to accelerating transformative medicines for patients living with cancers that have yet to be fully addressed."
The approval by the Japan Ministry of Health, Labour and Welfare (MHLW) is based on positive results from the Phase 2 CodeBreaK 100 clinical trial in NSCLC, the largest trial conducted to date for patients with the KRAS G12C mutation. Based on the approved label in Japan, LUMAKRAS 960 mg, orally administered once-daily, demonstrated an objective response rate (ORR) of 37% (95% CI: 28.8-46.6) in 123 evaluable patients (including 10 Japanese patients* with a data cutoff date: Sept. 1, 2020). Adverse reactions were observed in 128 (67%) of 190 patients† (including 13 Japanese patients). The most common adverse reactions (incidence ≥ 5%) were diarrhea (28%), nausea, increased alanine aminotransferase (ALT) and increased aspartate aminotransferase (AST) (16% each), fatigue (11%), increased blood alkaline phosphatase (8%), vomiting (7%) and abdominal pain (5%).
Results from the Phase 2 CodeBreaK clinical trial in NSCLC were published in The New England Journal of Medicine.
"KRAS gene mutations are one of the oldest known cancer driver gene mutations," said Steve Sugino, president and representative director, Amgen K.K. "However, it has proven to be very difficult to develop drugs for the treatment of KRAS gene mutations. For nearly 40 years, researchers have said that the mutation was 'undruggable.' I am very pleased that LUMAKRAS is now approved as a new treatment option for patients in Japan."
"The prognosis for patients with non-small cell lung cancer who have distant metastases or whose disease has relapsed after surgery, is generally poor," said Tetsuya Mitsudomi, M.D., professor, Department of Surgery, Division of Thoracic Surgery at Kindai University School of Medicine, past-president of the International Association for the Study of Lung Cancer (IASLC) and past-president of the Japan Lung Cancer Society (JLCS). "Recent developments in molecular-targeted drugs and immunotherapy have dramatically improved the prognosis for these patients. However, despite the relatively high frequency of the KRAS G12C mutation, no drugs specifically targeting this mutation have been available until recently. Therefore, the approval of LUMAKRAS in Japan is a major milestone in the treatment of non-small cell lung cancer patients with KRAS G12C mutations."
On March 11, 2021, the MHLW designated sotorasib as an orphan drug.
*3 subjects (including 1 Japanese subject) without measurable lesions at baseline as determined by the central review were excluded.
†Patients with non-small cell lung cancer who received at least 1 dose of this drug 960 mg in the phase I and II parts.
About LUMAKRAS®/LUMYKRAS® (sotorasib)
Amgen took on one of the toughest challenges of the last 40 years in cancer research by developing LUMAKRAS/LUMYKRAS, a KRASG12C inhibitor.1 LUMAKRAS/LUMYKRAS has demonstrated a positive benefit-risk profile with rapid, deep and durable anticancer activity in patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) harboring the KRAS G12C mutation with a once daily oral formulation.2
Amgen is progressing the largest and broadest global KRASG12C inhibitor development program with unparalleled speed and exploring more than 10 sotorasib combination regimens, including triplets, with clinical trial sites spanning five continents. To date, over 4,000 patients around the world have received LUMAKRAS/LUMYKRAS through the clinical development program and commercial use.
In May 2021, LUMAKRAS was the first KRASG12C inhibitor to receive regulatory approval anywhere in the world with its approval in the U.S., under accelerated approval. LUMAKRAS/LUMYKRAS® is also approved in the United Arab Emirates, the European Union and Switzerland, and in Canada and Great Britain under the FDA's Project Orbis. Through Project Orbis, Amgen also has Marketing Authorization Applications (MAAs) for sotorasib in review in Australia, Brazil, Singapore and Israel. Additionally, Amgen has submitted MAAs in South Korea, Turkey, Taiwan, Colombia, Thailand, Mexico, Hong Kong, Saudi Arabia, Argentina, Kuwait and Qatar.
LUMAKRAS/LUMYKRAS is also being studied in multiple other solid tumors.3
About CodeBreaK
The CodeBreaK clinical development program for Amgen's drug sotorasib is designed to study patients with an advanced solid tumor with the KRAS G12C mutation and address the longstanding unmet medical need for these cancers.
CodeBreaK 100, the Phase 1 and 2, first-in-human, open-label multicenter study, enrolled patients with KRAS G12C-mutant solid tumors.2,3 Eligible patients must have received a prior line of systemic anticancer therapy, consistent with their tumor type and stage of disease. The primary endpoint for the Phase 2 study was centrally assessed objective response rate. The Phase 2 trial in NSCLC enrolled 126 patients, 124 of whom had centrally evaluable lesions by RECIST at baseline.2 The Phase 2 trial in colorectal cancer (CRC) is fully enrolled and results have been published.11
CodeBreaK 200, the global Phase 3 randomized active-controlled study comparing sotorasib to docetaxel in KRAS G12C-mutated NSCLC completed enrollment of 345 patients. Eligible patients had previously treated, locally-advanced and unresectable or metastatic KRAS G12C-mutated NSCLC. The primary endpoint is progression-free survival and key secondary endpoints include overall survival, objective response rate, and patient-reported outcomes.
Amgen also has several Phase 1b studies investigating sotorasib monotherapy and sotorasib combination therapy across various advanced solid tumors (CodeBreaK 101) open for enrollment. A Phase 2 randomized study will evaluate sotorasib in patients with stage IV KRAS G12C-mutated NSCLC in need of first-line treatment (CodeBreaK 201).
For information, please visit www.hcp.codebreaktrials.com.
LUMAKRAS® (sotorasib) U.S. Indication
LUMAKRAS is indicated for the treatment of adult patients with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.
This indication is approved under accelerated approval based on overall response rate (ORR) and duration of response (DOR). Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
Please see LUMAKRAS full Prescribing Information.
For more information, visit www.amgen.com and follow us on www.twitter.com/amgen.
View original content to download multimedia:https://www.prnewswire.com/news-releases/lumakras-sotorasib-receives-approval-in-japan-for-patients-with-kras-g12c-mutated-advanced-non-small-cell-lung-cancer-301464697.html
SOURCE Amgen
Jan. 20, 2022 5:57 AM ETAmgen Inc. (AMGN)By: Niloofer Shaikh, SA News Editor

January 19, 2022 at 7:00 AM EST Back
TARRYTOWN, N.Y., Jan. 19, 2022 /PRNewswire/ --
Regulatory filing recently submitted in the European Union
Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) today announced that the U.S. Food and Drug Administration (FDA) has accepted for review the supplemental Biologics License Application (sBLA) for PD-1 inhibitor Libtayo® (cemiplimab-rwlc) in combination with chemotherapy as first-line treatment in advanced non-small cell lung cancer (NSCLC). The target action date for the FDA decision is September 19, 2022.
The sBLA is supported by results from a randomized, multicenter Phase 3 trial that investigated Libtayo in combination with a physician's choice of platinum-doublet chemotherapy (Libtayo combination), compared to platinum-doublet chemotherapy alone. Enrolled patients (n=466) had locally advanced or metastatic NSCLC, irrespective of PD-L1 expression level or tumor histology, and with no ALK, EGFR or ROS1 aberrations. A regulatory filing has also been recently submitted to the European Medicines Agency.
The Phase 3 trial supporting the sBLA was stopped early after the Libtayo combination demonstrated a significant overall survival improvement compared to chemotherapy alone. Results were presented at the European Society for Medical Oncology Virtual Congress in 2021. Among patients in the Libtayo combination (n=312) and chemotherapy alone (n=154) groups, treatment discontinuations due to adverse events (AEs) occurred in 5% and 3% of patients, respectively. Immune-mediated AEs occurred in 19% of patients in the Libtayo combination group.
Notably, the Phase 3 trial was designed to include baseline characteristics seen in everyday clinical practice. Among those enrolled, 43% had tumors with squamous histology, 67% had tumors with <50% PD-L1 expression, 15% had inoperable locally advanced disease not eligible for definitive chemoradiation, and 7% had pretreated and clinically stable brain metastases. In addition, 84% of patients had an ECOG 1 performance status, which indicates the presence of cancer-related symptoms. ECOG performance status assesses patient ability to conduct daily living activities and prognosis on a scale of increasing severity ranging from 0 (no symptoms) to 5 (death).
In 2021, Libtayo was approved in the U.S. and European Union as first-line monotherapy treatment for adult patients with advanced NSCLC whose tumors have high PD-L1 expression (tumor proportion score ≥50%), as determined by an FDA-approved test. Patients must either have metastatic or locally advanced tumors that are not candidates for surgical resection or definitive chemoradiation, and the tumors must not have EGFR, ALK or ROS1 aberrations.
Libtayo, which was invented using Regeneron's proprietary VelocImmune® technology, is being jointly developed by Regeneron and Sanofi under a global collaboration agreement. The use of Libtayo in combination with chemotherapy for advanced NSCLC is investigational, and its safety and efficacy have not been fully evaluated by any regulatory authority.
About Libtayo
Libtayo is a fully human monoclonal antibody targeting the immune checkpoint receptor PD-1 on T-cells. By binding to PD-1, Libtayo has been shown to block cancer cells from using the PD-1 pathway to suppress T-cell activation. Libtayo is indicated in certain patients with advanced basal cell carcinoma, advanced cutaneous squamous cell carcinoma, and advanced NSCLC.
The extensive clinical program for Libtayo is focused on difficult-to-treat cancers. Libtayo is currently being investigated in trials as a monotherapy, as well as in combination with either conventional or novel therapeutic approaches for other solid tumors and blood cancers. These potential uses are investigational, and their safety and efficacy have not been evaluated by any regulatory authority.
U.S. FDA-approved Indications
Libtayo is a prescription medicine used to treat people with:
It is not known if Libtayo is safe and effective in children.
Please see full Prescribing Information, including Medication Guide.
For additional information about the company, please visit www.regeneron.com or follow @Regeneron on Twitter.
View original content:https://www.prnewswire.com/news-releases/fda-accepts-for-review-libtayo-cemiplimab-rwlc-in-combination-with-chemotherapy-for-first-line-treatment-of-advanced-nsclc-301463552.html
SOURCE Regeneron Pharmaceuticals, Inc.
Jan. 19, 2022 11:52 AM ET Regeneron Pharmaceuticals, Inc. (REGN)SNY By: Ravikash, SA News Editor
https://seekingalpha.com/symbol/SNY
https://seekingalpha.com/symbol/REGN
Jan 20, 2022 5:00 AM
BRUKINSA received the China NMPA approval for the treatment of patients with relapsed or refractory Waldenström’s macroglobulinemia (WM) in June 2021
The submission, supported by the ASPEN trial results, could potentially expand BRUKINSA to front-line care of WM
CAMBRIDGE, Mass., & BEIJING--(BUSINESS WIRE)-- BeiGene (NASDAQ: BGNE; HKEX: 06160; SSE: 688235), a global, science-driven biotechnology company focused on developing innovative and affordable medicines to improve treatment outcomes and access for patients worldwide, today announced that the Center for Drug Evaluation (CDE) of the China National Medical Products Administration (NMPA) has accepted a supplemental new drug application (sNDA) for BeiGene’s BTK inhibitor BRUKINSA® (zanubrutinib) as a treatment for adult patients with Waldenström’s macroglobulinemia (WM).
“The sNDA acceptance is welcoming news, and following BRUKINSA’s recent NMPA approval for patients with WM in the relapsed or refractory setting, this represents an opportunity to expand access to more WM patients in China, subject to NMPA approval. As demonstrated in the ASPEN trial, BRUKINSA can offer an efficacious treatment option with improved safety in regard to certain cardiovascular events, such as atrial fibrillation, for patients with WM,” commented Jane Huang, M.D., Chief Medical Officer, Hematology, BeiGene. “The ASPEN trial has supported BRUKINSA’s approval for patients with WM in the U.S., Canada, Australia, and the European Union. We look forward to continued discussions with the CDE and the opportunity to bring this potential best-in-class therapy to more people in the WM community in China.”
The sNDA is supported by clinical results from the randomized, open-label, multicenter Phase 3 ASPEN trial (NCT03053400) comparing BRUKINSA to ibrutinib in patients with relapsed or refractory (R/R) or treatment-naïve (TN) WM.
As assessed by an independent review committee (IRC) based on the modified Sixth International Workshop on Waldenström’s Macroglobulinemia (IWWM-6) response criteria (Treon 2015), the combined rate of complete response (CR) and very good partial response (VGPR) in the overall intention-to-treat (ITT) population was 28% with BRUKINSA (95% CI: 20, 38), compared to 19% with ibrutinib (95% CI: 12, 28). While this difference was not statistically significant (p=0.09), BRUKINSA did achieve numerically higher VGPR rates and trends towards increased response quality.1
In the ASPEN trial, BRUKINSA demonstrated a more favorable safety profile compared to ibrutinib with lower frequency of certain adverse events, including atrial fibrillation or flutter (2% vs. 15%) and major hemorrhage (6% vs. 9%). Of the 101 patients with WM treated with BRUKINSA, 4% of patients discontinued due to adverse events, and adverse events leading to dose reduction occurred in 14% of patients.4
About Waldenström’s Macroglobulinemia
Waldenström’s macroglobulinemia is a rare, slow-growing lymphoma, characterized by bone marrow infiltration with monoclonal immunoglobulin M (IgM) secreting lymphoplasmacytic cells, that occurs in less than two percent of patients with non-Hodgkin’s lymphoma (NHL).1 The disease usually affects older adults and is primarily found in the bone marrow, although it may also impact lymph nodes and the spleen.2 In China, there are an estimated 88,200 patients diagnosed with lymphoma each year. Approximately 91% of these cases are classified as NHL, amounting to ~1,000 newly diagnosed WM patients per year in China.3
About BRUKINSA
BRUKINSA is a small molecule inhibitor of Bruton’s tyrosine kinase (BTK) discovered by BeiGene scientists that is currently being evaluated globally in a broad clinical program as a monotherapy and in combination with other therapies to treat various B-cell malignancies. Because new BTK is continuously synthesized, BRUKINSA was specifically designed to deliver complete and sustained inhibition of the BTK protein by optimizing bioavailability, half-life, and selectivity. With differentiated pharmacokinetics compared to other approved BTK inhibitors, BRUKINSA has been demonstrated to inhibit the proliferation of malignant B cells within a number of disease relevant tissues.
BRUKINSA is approved in the following indications and regions:
To date, more than 20 marketing authorization applications have been submitted for BRUKINSA for various indications..
* This indication was approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
** This indication was approved under conditional approval. Complete approval for this indication may be contingent upon results from ongoing randomized, controlled confirmatory clinical trials.
Please see full U.S. Prescribing Information at www.beigene.com/PDF/BRUKINSAUSPI.pdf and Patient Information at www.beigene.com/PDF/BRUKINSAUSPPI.pdf.
To learn more about BeiGene, please visit www.beigene.com and follow us on Twitter at @BeiGeneGlobal.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220120005178/en/
Source: BeiGene
Jan. 20, 2022 5:41 AM ET
By: Niloofer Shaikh, SA News Editor
Exelixis Announces Detailed Results for Cabozantinib in Combination with Immunotherapies in Patients with Advanced Colorectal Cancer at ASCO GI 2022PDF Version
– Cabozantinib in combination with atezolizumab evaluated in cohort 16 of the phase 1b COSMIC-021 trial demonstrated encouraging clinical activity with a manageable safety profile in patients with previously treated metastatic colorectal cancer –
– Cabozantinib in combination with durvalumab evaluated in cohort 2 of the phase 2 CAMILLA trial demonstrated promising efficacy and was generally well tolerated with no new safety signals in chemotherapy-refractory patients with advanced mismatch repair proficient/micro satellite stable colorectal cancer –
ALAMEDA, Calif.--(BUSINESS WIRE)--Jan. 18, 2022-- Exelixis, Inc. (Nasdaq: EXEL) today announced results for cabozantinib (CABOMETYX®) in combination with immunotherapies in patients with advanced colorectal cancer, including encouraging data from cohort 16 of the phase 1b COSMIC-021 trial of cabozantinib in combination with atezolizumab in patients with metastatic colorectal cancer who were previously treated with fluoropyrimidine-containing chemotherapy. Results from cohort 2 of the phase 2 CAMILLA trial of cabozantinib in combination with durvalumab in patients with advanced mismatch repair proficient/micro satellite stable (pMMR/MSS) colorectal cancer patients who were chemotherapy-refractory were also announced. The data from these studies are being presented during Poster Session C: Cancers of the Colon, Rectum, and Anus on Saturday, January 22 at the 2022 American Society of Clinical Oncology’s Gastrointestinal Cancers Symposium (ASCO GI).
Abstract 121: A phase 1b multi-tumor cohort study of cabozantinib plus atezolizumab in advanced solid tumors (COSMIC-021): Results of the colorectal cancer cohort
At a median follow-up of 28.1 months, the primary endpoint of objective response rate (ORR) by investigator per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 in cohort 16 (n=31) was 10%. The disease control rate (DCR; complete response + partial response + stable disease) was 71%. Median progression-free survival (PFS) was 3.0 months (95% confidence interval [CI]: 2.7-5.4), and median overall survival (OS) was 14.0 months (95% CI: 5.5-16.7). Median duration of response was 7.6 months (95% CI: 4.2-not estimable [NE]).
A post-hoc exploratory analysis showed that patients with wild-type RAS (n=12) had longer PFS and OS compared with patients with RAS mutations (n=19): median PFS was 5.8 months (95% CI: 2.8-11.0) compared with 2.7 months (95% CI: 1.6-4.1), respectively, and median OS was 16.7 months (95% CI: 8.4-NE) compared with 8.7 months (95% CI: 4.7-15.9), respectively. ORR was 25% for patients with wild-type RAS and 0% for patients with RAS mutations.
Abstract 135: Phase II trial of cabozantinib (Cabo) plus durvalumab (Durva) in chemotherapy refractory patients with advanced mismatch repair proficient/microsatellite stable (pMMR/MSS) colorectal cancer (CRC): CAMILLA CRC cohort results
Of the 36 patients enrolled in cohort 2 of the CAMILLA trial, 29 were evaluable for the efficacy analysis. The primary outcome of investigator-assessed ORR per modified RECIST version 1.1 was 27.6%. The confirmed partial response rate was 20.7%, and the DCR was 86.2%. Median PFS was 3.8 months (95% CI: 3.4-6.3), with a 6-month PFS of 34.5% (95% CI: 17.9-54.3). Median OS was 9.1 months (95% CI: 5.8-21.8). In a subgroup analysis of those with wild-type RAS (n=12), ORR was 50.0%, and DCR was 83.3%. Median PFS was 6.3 months (95% CI: 1.8-NE), and median OS was 21.8 months (95% CI: 4.5-NE).
Patients eligible for CAMILLA cohort 2 had advanced pMMR/MSS colorectal cancer and had progressed on two or more lines of therapy. Ninety percent of patients had an ECOG Performance Score of 1, 41% had wild-type RAS and 79% had liver metastases. Approximately half (52%) of patients had received at least three prior lines of therapy.
Mismatch repair status and microsatellite instability status are considered prognostic factors in colorectal cancer and can impact treatment decisions.1 Patients with metastatic colorectal cancer who have microsatellite stable and/or mismatch repair-proficient tumors tend to have poor responses to immune checkpoint inhibitor monotherapy, meaning alternative treatment strategies are needed.2, 3
Among the 36 patients evaluable for safety, the most common treatment-related AEs were grade 1/2 fatigue (53%), nausea (42%), diarrhea (36%), anorexia (31%) and hand-foot syndrome (25%). Eleven patients (31%) experienced grade 3 or higher treatment-related AEs. Grade 3 or higher immune-related AEs occurred in 16.6% of patients. One patient discontinued durvalumab due to AEs; no patients discontinued cabozantinib.
CAMILLA is an investigator-sponsored trial conducted by Anwaar Saeed, M.D., GI medical oncologist and Associate Director of the Early Phase Program at The University of Kansas Cancer Center.
“After disease progression following prior treatment, people with advanced colorectal cancer are in need of additional treatment options that can help control their disease,” said Vicki L. Goodman, M.D., Executive Vice President, Product Development and Medical Affairs, and Chief Medical Officer, Exelixis. “We are pleased that the findings from cohort 16 of COSMIC-021 and cohort 2 of CAMILLA validate the potential of cabozantinib in combination with immunotherapies in advanced colorectal cancer, as this patient community often faces poor outcomes.”
About COSMIC-021
COSMIC-021 is a multicenter, phase 1b, open-label study that is divided into two parts: a dose-escalation phase and an expansion cohort phase. The dose-escalation phase was designed to enroll patients either with advanced renal cell carcinoma (RCC) with or without prior systemic therapy or with inoperable, locally advanced, metastatic or recurrent urothelial carcinoma (UC), (including renal, pelvis, ureter, urinary bladder and urethra) after prior platinum-based therapy. Ultimately, all 12 patients who enrolled in this stage of the trial were patients with advanced RCC. The dose-escalation phase of the study determined the recommended dose of cabozantinib to be 40 mg daily when given in combination with atezolizumab (1200 mg infusion once every three weeks).
In the expansion phase, the trial is enrolling 24 cohorts in 12 tumor types: RCC, UC, non-small cell lung cancer (NSCLC), castration-resistant prostate cancer (CRPC), hepatocellular carcinoma (HCC), triple-negative breast cancer, epithelial ovarian cancer, endometrial cancer, gastric or gastroesophageal junction adenocarcinoma, colorectal adenocarcinoma, head and neck cancer and differentiated thyroid cancer.
Three of the cohorts are exploratory single agent cohorts: one enrolled 31 patients with advanced NSCLC, one has been expanded to enroll up to 80 patients with advanced CRPC to be treated with cabozantinib as a single-agent, and one enrolled 10 patients with advanced CRPC to be treated with single-agent atezolizumab. Exploratory single agent cohorts have the option to be expanded up to 80 patients (cabozantinib) and 30 patients (atezolizumab) total.
Exelixis is the study sponsor of COSMIC-021. Both Ipsen Pharma SAS (Ipsen) and Takeda Pharmaceutical Company Limited (Takeda) have opted in to participate in the trial and are contributing to the funding for this study under the terms of the companies’ respective collaboration agreements with Exelixis. Roche is providing atezolizumab for the trial. More information on this trial is available on ClinicalTrials.gov.
About CAMILLA
CAMILLA is a phase 1b/2 study that is evaluating cabozantinib in combination with durvalumab with or without tremelimumab in patients with advanced gastroesophageal and gastrointestinal cancers. Upon completion of the phase 1b gastrointestinal basket trial, which included 30 patients, the trial was expanded to a phase 2, multicenter trial of 117 patients with four disease cohorts. Three cohorts are evaluating cabozantinib in combination with durvalumab in gastric and esophageal cancer, colorectal cancer and HCC. The fourth cohort is evaluating cabozantinib in combination with durvalumab and tremelimumab in HCC patients. Patients enrolled in cohort 2 (CRC) received cabozantinib 40 mg daily in combination with durvalumab 1500 mg infusion once every four weeks. CAMILLA is an investigator-sponsored trial; more information is available on ClinicalTrials.gov.
About CABOMETYX® (cabozantinib)
In the U.S., CABOMETYX tablets are approved for the treatment of patients with advanced RCC; for the treatment of patients with HCC who have been previously treated with sorafenib; for patients with advanced RCC as a first-line treatment in combination with nivolumab; and for adult and pediatric patients 12 years of age and older with locally advanced or metastatic differentiated thyroid cancer that has progressed following prior VEGFR-targeted therapy and who are radioactive iodine-refractory or ineligible. CABOMETYX tablets have also received regulatory approvals in the European Union and additional countries and regions worldwide. In 2016, Exelixis granted Ipsen exclusive rights for the commercialization and further clinical development of cabozantinib outside of the U.S. and Japan. In 2017, Exelixis granted exclusive rights to Takeda Pharmaceutical Company Limited for the commercialization and further clinical development of cabozantinib for all future indications in Japan. Exelixis holds the exclusive rights to develop and commercialize cabozantinib in the U.S.
CABOMETYX in combination with atezolizumab or in combination with durvalumab is not indicated for colorectal cancer.
Please see accompanying full Prescribing Information https://www.cabometyx.com/downloads/CABOMETYXUSPI.pdf.
For more information about Exelixis, please visit www.exelixis.com, follow @ExelixisInc on Twitter or like Exelixis, Inc. on Facebook.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220118006159/en/
Source: Exelixis, Inc.
Jan. 19, 2022 1:20 PM ET
By: Jonathan Block, SA News Editor
January 18, 2022 5:00 pm ET
First Presentation of OS Data From Phase 3 KEYNOTE-394 Trial at ASCO GI 2022
KEYNOTE-394 Is One of Seven Clinical Trials in Merck’s Global Development Program in HCC
KENILWORTH, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, today announced the final results from the Phase 3 KEYNOTE-394 trial investigating KEYTRUDA, Merck’s anti-PD-1 therapy, plus best supportive care (BSC) in patients in Asia with advanced hepatocellular carcinoma (HCC) previously treated with sorafenib. KEYNOTE-394 is the first trial with an anti-PD-1/L1 as a second-line monotherapy treatment to show an improvement in overall survival (OS), progression-free survival (PFS) and objective response rate (ORR) compared to placebo plus BSC for these patients. These data add to the body of evidence relating to the use of KEYTRUDA as a monotherapy in second-line HCC post sorafenib.
KEYTRUDA plus BSC demonstrated a statistically significant and clinically meaningful improvement in the primary endpoint of OS, reducing the risk of death by 21% (HR=0.79 [95% CI, 0.63-0.99]; p=0.0180) compared to placebo plus BSC for patients with previously treated advanced HCC. For patients treated with KEYTRUDA plus BSC, median OS was 14.6 months (95% CI, 12.6-18.0) compared to 13.0 months (95% CI, 10.5-15.1) for patients treated with placebo plus BSC. The percentage of patients who were alive at two years was 34.3% for KEYTRUDA plus BSC compared to 24.9% for placebo plus BSC. These data will be presented at the 2022 American Society of Clinical Oncology Gastrointestinal Cancers (ASCO GI) Symposium (Abstract #352788) on Friday, Jan. 21 at 10:15 a.m. ET.
“Hepatocellular carcinoma is a leading cause of cancer death across the world, and there are limited treatment options shown to extend survival for patients following treatment with sorafenib,” said Dr. Shukui Qin, director, Cancer Center of Jinling Hospital, and professor, Nanjing University of Chinese Medicine. “These overall survival data are very encouraging for patients with HCC previously treated with sorafenib and show the potential of KEYTRUDA to extend the lives of these patients.”
Treatment-related adverse events (TRAEs) occurred in 66.9% of patients in the KEYTRUDA plus BSC arm and 49.7% of patients in the placebo plus BSC arm, and Grade 3-5 TRAEs occurred in 14.4% of patients in the KEYTRUDA plus BSC arm and 5.9% of patients in placebo plus BSC arm. Immune-mediated adverse events (AEs) of any grade occurred in 18.1% of patients receiving KEYTRUDA plus BSC and 10.5% of patients receiving placebo plus BSC. Grade 3-5 immune-mediated AEs occurred in 3.0% of patients receiving KEYTRUDA plus BSC. There were three deaths (due to gastrointestinal hemorrhage, autoimmune hepatitis, and soft tissue infection) in the KEYTRUDA arm related to the study intervention.
“Patients with advanced HCC still have a high unmet medical need with low survival rates, reinforcing the need for treatment options that can improve overall survival,” said Dr. Scot Ebbinghaus, vice president, clinical research, Merck Research Laboratories. “We are pleased to share these new data from KEYNOTE-394 and are committed to advancing research for patients with this difficult-to-treat cancer through our broad global program in HCC.”
In the U.S., KEYTRUDA is indicated for the treatment of patients with HCC who have been previously treated with sorafenib based on ORR and duration of response (DOR) data from KEYNOTE-224. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. Data from KEYNOTE-394 are being discussed with global regulatory authorities and will be evaluated as a potential confirmatory study in the U.S.
Merck is dedicated to advancing research in HCC and has a global development program of seven clinical trials that have enrolled or are expected to enroll approximately 3,000 patients. In HCC, KEYTRUDA is being studied across multiple settings and lines of therapy as monotherapy and in combination with other treatments, including therapies through our collaborations.
Study Design and Additional Data From KEYNOTE-394
KEYNOTE-394 (ClinicalTrials.gov, NCT03062358) is a randomized, double-blind Phase 3 trial evaluating KEYTRUDA plus BSC versus placebo plus BSC in Asian patients with advanced HCC previously treated with sorafenib or oxaliplatin-based chemotherapy. The primary endpoint is OS. Additional endpoints include PFS, ORR, and DOR. The study enrolled 453 patients who were randomized to receive either KEYTRUDA (intravenously every three weeks for up to 35 cycles of treatment [up to approximately two years]) plus BSC (including pain management and management of other potential complications including ascites per local standards of care) or placebo plus BSC.
Additional efficacy endpoint results of the trial showed KEYTRUDA plus BSC reduced the risk of disease progression or death by 26% (HR=0.74 [95% CI, 0.60-0.92]; p=0.0032) compared to placebo plus BSC. Median PFS was 2.6 months (95% CI, 1.5-2.8) for KEYTRUDA plus BSC and 2.3 months (95% CI, 1.4-2.8) for placebo plus BSC. KEYTRUDA plus BSC showed an ORR of 12.7% (95% CI, 9.1-17.0) and a median DOR of 23.9 months (range, 2.8 to 32.0+), where placebo plus BSC showed an ORR of 1.3% (95% CI, 0.2-4.6) and a median DOR of 5.6 months (range, 3.0+ to 5.6).
About Liver Cancer and Hepatocellular Carcinoma
Liver cancer is one of the leading causes of cancer-related deaths worldwide. It is estimated there were more than 905,000 new cases of liver cancer and more than 830,000 deaths from the disease globally in 2020. In the U.S., it is estimated there will be more than 41,000 new cases of liver cancer diagnosed and more than 30,000 deaths from this disease in 2022. An estimated 75% to 90% of primary liver cancer cases are hepatocellular carcinoma (HCC). Worldwide, major factors that increase the risk of HCC are chronic hepatitis B and chronic hepatitis C, alcohol use, and metabolic syndrome.
About KEYTRUDA® (pembrolizumab) Injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of adult and pediatric (12 years and older) patients with stage IIB, IIC, or III melanoma following complete resection.
Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [tumor proportion score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [combined positive score (CPS) ≥1] as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy.
KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Carcinoma
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
Non-muscle Invasive Bladder Cancer
KEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin (BCG) -unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with MSI-H central nervous system cancers have not been established.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC).
Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or gastroesophageal (GEJ) (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
KEYTRUDA, in combination with chemotherapy, with or without bevacizumab, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
KEYTRUDA is indicated for the adjuvant treatment of patients with RCC at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions.
Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf .
Source: Merck & Co., Inc.
Jan. 18, 2022 6:05 PM ET Merck & Co., Inc. (MRK)GILD By: Jonathan Block, SA News Editor4 Comments
Tuesday, January 18, 2022 - 06:45am
NEW YORK, January 18, 2022 -- Pfizer Inc. (NYSE: PFE) today shared results from multiple studies demonstrating that the in vitro efficacy of nirmatrelvir, the active main protease (Mpro) inhibitor of PAXLOVID™ (nirmatrelvir [PF-07321332] tablets and ritonavir tablets), is maintained against the SARS-CoV-2 variant Omicron. Taken together, these in vitro studies suggest that PAXLOVID has the potential to maintain plasma concentrations many-fold times higher than the amount required to prevent Omicron from replicating in cells.
“We specifically designed PAXLOVID to retain its activity across coronaviruses, as well as current variants of concern with predominantly spike protein mutations. Following the clinical findings – showing PAXLOVID reduced risk of hospitalization or death by nearly 90% compared to placebo for high-risk patients when treated within five days of symptom onset – we are encouraged by these initial laboratory findings,” said Mikael Dolsten, M.D., Ph.D., Chief Scientific Officer and President, Worldwide Research, Development and Medical of Pfizer. “These data suggest that our oral COVID-19 therapy can be an important and effective tool in our continued battle against this devastating virus and current variants of concern, including the highly transmissible Omicron. We will continue to monitor the treatment’s activity in real-world settings and believe that these in vitro findings will continue to be validated.”
In the first of these in vitro studies conducted by Pfizer, nirmatrelvir was tested against the Mpro – an enzyme that the coronavirus needs to replicate – from several SARS-CoV-2 variants of concern (VoCs), including Omicron, in a biochemical assay. The results showed in all cases that nirmatrelvir was a potent inhibitor of its target. Nirmatrelvir’s Ki – a measure of its ability to bind to an enzyme – was approximately 1 nanomolar (nM) (or Ki fold change <1) for both the Omicron and the original Washington variant (USA-WA1/2020) in this assay, indicating its continued ability to prevent in vitro viral replication. These results, together with a crystal structure demonstrating how nirmatrelvir binds to the Omicron variant, have been submitted to the online preprint server bioRxiv.
In a second in vitro study conducted by Pfizer, nirmatrelvir was tested against several SARS-CoV-2 VoCs, including Omicron, in an antiviral, cell-based assay. Reduction in viral load was measured through polymerase chain reaction (PCR) analysis, a test designed to detect the virus. Nirmatrelvir’s EC50 – a measure of drug potency showing a concentration that is effective in producing 50% of the maximal response – was 16 nM for the Omicron variant, compared to 38 nM for the USA-WA1/2020 variant, reaffirming its robust in vitro antiviral activity. These results are in line with the values that have been observed for other VoCs (Alpha, Beta, Gamma, Delta, Lambda, and Mu) in this assay, with EC50 measures ranging from 16 to 127 nM compared to USA-WA1/2020, where EC50 was 37 nM. Results from this study have been submitted to the online preprint server bioRxiv and will be submitted to a peer-reviewed journal.
An additional study, conducted by the Icahn School of Medicine at Mount Sinai (Icahn Mount Sinai) in collaboration with Pfizer, used a SARS-CoV-2-specific immunofluorescence-based assay to similarly detect the virus and measure the in vitro potency of nirmatrelvir, as well as some other authorized/approved COVID-19 therapeutics, against VoCs. In this assay, treatments were tested against the Alpha, Beta, Delta, and Omicron variants in two cell lines. IC50 values – a measure of drug efficacy indicating the concentration needed to inhibit infection by half – ranging from 22 to 225 nM for nirmatrelvir compared to USA-WA1/2020, where the IC50 was 38 to 207 nM, were observed. Results from this study have been submitted to the online preprint server bioRxiv and will be submitted to a peer-reviewed journal. These findings are consistent with data made available to the public on December 28, 2021 by the Rega Institute at KU Leuven in Belgium via the online preprint server, bioRxiv, and provide confirmation to Pfizer’s findings that nirmatrelvir is the only orally administrable, authorized/approved compound which, to date, has been shown to have low nanomolar in vitro activity against Omicron.
“Omicron is proving itself to be a formidable and highly transmissible variant of an already detrimental virus,” said Kris White, Ph.D., Assistant Professor in the Department of Microbiology at Icahn Mount Sinai). “We are heartened to see early data showing that this oral treatment is maintaining robust in vitro antiviral activity against it, as well as other variants of concern.”
Current VoCs can be resistant to treatments that work by binding to the spike protein found on the surface of the SARS-CoV-2 virus. PAXLOVID, however, works intracellularly by binding to the highly conserved Mpro of the SARS-CoV-2 virus. Previous data have also indicated that PAXLOVID maintains in vitro efficacy against earlier and current VoCs, including Alpha, Beta, Gamma, Delta, Lambda, and Mu.
PAXLOVID is currently authorized for conditional or emergency use in several countries across the globe. Pfizer has submitted applications for regulatory approval or authorization to multiple regulatory agencies and anticipates further regulatory decisions to follow.
Please see Full Emergency Use Authorization (EUA) Prescribing Information available at www.fda.gov and www.COVID19oralRx.com.
About PAXLOVID™ (nirmatrelvir [PF-07321332] tablets and ritonavir tablets)
PAXLOVID is a SARS-CoV-2 main protease (Mpro) inhibitor (also known as SARS-CoV-2 3CL protease inhibitor) therapy. It was developed to be administered orally so that it can be prescribed at the first sign of infection or, pending clinical success of the rest of the EPIC development program and subject to regulatory authorization, at first awareness of an exposure – potentially helping patients avoid severe illness (which can lead to hospitalization and death) or avoid disease development following contact with a household member who contracts COVID-19. Nirmatrelvir [PF-07321332], which originated in Pfizer laboratories, is designed to block the activity of the Mpro, an enzyme that the coronavirus needs to replicate. Co-administration with a low dose of ritonavir helps slow the metabolism, or breakdown, of nirmatrelvir in order for it to remain active in the body for longer periods of time at higher concentrations to help combat the virus.
Nirmatrelvir is designed to inhibit viral replication at a stage known as proteolysis, which occurs before viral RNA replication. In preclinical studies, nirmatrelvir did not demonstrate evidence of mutagenic DNA interactions.
PAXLOVID is authorized to be administered at a dose of 300 mg (two 150 mg tablets) of nirmatrelvir with one 100 mg tablet of ritonavir, given twice-daily for five days. One carton contains five blister packs of PAXLOVID, as co-packaged nirmatrelvir tablets with ritonavir tablets, providing all required doses for a full five-day treatment course.
Under the Emergency Use Authorization, the U.S. Government and State Governments decide how PAXLOVID is distributed among pharmacies, hospitals, urgent cares, and other entities. Healthcare providers and healthcare facilities should contact their state health department to determine how to access product. Additional information about how the U.S. Department of Health and Human Services will supply PAXLOVID can be found at www.PHE.gov and https://www.hhs.gov/coronavirus/covid-19-treatments-therapeutics/index.html. Locations of publicly available COVID-19 Therapeutics can be found at COVID-19 Public Therapeutic Locator | HealthData.gov.
Emergency Use Authorization Statement
PAXLOVID has not been approved, but has been authorized for emergency use by FDA under an EUA, for the treatment of mild-to-moderate COVID-19 in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS CoV-2 viral testing, and who are at high-risk for progression to severe COVID-19, including hospitalization or death.
The emergency use of PAXLOVID is only authorized for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of drugs and biological products during the COVID-19 pandemic under Section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the declaration is terminated or authorization revoked sooner.
AUTHORIZED USE
The U.S. Food and Drug Administration (FDA) has issued an Emergency Use Authorization (EUA) for the emergency use of the unapproved product PAXLOVID for the treatment of mild-to-moderate coronavirus disease 2019 (COVID-19) in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death.
LIMITATIONS OF AUTHORIZED USE
• PAXLOVID is not authorized for initiation of treatment in patients requiring hospitalization due to severe or critical COVID-19
• PAXLOVID is not authorized for use as pre-exposure or post-exposure prophylaxis for prevention of COVID-19
• PAXLOVID is not authorized for use for longer than 5 consecutive days
PAXLOVID may only be prescribed for an individual patient by physicians, advanced practice registered nurses, and physician assistants that are licensed or authorized under state law to prescribe drugs in the therapeutic class to which PAXLOVID belongs (i.e., anti-infectives).
PAXLOVID is not approved for any use, including for use for the treatment of COVID-19.
PAXLOVID is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of PAXLOVID under 564(b)(1) of the Food Drug and Cosmetic Act unless the authorization is terminated or revoked sooner.
https://www.pfizer.com/products/product-detail/paxlovidtm
for Consumers:
for Healthcare professionals:
Jan. 18, 2022 1:35 PM ET Pfizer Inc. (PFE)
By: Jonathan Block, SA News Editor6 Comments
Mechelen, Belgium; 18 January 2022; 22.01 CET; Galapagos NV (Euronext & Nasdaq: GLPG) announced today that the Medicines and Healthcare products Regulatory Agency (MHRA) has granted a marketing authorization for Jyseleca® (filgotinib 200mg tablets), as a new treatment for ulcerative colitis (UC) for Great Britain.
The MHRA has licensed an additional indication for Jyseleca, an oral once-daily, JAK1 preferential inhibitor, for use in adult patients with moderately to severely active UC who have had an inadequate response with, lost response to, or were intolerant to either conventional therapy or a biologic agent. The submission was supported by data from the Phase 2b/3 SELECTION program, published in the Lancet1. The decision from the MHRA follows authorisation from the European Commission (EC) for use in the same patient population.
Michele Manto, Chief Commercial Officer at Galapagos said: “At Galapagos we are committed to bringing new and innovative medicines to healthcare professionals who are treating patients with UC and today we are one step closer to offering a new treatment option to thousands of patients living in Great Britain with UC, a chronic and debilitating disease. Together with the EC decision, this decision represents an important milestone in our plans to make Jyseleca available to eligible adult patients with UC across Europe.”
Dr Ian Beales, Consultant in Gastroenterology and General Medicine, Norfolk and Norwich University Hospital and Chief Investigator for the SELECTION study in the UK said: “The prevalence of UC in the UK is increasing. 1 in every 420 people are currently estimated to have the disease. Despite available treatments there is still a need for innovative new therapies to provide relief from the symptoms that can have debilitating physical consequences for patients. In the SELECTION study when compared to placebo, more patients on filgotinib 200mg demonstrated corticosteroid-free remission from clinical symptoms with improvements in measures of health-related quality of life and was well-tolerated by patients. We welcome having a new treatment option available to help us with managing this disease.”
UC is a life-long condition characterized by inflammation of the mucosal lining of the colon and rectum. Current estimates suggest that in the UK more than 146,0002 people are currently living with UC. The prevalence of inflammatory bowel diseases, which includes UC, is rising in the UK with peak diagnosis in late adolescence or early adulthood3.
Ruth Wakeman, Director of Services, Advocacy & Evidence, Crohn’s & Colitis UK, said: “Ulcerative Colitis can be an extremely debilitating condition, affecting many parts of the body and many aspects of life. It can affect people in very individual ways, so effective and appropriate treatment based on personalized care and shared decision making is really important. For some people with UC, existing treatments may not work, so additional treatment options are welcome.”
About filgotinib
Filgotinib is licensed and marketed as Jyseleca (200mg and 100mg tablets) in Great Britain, the European Union and Japan for the treatment of adults with moderate to severe active rheumatoid arthritis (RA) who have responded inadequately or are intolerant to one or more disease modifying anti-rheumatic drugs (DMARDs). Filgotinib may be used as monotherapy or in combination with methotrexate (MTX). Filgotinib is also licensed and marketed as Jyseleca in Great Britain and the European Union for the treatment of adult patients with moderately to severely active ulcerative colitis who have had an inadequate response with, lost response to, or were intolerant to either conventional therapy or a biologic agent. An application has been submitted to the Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) for the treatment of adults with moderately to severely active UC and is currently under review. The Great Britain Summary of Product Characteristics for filgotinib can be found at www.medicines.org.uk/emc and the Northern Ireland Summary of Product Characteristics for filgotinib can be found at www.emcmedicines.com/en-GB/northernireland. The European Summary of Product Characteristics for filgotinib, which includes contraindications and special warnings and precautions, is available at www.ema.europa.eu. The interview form from the Japanese Ministry of Health, Labour and Welfare is available at www.info.pmda.go.jp. A global Phase 3 program with filgotinib is ongoing in Crohn’s Disease. More information about clinical trials can be accessed at https://www.clinicaltrials.gov.
Jyseleca® is a trademark of Galapagos NV and Gilead Sciences, Inc. or its related companies.
About the filgotinib collaboration
Gilead and Galapagos NV are collaborative partners in the global development and commercialization of filgotinib. Galapagos is responsible for the commercialization of filgotinib in Europe, while Gilead remains responsible for filgotinib outside of Europe, including in Japan, where filgotinib is co-marketed with Eisai.
Jan. 18, 2022 4:29 PM ET
By: Jonathan Block, SA News Editor
Wednesday, January 12, 2022 - 06:45am
NEW YORK--(BUSINESS WIRE)-- Pfizer Inc. (NYSE:PFE) today announced positive top-line results from a Phase 3 study (B74710126) describing the safety and immunogenicity of PREVNAR 20™ (Pneumococcal 20-valent Conjugate Vaccine) in 570 adults in the United States 65 years of age or older when administered at the same time as the Pfizer-BioNTech COVID-19 Vaccine or when each vaccine was given with placebo.
Responses elicited by PREVNAR 20 for all 20 serotypes were similar whether given with a dose of the Pfizer-BioNTech COVID-19 Vaccine (n=190) or with placebo (n=191). Responses to a booster dose of the Pfizer-BioNTech COVID-19 Vaccine were also similar when given with PREVNAR 20 or given with placebo (n=189). The safety profile of co-administering PREVNAR 20 with a booster dose of the Pfizer-BioNTech COVID-19 Vaccine generally reflected that observed with the Pfizer-BioNTech COVID-19 Vaccine booster dose.
“Pfizer is steadfast in its commitment to address the burden of certain respiratory diseases while raising awareness of the importance of adult immunizations,” said Kathrin U. Jansen, Ph.D., Senior Vice President and Head of Vaccine Research & Development, Pfizer. “These new safety and immunogenicity data provide further evidence supporting the potential to administer PREVNAR 20 and the Pfizer-BioNTech COVID-19 Vaccine at the same time, thereby reducing the number of visits adults make to their doctor’s office or pharmacy for recommended immunization. As the COVID-19 vaccines and booster doses continue to be administered, we believe that healthcare providers have an opportunity to talk to their adult patients about other recommended vaccines in line with CDC guidance.”
The initiation of the study exploring the coadministration of PREVNAR 20 along with a booster dose of the Pfizer-BioNTech COVID-19 Vaccine in older adults was announced in May 2021. The study recruited adults from the pivotal Phase 3 Pfizer-BioNTech COVID-19 Vaccine clinical trial and included adults who received their second dose of the vaccine at least six months prior to entering the coadministration study. Pfizer will seek to present and publish detailed outcomes from this clinical trial at a future date. At this time no coadministration data are included in the PREVNAR 20 or Pfizer-BioNTech COVID-19 Vaccine prescribing information.
About PREVNAR 20
PREVNAR 20 is Pfizer’s next-generation pneumococcal conjugate vaccine that includes capsular polysaccharide conjugates for the 13 serotypes (1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F and 23F) already included in PREVNAR 13® (Pneumococcal 13-valent Conjugate Vaccine [Diphtheria CRM197 Protein]). The vaccine also contains capsular polysaccharide conjugates for seven additional serotypes (8, 10A, 11A, 12F, 15B, 22F and 33F) that cause invasive pneumococcal disease (IPD),1,2,3,4,5 and have been associated with high case-fatality rates,6,7,8,9 antibiotic resistance,10,11,12 and/or meningitis.13,14 PREVNAR 20 contains the broadest conjugate serotype coverage and helps protect against more strains of the bacteria that cause pneumococcal pneumonia than any other conjugate vaccine available.
On June 8, 2021, Pfizer announced the U.S. Food and Drug Administration (FDA) approved PREVNAR 20, which is the U.S. trade name, for the prevention of invasive disease and pneumonia in adults age 18 years or older. On December 16, 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) issued a positive opinion to recommend the granting of a marketing authorization for the 20-valent pneumococcal conjugate vaccine candidate for the prevention of invasive disease and pneumonia caused by 20 Streptococcus pneumoniae (pneumococcus) serotypes in adults ages 18 years and older in the 27 European Union (EU) member states plus Iceland, Lichtenstein and Norway. The EMA had previously accepted review of Pfizer’s Marketing Authorization Application (MAA) for the 20-valent pneumococcal conjugate vaccine candidate on February 26, 2021.
Pfizer has recently submitted to the FDA a supplemental Biologics License Application to include data in the PREVNAR 20 prescribing information for adults age 18 years or older regarding coadministration of PREVNAR 20 with a seasonal inactivated influenza vaccine.
Pivotal Phase 3 studies of the 20-valent pneumococcal conjugate vaccine candidate in infants are expected to read out in the second half of 2022 and, if positive, form the basis of potential regulatory submissions to the FDA and EMA later this year.
U.S. INDICATIONS FOR PREVNAR 20™
Please see full prescribing information for PREVNAR 20™.
COMIRNATY® U.S. Indication & Authorized Use
HOW IS THE VACCINE GIVEN?
The vaccine will be given to you as an injection into the muscle.
Primary Series:
In individuals 5 years of age and older, the vaccine is administered as a 2-dose series, 3 weeks apart.
In individuals 5 years of age and older, a third primary series dose may be administered at least 28 days after the second dose to individuals who are determined to have certain kinds of immunocompromise.
Booster Dose:
WHAT IS THE INDICATION AND AUTHORIZED USE?
The Pfizer-BioNTech COVID-19 Vaccine has received EUA from FDA to provide:
COMIRNATY® (COVID-19 Vaccine, mRNA) is an FDA-approved COVID-19 vaccine made by Pfizer for BioNTech.
Click for
Fact Sheets and Prescribing Information for individuals 12 years of age and older
Full Prescribing Information (16 years of age and older) DILUTE BEFORE USE, Purple Cap
Full Prescribing Information (16 years of age and older) DO NOT DILUTE, Gray Cap
EUA Fact Sheet for Vaccination Providers (12 years of age and older), Purple Cap
EUA Fact Sheet for Vaccination Providers (12 years of age and older), Gray Cap
Recipients and Caregivers Fact Sheet (12 years of age and older)
Fact Sheets for individuals 5 through 11 years of age
EUA Fact Sheet for Vaccination Providers (5 through 11 years of age), Orange Cap
Recipients and Caregivers Fact Sheet (5 through 11 years of age)
In addition, to learn more, please visit us on www.Pfizer.com
Jan. 12, 2022 7:01 AM ET Pfizer Inc. (PFE)By: Mamta Mayani, SA News Editor2 Comments
In addition, to learn more, please visit us on www.pfizer.com
Friday, January 14, 2022 - 03:30pm
CIBINQO is a once-daily oral treatment with proven efficacy to manage symptoms for adults who have not yet found relief with current options
NEW YORK--(BUSINESS WIRE)-- Pfizer Inc. (NYSE: PFE) announced today that the United States (U.S.) Food and Drug Administration (FDA) approved CIBINQO® (abrocitinib), an oral, once-daily, Janus kinase 1 (JAK1) inhibitor, for the treatment of adults living with refractory, moderate-to-severe atopic dermatitis (AD) whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies is inadvisable.
CIBINQO is approved at the recommended doses of 100 mg and 200 mg, with the 200 mg dose being recommended for patients who are not responding to the 100 mg dose. Additionally, a 50 mg dose was approved to treat moderate-to-severe AD specifically in patients with moderate renal impairment (kidney failure), certain patients receiving treatment with inhibitors of cytochrome P450 (CYP) 2C19, or patients who are known or suspected to be poor metabolizers of CYP2C19. For patients with moderate renal impairment who are not responding to 50 mg once daily, 100 mg once daily may also be prescribed.
“The reality for patients living with chronic inflammatory skin disease such as moderate-to-severe atopic dermatitis is that many experience debilitating symptoms that are not managed by current treatment options. Today’s approval of CIBINQO will provide an important new oral option that could help those who have yet to find relief,” said Jonathan Silverberg, MD, PhD, MPH, Department of Dermatology, The George Washington University School of Medicine and Health Sciences. “In multiple large-scale clinical trials, CIBINQO demonstrated strong efficacy at clearing skin, improving itch, and managing the extent and severity of eczema, offering a benefit-risk profile that supports the use of this treatment in the FDA-approved patient population.”
The FDA approval was based on results of five clinical trials from a large-scale clinical trial program of more than 1,600 patients. The safety and efficacy of CIBINQO was evaluated in three randomized, placebo-controlled, Phase 3 trials. Additionally, safety was evaluated through a randomized, placebo-controlled, dose-ranging trial and an ongoing long-term open-label extension trial. Across the trials, CIBINQO demonstrated a consistent safety profile and profound improvements in skin clearance, extent of disease, and severity, as well as rapid improvement in itch after two weeks, for some people living with AD versus placebo. In addition, a higher proportion of subjects treated with CIBINQO in two monotherapy trials achieved improvement in itching at week 12 compared to placebo.
“The FDA’s approval offers hope to the millions of patients across the U.S. who are suffering daily with an immuno-inflammatory condition that can cause intense and persistent itching, pain, discomfort, and distress if left uncontrolled,” said Mike Gladstone, Global President of Pfizer Inflammation & Immunology. “CIBINQO, an efficacious once-daily pill, is a medical breakthrough made possible by Pfizer researchers and the people living with moderate-to-severe atopic dermatitis who participated in our clinical trials.”
“Atopic dermatitis is so much more than just a rash, and it goes beyond the surface of the skin. It’s a chronic condition that can both significantly disrupt patients’ daily lives and negatively impact their emotional well-being,” said Julie Block, President and CEO, National Eczema Association. “We appreciate Pfizer’s commitment to this resilient patient community and eagerly await the positive impact CIBINQO could have on the treatment landscape for moderate-to-severe atopic dermatitis.”
The most common adverse events reported in ≥5% of patients with CIBINQO included nasopharyngitis (12.4% with CIBINQO 100 mg, 8.7% with CIBINQO 200 mg, and 7.9%, with placebo), nausea (6%, 14.5%, and 2.1%, respectively), and headache (6%, 7.8%, and 3.5%, respectively).
The full prescribing information for CIBINQO can be found here. CIBINQO will be made available in the coming weeks.
Indication
CIBINQO is indicated for the treatment of adults with refractory, moderate to severe atopic dermatitis whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies is inadvisable.
Limitations of Use: CIBINQO is not recommended for use in combination with other JAK inhibitors, biologic immunomodulators, or with other immunosuppressants.
About CIBINQO® (abrocitinib)
CIBINQO is an oral small molecule that selectively inhibits Janus kinase (JAK) 1. Inhibition of JAK1 is thought to modulate multiple cytokines involved in pathophysiology of AD, including interleukin IL-4, IL-13, IL-31, IL-22, and thymic stromal lymphopoietin (TSLP).
In addition to receiving regulatory approval in the U.S., CIBINQO has received marketing authorization in the European Union, Great Britain, Japan, Korea, the United Arab Emirates, Norway, Iceland, and Singapore.
Source: Pfizer Inc.
Jan. 14, 2022 4:14 PM ET Pfizer Inc. (PFE)ABBV
By: Dulan Lokuwithana, SA News Editor4 Comments
January 14, 2022
- Approval of two dose strengths (15 mg and 30 mg) supported by efficacy and safety data from one of the largest registrational Phase 3 programs in atopic dermatitis, with more than 2,500 patients evaluated across three studies[1]
- RINVOQ (upadacitinib) monotherapy or with topical corticosteroids met all primary and ranked secondary endpoints in atopic dermatitis pivotal studies[1-3]
- RINVOQ demonstrated significant improvement in itch (Worst Pruritus NRS ≥4) as early as week one, as well as significant improvements in skin clearance (EASI 75 and vIGA-AD 0/1) at 16 weeks, compared to placebo[1-3]
- RINVOQ also demonstrated significantly higher levels of skin clearance (EASI 90 and 100) at 16 weeks, compared to placebo[1-3]
NORTH CHICAGO, Ill., Jan. 14, 2022 /PRNewswire/ -- AbbVie (NYSE: ABBV) today announced the U.S. Food and Drug Administration (FDA) has approved RINVOQ® (upadacitinib) for the treatment of moderate to severe atopic dermatitis in adults and children 12 years of age and older whose disease did not respond to previous treatment and is not well controlled with other pills or injections, including biologic medicines, or when use of other pills or injections is not recommended.1 RINVOQ 15 mg once daily can be initiated in adults and children 12 years of age and older weighing at least 40 kg.1 In these children and adults less than 65 years of age who do not achieve an adequate response, the dose may be increased to 30 mg once daily.1
"Early in my career as an allergist, I saw how relentless the itch and rash could be for my patients with moderate to severe atopic dermatitis yet had limited options to offer those whose disease could not be adequately controlled with systemic therapy," said Thomas Hudson, M.D., senior vice president, research and development, chief scientific officer, AbbVie. "This additional approval for RINVOQ provides a once-daily oral option that can significantly improve the debilitating itch and skin symptoms of atopic dermatitis. It's also a proud moment for AbbVie as we continue our efforts to improve care in this disease state and other chronic, immune-mediated conditions."
The FDA approval is supported by efficacy and safety data from one of the largest registrational Phase 3 programs for atopic dermatitis with more than 2,500 patients evaluated across three studies. Approximately 52 percent of the patients had prior exposure to systemic atopic dermatitis treatment. These studies evaluated the efficacy and safety of RINVOQ monotherapy (Measure Up 1 and 2) and with topical corticosteroids (AD Up), compared to placebo, in adults and children 12 years of age and older with moderate to severe atopic dermatitis.2-3
"Despite available therapies, many people with moderate to severe atopic dermatitis are caught in an endless cycle of itching and scratching," said Emma Guttman-Yassky, M.D., Ph.D., Waldman Professor and System Chair of Dermatology at the Icahn School of Medicine at Mount Sinai in New York City.* "In clinical trials, upadacitinib showed a robust response across skin and itch symptoms that may help evolve treatment goals for those who have not achieved adequate control of their disease. And as an oral pill with two dose strengths, upadacitinib is a welcome addition to the toolbox of clinicians who are striving to make a significant difference for their patients with moderate to severe atopic dermatitis."
About RINVOQ® (upadacitinib)
Discovered and developed by AbbVie scientists, RINVOQ is a selective JAK inhibitor that is being studied in several immune-mediated inflammatory diseases.10 Based on enzymatic and cellular assays, RINVOQ demonstrated greater inhibitory potency for JAK-1 vs JAK-2, JAK-3, and TYK-2.1 The relevance of inhibition of specific JAK enzymes to therapeutic effectiveness and safety is not currently known.
In the U.S., RINVOQ 15 mg and 30 mg is approved for use in adults and pediatric patients 12 years of age and older with refractory, moderate to severe atopic dermatitis whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies are inadvisable.1 RINVOQ 15 mg is also approved in the U.S. for adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more TNF blockers as well as adults with active psoriatic arthritis who have had an inadequate response or intolerance to one or more TNF blockers. In the EU, RINVOQ 15 mg is approved for the treatment of adults with moderate to severe active rheumatoid arthritis, adults with active psoriatic arthritis and adults with active ankylosing spondylitis. RINVOQ is also approved in the EU for adults (15 mg and 30 mg) and adolescents (15 mg) with moderate to severe atopic dermatitis.
Phase 3 trials of RINVOQ in rheumatoid arthritis, atopic dermatitis, psoriatic arthritis, axial spondyloarthritis, Crohn's disease, ulcerative colitis, giant cell arteritis and Takayasu arteritis are ongoing. 11-18
RINVOQ® (upadacitinib) U.S. Use and Important Safety Information1
RINVOQ is a prescription medicine used to treat:
It is not known if RINVOQ is safe and effective in children under 18 years of age with juvenile idiopathic arthritis or psoriatic arthritis.
RINVOQ is safe and effective in children 12 years of age and older weighing at least 88 pounds (40 kg) with atopic dermatitis.
It is not known if RINVOQ is safe and effective in children under 12 years of age with atopic dermatitis.
SOURCE AbbVie
Jan. 14, 2022 2:40 PM ET AbbVie Inc. (ABBV)
By: Dulan Lokuwithana, SA News Editor6 Comments
AbbVie (ABBV +1.3%) pared early losses to rise in afternoon hours on the announcement of FDA approval for the company’s JAK inhibitor RINVOQ (upadacitinib) for moderate to severe atopic dermatitis (AD).
Jan. 13, 2022 3:09 PM ETEli Lilly and Company (LLY)
TORONTO, Jan. 13, 2022 /CNW/ - Lilly Canada is pleased to announce that as of January 1st, 2022, Baqsimi (glucagon nasal powder) is now covered by all Federal health benefit programs, which includes the Non-Insured Health Benefits (NIHB) Program, Veterans Affairs Canada (VAC), and Correctional Services of Canada (CSC).
Baqsimi is indicated for the treatment of severe hypoglycemic reactions in people with insulin-treated diabetes mellitus 4 years old and above, when impaired consciousness precludes oral carbohydrates.
"Diabetes affects one in three Canadians, and we know that many factors contribute to the successful management of the disease, including access to new treatments, supplies, and technology," says Rhonda Pacheco, General Manager, Lilly Canada. "We're so thrilled that at-need Canadians who can benefit from this important rescue treatment are able to access it, providing patients and their caregivers with protection, if they experience a severe blood sugar low."
"Expanding Baqsimi's access to Canadian patients who are at risk from complications of diabetes is so critical," says Dr. Stuart Ross, Endocrinologist, Clinical Professor of Medicines at the University of Calgary. "The threat of a severe hypoglycemic event is present every day for someone with diabetes, and for those patient populations that don't have access to emergency services and immediate assistance, such as remote First Nation's communities, this rescue treatment dramatically improves safety and care."
With the Federal benefit programs now covering Baqsimi for eligible patients, Lilly is excited to be working with the provinces to make Baqsimi available in those jurisdictions.
About Baqsimi
Baqsimi is a portable, ready-to-use form of glucagon which requires no reconstitution or priming. It is administered in a single fixed dose of 3 mg which is sprayed in the nose, where it is absorbed. Baqsimi does not need to be refrigerated and can be stored at temperatures up to 30°C/86°F in its shrink-wrapped tube. Baqsimi is indicated for the treatment of severe hypoglycemic reactions in people with insulin-treated diabetes mellitus 4 years old and above when impaired consciousness precludes oral carbohydrates. The formulation was discovered in Canada.
To learn more about Lilly Canada, please visit us at www.lilly.ca.
Jan. 13, 2022 3:28 PM ET Eli Lilly and Company (LLY)
By: Ravikash, SA News Editor
https://seekingalpha.com/symbol/LLY
PUBLISHED13 January 2022
WILMINGTON, Del., January 13, 2022 – AstraZeneca and Amgen today announced TEZSPIRE™ (tezepelumab-ekko) is now available for shipment to wholesalers in the US. TEZSPIRE was approved by the US Food and Drug Administration (FDA) on December 17, 2021 for the add-on maintenance treatment of adult and pediatric patients aged 12 years and older with severe asthma.1
Mina Makar, Senior Vice President, US Respiratory & Immunology, AstraZeneca, said: “Due to the complexity of severe asthma, many patients living with the disease continue to experience frequent exacerbations, an increased risk of hospitalization and a reduced quality of life. With TEZSPIRE now available, this important new treatment has the potential to transform care for a broad population of patients living with severe asthma.”
TEZSPIRE is a first-in-class biologic for severe asthma that acts at the top of the inflammatory cascade by targeting thymic stromal lymphopoietin (TSLP), an epithelial cytokine.2 TEZSPIRE is the first and only biologic approved for severe asthma with no phenotype (e.g. eosinophilic or allergic) or biomarker limitation within its approved label.1,3-8 TEZSPIRE consistently and significantly reduced asthma exacerbations across Phase II and III clinical trials which included a broad population of severe asthma patients irrespective of key biomarkers, including blood eosinophil counts, allergic status and fractional exhaled nitric oxide (FeNO).2,9
In clinical studies, the most common adverse reactions in patients who received TEZSPIRE were pharyngitis, arthralgia and back pain.1
AstraZeneca and Amgen are committed to providing appropriate patients who are prescribed TEZSPIRE with affordable access to the medicine. The Tezspire Together Program offers provider and patient product resources and support, including information related to coverage, reimbursement and distribution. For more information about the Tezspire Together Program, call 1-888-TZSPIRE (1-888-897-7473) or visit Tezspire.com.
AstraZeneca and Amgen also provide patient assistance for TEZSPIRE for qualifying individuals with no or limited drug coverage by providing free medicines through the Tezspire Patient Assistance Program. For additional information, patients and caregivers may contact Tezspire Together.
TEZSPIRE™ (tezepelumab-ekko) US Indication
TEZSPIRE is indicated for the add-on maintenance treatment of adult and pediatric patients aged 12 years and older with severe asthma.
TEZSPIRE is not indicated for the relief of acute bronchospasm or status asthmaticus.
TEZSPIRE™ (tezepelumab-ekko) Important Safety Information
CONTRAINDICATIONS
Known hypersensitivity to tezepelumab-ekko or excipients.
Please see the TEZSPIRE full Prescribing Information.
Clinical trials
In addition to the Phase IIb PATHWAY trial, the PATHFINDER program included two Phase III trials, NAVIGATOR9,21 and SOURCE.22,23 The program also includes additional mechanistic and long-term safety trials.24,25
NAVIGATOR is a Phase III, randomized, double-blinded, placebo-controlled trial in adults (18–80 years old) and adolescents (12–17 years old) with severe, uncontrolled asthma, who were receiving standard of care (SoC). SoC was treatment with medium- or high-dose inhaled corticosteroids plus at least one additional controller medication with or without daily OCS treatment. The trial population included approximately equal proportions of patients with high (≥300 cells per microliter) and low (<300 cells per microliter) blood eosinophil counts. The trial comprised a five-to-six-week screening period, a 52-week treatment period and a 12-week post-treatment follow-up period. All patients received their prescribed controller medications without change throughout the trial.9
The primary efficacy endpoint was the annualized asthma exacerbation rate (AAER) during the 52-week treatment period. Key secondary endpoints included the effect of TEZSPIRE on lung function, asthma control and health-related quality of life.9
As part of prespecified analyses, the AAER over 52 weeks was also assessed in patients grouped by baseline blood eosinophil count, FeNO level and serum specific immunoglobin E (IgE) status (perennial aeroallergen sensitivity positive or negative).9 These are inflammatory biomarkers used by clinicians to inform treatment options and involve tests analyzing a patient’s blood (eosinophils/IgE) and exhaled air (FeNO).
There were no clinically meaningful differences in safety results between the TEZSPIRE and placebo groups in the NAVIGATOR trial.9 The most frequently reported adverse events for TEZSPIRE were nasopharyngitis, upper respiratory tract infection and headache.9
NAVIGATOR is the first Phase III trial to show benefit in severe asthma irrespective of eosinophils by targeting TSLP.9 These results support the FDA Breakthrough Therapy Designation granted to TEZSPIRE in September 2018 for patients with severe asthma, without an eosinophilic phenotype. In July 2021, TEZSPIRE was the first and only biologic to be granted Priority Review in the US for the treatment of asthma by the FDA.
Patients who participated in our Phase III trials were eligible to continue in DESTINATION, a Phase III extension trial assessing long-term safety and efficacy.24
TEZSPIRE
TEZSPIRE™ (tezepelumab) is being developed by AstraZeneca in collaboration with Amgen as a first-in-class human monoclonal antibody that inhibits the action of TSLP, a key epithelial cytokine that sits at the top of multiple inflammatory cascades and is critical in the initiation and persistence of allergic, eosinophilic and other types of airway inflammation associated with severe asthma, including airway hyperresponsiveness.2,26 TSLP is released in response to multiple triggers associated with asthma exacerbations, including allergens, viruses and other airborne particles.2,26 Expression of TSLP is increased in the airways of patients with asthma and has been correlated with disease severity.2,27 Blocking TSLP may prevent the release of pro-inflammatory cytokines by immune cells, resulting in the prevention of asthma exacerbations and improved asthma control.1,2,9,27 TEZSPIRE acts at the top of the inflammation cascade and has the potential to help address a broad population of severe asthma patients irrespective of biomarker levels.2,9
TEZSPIRE is also in development for other potential indications including chronic obstructive pulmonary disease, chronic rhinosinusitis with nasal polyps, chronic spontaneous urticaria and eosinophilic esophagitis (EoE). In October 2021, tezepelumab was granted Orphan Drug Designation by the FDA for the treatment of EoE.
Amgen collaboration
In 2020, AstraZeneca and Amgen updated a 2012 collaboration agreement for TEZSPIRE. Both companies will continue to share costs and profits equally after payment by AstraZeneca of a mid single-digit inventor royalty to Amgen. AstraZeneca continues to lead development and Amgen continues to lead manufacturing. All aspects of the collaboration are under the oversight of joint governing bodies. Under the amended agreement, AstraZeneca and Amgen will jointly commercialize TEZSPIRE in North America. Amgen will record product sales in the US, with AZ recording its share of US profits as Collaboration Revenue. Outside of the US, AstraZeneca will record product sales, with Amgen recording profit share as Other/Collaboration revenue.
For more information, please visit www.astrazeneca-us.com
Jan. 13, 2022 8:19 AM ET Amgen Inc. (AMGN), AZN
By: Mamta Mayani, SA News Editor
PUBLISHED13 January 2022
Positive results from a preliminary analysis of an ongoing safety and immunogenicity trial (D7220C00001) showed that Vaxzevria (ChAdOx1-S [Recombinant]), when given as a third dose booster, increased the immune response to Beta, Delta, Alpha and Gamma SARS-CoV-2 variants, while a separate analysis of samples from the trial showed increased antibody response to the Omicron variant.
The results were observed among individuals previously vaccinated with either Vaxzevria or an mRNA vaccine.
A separate Phase IV trial reported in a preprint with The Lancet on SSRN showed that a third dose of Vaxzevria substantially increased antibody levels following a primary vaccine series with CoronaVac (Sinovac Biotech).1
These data add to the growing body of evidence supporting Vaxzevria as a third dose booster irrespective of the primary vaccination schedules tested.2,3 The Company is submitting these additional data to health authorities around the world given the urgent need for third dose boosters.
Sir Mene Pangalos, Executive Vice President, BioPharmaceuticals R&D, AstraZeneca, said: “Vaxzevria has protected hundreds of millions of people from COVID-19 around the world and these data show that it has an important role to play as a third dose booster, including when used after other vaccines. Given the ongoing urgency of the pandemic and Vaxzevria's increased immune response to the Omicron variant, we will continue to progress regulatory submissions around the world for its use as a third dose booster.”
Professor Sir Andrew J Pollard, chief investigator and director of the Oxford Vaccine Group at the University of Oxford, said: “These important studies show that a third dose of Vaxzevria after two initial doses of the same vaccine, or after mRNA or inactivated vaccines, strongly boosts immunity against COVID-19. The Oxford-AstraZeneca vaccine is suitable as an option to enhance immunity in the population for countries considering booster programmes, adding to the protection already demonstrated with the first two doses.”
The D7220C00001 safety and immunogenicity trial showed that Vaxzevria continued to be generally well tolerated. Further analyses from the trial are expected in the first half of 2022.
Previous studies support Vaxzevria as a third dose booster as part of a homologous or heterologous schedule.2,3 In a sub-analysis from the COV001 and COV002 trials, a third dose of Vaxzevria given at least six months after a second dose significantly boosted antibody levels and maintained T cell response.2 It also resulted in higher neutralising activity against the Alpha, Beta, and Delta variants, compared with a two-dose regimen.2 The COV-BOOST trial also showed that a third dose booster of Vaxzevria induced significantly higher immune responses compared with controls against the Delta variant and original strain following a primary vaccine series of Vaxzevria or Pfizer BioNtech (BNT162b2).3
Notes
D7220C00001 Trial
D7220C00001 is an ongoing partially double-blind, randomised, multinational, active-controlled trial in both previously vaccinated and unvaccinated adults to determine the safety and immunogenicity of Vaxzevria and AZD2816, a vaccine developed for the prevention of COVID-19 caused by the Beta variant of the SARS-CoV-2 virus.4
Immunogenicity data for the preliminary analysis were from samples taken 28 days after the third dose booster vaccination. Individuals included in the preliminary analysis of the trial had been fully vaccinated with two doses of Vaxzevria (n=700), or an mRNA-based vaccine (n=600), prior to being given a third dose booster vaccination of Vaxzevria or AZD2816 at least three months after their last injection.
RHH-001 Phase IV Trial
The Phase IV randomized, single-blind, two-centre trial assessed the safety and immunogenicity of a third heterologous booster dose of either Vaxzevria, an mRNA vaccine (BNT162b2, Pfizer/BioNTech), or a recombinant adenoviral vectored vaccine (AD26.COV2-S, Janssen), compared with a third homologous booster dose of CoronaVac in Brazilian adults who had received two doses of CoronaVac six months previously. Between 16 August 2021 and 1 September 2021, 1,240 participants were randomised to receive a third dose booster in São Paulo and Salvador, of whom 1,239 were vaccinated.1
The primary outcome of the trial was non-inferiority of anti-spike IgG antibodies 28 days after the booster dose in the heterologous boost groups compared with homologous regimen.1
Vaxzevria, (ChAdOx1-S [Recombinant], formerly AZD1222)
Vaxzevria was invented by the University of Oxford. It uses a replication-deficient chimpanzee viral vector based on a weakened version of a common cold virus (adenovirus) that causes infections in chimpanzees and contains the genetic material of the SARS-CoV-2 virus spike protein. After vaccination, the surface spike protein is produced, priming the immune system to attack the SARS-CoV-2 virus if it later infects the body.
The vaccine has been granted a conditional marketing authorisation or emergency use in more than 90 countries. It also has Emergency Use Listing from the World Health Organization, which accelerates the pathway to access in up to 144 countries through the COVAX Facility.
Under a sub-license agreement with AstraZeneca, the vaccine is manufactured and supplied by the Serum Institute of India under the name COVISHIELD.
Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Jan. 13, 2022 6:48 AM ETAstraZeneca PLC (AZN)By: Mamta Mayani, SA News Editor2 Comments

BackPDF VersionJan 11, 2022
- More than 1,500 children now eligible to receive a treatment targeting the underlying cause of cystic fibrosis for the first time -
LONDON--(BUSINESS WIRE)--Jan. 11, 2022-- Vertex Pharmaceuticals Incorporated (Nasdaq: VRTX) today announced that the European Commission has granted approval for the label extension of KAFTRIO® (ivacaftor/tezacaftor/elexacaftor) in a combination regimen with ivacaftor for the treatment of cystic fibrosis (CF) in patients ages 6 through 11 years old who have at least one F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene.
“We are delighted that KAFTRIO (ivacaftor/tezacaftor/elexacaftor) in a combination regimen with ivacaftor is now approved for these young patients in the European Union. It provides a new treatment option for physicians to help treat the underlying cause of this devastating disease early in life,” said Reshma Kewalramani, M.D., Chief Executive Officer and President at Vertex. “This important milestone brings us one step closer to our ultimate goal of developing treatments for all patients living with CF.”
“Ivacaftor/tezacaftor/elexacaftor plus ivacaftor has shown clinical benefit since its availability last year for people with CF ages 12 and above,” said Professor Marcus A. Mall, M.D., Head of the Department of Pediatric Respiratory Medicine, Immunology and Critical Care Medicine at Charité University Medical Center Berlin. “CF is a progressive disease, in which symptoms and organ damage manifest very early in life. As a physician, I welcome the approval of this medicine for this younger age group, as it will help us treat eligible children with CF as early as 6 years old.”
As a result of long-term reimbursement agreements in Austria, Northern Ireland and Denmark, and provisions for access in health care systems such as Germany, eligible patients in these countries will have access to the expanded indication for KAFTRIO® (ivacaftor/tezacaftor/elexacaftor) in a combination regimen with ivacaftor shortly following regulatory approval by the European Commission. Vertex will continue to work with reimbursement bodies across the European Union to ensure access for all eligible patients.
This medicine has also been approved by regulatory authorities in New Zealand and in Switzerland, where it is known as TRIKAFTA® (elexacaftor/tezacaftor/ivacaftor and ivacaftor), for people with CF ages 6 and above, and we continue to work closely with reimbursement bodies in these countries to ensure access for all eligible patients.
About KAFTRIO® (ivacaftor/tezacaftor/elexacaftor) in A Combination Regimen With Ivacaftor
In people with certain types of mutations in the CFTR gene, the CFTR protein is not processed or folded normally within the cell, and this can prevent the CFTR protein from reaching the cell surface and functioning properly. KAFTRIO® (ivacaftor/tezacaftor/elexacaftor) in combination with ivacaftor is an oral medicine designed to increase the quantity and function of the CFTR protein at the cell surface. Elexacaftor and tezacaftor work together to increase the amount of mature protein at the cell surface by binding to different sites on the CFTR protein. Ivacaftor, which is known as a CFTR potentiator, is designed to facilitate the ability of CFTR proteins to transport salt and water across the cell membrane. The combined actions of ivacaftor, tezacaftor and elexacaftor help hydrate and clear mucus from the airways.
For complete product information, please see the Summary of Product Characteristics that can be found on www.ema.europa.eu.
For company updates and to learn more about Vertex's history of innovation, visit https://global.vrtx.com/ or follow us on Twitter and LinkedIn.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220110005889/en/
Source: Vertex Pharmaceuticals Incorporated
Jan. 11, 2022 5:14 AM ET Vertex Pharmaceuticals Incorporated (VRTX) By: Mamta Mayani, SA News Editor

SINGLE DOSE OF MESOBLAST’S ALLOGENEIC CELL THERAPY PROVIDES DURABLE PAIN REDUCTION FOR AT LEAST THREE YEARS IN PATIENTS WITH DEGENERATIVE DISC DISEASE 36-Month Results of Phase 3 Trial in Chronic Low Back Pain Presented at 2022 Biotech Showcase Melbourne, Australia; January 12, and New York, USA; January 11, 2022: Mesoblast Limited (ASX:MSB; Nasdaq:MESO), global leader in allogeneic cellular medicines for inflammatory diseases, today announced 36-month follow-up results from the 404-patient Phase 3 trial of its allogeneic cell therapy rexlemestrocel-L (MPC-06-ID) in patients with chronic low back pain (CLBP) associated with degenerative disc disease (DDD). Mesoblast Chief Executive Dr Silviu Itescu presented results from the three-arm trial at the 2022 Biotech Showcase event being held this week, which showed durable reduction in back pain lasting at least three years from a single intra-discal injection of rexlemestrocelL+hyaluronic acid (HA) carrier. There is a significant need for a safe, effective, and durable opioid-sparing treatment in patients with CLBP associated with degenerative disc disease. Results presented from this trial showed that: • Durable reduction in pain through 36 months was greatest in the pre-specified population with CLBP of shorter duration than the study median of 68 months (n=194), suggesting that greatest benefits may be seen when the therapy is administered earlier in the disease process when there is active inflammation and before irreversible fibrosis of the intervertebral disc has occurred • Pain reduction through 36 months was also seen in the subset of patients using opioids at baseline (n=168) with the rexlemestrocel-L+HA group having substantially greater reduction at all time points compared with saline controls • Among patients on opioids at baseline, despite instructions to maintain existing therapies throughout the trial, at 36 months 28% who received rexlemestrocel-L + HA were not taking an opioid compared with 8% of saline treated controls (nominal p value 0.0075). Mesoblast recently received feedback from the US Food & Drug Administration’s (FDA) Office of Tissues and Advanced Therapies (OTAT) on the Phase 3 program for CLBP and plans to conduct an additional US Phase 3 trial which may support submissions for potential approval in both the US and EU. Following review of the completed Phase 3 trial data, OTAT agreed with Mesoblast’s proposal for pain reduction at 12 months as the primary endpoint of the next trial, with functional improvement and reduction in opioid use as secondary endpoints. The Biotech Showcase presentation materials have been lodged with the ASX
For more information, please see www.mesoblast.com
Jan. 12, 2022 2:07 AM ET
By: Mamta Mayani, SA News Editor1 Comment

01/11/22 at 7:12 AM ESTPDF Version
– 600,000 additional doses to be supplied to the US government for distribution in Q1 2022, enabling further access to sotrovimab nationwide –
– Brings total number of doses secured to date through binding agreements to approximately 1.7 million globally –
– Preclinical data generated through both pseudo-virus and live virus testing demonstrate sotrovimab retains activity against all tested SARS-CoV-2 variants of concern including Delta and Omicron –
LONDON and SAN FRANCISCO, Jan. 11, 2022 (GLOBE NEWSWIRE) -- GlaxoSmithKline plc (LSE/NYSE: GSK) and Vir Biotechnology, Inc. (Nasdaq: VIR) today announced that the US government will purchase an additional 600,000 doses of sotrovimab, an investigational monoclonal antibody for the early treatment of COVID-19, enabling further nationwide access to sotrovimab for patients. The additional 600,000 doses will be delivered throughout the first quarter of 2022. This agreement is an amendment to earlier commitments announced with the US government in November 2021.
Including the commitments announced today, GSK and Vir have received binding agreements for the sale of approximately 1.7 million doses of sotrovimab worldwide. In addition, today’s agreement also includes the option for the US government to purchase further additional doses in the second quarter of 2022.
Sotrovimab, which was granted Emergency Use Authorization (EUA) by the US Food and Drug Administration (FDA) in May 2021, is an investigational single-dose intravenous (IV) infusion SARS-CoV-2 monoclonal antibody. Under the EUA, sotrovimab can be used for the treatment of mild-to-moderate COVID-19 in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death.
GSK and Vir expect to manufacture approximately 2 million doses globally in the first half of 2022 and additional doses in the second half of the year.
GSK and Vir are committed to the ongoing evaluation of sotrovimab as the COVID-19 landscape continues to evolve at different rates across the globe and new variants of concern and interest emerge. Preclinical pseudovirus data, published in bioRxiv, demonstrate that sotrovimab retains activity against all tested variants of concern and interest of the SARS-CoV-2 virus as defined by the World Health Organization, including, but not limited to, Omicron (B.1.1.529), Delta (B.1.617.2), Delta Plus (AY.1 or AY.2) and Mu (B.1.621). Preclinical live virus testing has also been completed with data, recently published in bioRxiv, further demonstrating that sotrovimab retains activity against the Omicron variant.
About Sotrovimab
Sotrovimab is an investigational SARS-CoV-2 neutralizing monoclonal antibody. The antibody binds to an epitope on SARS-CoV-2 shared with SARS-CoV-1 (the virus that causes SARS), indicating that the epitope is highly conserved, which may make it more difficult for resistance to develop. Sotrovimab, which incorporates Xencor, Inc.’s Xtend™ technology, has also been designed to achieve high concentration in the lungs to ensure optimal penetration into airway tissues affected by SARS-CoV-2 and to have an extended half-life.
About Global Access to Sotrovimab
Sotrovimab is authorized for emergency use in the US and has been granted a marketing authorization in the EU, conditional marketing authorization in Great Britain, provisional marketing authorization in Australia, and conditional marketing authorization in Saudi Arabia. It has also been approved via Japan’s Special Approval for Emergency Pathway. Temporary authorizations for sotrovimab have also been granted in 12 other countries.
Sotrovimab is supplied in several countries worldwide, including through national agreements in the US, UK, Japan, Australia, Canada, Singapore, Switzerland, and the United Arab Emirates. The companies are also supplying sotrovimab to participating Member States of the EU through a Joint Procurement Agreement with the European Commission. Additional agreements are yet to be disclosed due to confidentiality or regulatory requirements.
Sotrovimab in the United States
The following is a summary of information for sotrovimab. Healthcare providers in the US should review the Fact Sheets for information about the authorized use of sotrovimab and mandatory requirements of the EUA. Please see the Food and Drug Administration (FDA) Letter of Authorization, full Fact Sheet for Healthcare Providers and full Fact Sheet for Patients, Parents, and Caregivers.
Sotrovimab has been authorized by the US FDA for the emergency use described below. Sotrovimab is not FDA-approved for this use.
Sotrovimab is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of sotrovimab under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.
Authorized Use
The US FDA has issued an EUA to permit the emergency use of the unapproved product sotrovimab for the treatment of mild-to-moderate coronavirus disease 2019 (COVID-19) in adults and pediatric patients (12 years of age and older weighing at least 40kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death.
Limitations of Authorized Use
Sotrovimab is not authorized for use in patients:
Benefit of treatment with sotrovimab has not been observed in patients hospitalized due to COVID-19. SARS-CoV-2 monoclonal antibodies may be associated with worse clinical outcomes when administered to hospitalized patients with COVID-19 requiring high flow oxygen or mechanical ventilation.
Important Safety Information
CONTRAINDICATIONS
Sotrovimab is contraindicated in patients who have a history of anaphylaxis to sotrovimab or to any of the excipients in the formulation.
For further information please visit www.gsk.com/aboutus. About the GSK and Vir Collaboration
In April 2020, GSK and Vir entered into a collaboration to research and develop solutions for coronaviruses, including SARS-CoV-2, the virus that causes COVID-19. The collaboration uses Vir’s proprietary monoclonal antibody platform technology to accelerate existing and identify new anti-viral antibodies that could be used as therapeutic or preventive options to help address the current COVID-19 pandemic and future outbreaks. The companies will leverage GSK’s expertise in functional genomics and combine their capabilities in CRISPR screening and artificial intelligence to identify anti-coronavirus compounds that target cellular host genes. They will also apply their combined expertise to research SARS-CoV-2 and other coronavirus vaccines.
For more information, please visit www.vir.bio.
Jan. 11, 2022 8:47 AM ET Vir Biotechnology, Inc. (VIR), GSK By: Jonathan Block, SA News Editor2 Comments
First Targeted Therapy for Patients With the KRAS G12C Mutation Approved in the European Union
Approval Based on Pivotal CodeBreaK 100 Data Demonstrating Durable Responses and a Favorable Benefit-Risk Profile With LUMYKRAS
LUMYKRAS Now Approved in 35 Countries Around the World Through Most Advanced KRAS G12C Clinical Development Program
THOUSAND OAKS, Calif., Jan. 9, 2022 /PRNewswire/ -- Amgen (NASDAQ: AMGN) today announced that the European Commission (EC) has granted conditional marketing authorization for LUMYKRAS® (sotorasib), a first-in-class KRASG12C inhibitor, for the treatment of adults with advanced non-small cell lung cancer (NSCLC) with KRAS G12C mutation and who have progressed after at least one prior line of systemic therapy. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
"The approval of LUMYKRAS, the first and only targeted therapy for KRAS G12C-mutated NSCLC with proven efficacy, has the potential to transform treatment outcomes for people in the European Union living with this notoriously difficult-to-treat cancer," said David M. Reese, M.D., executive vice president of Research and Development at Amgen. "Amgen's landmark scientific discovery allowed investigators to advance the first KRASG12C inhibitor into the clinic, and we look forward to bringing this critical innovation to more patients across the globe."
The EC decision follows the recommendation for approval by the Committee for Medicinal Products for Human Use (CHMP) and is based on the positive results from the Phase 2 CodeBreaK 100 clinical trial in NSCLC, the largest trial conducted to date for patients with the KRAS G12C mutation. LUMYKRAS 960 mg, administered orally once-daily, demonstrated an objective response rate of 37.1% (95% CI: 28.6-46.2) and a median duration of response (DoR) of 11.1 months. The most common adverse reactions were diarrhea (34%), nausea (25%), and fatigue (21%). The most common severe (grade ≥ 3) adverse reactions were increased alanine aminotransferase level (ALT; 5%), increased aspartate aminotransferase (AST; 4%), and diarrhea (4%).
NSCLC accounts for approximately 84% of the 2.2 million new lung cancer diagnoses globally each year, including approximately 400,000 new cases in Europe 1,2 KRAS G12C is one of the most prevalent driver mutations in NSCLC, with about 13-15% of European patients with non-squamous NSCLC having the KRAS G12C mutation.3,4 With EC approval, and subject to local reimbursement applications, clinicians in all European Union member countries, as well as Norway, Iceland, and Liechtenstein, will be able to offer LUMYKRAS to appropriate patients with NSCLC.
About LUMAKRAS®/LUMYKRAS® (sotorasib)
Amgen took on one of the toughest challenges of the last 40 years in cancer research by developing LUMAKRAS/LUMYKRAS, a KRASG12C inhibitor.5 LUMAKRAS/LUMYKRAS has demonstrated a positive benefit-risk profile with rapid, deep and durable anticancer activity in patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) harboring the KRAS G12C mutation with a once daily oral formulation.6
Amgen is progressing the largest and broadest global KRASG12C inhibitor development program with unparalleled speed and exploring more than 10 sotorasib combination regimens, including triplets, with clinical trial sites spanning five continents. To date, over 4,000 patients around the world have received LUMAKRAS/LUMYKRAS through the clinical development program and commercial use.
In May 2021, LUMAKRAS was the first KRASG12C inhibitor to receive regulatory approval anywhere in the world with its approval in the U.S., under accelerated approval.
Regulatory approvals have also been received in the United Arab Emirates (LUMAKRAS), Switzerland (LUMYKRAS), and under the FDA's Project Orbis in Canada (LUMAKRAS) and Great Britain (LUMYKRAS). Through Project Orbis, Amgen also has Marketing Authorization Applications (MAAs) for sotorasib in review in Australia, Brazil, Singapore and Israel. Additionally, Amgen has submitted MAAs in Japan, South Korea, Turkey, Taiwan, Colombia, Thailand, Mexico, Hong Kong, Saudi Arabia, Argentina, Kuwait and Qatar.
LUMAKRAS/LUMYKRAS is also being studied in multiple other solid tumors.7
About CodeBreaK
The CodeBreaK clinical development program for Amgen's drug sotorasib is designed to study patients with an advanced solid tumor with the KRAS G12C mutation and address the longstanding unmet medical need for these cancers.
CodeBreaK 100, the Phase 1 and 2, first-in-human, open-label multicenter study, enrolled patients with KRAS G12C-mutant solid tumors.11 Eligible patients must have received a prior line of systemic anticancer therapy, consistent with their tumor type and stage of disease. The primary endpoint for the Phase 2 study was centrally assessed objective response rate. The Phase 2 trial in NSCLC enrolled 126 patients, 124 of whom had centrally evaluable lesions by RECIST at baseline.7 The Phase 2 trial in colorectal cancer (CRC) is fully enrolled and results have been submitted for publication.12
CodeBreaK 200, the global Phase 3 randomized active-controlled study comparing sotorasib to docetaxel in KRAS G12C-mutated NSCLC completed enrollment of 345 patients. Eligible patients had previously treated, locally-advanced and unresectable or metastatic KRAS G12C-mutated NSCLC. The primary endpoint is progression-free survival and key secondary endpoints include overall survival, objective response rate, and patient-reported outcomes.
Amgen also has several Phase 1b studies investigating sotorasib monotherapy and sotorasib combination therapy across various advanced solid tumors (CodeBreaK 101) open for enrollment. A Phase 2 randomized study will evaluate sotorasib in patients with stage IV KRAS G12C-mutated NSCLC in need of first-line treatment (CodeBreaK 201).
For information, please visit www.hcp.codebreaktrials.com.
Important EU/EEA Product Information
LUMYKRAS® (sotorasib) as monotherapy is indicated for the treatment of adults with advanced non-small cell lung cancer (NSCLC) with KRAS G12C mutation and who have progressed after at least one prior line of systemic therapy.
LUMAKRAS® (sotorasib) U.S. Indication
LUMAKRAS is indicated for the treatment of adult patients with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.
This indication is approved under accelerated approval based on overall response rate (ORR) and duration of response (DOR). Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
Please see LUMAKRAS full Prescribing Information.
For more information, visit www.amgen.com and follow us on www.twitter.com/amgen.
View original content to download multimedia:https://www.prnewswire.com/news-releases/european-commission-approves-lumykras-sotorasib-for-patients-with-kras-g12c-mutated-advanced-non-small-cell-lung-cancer-301456869.html
SOURCE Amgen
Jan. 10, 2022 2:19 AM ET Amgen Inc. (AMGN) By: Mamta Mayani, SA News Editor
Jan 10, 2022
Basel, January 10, 2022 — Novartis and Molecular Partners today announced that Part A of the EMPATHY clinical trial1 that compared single intravenous doses of ensovibep, a DARPin antiviral therapeutic candidate vs. placebo to treat COVID-19, met the primary endpoint of viral load reduction over eight days. The two secondary endpoints also showed clinically meaningful benefit over placebo – (1) composite endpoint of hospitalization and/or Emergency Room (ER) visits or death, and (2) time to sustained clinical recovery. Novartis confirms it will now exercise its option to in-license ensovibep from Molecular Partners and, following exercise of the option, will seek expedited access globally, first via the FDA’s EUA process.
The global EMPATHY clinical trial, which is being conducted by Novartis, with Molecular Partners as sponsor, is a randomized, double-blind, placebo controlled study in ambulatory (non-hospitalized) adult patients with COVID-19. EMPATHY Part A enrolled 407 patients to identify a dose of ensovibep with optimal safety and efficacy and recruited patients in the USA, South Africa, India, the Netherlands and Hungary to explore three doses: 75mg, 225mg and 600mg.
Results from the study showed that the primary endpoint was met with a statistically significant reduction in viral load over eight days, compared to placebo, for all three dosing arms. The secondary endpoint of hospitalization and/or ER visits related to COVID-19, or death showed an overall 78% reduction in risk of events across ensovibep arms compared to placebo. Treatment arms were generally balanced in terms of demographic, baseline and disease characteristics. The placebo arm with 99 patients had a total of six events (event rate of 6.0%); five patients were hospitalized, two of whom died due to worsening of COVID-19 and one patient had an ER visit only. In the 301 patients treated with ensovibep, there were four events, hospitalizations occurred in two patients and two needed to visit ER (event rate of 1.3%). No deaths occurred in any of the patients treated with ensovibep. All doses were well-tolerated and no unexpected safety issues were identified for any of the doses2 . The lowest dose of 75mg is the planned dose for further development. The data will now undergo further review so that Novartis and Molecular Partners can determine the appropriate next steps for the program.
“We are pleased that the results from the EMPATHY trial demonstrate the positive therapeutic effect of ensovibep, with the potential to be an important new treatment option to combat the rapidly evolving SARS-CoV-2 pandemic,” said Vas Narasimhan, CEO of Novartis. “As COVID-19 continues to burden healthcare systems across the globe, a range of treatments will be needed, and Novartis is proud to continue our collaboration with Molecular Partners on this unique treatment for COVID-19 and contribute ensovibep to this suite of options.”
With the decision made to exercise the option, Novartis will become responsible for development, manufacturing, distribution and commercialization activities of ensovibep. Novartis has already initiated scale-up activities in its large-scale biologics production facilities.
As the SARS-CoV-2 virus evolves, a multi-solution strategy is needed to combat the pandemic and there will be a need for antiviral treatments to complement the global vaccination efforts. Despite availability of vaccinations, there continues to be disease transmission, either through pockets of unvaccinated populations, in patients with compromised immune systems and co-morbidities or through emerging variants, and breakthrough infections are likely to continue. A recent in vitro analysis3 also showed that ensovibep maintains full neutralization of the pseudoviruses containing the mutations identical to the Omicron variant of concern.
“These encouraging results come at a time when the need for therapies with pan-variant activity, such as ensovibep, has never been greater. We are incredibly excited about the opportunity to provide a potential therapeutic option for patients around the world who require access to effective COVID-19 treatments,” said Patrick Amstutz, Ph.D., CEO of Molecular Partners. “Today’s data are a culmination of a persistent team effort, between ourselves and Novartis, to deliver a tailored antiviral with demonstrated safety and efficacy in global clinical trials. As pioneers of DARPin therapeutics, our team has the unique ability to rapidly generate and develop multi-specific DARPin therapeutics. We look forward to continue to demonstrate our capabilities and the potential of our pipeline in oncology, virology and for patients in need.”
Given the pressing public health emergency and the rapid spread of the Omicron variant across the world, Novartis and Molecular Partners are in close liaison with regulatory bodies to seek expedited review and approval of ensovibep as soon as possible. If approved, ensovibep will be the first multi-specific antiviral molecule for the treatment of COVID-19.
About ensovibep
Ensovibep is a DARPin therapeutic candidate, designed specifically to inactivate
SARS-CoV-2, the virus that causes COVID-19. DARPins (Designed Ankyrin Repeat Proteins) are mono or multi-specific protein-based therapies, designed to specifically engage their targets for various effects. Ensovibep was designed to include three individual DARPin domains, each highly neutralizing to SARS-CoV-2. With these domains constructed into a single molecule, ensovibep can block the receptor-binding domain (RBD) of the SARS-CoV-2 spike protein through highly potent and cooperative binding. This design ensures strong neutralization, even in the presence of mutations of the spike protein and limits the development of escape mutants. Several characteristics of DARPin therapeutics make them suitable for COVID-19 treatment, including multi-specific binding, the rapid onset of action, and scalable bacterial production.
In vitro testing has shown high neutralization activity of ensovibep against all known SARS-CoV-2 variants, including the variants of concern: Alpha, Beta, Gamma, Delta and Omicron.3
About the EMPATHY clinical trial program2
Following promising Phase 1 clinical data for ensovibep the global EMPATHY clinical trial was initiated by Novartis, with Molecular Partners as sponsor, in May 2021. EMPATHY is a Phase 2 and 3 study looking at the safety and efficacy of ensovibep in symptomatic COVID-19 patients in the ambulatory (non-hospitalized) setting. Ensovibep is administered via a single dose IV infusion.
The EMPATHY clinical trial plans to enroll 2,100 patients. 407 patients were randomized into four arms of Part A of the study to identify a dose with optimal safety and efficacy. The clinical efficacy and safety of this dose vs. placebo will be further evaluated in Part B, the Phase 3 component of the EMPATHY study which will enroll an additional 1,700 patients globally.
The EMPATHY clinical trial enrolled both vaccinated and unvaccinated adult patients who have experienced at least two mild/ moderate symptoms of COVID-19 within seven days of onset and had a positive rapid antigen test on the day of dosing, confirmed by a PCR test at baseline. The COVID-19 symptoms include fever, cough, sore throat, low energy, tiredness, headache, muscle or body aches, chills and/ or shortness of breath.
The collaboration with Molecular Partners
Novartis is proud to collaborate with Molecular Partners on the development of ensovibep, a DARPin therapeutic candidate designed for potential use against COVID-19. With the decision made to exercise the option, Novartis will be responsible for further development, manufacturing, distribution and commercialization activities of ensovibep. Under the terms of the agreement, Molecular Partners had received an upfront payment of CHF 60 million, including equity. Molecular Partners will receive a further payment of CHF 150 million, upon Novartis electing to take up the option on the therapeutic candidate, and significant royalties on sales. Molecular Partners has agreed to forgo royalties in lower income countries, and is aligned with Novartis’ plans to ensure affordability based on countries’ needs and capabilities.
More information about the Novartis response to COVID-19 is available at www.novartis.com/COVID-19.
Jan. 10, 2022 1:58 AM ETMolecular Partners AG (MOLN), NVSBy: Mamta Mayani, SA News Editor
January 10, 2022Download PDF
https://www.molecularpartners.com/
BioMarin Announces Stable and Durable Annualized Bleed Control in the Largest Phase 3 Gene Therapy Study in Adults with Severe Hemophilia A; 134-Participant Study Met All Primary and Secondary Efficacy Endpoints at Two Year Analysis
- Annualized Bleeding Rate (ABR) Reduced by 85% from Baseline, Demonstrating Superiority to Factor VIII Prophylaxis- Mean Factor VIII Activity of 23 IU/dL (Chromogenic Assay); 36 IU/dL (One-Stage Assay) Observed at Year 2- Company Plans to Present Additional Data at Upcoming Medical Meetings- Marketing Authorization Application under review with the European Medicines Agency (EMA) and Regulatory Submission to FDA Expected in 2Q 2022- Conference Call and Webinar to be Held Today, Sunday, January 9, 2022 at 5:00 PM Eastern TimeJan 9, 2022
SAN RAFAEL, Calif., Jan. 9, 2022 /PRNewswire/ -- BioMarin Pharmaceutical Inc. (NASDAQ: BMRN) today announced positive results from its ongoing global Phase 3 GENEr8-1 study of valoctocogene roxaparvovec, an investigational gene therapy for the treatment of adults with severe hemophilia A. This is the largest global Phase 3 study to date for any gene therapy in hemophilia, with 134 participants.
In the GENEr8-1 Phase 3 study, Annualized Bleeding Rate (ABR) was significantly reduced by 4.1 treated bleeds per year (p-value <0.0001), or 85% from a baseline mean of 4.8 (median 2.8), in the pre-specified primary analysis in participants from a prior non-interventional study (rollover population; N=112; median follow-up of 110 weeks). The mean ABR was 0.8 (median 0.0) through the entire efficacy evaluation period, 0.9 (median 0.0) during year one, and 0.7 (median 0.0) during year two.
Valoctocogene roxaparvovec also significantly reduced the mean annualized Factor VIII infusion rate in the rollover population by 133 infusions per year (p-value <0.0001) or 98% from baseline. The mean annualized infusion rate was 2.6 (median 0.0) through the entire efficacy evaluation period, 1.5 (median 0.0) during year one, and 3.4 (median 0.0) during year two.
At the end of the second year post-infusion with valoctocogene roxaparvovec, participants in the modified intent-to-treat (mITT) population (N=132) had a mean endogenous Factor VIII activity level of 23.0 (median 11.8) IU/dL, as measured by the chromogenic substrate (CS) assay and 36.1 (median 21.6) IU/dL, as measured by the one-stage (OS) assay.
In a subset of the mITT population that had been dosed at least three years prior to the data cut (N=17), mean Factor VIII activity was 16.8 (median 9.3) IU/dL by CS assay and 27.0 (median 19.1) IU/dL by OS assay at the end of year three. The mean cumulative ABR for this subpopulation was 0.7 (median 0.0) through the entire efficacy evaluation period (median follow up 174 weeks) and 0.6 (median 0.0) during year three.
For comparison, the table below provides results from both the Phase 3 GENEr8-1 Study and the Phase 1/2 Study (201), by Study Year, for Factor VIII activity (by CS assay), ABR, and annualized Factor VIII utilization (infusions per year).
Two-Year Results Demonstrate Consistent Clinical Benefit in ABR and Factor VIII Utilization Across Phase 3 and Phase 1/2 Studies with Valoctocogene Roxaparvovec
"We are delighted that our perseverance on behalf of people with hemophilia A has led to today's transformative results in the largest gene therapy study for hemophilia A," said Hank Fuchs, M.D., President of Worldwide Research and Development at BioMarin. "We are grateful for the support of the bleeding disorders community to conduct this clinical program. These results show that valoctocogene roxaparvovec could profoundly change the way hemophilia A is treated. We are looking forward to continuing to work with health authorities to bring this therapy to patients with hemophilia A."
Valoctocogene Roxaparvovec Safety
Overall, in the Phase 3 study, valoctocogene roxaparvovec has continued to be well tolerated. All participants received a single 6e13 vg/kg dose. No participants developed inhibitors to Factor VIII, malignancy, or thromboembolic events. During year two, no new safety signals emerged, and no treatment-related serious adverse events (SAE) were reported. Most patients had discontinued any corticosteroid (CS) use in year one, and there were no CS-related SAEs in the remaining patients being tapered off CS in year two. Overall, the most common adverse events (AE) associated with valoctocogene roxaparvovec occurred early and included transient infusion associated reactions and mild to moderate rise in liver enzymes with no long-lasting clinical sequelae. Alanine aminotransferase (ALT) elevation (119 participants, 89%), a laboratory test of liver function, remained the most common AE. Other common adverse events were headache (55 participants, 41%), arthralgia (53 participants, 40%), nausea (51 participants, 38%), aspartate aminotransferase (AST) elevation (47 participants, 35%), and fatigue (40 participants, 30%).
For additional information, please visit www.biomarin.com.
Jan. 10, 2022 4:35 AM ET BioMarin Pharmaceutical Inc. (BMRN)
By: Mamta Mayani, SA News Editor1 Comment
January 10, 2022 6:45 am ET
First Positive Study for KEYTRUDA in Adjuvant Stage IB-IIIA NSCLC
KENILWORTH, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, the European Organisation for Research and Treatment of Cancer (EORTC) and the European Thoracic Oncology Platform (ETOP) today announced that the Phase 3 KEYNOTE-091 trial, also known as EORTC-1416-LCG/ETOP-8-15 – PEARLS, investigating KEYTRUDA, Merck’s anti-PD-1 therapy, met one of its dual primary endpoints of disease-free survival (DFS) for the adjuvant treatment of patients with stage IB-IIIA non-small cell lung cancer (NSCLC) following surgical resection regardless of PD-L1 expression. Based on an interim analysis review conducted by an independent Data Monitoring Committee, adjuvant treatment with KEYTRUDA resulted in a statistically significant and clinically meaningful improvement in DFS compared with placebo in the all-comer population of patients with stage IB-IIIA NSCLC.
At the interim analysis, there was also an improvement in DFS for patients whose tumors express PD-L1 (tumor proportion score [TPS] ≥50%) treated with KEYTRUDA compared to placebo; however, this dual primary endpoint did not meet statistical significance per the pre-specified statistical plan. The trial will continue to analyze DFS in patients whose tumors express high levels of PD-L1 (TPS ≥50%) and evaluate overall survival (OS), a key secondary endpoint.
The safety profile of KEYTRUDA in this study was consistent with that observed in previously reported studies. Results will be presented at an upcoming medical meeting and will be submitted to regulatory authorities.
“KEYTRUDA has become foundational in the treatment of metastatic non-small cell lung cancer and we continue to advance research to explore its potential to help fight cancer earlier,” said Dr. Roy Baynes, senior vice president and head of global clinical development, chief medical officer, Merck Research Laboratories. “We are encouraged by these results supporting the potential role of KEYTRUDA in stage IB-IIIA non-small cell lung cancer. We thank the patients, investigators and our partners at EORTC and ETOP for their important contributions to this study and look forward to sharing these results with the medical community as soon as possible.”
“Surgery is widely considered the first and most important intervention for most patients with early-stage non-small cell lung cancer; however, an estimated 43% of those who undergo surgery will see their disease return,” said Professor Mary O’Brien, The Royal Marsden, NHS Foundation Trust, and Imperial College, London, UK and co-principal investigator. “Data from KEYNOTE-091 suggest adjuvant KEYTRUDA reduced the risk of disease recurrence or death after surgery in the overall population of patients with stage IB-IIIA non-small cell lung cancer.”
“Globally, lung cancer is the leading cause of cancer death and it remains critically important to detect and treat lung cancer early,” said Dr. Luis Paz-Ares, chair of the medical oncology department, Hospital Universitario Doce de Octubre, Madrid, Spain and co-principal investigator. “The goal of adjuvant treatment is to lower the risk of cancer returning after surgery. By moving KEYTRUDA into earlier stages of non-small cell lung cancer, we may be able to reduce the risk of disease recurrence after surgery for patients with stage IB-IIIA non-small cell lung cancer.”
Merck has an extensive clinical development program in lung cancer and is advancing multiple registration-enabling studies with KEYTRUDA in combination with other treatments and as monotherapy for the treatment of lung cancer. The results from KEYNOTE-091 are a first for KEYTRUDA in the adjuvant treatment setting in patients with stage IB-IIIA NSCLC.
About KEYNOTE-091/EORTC-1416-LCG – PEARLS
KEYNOTE-091, also known as EORTC-1416-LCG/ETOP-8-15 – PEARLS, is a randomized, Phase 3 trial (ClinicalTrials.gov, NCT02504372) sponsored by Merck and conducted in collaboration with EORTC and ETOP evaluating KEYTRUDA compared to placebo for the adjuvant treatment of patients with stage IB-IIIA NSCLC following surgical resection (lobectomy or pneumonectomy) with or without adjuvant chemotherapy. The dual primary endpoints are DFS in the overall population and in patients whose tumors express PD-L1 (TPS ≥50%). DFS is calculated as the time from randomization to the date of disease recurrence, occurrence of second lung cancer primary, occurrence of second malignancy, or death from any cause, whichever occurs first. The secondary endpoints include OS and lung cancer-specific survival (the time from randomization to date of death due to lung cancer specifically). The study enrolled 1,177 patients who were randomly assigned (1:1) to receive either:
About EORTC
The European Organisation for Research and Treatment of Cancer (EORTC) is a non-governmental, non-profit organisation, which unites clinical cancer research experts, throughout Europe, to define better treatments for cancer patients to prolong survival and improve quality of life. Spanning from translational to large, prospective, multi-centre, phase III clinical trials that evaluate new therapies and treatment strategies as well as patient quality of life, its activities are coordinated from EORTC Headquarters, a unique international clinical research infrastructure, based in Brussels, Belgium.
For further information, please visit the EORTC website: www.eortc.org.
About KEYTRUDA® (pembrolizumab) Injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of adult and pediatric (12 years and older) patients with stage IIB, IIC, or III melanoma following complete resection.
Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [tumor proportion score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [combined positive score (CPS ≥1)] as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy. KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Carcinoma
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
Non-muscle Invasive Bladder Cancer
KEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin (BCG)-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ (CIS) with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with MSI-H central nervous system cancers have not been established.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC).
Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or GEJ (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
KEYTRUDA, in combination with chemotherapy, with or without bevacizumab, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
KEYTRUDA is indicated for the adjuvant treatment of patients with renal cell carcinoma (RCC) at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions.
Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf.
Jan. 10, 2022 9:13 AM ETMerck & Co., Inc. (MRK)By: Dulan Lokuwithana, SA News Editor
Jan 07, 2022Download PDF
Evkeeza is a first-in-class medicine approved by the U.S. Food and Drug Administration (FDA) and European Commission (EC) to treat an ultra-rare inherited form of high cholesterol
TARRYTOWN, N.Y. and NOVATO, C.A., January 7, 2022 – Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) and Ultragenyx Pharmaceutical Inc. today announced a license and collaboration agreement for Ultragenyx to clinically develop, commercialize and distribute Evkeeza® (evinacumab) in countries outside of the U.S. This includes the European Economic Area, where Evkeeza was approved in June 2021 as a first-in-class therapy for use together with diet and other low-density lipoprotein-cholesterol (LDL-C) lowering therapies to treat adults and adolescents aged 12 years and older with homozygous familial hypercholesterolemia (HoFH). Regeneron discovered and developed Evkeeza, and launched the medicine in the U.S. in February 2021 when it was approved by the FDA.
“Evkeeza is a transformational medicine for those living with homozygous familial hypercholesterolemia, as previous therapies were insufficient for many patients who still faced extremely high LDL cholesterol levels and treatment-related tolerability issues,” said George D. Yancopoulos, M.D., Ph.D., Chief Scientific Officer and President of Regeneron. “With its focus on rare, debilitating genetic conditions, Ultragenyx is an ideal partner for us, and we look forward to working together to bring this much needed medicine to patients around the world.”
Under the terms of the agreement, Regeneron will receive a $30 million upfront payment and is eligible to receive up to $63 million in additional potential regulatory and sales milestones. Ultragenyx will receive the rights to develop, commercialize and distribute the medicine in countries outside of the U.S. and make payments to Regeneron based on net sales. Ultragenyx will share in certain costs for global trials led by Regeneron and also have the right to continue to clinically develop Evkeeza in countries outside of the U.S. for HoFH and other potential indications.
Regeneron has also granted Ultragenyx an exclusive option to negotiate a separate agreement to collaborate on the development and commercialization outside of the U.S. of Regeneron’s investigational antibody currently in Phase 2/3 development for the treatment of the ultra-rare disease, fibrodysplasia ossificans progressiva (FOP) under terms to be agreed upon by both companies.
“Evkeeza utilizes a novel, potent biological mechanism to significantly reduce LDL cholesterol levels beyond historical standard of care for people with HoFH. This is a highly complementary partnership that combines Regeneron’s bold science with our proficiency in rare disease,” said Emil D. Kakkis, M.D., Ph.D., Chief Executive Officer and President of Ultragenyx. “We have developed our commercial expertise to support patient identification and access across regions and we believe we can make a real difference for the HoFH community outside of the U.S.”
Regeneron will continue to solely commercialize Evkeeza in the U.S., where more patients with HoFH are now being treated with Evkeeza than the prior standard-of-care. In countries outside of the U.S., Ultragenyx will be responsible for commercialization efforts.
About Homozygous Familial Hypercholesterolemia (HoFH)
HoFH, also known as homozygous FH, is an ultra-rare inherited condition that affects 1 in 160,000 to 300,000 people worldwide. HoFH occurs when two copies of the familial hypercholesterolemia (FH)-causing genes are inherited, one from each parent, resulting in dangerously high levels (>400 mg/dL) of LDL-C, or bad cholesterol. Patients with HoFH are at risk for premature atherosclerotic disease and cardiac events as early as their teenage years.
About Evkeeza® (evinacumab)
Evkeeza is a fully human monoclonal antibody that binds to and blocks the function of angiopoietin-like 3 (ANGPTL3), a protein that plays a key role in lipid metabolism. Regeneron scientists discovered the angiopoietin gene family more than two decades ago. Human genetics research published in New England Journal of Medicine (NEJM) in 2017 by scientists from the Regeneron Genetics Center found that patients whose ANGPTL3 gene did not function properly (called a "loss-of function mutation") have significantly lower levels of key blood lipids, including LDL-C, and that this is associated with a significantly lower risk of coronary artery disease.
In the U.S., Evkeeza is indicated as an adjunct to other LDL-C lowering therapies for the treatment of adult and pediatric patients, aged 12 years and older with HoFH. The safety and effectiveness of Evkeeza have not been established in patients with other causes of hypercholesterolemia, including those with heterozygous familial hypercholesterolemia (HeFH). The effect of Evkeeza on cardiovascular morbidity and mortality has not been determined.
Evkeeza was invented using Regeneron's VelocImmune® technology and is the first ANGPTL3-targeted therapy approved by the European Commission and U.S. FDA. The approvals were based on results from the Phase 3 ELIPSE HoFH trial, which was published in the NEJM in August 2020.
The generic name for Evkeeza in its approved U.S. indications is evinacumab-dgnb, with dgnb the suffix designated in accordance with Nonproprietary Naming of Biological Products Guidance for Industry issued by the U.S. FDA.
For additional information about the company, please visit www.regeneron.com or follow @Regeneron on Twitter.
For more information on Ultragenyx, please visit the company's website at: www.ultragenyx.com.
Jan. 07, 2022 7:08 AM ET
Regeneron Pharmaceuticals, Inc. (REGN), RARE
By: Mamta Mayani, SA News Editor
January 7, 2022
- Submissions supported by Phase 3 study in which upadacitinib (RINVOQ®) demonstrated significant improvements in signs and symptoms as well as physical function and disease activity versus placebo
- No new safety risks were observed compared to the known safety profile of upadacitinib
- AbbVie has also submitted results from two studies of upadacitinib in adult patients with ankylosing spondylitis (AS) to request label enhancements in the European Union (EU)
- RINVOQ is approved for use in active psoriatic arthritis (PsA), moderate to severe active rheumatoid arthritis (RA), moderate to severe atopic dermatitis (AD) and active AS in the EU, and for PsA and RA in the US
NORTH CHICAGO, Ill., Jan. 7, 2022 /PRNewswire/ -- AbbVie (NYSE: ABBV) today announced that it has submitted applications seeking approvals for upadacitinib (RINVOQ®, 15 mg once daily) to the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the treatment of adults with active non-radiographic axial spondyloarthritis (nr-axSpA) with objective signs of inflammation who have responded inadequately to nonsteroidal anti-inflammatory drugs (NSAIDs). The applications are supported by the Phase 3 SELECT-AXIS 2 (Study 2) clinical trial.
In the SELECT-AXIS 2 trial (Study 2), evaluating the efficacy and safety of upadacitinib in adult patients with nr-axSpA, upadacitinib met its primary and most ranked secondary endpoints.1 Treatment with upadacitinib 15 mg once daily resulted in reductions in signs and symptoms of nr-axSpA, including back pain and inflammation, as well as improvements in physical function and disease activity at week 14 versus placebo.1
"Axial spondyloarthritis is a chronic inflammatory disease affecting the spine and can cause patients, who tend to be younger adults living active lives, to suffer from debilitating pain and significantly decrease their quality of life,"2 said Thomas Hudson, M.D., senior vice president of research and development, chief scientific officer, AbbVie. "AbbVie is committed to working with the FDA and EMA to make upadacitinib available as a treatment option for patients living with this disease."
In addition, AbbVie has requested label enhancements for upadacitinib in the European Union (EU) to include adult patients with active ankylosing spondylitis (AS) who had an inadequate response to biologic disease-modifying anti-rheumatic drugs (bDMARDs), based on the results of the Phase 3 SELECT-AXIS 2 clinical trial (Study 1), as well as two-year results of the Phase 2/3 SELECT-AXIS 1 clinical trial.1,3 AbbVie also provided these data to the FDA in support of the agency's ongoing review of the supplemental New Drug Application (sNDA) for upadacitinib in AS.
The safety data observed in these patients with AS or nr-axSpA were generally consistent with the known safety profile of upadacitinib. No new safety risks were identified.1,3,4,5
About axSpA
Axial spondyloarthritis is a chronic inflammatory disease that affects the spine, causing back pain, limited mobility, and structural damage.6 It consists of two subsets that have been clinically defined as ankylosing spondylitis (AS) and non-radiographic axial spondyloarthritis (nr-axSpA).6 In ankylosing spondylitis, patients have definitive structural damage of the sacroiliac joints visible on x-rays.6 Non-radiographic axial spondyloarthritis is clinically defined by the absence of definitive x-ray evidence of structural damage to the sacroiliac (SI) joint by plain x-ray.6
About SELECT-AXIS 1 and SELECT-AXIS 2 trial programs1,4,5,7
SELECT-AXIS 1 is a Phase 2/3, multicenter, randomized, double-blind, parallel-group, placebo-controlled study designed to evaluate the safety and efficacy of RINVOQ in adult patients with active ankylosing spondylitis who are bDMARD-naïve and had inadequate response to at least two NSAIDs or intolerance to/contraindication for NSAIDs. Period 2 is an open-label extension period to evaluate the long-term safety, tolerability and efficacy of RINVOQ in subjects who completed Period 1. Primary results from SELECT-AXIS 1 were previously announced in November 2019 with 2-year results presented in November 2021. More information on this trial can be found at www.clinicaltrials.gov (NCT03178487).
SELECT-AXIS 2 was conducted as a master study protocol that contains two standalone studies with randomization, data collection, analysis and reporting conducted independently. The Phase 3, randomized, placebo-controlled, double-blind studies are evaluating the efficacy and safety of upadacitinib compared with placebo on reduction of signs and symptoms in adult participants with active axSpA including bDMARD-IR AS (Study 1) and nr-axSpA (Study 2). More information on this trial can be found at www.clinicaltrials.gov (NCT04169373).
In all studies, the primary endpoint was the percentage of subjects achieving an ASAS40 response after 14 weeks of treatment.
About RINVOQ® (upadacitinib)
Discovered and developed by AbbVie scientists, RINVOQ is a selective JAK inhibitor that is being studied in several immune-mediated inflammatory diseases. Based on enzymatic and cellular assays, RINVOQ demonstrated greater inhibitory potency for JAK-1 vs JAK-2, JAK-3, and TYK-2.8 The relevance of inhibition of specific JAK enzymes to therapeutic effectiveness and safety is not currently known. RINVOQ 15 mg is approved by the U.S. Food and Drug Administration (FDA) for adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more TNF blockers and adults with active psoriatic arthritis who have had an inadequate response or intolerance to one or more TNF blockers. RINVOQ 15 mg is approved by the European Commission for adults with moderate to severe active rheumatoid arthritis, adults with active psoriatic arthritis and adults with active ankylosing spondylitis. RINVOQ is approved by the European Commission for adults (15 mg and 30 mg) and adolescents (15 mg) with moderate to severe atopic dermatitis. Phase 3 trials of RINVOQ in rheumatoid arthritis, atopic dermatitis, psoriatic arthritis, axial spondyloarthritis, Crohn's disease, ulcerative colitis, giant cell arteritis and Takayasu arteritis are ongoing.9-16 Use of RINVOQ in AS is not approved by FDA and its safety and efficacy remain under review. Use of RINVOQ in nr-axSpA is not approved and its safety and efficacy have not been evaluated by regulatory authorities.
RINVOQ U.S. Uses and Important Safety Information7
RINVOQ is a prescription medicine used to treat adults with:
It is not known if RINVOQ is safe and effective in children under 18 years of age.
EU Indications and Important Safety Information about RINVOQ™ (upadacitinib)
For more information about AbbVie, please visit us at www.abbvie.com.
Jan. 07, 2022 9:30 AM ET AbbVie Inc. (ABBV) By: Mamta Mayani, SA News Editor1 Comment

Jan 06, 2022 3:30 AM
Tislelizumab is now approved in six indications in China
This marks tislelizumab’s third approved lung cancer indication in China and first in a previously treated patient population
The approval was supported by results from the global Phase 3 trial RATIONALE 303, in which tislelizumab significantly prolonged overall survival in these patients compared to docetaxel and was generally well-tolerated, consistent with known risks
CAMBRIDGE, Mass. & BEIJING--(BUSINESS WIRE)-- BeiGene (NASDAQ: BGNE; HKEX: 06160; SSE: 688235), a global, science-driven biotechnology company focused on developing innovative and affordable medicines to improve treatment outcomes and access for patients worldwide, today announced that the China National Medical Products Administration (NMPA) has approved BeiGene’s anti-PD-1 antibody tislelizumab as a second- or third-line treatment for patients with locally advanced or metastatic non-small cell lung cancer (NSCLC). A supplemental biologics license application for tislelizumab in this indication was previously accepted for review by the China NMPA in March 2021.
“This latest approval for tislelizumab demonstrates BeiGene’s commitment to bringing innovative, impactful treatments to patients in need. With six approved indications in China, tislelizumab has the potential to reach and help the country’s large patient community, and our science-based commercial team of nearly 3,000 people in China is working to make it more broadly available to those who may benefit from this important immunotherapy,” commented Xiaobin Wu, Ph.D., President, Chief Operating Officer, and General Manager of China at BeiGene. “As our strategic oncology collaboration with Novartis deepens, we look forward to continued opportunities to expand access to tislelizumab globally and further explore its therapeutic potential.”
“As its third approved lung cancer indication in China, today’s approval represents an important milestone, with tislelizumab now available in both front-line and second- or third-line care of NSCLC. We hope, as demonstrated in the promising results from the global RATIONALE 303 trial, that tislelizumab will become an important treatment option for these patients in China,” commented Yong (Ben) Ben, M.D., Chief Medical Officer, Solid Tumors at BeiGene. “Tislelizumab’s broad global development program of 13 Phase 3 trials and four pivotal Phase 2 trials is providing a growing body of clinical evidence on its efficacy and safety and establishing its therapeutic impact across multiple cancer types.”
“In the global Phase 3 trial, tislelizumab demonstrated a significant improvement in overall survival and was well-tolerated in patients with previously treated NSCLC,” said Caicun Zhou, M.D., Ph.D., Director of the Department of Oncology at Shanghai Pulmonary Hospital, Director of Cancer Institute of Tongji University, and the principal investigator of the trial. “The NMPA’s approval of tislelizumab is welcoming news to the lung cancer community in China, and we hope this immunotherapy will help address the unmet needs in second- or third-line treatment of NSCLC.”
The approval of tislelizumab was supported by clinical results from a randomized, open-label, global Phase 3 trial RATIONALE 303 (NCT03358875) comparing tislelizumab to docetaxel in the second- or third-line setting in patients with locally advanced or metastatic NSCLC who have progressed on prior platinum-based chemotherapy. A total of 805 patients in 10 countries across Asia, Europe, the Americas, and Oceania were enrolled in the trial, randomized 2:1 to either the tislelizumab arm or the docetaxel arm.
As announced in November 2020, the trial met the primary endpoint of overall survival (OS) at the planned interim analysis, as recommended by the independent Data Monitoring Committee (IDMC). Tislelizumab was generally well-tolerated, consistent with known safety risks from previously reported results across different tumor types, with no new safety signals identified. The results of the interim analysis of the trial were presented at the American Association for Cancer Research (AACR) Annual Meeting and announced by BeiGene in April 2021.
About Tislelizumab
Tislelizumab (BGB-A317) is a humanized IgG4 anti-PD-1 monoclonal antibody specifically designed to minimize binding to FcγR on macrophages. In pre-clinical studies, binding to FcγR on macrophages has been shown to compromise the anti-tumor activity of PD-1 antibodies through activation of antibody-dependent macrophage-mediated killing of T effector cells. Tislelizumab is the first drug from BeiGene’s immuno-oncology biologics program and is being developed internationally as a monotherapy and in combination with other therapies for the treatment of a broad array of both solid tumor and hematologic cancers.
The China National Medical Products Administration (NMPA) has approved tislelizumab in six indications, including full approval for first-line treatment of patients with advanced squamous non-small cell lung cancer (NSCLC) in combination with chemotherapy, for first-line treatment of patients with advanced non-squamous NSCLC in combination with chemotherapy, and for second- or third-line treatment of patients with locally advanced or metastatic NSCLC who progressed on prior platinum-based chemotherapy. NMPA also granted conditional approval for the treatment of patients with classical Hodgkin’s lymphoma (cHL) who received at least two prior therapies, for the treatment of patients with locally advanced or metastatic urothelial carcinoma (UC) with PD-L1 high expression whose disease progressed during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy, and for the treatment of patients with hepatocellular carcinoma (HCC) who have received at least one systemic therapy. Full approval for these indications is contingent upon results from ongoing randomized, controlled confirmatory clinical trials.
In addition, three supplemental Biologics License Applications for tislelizumab are under review by the Center for Drug Evaluation (CDE) of the NMPA, including for patients with previously treated, locally advanced unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair-deficient (dMMR) solid tumors, for the treatment of patients with locally advanced or metastatic esophageal squamous cell carcinoma (ESCC) who have disease progression following or are intolerant to first-line standard chemotherapy, and for first-line treatment of patients with recurrent or metastatic nasopharyngeal cancer (NPC).
In the U.S., a Biologics License Application for tislelizumab as a treatment for patients with unresectable recurrent locally advanced or metastatic ESCC after prior systemic therapy is currently under review by the U.S. Food and Drug Administration with a PDUFA target action date of July 12, 2022.
BeiGene has initiated or completed 17 potentially registration-enabling clinical trials in China and globally, including 13 Phase 3 trials and four pivotal Phase 2 trials.
In January 2021, BeiGene and Novartis entered into a collaboration and license agreement granting Novartis rights to develop, manufacture, and commercialize tislelizumab in North America, Europe, and Japan.
Tislelizumab is not approved for use outside of China.
About the Tislelizumab Clinical Program
Clinical trials of tislelizumab include:
To learn more about BeiGene, please visit www.beigene.com.
View source version on businesswire.com: https://www.businesswire.com/news/home/20220106005053/en/
Source: BeiGene
Jan. 06, 2022 4:24 AM ET BeiGene, Ltd. (BGNE) By: Mamta Mayani, SA News Editor
Trastuzumab Deruxtecan Type II Variation Application Validated by EMA for Patients with HER2 Positive Metastatic Breast Cancer Treated with a Prior Anti-HER2-Based Regimen • Application based on DESTINY-Breast03 results showing Daiichi Sankyo and AstraZeneca’s trastuzumab deruxtecan reduced risk of disease progression or death by 72% versus trastuzumab emtansine (T-DM1) Tokyo and Munich – (December 28, 2021) – Daiichi Sankyo Company, Limited (hereafter, Daiichi Sankyo) today announced that the European Medicines Agency (EMA) has validated the Type II Variation application for trastuzumab deruxtecan for the treatment of adult patients with unresectable or metastatic HER2 positive breast cancer who have received one or more prior anti-HER2-based regimens. Trastuzumab deruxtecan is a HER2 directed antibody drug conjugate (ADC) being jointly developed by Daiichi Sankyo and AstraZeneca (LSE/STO/Nasdaq: AZN). Validation confirms that the application is complete and commences the scientific review process by the EMA’s Committee for Medicinal Products for Human Use (CHMP). This application is based on data from the DESTINY-Breast03 phase 3 trial presented at the 2021 European Society for Medical Oncology (ESMO) Congress. Breast cancer is the most common cancer worldwide, with more than two million cases diagnosed in 2020, resulting in nearly 685,000 deaths globally.1 In Europe, approximately 531,000 cases of breast cancer are diagnosed annually.2 Approximately one in five cases of breast cancer are considered HER2 positive.3 Despite initial treatment with trastuzumab and a taxane, patients with HER2 positive metastatic breast cancer will often experience disease progression.4 More treatment options are needed to further delay progression and extend survival. 4,5,6 “We are excited to have submitted a second application this year seeking approval for trastuzumab deruxtecan for a potential third indication in Europe,” said Gilles Gallant, BPharm, PhD, FOPQ, Senior Vice President, Global Head, Oncology Development, Oncology R&D, Daiichi Sankyo. “With this specific application, we look forward to working closely with the EMA to support the review of trastuzumab deruxtecan to be used in an earlier setting for patients with HER2 positive metastatic breast cancer.” 2 In DESTINY-Breast03, trastuzumab deruxtecan demonstrated a 72% reduction in the risk of disease progression or death compared to trastuzumab emtansine (T-DM1) (hazard ratio [HR] = 0.28; 95% confidence interval [CI]: 0.22-0.37; p=7.8x10-22) in patients with HER2 positive unresectable and/or metastatic breast cancer previously treated with trastuzumab and a taxane. The median progression-free survival (PFS) for patients treated with trastuzumab deruxtecan was not reached (95% CI: 18.5-NE) compared to 6.8 months for T-DM1 (95% CI: 5.6-8.2) as assessed by blinded independent central review (BICR). In the secondary endpoint analysis of PFS assessed by investigators, patients treated with trastuzumab deruxtecan experienced an improvement in PFS of 25.1 months (95% CI: 22.1-NE) compared to 7.2 months (95% CI: 6.8-8.3) for T-DM1 (HR=0.26; 95% CI: 0.20-0.35). There was a strong trend towards improved overall survival (OS) with trastuzumab deruxtecan (HR=0.56; 95% CI: 0.36-0.86; p=0.007172), however this analysis is not yet mature and is not statistically significant. Nearly all patients treated with trastuzumab deruxtecan were alive at one year (94.1%; 95% CI: 90.3-96.4) compared to 85.9% of patients treated with T-DM1 (95% CI: 80.9-89.7). Confirmed objective response rate (ORR) was more than doubled in the trastuzumab deruxtecan arm versus the T-DM1 arm (79.7% [n=208; 95% CI: 74.3-84.4] versus 34.2% [n=90; 95% CI: 28.5-40.3]). The safety profile of the most common adverse events with trastuzumab deruxtecan in DESTINY-Breast03 was consistent with previous clinical trials with no new safety concerns identified. The most common grade 3 or higher drug-related treatment emergent adverse events in the trastuzumab deruxtecan arm were neutropenia (19.1%), thrombocytopenia (7.0%), leukopenia (6.6%), nausea (6.6%), anemia (5.8%), fatigue (5.1%), vomiting (1.6%), increase in ALT (1.6%), decreased appetite (1.2%), increase in AST (0.8%), diarrhea (0.4%) and alopecia (0.4%). Overall, 10.5% of patients had interstitial lung disease (ILD) or pneumonitis related to treatment as determined by an independent adjudication committee. The majority of ILD events (9.7%) were low grade (grade 1 (2.7%) or grade 2 (7.0%)) with two grade 3 (0.8%) events reported. No grade 4 or grade 5 ILD or pneumonitis events occurred.
About DESTINY-Breast03 DESTINY-Breast03 is a global, head-to-head, randomized, open-label, pivotal phase 3 trial evaluating the efficacy and safety of trastuzumab deruxtecan (5.4 mg/kg) versus T-DM1 in patients with HER2 positive unresectable and/or metastatic breast cancer previously treated with trastuzumab and a taxane. The primary efficacy endpoint of DESTINY-Breast03 is PFS based on BICR. Secondary efficacy endpoints include OS, ORR, duration of response, PFS based on investigator assessment and safety. DESTINY-Breast03 enrolled 524 patients at multiple sites in Asia, Europe, North America, Oceania and South America. For more information about the trial, visit ClinicalTrials.gov. About Trastuzumab Deruxtecan Trastuzumab deruxtecan (fam-trastuzumab deruxtecan-nxki in the U.S. only) is a HER2 directed ADC. Designed using Daiichi Sankyo’s proprietary DXd ADC technology, trastuzumab deruxtecan is the lead ADC in the oncology portfolio of Daiichi Sankyo and the most advanced program in AstraZeneca’s ADC scientific platform. Trastuzumab deruxtecan consists of a HER2 monoclonal antibody attached to a topoisomerase I inhibitor payload, an exatecan derivative, via a stable tetrapeptide-based cleavable linker. Trastuzumab deruxtecan (5.4 mg/kg) is approved in more than 30 countries for the treatment of adult patients with unresectable or metastatic HER2 positive breast cancer who have received two or more prior antiHER2-based regimens based on the results from the DESTINY-Breast01 trial. A supplemental New Drug Application is under review in Japan for the treatment of adult patients with HER2 positive unresectable or recurrent breast cancer previously treated with trastuzumab and a taxane, based on the results from the DESTINY-Breast03 trial. Trastuzumab deruxtecan (6.4 mg/kg) is also approved in several countries for the treatment of adult patients with locally advanced or metastatic HER2 positive gastric or gastroesophageal junction (GEJ) adenocarcinoma who have received a prior trastuzumab-based regimen based on the results from the DESTINY-Gastric01 trial. A Type II Variation is currently under review by the EMA for the treatment of adult patients with locally advanced or metastatic HER2 positive gastric or GEJ adenocarcinoma who have received a prior anti-HER2- based regimen.
About the Trastuzumab Deruxtecan Clinical Development Program A comprehensive global development program is underway evaluating the efficacy and safety of trastuzumab deruxtecan monotherapy across multiple HER2 targetable cancers including breast, gastric, lung and colorectal cancers. Trials in combination with other anticancer treatments, such as immunotherapy, are also underway. Trastuzumab deruxtecan was highlighted in the Clinical Cancer Advances 2021 report as one of two significant advancements in the “ASCO Clinical Advance of the Year: Molecular Profiling Driving Progress in GI Cancers,” based on data from both the DESTINY-Gastric01 and DESTINY-CRC01 trials, as well as one of the targeted therapy advances of the year in NSCLC based on the interim results of the HER2 mutant cohort of the DESTINY-Lung01 trial. Trastuzumab deruxtecan recently received its fourth Breakthrough Therapy Designation in the U.S. for the treatment of adult patients with unresectable or metastatic HER2 positive breast cancer who have received one or more prior anti-HER2-based regimens. About the Daiichi Sankyo and AstraZeneca Collaboration Daiichi Sankyo and AstraZeneca entered into a global collaboration to jointly develop and commercialize trastuzumab deruxtecan in March 2019 and datopotamab deruxtecan (Dato-DXd) in July 2020, except in Japan where Daiichi Sankyo maintains exclusive rights for each ADC. Daiichi Sankyo is responsible for the manufacturing and supply of trastuzumab deruxtecan and datopotamab deruxtecan.
For more information, please visit: www.daiichisankyo.com.
Dec. 28, 2021 2:09 AM ET AstraZeneca PLC (AZN), DSKYFDSNKY
By: Mamta Mayani, SA News Editor
January 6, 2022 at 7:30 AM ESTPDF Version
SHANGHAI and SAN FRANCISCO and CAMBRIDGE, Mass., Jan. 06, 2022 (GLOBE NEWSWIRE) -- Zai Lab Limited (NASDAQ: ZLAB; HKEX: 9688), a patient-focused, innovative, commercial-stage, global biopharmaceutical company, today announced that the China National Medical Products Administration (NMPA) has accepted the New Drug Application (NDA) for margetuximab, an investigational, Fc-engineered monoclonal antibody that targets HER2. The margetuximab NDA is for the treatment of adult patients with metastatic HER2-positive breast cancer who have received two or more prior anti-HER2 regimens, at least one of which was for metastatic disease, in combination with chemotherapy.
“We are pleased to have NMPA’s acceptance of our NDA for margetuximab, which is the only HER2-targeted agent to have shown a progression-free survival (PFS) improvement versus trastuzumab in SOPHIA, a head-to-head global Phase 3 clinical trial,” said Alan Sandler, MD, President and Head of Global Development, Oncology, at Zai Lab. “Both SOPHIA and Zai Lab’s registrational bridging trial support the potential use of margetuximab as an important new treatment option for a very difficult-to-treat patient population. The potential approval of margetuximab will also be an important addition to our growing women’s oncology franchise and marks Zai Lab’s sixth NDA acceptance by the NMPA.”
“Early detection and treatment of breast cancer have had a positive impact on patient survival. However, we still need to improve the prognosis for people diagnosed with HER2-positive metastatic breast cancer, and additional anti-HER2 targeted therapies are needed,” said Professor Zefei Jiang, Chairman of Chinese Society of Clinical Oncology (CSCO) Breast Cancer Expert Committee and Deputy Director of Department of Oncology, Chinese PLA General Hospital. “Zai Lab’s bridging study confirmed the clinical benefit of margetuximab in Chinese patients. We are excited to see this potential new treatment option for patients living with metastatic breast cancer in China."
In December 2020, MacroGenics, Inc. announced that the U.S. Food and Drug Administration (FDA) approved margetuximab (brand name MARGENZA®) in combination with chemotherapy for the treatment of adult patients with metastatic HER2-positive breast cancer who received two or more prior anti-HER2 regimens, at least one of which was for metastatic disease. The approval was based on efficacy and safety results from the pivotal Phase 3 SOPHIA trial.
In October 2021, Zai Lab announced that the bridging study of margetuximab plus chemotherapy as compared with trastuzumab plus chemotherapy in advanced, previously treated HER2-positive breast cancer patients met its primary endpoint, median PFS evaluated by blinded independent central review (BICR). The safety profile was consistent with that seen in the SOPHIA study. Zai Lab is planning to present the detailed study results at an upcoming medical conference.
About Margetuximab
MARGENZA (margetuximab-cmkb) is an Fc-engineered monoclonal antibody that targets the HER2 oncoprotein. HER2 is expressed by tumor cells in breast, gastroesophageal and other solid tumors. Similar to trastuzumab, margetuximab-cmkb inhibits tumor cell proliferation, reduces shedding of the HER2 extracellular domain and mediates antibody-dependent cellular cytotoxicity (ADCC). However, through MacroGenics’ Fc Optimization technology, margetuximab-cmkb has been engineered to enhance the engagement of the immune system. In vitro, the modified Fc region of margetuximab-cmkb increased binding to the activating Fc receptor FCGR3A (CD16A) and decreased binding to the inhibitory Fc receptor FCGR2B (CD32B). These changes led to greater in vitro ADCC and NK cell activation. The clinical significance of in vitro data is unknown.
For additional information about Zai Lab, please visit www.zailaboratory.com or follow us at www.twitter.com/ZaiLab_Global.
For more investor-related information about Zai Lab, please go to www.SEC.gov or visit www.zailaboratory.com.
Jan. 06, 2022 8:31 AM ET
By: Mamta Mayani, SA News Editor
Dec 23, 2021
Basel, December 22, 2021 — Novartis, a leader in rheumatology and immuno-dermatology, today announced the US Food and Drug Administration (FDA) has approved Cosentyx® (secukinumab) for the treatment of active enthesitis-related arthritis (ERA) in four years and older, and active juvenile psoriatic arthritis (JPsA) in patients two years and older1. Cosentyx is now the first biologic indicated for ERA, and the only biologic treatment approved for both ERA and PsA in pediatric patients in the US. These are the second and third approvals for Cosentyx in a pediatric population in the US, and Cosentyx now has a total of five indications across rheumatology and dermatology1.
“Prior research suggests that despite receiving treatment, some children and adolescents with PsA or ERA can continue to experience symptoms,” said Hermine Brunner, M.D., Cincinnati Children’s Hospital. “The findings from the Phase III JUNIPERA trial show that pediatric patients treated with secukinumab demonstrated marked responses throughout the treatment period. This approval is positive news for some patients who continue to struggle with painful symptoms like inflammation of the joints and swollen fingers and toes.”
ERA and JPsA, subtypes of juvenile idiopathic arthritis (JIA), are autoimmune diseases3-5. ERA is characterized by joint swelling and pain where tendons and ligaments attach to bone and may present with low back pain or tenderness at the palpation of the hips5. JPsA is characterized by joint swelling and skin psoriasis and may present with nail changes, inflammation of fingers and/or toes or psoriatic skin changes in a first-degree relative6. If left untreated, they can lead to high levels of pain and disability3-5.
“This marks the second and third US pediatric approval this year for Cosentyx, following pediatric psoriasis approval and further reinforces the proven efficacy and safety of the therapy. With more than 500,000 adult and pediatric patients treated worldwide since launch, healthcare professionals and patients can feel confident in Cosentyx,” said Todd Fox, Global Head of Medical Affairs Immunology, Hepatology and Dermatology at Novartis. “Furthermore, we are pleased to build on our strong heritage of bringing innovative treatments to young people living with rheumatic diseases, which began with the FDA approval of Ilaris. We are committed to bringing Cosentyx to this pediatric community globally as part of our ambition to expand Cosentyx to 10 indications in areas of high unmet need.”
The approved pediatric dosing for Cosentyx in children and adolescents is 75 mg (15 kg or more to less than 50 kg) or 150 mg (50 kg or more)1. It is administered as a subcutaneous injection by a pre-filled syringe or Sensoready® pen every 4 weeks after initial loading doses1. With appropriate guidance/instruction from a healthcare professional, Cosentyx can be administered by an adult caregiver outside of a healthcare provider’s office via a single-dose prefilled syringe or Sensoready® pen.
The approval is based on data from the Phase III JUNIPERA study, a two-year, three-part, double-blind, placebo-controlled, randomized-withdrawal trial that enrolled 86 children and adolescents aged 2 to 18 years old with a confirmed diagnosis of ERA or JPsA according to a modified International League of Associations for Rheumatology classification criteria7. The primary endpoint of the study was time to flare in the treatment period 2 (Week 12 to Week 104)7. In children and adolescents aged 2 to 18 years old, the study demonstrated that patients with active JPsA (n = 34; mean age: 12.2) treated with Cosentyx had a longer time to flare, showing an 85% reduction in the risk of flare (P<0.001) versus placebo1,8. The study also demonstrated that patients with active ERA (n = 52; mean age: 13.7) treated with Cosentyx had a significantly longer time to flare, showing a 53% reduction in the risk of flare versus placebo1,8. Safety in this pediatric population was consistent with the known safety profile of Cosentyx for the treatment of plaque psoriasis, PsA, non-radiographic axial spondyloarthritis and ankylosing spondylitis2.
“The symptoms of PsA and ERA can be debilitating for children and adolescents living with these chronic conditions, impacting their daily lives,” said Tiffany Westrich-Robertson, CEO, International Foundation for Autoimmune & Autoinflammatory Arthritis (AiArthritis). “It is encouraging to see an additional treatment option for these underserved patient populations.”
In July 2020, Cosentyx received EU approval as a first-line systemic treatment for pediatric psoriasis in patients aged 6 to 18 years old and recently received approval in the US and China1,9,10. In 2021, Cosentyx was also approved in Japan to treat both PsA and psoriasis in pediatric patients aged 6 years or older, as well as those with generalized pustular psoriasis11.
Novartis has filed a regulatory submission for Cosentyx in ERA and JPsA in Europe with a decision anticipated in the coming months.
About Cosentyx® (secukinumab)
Cosentyx is the first and only fully human biologic that directly inhibits interleukin-17A (IL-17A), an important cytokine involved in the inflammation of psoriatic arthritis (PsA), moderate to severe plaque psoriasis, ankylosing spondylitis (AS) and non-radiographic axial spondyloarthritis (nr-axSpA)12,13. Cosentyx is a proven medicine and has been studied clinically for more than 14 years. The medicine is backed by robust evidence, including 5 years of clinical data in adults supporting long-term safety and efficacy across moderate to severe plaque psoriasis, PsA and AS14-20. These data strengthen the position of Cosentyx as a treatment across AS, nr-axSpA, PsA and moderate to severe plaque psoriasis, supported by more than 500,000 patients treated worldwide since launch in 20151,21,22.
Find out more at https://www.novartis.com.
Dec. 23, 2021 2:22 AM ET
By: Mamta Mayani, SA News Editor

January 6, 2022
–Once daily linzagolix 200mg with ABTmet both co-primary efficacy objectives, demonstrating reductions in dysmenorrhea and non-menstrual pelvic pain versus placebo at 3 months; showed statistically significant and clinically meaningful improvements versus placebo in ranked secondary endpoints of dysmenorrhea, non-menstrual pelvic pain, dyschezia, overall pelvic pain, and ability to perform daily activities at 6 months–
-Once daily linzagolix 75 mg without ABT demonstrated statistically significant improvement for dysmenorrhea versus placebo and showed improvement but did not meet the co-primary objective of reduction in non-menstrual pelvic pain at 3 months; also showed improvement in secondary endpoints at 6 months-
-Both doses of linzagolix were well-tolerated with minimal bone mineral density (BMD) decrease and few adverse events occurring in over 5% of patients in either active treatment arm –
-Results support continued development of linzagolix with ABT and non-ABT doses for the treatment of endometriosis-
– Additional data from the post-treatment follow-up of EDELWEISS 3 are expected in 2Q 2022 and from the post-treatment follow-up of the extension study in 4Q 2022-
-Conference call and webcast with Dr. Hugh Taylor to be held today at 8:00 a.m. EST-
Ad hoc announcement pursuant to Art. 53 LR of the SIX Swiss Exchange
GENEVA, Switzerland January 6, 2022 – ObsEva SA (NASDAQ: OBSV; SIX: OBSN), a biopharmaceutical company developing and commercializing novel therapies for women’s health, today announced positive topline results from the Phase 3 EDELWEISS 3 trial of linzagolix, an oral GnRH antagonist, in women with moderate-to-severe endometriosis-associated pain (EAP). Two doses were tested, a 200 mg once-daily dose of linzagolix in combination with add-back therapy (ABT) and a 75 mg dose of linzagolix without ABT.
The 200 mg dose met the co-primary efficacy objectives, demonstrating reductions in dysmenorrhea (DYS) and non-menstrual pelvic pain (NMPP) at 3 months. There were statistically significant and clinically meaningful improvements in the first five ranked secondary endpoints at 6 months: dysmenorrhea, non-menstrual pelvic pain, dyschezia, overall pelvic pain, and ability to do daily activities. The 75mg dose without ABT demonstrated a statistically significant reduction versus placebo in DYS at 3 months. Although it showed improvement in NMPP at 3 months, it did not reach statistical significance versus placebo, and thus did not meet the co-primary efficacy objective. Improvements were also observed at 6 months in the first five ranked secondary endpoints, as for the 200 mg plus ABT dose. Both linzagolix doses were generally well-tolerated with minimal BMD decrease and few adverse events occurring in more than 5% of patients up to 6 months.
“While there have been recent advances in non-surgical endometriosis treatment, there is still a critical need for therapeutic options for women who suffer from this chronic condition,” said Hugh Taylor, MD, Professor and Chair of Obstetrics and Gynecology at Yale University. “Once daily linzagolix 200 mg with ABT demonstrated excellent efficacy along with minimal changes in bone mineral density, suggesting this dose may be used for long-term treatment. Availability of medical therapies that can be used long-term is important for this typically younger patient population.. The demonstration of improvement in dyschezia is of particular interest as this is the first Phase 3 trial of an oral GnRH antagonist to report efficacy in this common and debilitating symptom of endometriosis.”
Elizabeth Garner, MD, MPH, Chief Medical Officer of ObsEva, commented, “We are extremely pleased by the results highlighting the promising clinical profile of linzagolix 200 mg once-daily dose with ABT for women with moderate-to-severe EAP and the potential to be a leading GnRH option that balances safety and efficacy. Furthermore, by effectively addressing debilitating pain symptoms, linzagolix may improve overall quality of life and the ability to perform daily activities. While the 75 mg dose did not meet the NMPP endpoint, the statistically significant and clinically meaningful responder rates versus placebo for dysmenorrhea at 3 months and the evidence of clinical activity and tolerability at 6 months are encouraging. We plan to complete the Phase 3 program for this important indication and intend to further explore a non-ABT dose option as we remain dedicated to meeting the individual treatment needs and preferences of all women.”
Top-line Efficacy Analysis
Co-Primary Endpoints at 3 Months
About the Phase 3 EDELWEISS Program in Endometriosis
EDELWEISS 3 (Europe and the U.S.) was a randomized, double-blind, placebo-controlled, Phase 3 trial that analyzed 484 women with moderate-to-severe EAP. The study was designed to evaluate the long-term efficacy and safety of linzagolix, with a co-primary endpoint of reduction in both dysmenorrhea and non-menstrual pelvic pain at 3 months, along with stable or decreased use of analgesics for EAP. The study included a 200 mg once-daily dose in combination with ABT (1 mg estradiol / 0.5 mg norethindrone acetate), and a 75 mg once-daily dose without ABT. Subjects who completed the initial 6-month treatment period were offered to enter a 6-month treatment extension under the Edelweiss 6 protocol or to enter a 6-month post-treatment follow-up period. Results from the post-treatment follow-up of EDELWEISS 3 are expected in 2Q 2022 and from the post-treatment follow-up of the extension study in 4Q 2022. Additional information about this study can be found here.
About Endometriosis
The World Endometriosis Research Foundation estimates that endometriosis affects one in ten women during their reproductive years, representing approximately 176 million women worldwide between the ages of 15 and 49.
Endometriosis is a disease in which the endometrium (tissue lining the inside of the uterus) is found outside the uterus, where it induces a chronic inflammatory reaction that may result in scar tissue. It is primarily found on the pelvic peritoneum, on the ovaries, in the rectovaginal septum, on the bladder and bowel. The most common symptom of endometriosis is pelvic pain, which often correlates to the menstrual cycle. Patients may also experience painful ovulation, pain during or after sexual intercourse (dyspareunia), dyschezia (difficult or painful defecation), heavy bleeding, fatigue, and infertility. Endometriosis pain can be so severe and debilitating that it affects day-to-day activities and has a negative impact on general, physical, mental, and social well-being. Endometriosis treatments aim first to alleviate pain, then to remove or decrease the size and number of endometrial lesions, and possibly improve fertility.
About Linzagolix
Linzagolix is a novel, once daily, oral GnRH receptor antagonist with a potentially best-in-class profile.1,2,3 Linzagolix has completed clinical trial development for the treatment of heavy menstrual bleeding associated with uterine fibroids and is currently in late-stage clinical development for the treatment of pain associated with endometriosis. ObsEva licensed linzagolix from Kissei in late 2015 and retains worldwide commercial rights, excluding Asia, for the product. Linzagolix is not currently approved anywhere in the world.
About ObsEva
ObsEva is a biopharmaceutical company developing and commercializing novel therapies to improve women’s reproductive health and pregnancy. Through strategic in-licensing and disciplined drug development, ObsEva has established a late-stage clinical pipeline with development programs focused on new therapies for the treatment of uterine fibroids, endometriosis, and preterm labor. ObsEva is listed on the Nasdaq Global Select Market and is traded under the ticker symbol “OBSV” and on the SIX Swiss Exchange where it is traded under the ticker symbol “OBSN”. For more information, please visit www.ObsEva.com
About Kissei Pharmaceutical Co., Ltd.
Kissei is a Japanese pharmaceutical company based on the management philosophy “contributing to society through high-quality, innovative pharmaceutical products” and “serving society through our employees.” As a strong R&D-oriented corporation, it concentrates on providing innovative pharmaceuticals to patients worldwide in the focus fields of urology, nephrology/dialysis, gynecology and rare/intractable diseases.
Jan. 06, 2022 6:55 AM ET
By: Mamta Mayani, SA News Editor4 Comments
NEWS PROVIDED BY
Dec 23, 2021, 15:13 ET
MISSISSAUGA, ON, Dec. 23, 2021 /CNW/ -
GSK announces today that it has been granted Notice of Compliance with conditions for its endometrial cancer treatment JEMPERLI (dostarlimab for injection) by Health Canada.
As an anti-PD-1 therapy for recurrent or advanced endometrial cancer approved in Canada, JEMPERLI is indicated as a monotherapy for the treatment of adult patients with mismatch repair deficient (dMMR) or microsatellite instability-high (MSI-H) recurrent or advanced endometrial cancer, that has progressed on or following prior treatment with a platinum containing regimen.
"Canadian women with recurrent endometrial cancer, or advanced disease that has progressed on or after chemotherapy, currently have few options for treatment and face a poor prognosis," said Marni Freeman, Country Medical Director at GSK Canada. "This critical approval has the potential to noticeably improve the treatment landscape for these women and demonstrates GSK's continued commitment to fill an unmet need of patients with gynaecologic cancers."
"Women with endometrial cancer have few therapeutic options. We at the Society of Gynecologic Oncology of Canada (GOC) welcome any approval that paves the way for women with endometrial cancer to access immunotherapy as part of their standard of care therapeutic options" expressed Dr. Helen MacKay, President, The Society of Gynecologic Oncology of Canada and Head, Division of Medical Oncology & Hematology, Sunnybrook Odette Cancer Centre.
About endometrial cancer
Endometrial cancer is the most common gynecologic cancer in Canada. The disease is found in the inner lining of the uterus, known as the endometrium, and represents 95% of all uterine cancers. It is estimated that 7,600 Canadian women will be diagnosed with endometrial cancer and 1,235 will die of the disease in 2021.
There are limited treatment options for women whose disease progresses on or after first-line therapy. Approximately 25% of women with endometrial cancer will be diagnosed with advanced disease and 20% will have a recurrence of the disease. The 5-year overall survival is estimated to be 83%, however is below 20% for those with metastatic disease, thus research and new treatment availability is of utmost importance.
About JEMPERLI (dostarlimab for injection)
JEMPERLI (dostarlimab for injection) is indicated as monotherapy for the treatment of adult patients with mismatch repair deficient (dMMR) or microsatellite instability-high (MSI-H) recurrent or advanced endometrial cancer that has progressed on or following prior treatment with a platinum containing regimen.
JEMPERLI is an anti-programmed death receptor-1 (PD-1) antibody that binds to the PD-1 receptor and blocks its interaction with the ligands PD-L1 and PD-L2. This conditional approval of JEMPERLI is based on results from the multi-cohort GARNET study. In addition to GARNET, JEMPERLI is being investigated as monotherapy and as part of combination regimens for women with recurrent or primary advanced endometrial cancer, stage III or IV non-mucinous epithelial ovarian cancer, and for patients with advanced solid tumours or metastatic cancer.
Please consult the Product Monograph at www.gsk.ca for complete safety information. The Product Monograph is also available by calling 1-800-387-7374.
Dec. 23, 2021 4:18 PM ET
By: Jonathan Block, SA News Editor1 Comment
DECEMBER, 20, 2021 DOWNLOAD(OPENS IN NEW WINDOW)
Authorized booster (50 µg of mRNA-1273) increases Omicron neutralizing antibody levels approximately 37-fold; a 100 µg booster dose of mRNA-1273 increases Omicron neutralizing antibody levels approximately 83-fold
M oderna will continue to advance an Omicron-specific booster (mRNA-1273.529) to clinical trials
C onference call to be held today at 8:00 a.m. ET
CAMBRIDGE, Mass.--(BUSINESS WIRE)-- Moderna, Inc. (Nasdaq: MRNA), a biotechnology company pioneering messenger RNA (mRNA) therapeutics and vaccines, today announced preliminary neutralizing antibody data against the Omicron variant following the Company’s booster candidates at 50 µg and 100 µg dose levels. The currently authorized 50 µg booster of mRNA-1273 increased neutralizing antibody levelsagainst Omicron approximately 37-fold compared to pre-boost levels and a 100 µg dose of mRNA-1273 increased neutralizing antibody levelsapproximately 83-fold compared to pre-boost levels.
“The dramatic increase in COVID-19 cases from the Omicron variant is concerning to all. However, these data showing that the currently authorized Moderna COVID-19 booster can boost neutralizing antibody levels 37-fold higher than pre-boost levels are reassuring,” said Stéphane Bancel, Chief Executive Officer of Moderna. “To respond to this highly transmissible variant, Moderna will continue to rapidly advance an Omicron-specific booster candidate into clinical testing in case it becomes necessary in the future. We will also continue to generate and share data across our booster strategies with public health authorities to help them make evidence-based decisions on the best vaccination strategies against SARS-CoV-2.”
As previously described, the Company is continuously advancing booster candidates to address emerging variants of concern (VOC). The strategy includes evaluating the prototype vaccine (mRNA-1273) at the authorized booster dose (50 µg) and a higher dose (100 µg), multivalent candidates that incorporate previous VOCs (mRNA-1273.211, mRNA-1273.213) also at 50 µg and 100 µg, and VOC-specific booster candidates (Delta, Omicron). Booster candidates are being evaluated in ongoing Phase 2/3 studies of approximately 300-600 participants per arm.
Today’s data includes sera from 20 booster recipients each of mRNA-1273 at the 50 µg and 100 µg dose levels, multivalent candidate mRNA-1273.211 at the 50 µg and 100 µg dose levels, and multivalent candidate mRNA-1273.213 at the 100 µg dose level. Neutralizing antibodies against Omicron were assessed in a pseudovirus neutralization titer (ID50) assay (PsVNT) conducted at laboratories established by the National Institute of Allergy and Infectious Diseases’ (NIAID) Vaccine Research Center and Duke University Medical Center. A preprint submission is being prepared based on the data.
All groups had low neutralizing antibody levels in the Omicron PsVNT assay prior to boosting. At day 29 post boost, the authorized 50 µg booster of mRNA-1273 increased neutralizing geometric mean titers (GMT) against Omicron to 850, which is approximately 37-fold higher than pre-boost levels. At day 29 post boost, the 100 µg dose booster of mRNA-1273 increased neutralizing GMT to 2228, which is approximately 83-fold higher than pre-boost levels. The multivalent candidates boosted Omicron specific neutralizing antibody levels to similarly high levels at both the 50 µg and 100 µg levels. Based on the strength of neutralizing titers generated by mRNA-1273, the rapid pace of Omicron expansion, and the increased complexity of deploying a new vaccine, the Company will focus its near-term efforts to address Omicron on the mRNA-1273 booster. The Company will continue to assess the breadth and durability of neutralizing antibodies from the multivalent booster candidates in the months ahead.
The Company also announced the safety and tolerability data from the Phase 2/3 study of the 100 µg booster dose of mRNA-1273 (N=305). A 100 µg booster dose of mRNA-1273 was generally safe and well tolerated. The frequency and nature of solicited systemic and local adverse events 7 days after receiving a booster were generally comparable to those seen after the two-dose primary series. There was a trend toward slightly more frequent adverse reactions following the 100 µg booster dose relative to the authorized 50 µg booster dose of mRNA-1273.
Moving forward, given the strength of the mRNA-1273 and the speed at which the Omicron variant is spreading, Moderna’s first line of defense against Omicron will be a booster dose of mRNA-1273. Given the long-term threat demonstrated by Omicron’s immune escape, Moderna will also continue to develop an Omicron-specific variant vaccine (mRNA-1273.529) that it expects to advance into clinical trials in early 2022 and will evaluate including Omicron in its multivalent booster program.
To learn more, visit www.modernatx.com.
Dec. 20, 2021 6:04 AM ET Moderna, Inc. (MRNA) By: Mamta Mayani, SA News Editor7 Comments
PUBLISHED21 December 2021
The supplemental Biologics License Application (sBLA) for Ultomiris (ravulizumab-cwvz) in adults with generalised myasthenia gravis (gMG) has been accepted for Priority Review by the US Food and Drug Administration (FDA).
The FDA set a Prescription Drug User Fee Act date during the second quarter of 2022, following use of a rare disease priority review voucher by Alexion, AstraZeneca’s Rare Disease group.
gMG is a rare, debilitating, chronic, autoimmune neuromuscular disease that leads to a loss of muscle function and severe weakness.1 The diagnosed prevalence of gMG in the US is estimated at 64,000.2-10
Marc Dunoyer, Chief Executive Officer, Alexion, said: “Soliris was the first new treatment approved for this devastating disease in approximately 60 years, and this filing for Ultomiris demonstrates Alexion’s continued commitment to improve outcomes for patients living with gMG. The Phase III trial shows that Ultomiris may help a broader range of patients including those with milder symptoms or who are earlier in their treatment journey.”
The sBLA submission in the US is based on results from the Phase III trial of Ultomiris in gMG, which were announced by Alexion in July 2021, and showed efficacy as early as Week 1 and sustained for 52 weeks (26 weeks randomised controlled period + 26 weeks of open-label extension). In the trial, the safety profile of Ultomiris was consistent with that observed in Phase III trials of Ultomiris in paroxysmal nocturnal haemoglobinuria (PNH) and atypical haemolytic uraemic syndrome (aHUS).
Regulatory submissions for Ultomiris for the treatment of gMG are also currently under review with health authorities in the European Union (EU) and Japan.
Clinical trial
The global Phase III randomised, double-blind, placebo-controlled, multicentre 26-week trial evaluated the safety and efficacy of Ultomiris in adults with gMG who were not previously treated with a complement inhibitor medicine. The trial enrolled 175 patients across North America, Europe, Asia-Pacific and Japan. Participants were required to have a confirmed myasthenia gravis diagnosis at least six months prior to the screening visit with a positive serologic test for anti-AChR antibodies, Myasthenia Gravis-Activities of Daily Living Profile (MG-ADL) total score of at least 6 at trial entry and Myasthenia Gravis Foundation of America Clinical Classification Class II to IV at screening. There was no requirement for prior treatment failure, and patients could stay on stable standard of care medicines, with a few exceptions, for the duration of the trial.16
Patients were randomised 1:1 to receive Ultomiris or placebo for a total of 26 weeks. Patients received a single weight-based loading dose on Day 1, followed by regular weight-based maintenance dosing beginning on Day 15, every eight weeks. The primary endpoint of change from baseline in the MG-ADL total score at Week 26 was assessed along with multiple secondary endpoints evaluating improvement in disease-related and quality-of-life measures.
Patients who completed the randomised controlled period were eligible to continue into an open-label extension period evaluating the safety and efficacy of Ultomiris for up to two years, which is ongoing. At the time of the preliminary analysis of the open-label extension period, 75 patients had completed 26 weeks of treatment, for a total of 52 weeks of treatment.
Ultomiris
Ultomiris (ravulizumab), the first and only long-acting C5 complement inhibitor, offers immediate, complete, and sustained complement inhibition. The medication works by inhibiting the C5 protein in the terminal complement cascade, a part of the body’s immune system. When activated in an uncontrolled manner, the complement cascade over-responds, leading the body to attack its own healthy cells. Ultomiris is administered intravenously every eight weeks or, for paediatric patients less than 20kg, every four weeks, following a loading dose. Ultomiris is approved in the US for the treatment of adults and children (one month of age and older) with PNH; in the EU for adults, as well as for children (with a body weight of 10kg or above) and adolescents with PNH who experience haemolysis with clinical symptom(s) indicative of high disease activity, as well as for individuals who are clinically stable after having been treated with Soliris for at least the past six months; and in Japan as a treatment for adults with PNH. It is also approved in the US for aHUS to inhibit complement-mediated thrombotic microangiopathy in adult and paediatric (one month of age and older) patients, in the EU for the treatment of adults and children with a body weight of at least 10kg with aHUS, as well as in Japan for adults and children with aHUS.
Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Dec. 21, 2021 6:40 AM ET
By: Mamta Mayani, SA News Editor
December 21, 2021
-- Approval Based on Phase 3 Data Showing Veklury Significantly Reduced Risk of Hospitalization Compared with Placebo --
-- Expanded Indication Will Enable Use at Earlier Stages of the Disease to Help Prevent Disease Progression of COVID-19 in High-Risk Patients --
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the European Commission (EC) has approved a variation to the Conditional Marketing Authorization for Veklury® (remdesivir) to include adults who do not require supplemental oxygen and are at an increased risk of progressing to severe COVID-19. This decision follows the positive recommendation of the Committee for Medicinal Products for Human Use (CHMP), the scientific committee of the European Medicines Agency (EMA), to expand the indication for Veklury on December 16.
“As the rates of COVID-19 climb again and new variants like Omicron emerge, we need effective tools like Veklury to treat various stages of the disease,” said Roger Paredes, MD, PhD, Chief Infectious Diseases Department, and IrsiCaixa AIDS Research Institute Hospital Universitari Germans Trias I Pujol Badalona, Spain. “We can now use Veklury to help prevent high-risk patients from progressing to more severe disease, even when they do not require oxygen, as well as continue to utilize Veklury as a key tool in the treatment of severe disease. This latest approval will also help to relieve some of the pressure on healthcare systems that are already under significant strain from the burden of COVID-19.”
The EC’s decision is supported by results from a Phase 3 randomized, double-blind, placebo-controlled trial to evaluate the efficacy and safety of a three-day course of Veklury for intravenous (IV) use for the treatment of COVID-19 in non-hospitalized patients at high risk for disease progression. In an analysis of 562 participants randomly assigned in a 1:1 ratio to receive Veklury or placebo, Veklury demonstrated a statistically significant 87% reduction in risk for the composite primary endpoint of COVID-19 related hospitalization or all-cause death by Day 28 (0.7% [2/279]) compared with placebo (5.3% [15/283]) p=0.008. In the study, no deaths were observed in either arm by Day 28. The safety profile was similar between Veklury and placebo across the variety of outpatient settings in this trial, with the most common treatment emergent adverse events (≥5%) in patients taking Veklury being nausea and headache. These data have also been shared with other regulatory agencies around the world and submitted for scientific peer-reviewed publication.
This expanded indication in the EU adds to the previous Conditional Marketing Authorization of Veklury enabling the treatment of COVID-19 in adults and adolescents (aged 12 to less than 18 years and weighing at least 40 kg) with pneumonia requiring supplemental oxygen (low- or high-flow oxygen or other non-invasive ventilation at start of treatment).
“The swift action of the European Commission is a testament to the need for effective treatments that can be used earlier in the course of disease to help people with COVID-19 across Europe,” said Merdad Parsey, M.D., Ph.D., Chief Medical Officer, Gilead Sciences. “As we learn more about how COVID-19 disease progresses, it is clear that an antiviral like Veklury can have a significant impact if used early in the course of disease. As the antiviral standard of care for patients hospitalized with COVID-19, we are proud of the role Veklury continues to play on the front lines of the pandemic, and we believe Veklury will now be able to help more patients decrease their time to recovery from COVID-19 in Europe.”
In the United States, Veklury is indicated for adults and pediatric patients (12 years of age and older and weighing at least 40 kg) for the treatment of COVID-19 requiring hospitalization. The use of Veklury for the treatment of non-hospitalized patients with three days of dosing in the U.S. is investigational, and the safety and efficacy for this use and dosing duration have not been approved by the U.S. Food and Drug Administration (FDA). Veklury is contraindicated in patients who are allergic to Veklury or any of its components; please see below for additional Important Safety Information for Veklury.
As new SARS-CoV-2 variants of concern emerge around the world, Gilead continuously evaluates the effectiveness of Veklury against viral variants. An initial analysis of genetic information from the Omicron variant suggests that Veklury will continue to be active against this variant. Gilead will conduct laboratory testing to confirm this analysis. To date, no major genetic changes have been identified in any of the known variants of concern that would significantly alter the viral RNA polymerase targeted by Veklury. Veklury’s antiviral activity has been tested in vitro against isolates of variants of SARS-CoV-2, including Alpha, Beta, Gamma, Delta and Epsilon. These laboratory findings suggest that Veklury will continue to be active against the currently identified variations in the SARS-CoV-2 virus, including the Omicron variant.
About Veklury
Veklury (remdesivir) is a nucleotide analog invented by Gilead, building on more than a decade of the company’s antiviral research. Veklury is the antiviral standard of care for the treatment of hospitalized patients with COVID-19. At this time, more than half of patients hospitalized with COVID-19 in the United States are treated with Veklury. It can help reduce disease progression across the spectrum of disease severity and enable hospitalized patients to recover faster, freeing up limited hospital resources and saving healthcare systems money.
Veklury is approved or authorized for temporary use in approximately 50 countries worldwide. To date, Veklury and generic remdesivir have been made available to nine million patients around the world, including 6.5 million people in 127 middle- and low-income countries through Gilead’s voluntary licensing program. These licenses currently remain royalty-free, reflecting Gilead’s existing commitment to enabling broad patient access to remdesivir.
U.S. Indication for Veklury
Veklury® (remdesivir 100 mg for injection) is indicated for adults and pediatric patients (12 years of age and older and weighing at least 40 kg) for the treatment of COVID-19 requiring hospitalization. Veklury should only be administered in a hospital or in a healthcare setting capable of providing acute care comparable to inpatient hospital care. Veklury is contraindicated in patients who are allergic to Veklury or any of its components. For more information, please see the U.S. full Prescribing Information available at www.gilead.com.
U.S. full Prescribing Information for Veklury is available at www.gilead.com.
Veklury, Gilead and the Gilead logo are registered trademarks of Gilead Sciences, Inc., or its related companies.
For more information about Gilead, please visit the company’s website at www.gilead.com , follow Gilead on Twitter (@Gilead Sciences) or call Gilead Public Affairs at 1-800-GILEAD-5 or 1-650-574-3000.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211216006213/en/
Source: Gilead Sciences, Inc.
Dec. 21, 2021 4:52 AM ET
By: Mamta Mayani, SA News Editor3 Comments
FDA Approves Two New Indications for XARELTO® (rivaroxaban) to Help Prevent and Treat Blood Clots in Pediatric PatientsXARELTO® is available as both an oral tablet and oral suspension formulation for use in appropriate children less than 18 years of ageConvenient oral suspension formulation advances standard of care for children; alleviates administration challenges found with injectable alternativesXARELTO® now has 11 indications, the most of any direct oral anticoagulant (DOAC), and is the only Factor Xa anticoagulant to offer flexible weight-based dosing for pediatric patients
RARITAN, NJ, Dec. 20, 2021 – The Janssen Pharmaceutical Companies of Johnson & Johnson announced today that the U.S. Food and Drug Administration (FDA) has approved two pediatric indications for XARELTO® (rivaroxaban): the treatment of venous thromboembolism (VTE, or blood clots) and reduction in the risk of recurrent VTE in patients from birth to less than 18 years after at least five days of initial parenteral (injected or intravenous) anticoagulant treatment; and thromboprophylaxis (prevention of blood clots and blood-clot related events) in children aged two years and older with congenital heart disease who have undergone the Fontan procedure. XARELTO® is the only direct oral anticoagulant (DOAC) FDA-approved for primary prevention of clots in pediatric patients following the Fontan procedure and the only DOAC in the U.S. to offer an oral suspension formulation for flexible, body weight-adjusted dosing options for pediatric patients.
CLICK TO TWEET: #BREAKINGNEWS: @US_FDA approves @JanssenUS therapy for the treatment and prevention of #bloodclots in #pediatric patients, providing an alternative to injectable anticoagulants that have long been the standard of care. Learn more: https://bit.ly/3E9qAOk
While VTE more commonly occurs in adults, blood clots can still be a serious problem in children, affecting approximately 58 per 10,000 of those hospitalized in the U.S., with rates increasing. Children may be at greater risk of blood clots when suffering from other conditions, such as infectious diseases, active cancer, or after undergoing surgery, like the Fontan procedure, which is performed in children who have a single functioning heart ventricle to redirect blood flow from the lower body to the lungs.2,3
Current medical guidelines are limited and recommend that young patients with or at risk for developing blood clots be treated with standard anticoagulation therapy, such as warfarin or heparin.4 Often times, physicians have to adjust adult doses, based on limited data for these therapies for younger patients.4 Additionally, for some of these treatment options, that can mean painful injections, dietary restrictions and regular laboratory monitoring – all things that can be especially challenging for younger patients and their caregivers.4,5,6
“Historically, there has been limited guidance and options for healthcare providers on how to help reduce potentially serious, even fatal, blood clots and related events in young children,” said Andrew Van Bergen, M.D., Pediatric Cardiologist, Advocate Children’s Hospital.* “We have had to adjust adult doses of standard anticoagulation therapies, which are both burdensome and uncomfortable for patients, and require frequent monitoring. Now that XARELTO® is FDA-approved with weight-based dosing options, either as tablets or liquid formulation, a convenient option is available allowing flexibility to tailor the treatment for my patients. This is a major advancement in antithrombotic care for those patients under the age of 18.”
The oral suspension formulation will be administered through a color-coded dosing device that was designed to help minimize dosing errors and is expected to become available in the U.S. for pediatric patients in mid-January 2022. The oral tablets are currently available in the U.S. for appropriate pediatric patients.
“When a child is experiencing health challenges, learning that they are also at risk for a blood clot can feel overwhelming for the patient, and also their parents or caregivers,” said Andrea Baer, MS, BCPA, Executive Director of The Mended Hearts, Inc., a patient advocacy organization whose program Mended Little Hearts serves patients and families with congenital heart disease.** “Knowing that there is now an FDA-approved oral treatment option to reduce the risk of blood clots that’s easy and may be more comfortable than injections to administer may help ease that burden.”
XARELTO® now has 11 indications in the U.S. – the most of any DOAC – and is the most studied oral Factor Xa inhibitor in its class. This latest approval is based on two Phase 3 pediatric studies from the industry-leading EXPLORER clinical research program, EINSTEIN-Jr, the largest study to date evaluating pediatric patients from birth to <18 years of age with previously diagnosed VTE; and UNIVERSE, the first clinical trial to examine a DOAC for the prevention of VTE in pediatric patients after recently undergoing the Fontan procedure.7,8
“Today’s FDA approval marks two new XARELTO® indications for pediatric patients, an often underrecognized, but especially important patient population,” said James List, M.D., Ph.D., Global Therapeutic Area Head, Cardiovascular, Metabolism, & Retina, Janssen Research & Development, LLC. “At Janssen, we are committed to addressing unmet medical needs and the approval of the 10th and 11th indications for XARELTO® underscores its capability in reducing the risk of blood clots and cardiovascular events in patients from young to old and with a variety of conditions.”
In 2021, Janssen’s development partner, Bayer, received approval for XARELTO® in Canada, the European Union, the United Kingdom, Japan, Switzerland and in various Latin American countries for the treatment of VTE and prevention of VTE recurrence in the pediatric population, from birth to adolescents less than 18 years after at least five days of initial parenteral anticoagulation treatment.
About EINSTEIN-Jr
EINSTEIN-Jr was a randomized, multicenter, active-controlled, open-label Phase 3 study that evaluated the use of XARELTO® in 500 children, aged birth to 17 years, with previously diagnosed acute VTE who had started parenteral anticoagulation therapy. Participants were enrolled from November 2014 to September 2018, from 107 sites in 28 countries, and were assigned in a 2:1 ratio to receive either an open-label, body weight-adjusted dose of XARELTO® to approximate a 20-mg adult dose (tablets or a new oral suspension) (n=335) or standard anticoagulation therapy (n=165). EINSTEIN-Jr is the largest pediatric study completed to date in the entire pediatric age population for the treatment of VTE. It is part of the comprehensive EINSTEIN program, which also included four pivotal Phase 3 studies in adult populations: EINSTEIN-DVT, EINSTEIN-PE, EINSTEIN-EXT and EINSTEIN CHOICE.
About UNIVERSE
UNIVERSE was a randomized, multicenter, open-label, active-controlled, two-part, Phase 3 study that examined the use of a novel, oral suspension XARELTO® formulation in children 2-8 years old with single ventricle physiology who had the Fontan procedure within four months before enrollment. From November 2016 to June 2019, a total of 112 participants were enrolled across 36 sites in 10 countries.
Part A evaluated the single- and multiple-dose pharmacokinetic (PK) and pharmacodynamic (PD) properties of XARELTO® while Part B evaluated the comparative safety and efficacy of XARELTO® versus aspirin when used for thromboprophylaxis for 12 months.
About EXPLORER
A collaborative effort between Bayer and Janssen, our industry-leading EXPLORER program seeks to generate important clinical evidence on the safety and efficacy of XARELTO® and its potential role in addressing a wide range of critical medical needs. EXPLORER is unmatched by any oral anticoagulant in the Factor Xa inhibitor class in its size, scope and ambition.
WHAT IS XARELTO® (rivaroxaban)?
XARELTO® is a prescription medicine used to:
• reduce the risk of stroke and blood clots in adults who have a medical condition called atrial fibrillation that is not caused by a heart valve problem. With atrial fibrillation, part of the heart does not beat the way it should. This can lead to the formation of blood clots, which can travel to the brain, causing a stroke, or to other parts of the body
• treat blood clots in the veins of your legs (deep vein thrombosis or DVT) or lungs (pulmonary embolism or PE)
• reduce the risk of blood clots happening again in adults who continue to be at risk for DVT or PE after receiving treatment for blood clots for at least 6 months
• help prevent a blood clot in the legs and lungs of adults who have just had hip or knee replacement surgery
• help prevent blood clots in certain adults hospitalized for an acute illness and after discharge, who are at risk of getting blood clots because of the loss of or decreased ability to move around (mobility) and other risks for getting blood clots, and who do not have a high risk of bleeding
XARELTO® is used with low dose aspirin to:
• reduce the risk of serious heart problems, heart attack and stroke in adults with coronary artery disease (a condition where the blood supply to the heart is reduced or blocked)
• reduce the risk of a sudden decrease in blood flow to the legs, major amputation, serious heart problems or stroke in adults with peripheral artery disease (a condition where the blood flow to the legs is reduced) and includes adults who have recently had a procedure to improve blood flow to the legs.
XARELTO® is used in children to:
• treat blood clots or reduce the risk of blood clots from happening again in children from birth to less than 18 years, after receiving initial treatment with injectable or intravenous medicines used to treat blood clots.
• help prevent blood clots in children 2 years and older with congenital heart disease after the Fontan procedure.
XARELTO® was not studied and is not recommended in children less than 6 months of age who:
• were less than 37 weeks of growth (gestation) at birth
• had less than 10 days of oral feeding, or
• had a body weight of less than 5.7 pounds (2.6 kg)
Please read full Prescribing Information, including Boxed Warnings, and Medication Guide for XARELTO®.
Dec. 21, 2021 12:35 PM ET Johnson & Johnson (JNJ) By: Jonathan Block, SA News Editor1 Comment
December 21, 2021Download PDF-
Study met all primary and key secondary endpoints
- Safety profile consistent with prior lebrikizumab studies in atopic dermatitis
- Global regulatory submissions to occur next year based on data from the Phase 3 clinical trial program
INDIANAPOLIS, Dec. 21, 2021 /PRNewswire/ -- Lebrikizumab, an IL-13 inhibitor, significantly improved disease severity when combined with topical corticosteroids (TCS) in people with moderate-to-severe atopic dermatitis (AD) in Eli Lilly and Company's (NYSE: LLY) third pivotal Phase 3 trial (ADhere). By Week 16, the study met all primary and key secondary endpoints for patients on the lebrikizumab combination arm.
Lebrikizumab is a novel, investigational monoclonal antibody (mAb) that binds soluble IL-13 with high affinity, has high bioavailability, a long half-life and blocks IL-13 signaling.1-5 In people with AD, the IL-13 protein—a central pathogenic mediator in the disease—is overexpressed, driving multiple aspects of AD pathophysiology by promoting T-helper type 2 (Th2) cell inflammation and resulting in skin barrier dysfunction, itch, infection, flares and hard, thickened areas of skin.6,7
"AD is often complex and challenging to treat, as many patients need help controlling their symptoms when topical steroids alone are not enough," said Eric Simpson, M.D., M.C.R., Professor of Dermatology and Director of Clinical Research at Oregon Health & Science University in Portland and a principal investigator of the ADhere study. "I'm encouraged by the aggregate efficacy and safety data which have demonstrated the potential for lebrikizumab as both monotherapy and combination therapy to address unmet needs and improve care for people living with persistent itch and inflamed skin caused by AD."
The primary endpoints were Investigator Global Assessment (IGA) score of clear (0) or almost clear (1) skin with a reduction of at least two points from baseline and at least a 75 percent change from baseline in the Eczema Area and Severity Index (EASI) score, both at Week 16. Lebrikizumab in combination with TCS also achieved all key secondary endpoints versus placebo in combination with TCS in patients with AD, including skin improvement, itch relief, improvement in interference of itch on sleep, and quality of life. Key secondary endpoints were measured by EASI, the Pruritus Numeric Rating Scale, Sleep-Loss due to Pruritus, and the Dermatology Life Quality Index.
Safety results in the 16-week placebo-controlled ADhere study were consistent with the 16-week period of the two monotherapy studies in the lebrikizumab Phase 3 program for AD. The most common adverse events (AEs) included conjunctivitis and headache for lebrikizumab-treated patients.
In August 2021, Lilly announced top-line data from ADvocate 1 and ADvocate 2 showing lebrikizumab as a monotherapy met primary and all key secondary endpoints including itch, interference of itch on sleep and quality of life at Week 16.
"Physicians treating atopic dermatitis continue to need new options for their patients along with current standard of care, given the heterogeneity of disease and variable outcomes for patients' signs and symptoms," said Lotus Mallbris, M.D., Ph.D., vice president of global immunology development and U.S. and global medical affairs at Lilly. "These results add to the growing body of evidence from our robust Phase 3 clinical trial program for lebrikizumab and support the hypothesis that targeting the IL-13 pathway is critical in treating AD and helping improve outcomes for these patients. We look forward to continuing to evaluate lebrikizumab's clinical utility in the ongoing studies in the hopes of making this medicine available to those who still have unmet needs."
Additional data analyses from ADhere, along with results from two monotherapy Phase 3 trials, ADvocate 1 and ADvocate 2, are planned for future scientific congresses in 2022. Pending successful completion of the ongoing ADvocate 1 and ADvocate 2 monotherapy trials, Lilly and Almirall intend to begin U.S., EU and other regulatory submissions next year.
"These results validate the important role that IL-13 cytokine inhibitors play in AD treatment and the success of lebrikizumab in this study represents another key achievement in our journey to offer treatment advances in AD for patients and healthcare professionals," stated Karl Ziegelbauer, Ph.D., Almirall S.A.'s Chief Scientific Officer.
Lilly has exclusive rights for development and commercialization of lebrikizumab in the United States and rest of world outside Europe. Almirall has licensed the rights to develop and commercialize lebrikizumab for the treatment of dermatology indications, including AD, in Europe.
About ADhere and the Phase 3 Program
ADhere is a 16-week randomized, double-blind, placebo-controlled, parallel-group, Phase 3 study to evaluate the efficacy and safety of lebrikizumab in combination with TCS in adult and adolescent patients (aged 12 to less than 18 years of age and weighing at least 40 kg) with moderate-to-severe AD. In the study, patients' AD symptoms were inadequately controlled by TCS with or without topical calcineurin inhibitors (TCI).
The U.S. Food and Drug Administration (FDA) granted lebrikizumab Fast Track designation in AD in December 2019. The lebrikizumab Phase 3 program consists of five key ongoing, global studies including two monotherapy studies, today's ADhere combination study as well as long-term extension (ADjoin) and adolescent open label (ADore) trials.
About Lebrikizumab
Lebrikizumab is a novel, investigational, monoclonal antibody designed to bind IL-13 with high affinity to specifically prevent the formation of the IL-13Rα1/IL-4Rα heterodimer complex and subsequent signaling, thereby inhibiting the biological effects of IL-13 in a targeted and efficient fashion. IL-13 is a central pathogenic mediator that drives multiple aspects of the pathophysiology underlying the range of signs and symptoms of AD by promoting type 2 inflammation and mediating its effects on tissue, resulting in skin barrier dysfunction, itch, skin thickening and infection.6
To learn more about Lilly, please visit us at lilly.com and lilly.com/newsroom. P-LLY
View original content to download multimedia:https://www.prnewswire.com/news-releases/lillys-lebrikizumab-demonstrated-significant-skin-improvement-and-itch-relief-when-combined-with-topical-corticosteroids-in-people-with-atopic-dermatitis-in-third-phase-3-study-301448625.html
SOURCE Eli Lilly and Company
Dec. 21, 2021 4:33 AM ET
By: Mamta Mayani, SA News Editor4 Comments
Otezla is the First and Only Oral Therapy Approved in Adult Patients with Plaque Psoriasis Across all Severities, Including Mild, Moderate and Severe
Approximately 8 Million People in the U.S. Have Plaque Psoriasis
THOUSAND OAKS, Calif., Dec. 20, 2021 /PRNewswire/ -- Amgen (NASDAQ: AMGN) today announced that the U.S. Food and Drug Administration (FDA) has approved Otezla® (apremilast) for the treatment of adult patients with plaque psoriasis who are candidates for phototherapy or systemic therapy. With this expanded indication, Otezla is now the first and only oral treatment approved in adult patients with plaque psoriasis across all severities, including mild, moderate and severe.
"Plaque psoriasis can place a significant burden on the lives of patients, regardless of the severity of skin involvement. A substantial unmet need remains for mild to moderate plaque psoriasis patients for whom topical therapies may not be sufficient, especially for those with difficult-to-treat areas, like the scalp," said David M. Reese, M.D., executive vice president of Research and Development at Amgen. "With this expanded indication for Otezla, patients across all levels of disease severity now have an oral, systemic option that has already been used by more than 650,000 people worldwide and has no lab monitoring requirement."1
"Given that psoriasis is a systemic inflammatory disease, some patients may need more than surface level relief," said Dr. Linda Stein Gold, director of Dermatology Clinical Research at Henry Ford Health System, Detroit, and ADVANCE investigator. "For the first time, dermatologists can offer patients struggling with plaque psoriasis of any degree an effective oral treatment with an established safety profile."
The FDA approval is based on findings from the Phase 3 ADVANCE trial, in which five times as many adults with mild to moderate plaque psoriasis receiving oral Otezla 30 mg twice daily achieved the primary endpoint of Static Physician's Global Assessment (sPGA) response at week 16 compared to placebo (21.6% versus 4.1%, p<0.0001), a difference that was statistically significant. Otezla also demonstrated statistically significant improvements in key symptoms, such as Whole Body Itch NRS response (43.2% versus 18.6%), and a difficult-to-treat area, the scalp, measured by Scalp Physician's Global Assessment (ScPGA) response (44% versus 16.6%), at week 16 compared to placebo. Improvements in sPGA response, Whole Body Itch NRS and ScPGA response were observed as early as week 2 and maintained through week 32.
The adverse events observed in the trial were consistent with the known safety profile of Otezla. The most commonly reported (≥5%) treatment-emergent adverse events with Otezla treatment were diarrhea, headache, nausea and nasopharyngitis.
Approximately 8 million people in the U.S. have plaque psoriasis, and 5 million people in the U.S. have mild to moderate disease. Despite the prevalence and treatment advances in recent years, a significant unmet need remains, particularly for people with mild to moderate plaque psoriasis or those who experience persistent symptoms despite topical treatment.
"Plaque psoriasis often affects patients more severely than can be measured by Body Surface Area alone, particularly for those with manifestations in difficult-to-treat areas like the scalp. The location of plaques may make the area sensitive to topical treatments or challenging to apply them,"2 said Stacie Bell, Ph.D., chief scientific and medical officer at the National Psoriasis Foundation. "It's welcome news to finally have an oral systemic option with a well-established safety profile available for all adult plaque psoriasis patients."
Otezla is approved for three indications in the U.S., including adult patients with plaque psoriasis who are candidates for phototherapy or systemic therapy, adult patients with active psoriatic arthritis and for adult patients with oral ulcers associated with Behçet's Disease. Otezla is the most prescribed brand for plaque psoriasis patients starting systemic therapy.3 Amgen is committed to investigating the potential of Otezla across the continuum of psoriasis, including underserved patients with genital psoriasis, pediatric psoriasis, juvenile psoriatic arthritis and other areas of high burden.
About ADVANCE (PSOR-022)
ADVANCE (NCT03721172) is a Phase 3, multicenter, randomized, placebo-controlled, double-blind study evaluating the efficacy and safety of Otezla in patients with mild to moderate plaque psoriasis (defined as BSA involvement of 2% to 15%, Psoriasis Area and Severity Index [PASI] score of 2 to 15, Physician's Global Assessment [sPGA] score of 2 to 3). The study randomized 595 patients 1:1 to receive Otezla (n=297) 30 mg twice daily or placebo (n=298) for the first 16 weeks. All patients then received Otezla during an open-label extension phase through week 32.
The primary endpoint was the percentage of patients with sPGA response (defined as a sPGA score of clear [0] or almost clear [1] with at least a 2-point reduction from baseline) at week 16. Secondary endpoints include the percentage of patients with Whole Body Itch Numeric Rating Scale (NRS) score response (defined as at least a 4-point reduction from baseline, where 0 represents no itch and 10 represents the worst imaginable itch) and Scalp Physician Global Assessment (ScPGA) response (defined as an ScPGA score of clear [0] or almost clear [1] with at least a 2-point reduction from baseline) at week 16.
About Otezla® (apremilast)
OTEZLA® (apremilast) is an oral small-molecule inhibitor of phosphodiesterase 4 (PDE4) specific for cyclic adenosine monophosphate (cAMP). PDE4 inhibition results in increased intracellular cAMP levels, which is thought to indirectly modulate the production of inflammatory mediators. The specific mechanism(s) by which Otezla exerts its therapeutic action in patients is not well defined.
INDICATIONS
Otezla® (apremilast) is indicated for the treatment of adult patients with plaque psoriasis who are candidates for phototherapy or systemic therapy.
Otezla is indicated for the treatment of adult patients with active psoriatic arthritis.
Otezla is indicated for the treatment of adult patients with oral ulcers associated with Behçet's Disease.
Please click here for the full Prescribing Information for Otezla.
For more information, visit www.amgen.com and follow us on www.twitter.com/amgen.
View original content to download multimedia:https://www.prnewswire.com/news-releases/fda-approves-otezla-apremilast-for-the-treatment-of-adult-patients-with-plaque-psoriasis-regardless-of-severity-level-301448542.html
SOURCE Amgen
Dec. 20, 2021 6:08 PM ET Amgen Inc. (AMGN)
By: Jonathan Block, SA News Editor1 Comment
12/17/2021
- If approved, PADCEV would be the first medicine for patients in the EU who have received prior platinum-based chemotherapy and a PD-1/L1 inhibitor -
TOKYO & BOTHELL, Wash.--(BUSINESS WIRE)-- Astellas Pharma Inc. (TSE:4503, President and CEO: Kenji Yasukawa, Ph.D., “Astellas”) and Seagen Inc. (Nasdaq:SGEN) today announced that the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) has adopted a positive opinion, recommending approval of the antibody-drug conjugate (ADC) PADCEV™ (enfortumab vedotin) as monotherapy for the treatment of adult patients with locally advanced or metastatic urothelial cancer who have previously received platinum-containing chemotherapy and a PD-1/L1 inhibitor.1
Urothelial cancer is the most common type of bladder cancer.2 In Europe, an estimated 204,000 people were diagnosed with urothelial cancer in 2020, and more than 67,000 died as a result of the disease.3 If approved by the European Commission (EC), enfortumab vedotin will be the first ADC authorized in the European Union for people living with advanced urothelial cancer.
“People with advanced bladder cancer have few treatment options after platinum-based chemotherapy and immunotherapy,” said Ahsan Arozullah, M.D., M.P.H., Vice President, Medical Sciences-Oncology, Astellas. “The CHMP's positive opinion is an important step as we work to expand availability of enfortumab vedotin as quickly as possible.”
The CHMP recommendation is based on data from the global phase 3 EV-301 trial, which evaluated enfortumab vedotin versus chemotherapy in adult patients with locally advanced or metastatic urothelial cancer who were previously treated with platinum-based chemotherapy and a PD-1/L1 inhibitor. Results from the trial, which had a primary endpoint of overall survival, were published in the New England Journal of Medicine.
The positive opinion from the CHMP will now be reviewed by the EC. EC decisions are valid in the European Union Member States, as well as Iceland, Norway and Liechtenstein.4
About the EV-301 Trial
The EV-301 trial (NCT03474107) was a global, multicenter, open-label, randomized phase 3 trial designed to evaluate enfortumab vedotin versus physician's choice of chemotherapy (docetaxel, paclitaxel or vinflunine) in 608 patients with locally advanced or metastatic urothelial cancer who were previously treated with a PD-1/L1 inhibitor and platinum-based therapies.5 The primary endpoint was overall survival and secondary endpoints included progression-free survival, overall response rate, duration of response and disease control rate, as well as assessment of safety/tolerability and quality-of-life parameters.
About Enfortumab Vedotin
Enfortumab vedotin is an antibody-drug conjugate (ADC) that is directed against Nectin-4, a protein located on the surface of cells and highly expressed in bladder cancer.6,7 Nonclinical data suggest the anticancer activity of enfortumab vedotin is due to its binding to Nectin-4 expressing cells followed by the internalization and release of the anti-tumor agent monomethyl auristatin E (MMAE) into the cell, which result in the cell not reproducing (cell cycle arrest) and in programmed cell death (apoptosis).6
PADCEV (enfortumab vedotin-ejfv) U.S. Indication & Important Safety Information
For more information on our marketed products and robust pipeline, visit www.seagen.com and follow @SeagenGlobal on Twitter.
Indication
PADCEV® is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer (mUC) who:
About the Astellas and Seagen Collaboration
Astellas and Seagen are co-developing enfortumab vedotin under a 50:50 worldwide development and commercialization collaboration. In the United States, Astellas and Seagen co-promote enfortumab vedotin under the brand name PADCEV® (enfortumab vedotin-ejfv). In the Americas outside the US, Seagen holds responsibility for commercialization activities and regulatory filings. Outside of the Americas, Astellas holds responsibility for commercialization activities and regulatory filings.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211215006165/en/
Source: Seagen Inc.
Dec. 17, 2021 7:11 AM ET Seagen Inc. (SGEN), ALPMY
By: Mamta Mayani, SA News Editor
NewsDecember 17, 2021
argenx Announces U.S. Food and Drug Administration (FDA) Approval of VYVGART™ (efgartigimod alfa-fcab) in Generalized Myasthenia Gravis
Breda, the Netherlands—Dec. 17, 2021
Regulated Information/Inside Information
Breda, the Netherlands—Dec. 17, 2021—argenx SE (Euronext & Nasdaq: ARGX), a global immunology company committed to improving the lives of people suffering from severe autoimmune diseases and cancer, today announced that the U.S. Food and Drug Administration (FDA) has approved VYVGARTô (efgartigimod alfa-fcab) for the treatment of generalized myasthenia gravis (gMG) in adult patients who are anti-acetylcholine receptor (AChR) antibody positive. These patients represent approximately 85% of the total gMG population1. With this regulatory milestone, VYVGART is the first-and-only FDA-approved neonatal Fc receptor (FcRn) blocker.
This press release features multimedia. View the full release here.
Access and My VYVGART Path
argenx is committed to supporting affordable access to VYVGART. As part of this commitment, argenx is launching My VYVGART Path, a program designed to connect patients and clinicians to personalized support throughout the treatment journey. Program resources include disease and product education, access support and benefits verification, and financial assistance programs for eligible patients. Patients and healthcare providers can visit VYVGART.com for more information.
Adults living with gMG each experience the course of the disease differently, contributing to variability of disease severity and response to therapies. argenx prioritized early and active engagement with leading payers to address questions around broader budget predictability. The Company has reached agreements in principle with several national and regional commercial payers to structure a value-based agreement. The agreements are meant to provide predictability of cost for payers and appropriate access for patients.
“Generalized myasthenia gravis imposes a significant lifestyle and treatment burden on patients, families, and the overall healthcare system. This autoimmune disease affects each patient differently which can create variability in dosing and the resulting cost per patient,” said Steve Miller, M.D., Executive Vice President and Chief Clinical Officer at Cigna Corp. “The approval of VYVGART promises to address a treatment gap for patients suffering from this disease. argenx has put forth an innovative, value-based approach to contracting that will help payers with cost predictability as they face the challenge of ensuring real-world dosing remains affordable. This was a direct result of early engagement between argenx and Evernorth leading up to approval. Cigna looks forward to continued collaboration with argenx to enable appropriate access to the gMG patients who will benefit from VYVGART.”
Marketing Authorization Applications for efgartigimod for the treatment of gMG are currently under review with Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) and the European Medicines Agency (EMA), with anticipated decisions from each agency in the first quarter and second half of 2022, respectively.
argenx has an exclusive partnership agreement with Zai Lab for the development and commercialization of efgartigimod in Greater China. Zai Lab is on track to file for approval in Greater China by mid-2022. Under the terms of the strategic agreement with Zai Lab, argenx will receive a $25 million milestone payment with this U.S. approval of VYVGART. In addition, argenx has an exclusive partnership with Medison for the commercialization of efgartigimod in gMG in Israel. Medison is on track to file for approval in Israel in the second quarter of 2022.
See Important Safety Information and full Prescribing Information below for additional information.
What is VYVGARTTM (efgartigimod alfa-fcab)?
VYVGART is a prescription medicine used to treat a condition called generalized myasthenia gravis, which causes muscles to tire and weaken easily throughout the body, in adults who are positive for antibodies directed toward a protein called acetylcholine receptor (anti-AChR antibody positive).
About Phase 3 ADAPT Trial
The Phase 3 ADAPT trial was a 26-week randomized, double-blind, placebo-controlled, multi-center, global trial evaluating the safety and efficacy of VYVGART in adult patients with gMG. A total of 167 adult patients with gMG in North America, Europe and Japan enrolled in the trial. Patients were randomized in a 1:1 ratio to receive VYVGART or placebo, in addition to stable doses of their current gMG treatment. ADAPT was designed to enable an individualized treatment approach with an initial treatment cycle followed by subsequent treatment cycles based on clinical evaluation. The primary endpoint was the comparison of percentage of MG-ADL responders in the first treatment cycle between VYVGART and placebo treatment groups in the anti-AChR antibody positive population.
About VYVGART
VYVGART (efgartigimod alfa-fcab) is a human IgG1 antibody fragment that binds to the neonatal Fc receptor (FcRn), resulting in the reduction of circulating IgG. It is the first and only approved FcRn blocker. VYVGART is approved in the United States for the treatment of adults with generalized myasthenia gravis (gMG), who are anti-AChR antibody positive.
Dec. 17, 2021 5:06 PM ET ARGX
By: Jonathan Block, SA News Editor
First and Only Biologic to Consistently and Significantly Reduce Exacerbations in a Broad Population of Severe Asthma Patients
Only Biologic for Severe Asthma Approved With no Phenotype or Biomarker Limitations
THOUSAND OAKS, Calif., Dec. 17, 2021 /PRNewswire/ -- Amgen (NASDAQ:AMGN) today announced that the U.S. Food and Drug Administration (FDA) has approved Amgen and AstraZeneca's Tezspire™ (tezepelumab-ekko) for the add-on maintenance treatment of adult and pediatric patients aged 12 years and older with severe asthma.1
To view the Multimedia News Release, please visit: https://www.multivu.com/players/English/8812852-amgen-fda-approval-tezepelumab-severe-asthma-inflammation/
Tezspire was approved following a Priority Review by the FDA and based on results from the PATHFINDER clinical trial program. The application included results from the pivotal NAVIGATOR Phase 3 trial in which Tezspire demonstrated superiority across every primary and key secondary endpoint in patients with severe asthma, compared to placebo, when added to standard therapy.2
"Today's approval by the FDA marks the first time patients and their physicians will have a biologic option for severe asthma without phenotypic limitations and irrespective of biomarker levels," said David M. Reese, M.D., executive vice president of Research and Development at Amgen. "Asthma is a complex and chronic inflammatory disease that affects everyone differently. By working at the top of the inflammation cascade, Tezspire helps stop the inflammation that causes asthma attacks at the source and has the potential to treat a broad population of people with severe asthma, including those who have historically lacked effective treatment options."
Tezspire is a first-in-class biologic for severe asthma that acts at the top of the inflammatory cascade by targeting thymic stromal lymphopoietin (TSLP), an epithelial cytokine.3 It is the first and only biologic to consistently and significantly reduce asthma exacerbations across Phase 2 and 3 clinical trials, which included a broad population of severe asthma patients irrespective of key biomarkers, including blood eosinophil counts, allergic status and fractional exhaled nitric oxide (FeNO).2,3 Tezspire is the first and only biologic for severe asthma that does not have a phenotype—eosinophilic or allergic—or biomarker limitation within its approved label.4-11
"Due to the complex and heterogeneous nature of severe asthma and despite recent advances, many patients continue to experience frequent exacerbations, an increased risk of hospitalization and a significantly reduced quality of life," said Professor Andrew Menzies-Gow, director of the Lung Division, Royal Brompton Hospital, London, UK, and the principal investigator of the NAVIGATOR trial. "Tezspire represents a much-needed new treatment for the many patients who remain underserved and continue to struggle with severe, uncontrolled asthma."
"Severe asthma continues to have a debilitating impact on many of the 34 million people living with the disease worldwide, affecting their breathing and limiting aspects of day-to-day life. The approval of Tezspire is long-awaited positive news for the asthma community," said Tonya Winders, president and chief executive officer at the Allergy & Asthma Network (AAN) and president of the Global Allergy and Airways Patient Platform (GAAPP). "For the first time, many people living with severe asthma have the opportunity to receive treatment regardless of the cause of their inflammation."
Results from the NAVIGATOR Phase 3 trial were published in The New England Journal of Medicine in May 2021.2 In clinical studies of Tezspire, the most common adverse reactions were nasopharyngitis, upper respiratory tract infection and headache.2
Tezspire is under regulatory review in the EU, Japan and several other countries around the world.
Commitment to Patient Support
Amgen and AstraZeneca are committed to providing appropriate patients who are prescribed Tezspire with affordable access to the medicine. Patients, caregivers and physicians who need support or resources can contact the Tezspire Together program starting on Monday, Dec. 20 at 8:00 a.m. ET by calling 1-888-TZSPIRE (1-888-897-7473).
Tezspire™ (tezepelumab-ekko) U.S. Indication
Tezspire is a first-in-class medicine indicated for the add-on maintenance treatment of adult and pediatric patients aged 12 years and older with severe asthma.
Tezspire is not indicated for the relief of acute bronchospasm or status asthmaticus.
Please see the Tezspire full Prescribing Information.
About Tezspire™ (tezepelumab-ekko)
Tezspire is a first-in-class human monoclonal antibody that works on the primary source of inflammation: the airway epithelium, which is the first point of contact for viruses, allergens, pollutants and other environmental insults. Specifically, Tezspire targets and blocks TSLP, a key epithelial cytokine that sits at the top of multiple inflammatory cascades and initiates an overreactive immune response to allergic, eosinophilic and other types of airway inflammation associated with severe asthma.3,17 TSLP is released in response to multiple triggers associated with asthma exacerbations, including allergens, viruses and other airborne particles.3,17 Expression of TSLP is increased in the airways of patients with asthma and has been correlated with disease severity.3,18 Blocking TSLP may prevent the release of pro-inflammatory cytokines by immune cells, resulting in the prevention of asthma exacerbations and improved asthma control.3,18 By working at the top of the cascade, Tezspire helps stop inflammation at the source and has the potential to treat a broad population of severe asthma patients.3,18
Tezspire is also in development for other potential indications including chronic obstructive pulmonary disease, chronic rhinosinusitis with nasal polyps, chronic spontaneous urticaria and eosinophilic esophagitis (EoE). In October 2021, tezepelumab was granted Orphan Drug Designation by the FDA for the treatment of EoE.
About the Amgen and AstraZeneca Collaboration
In 2020, Amgen and AstraZeneca updated the 2012 collaboration agreement for Tezspire. Both companies will continue to share costs and profits equally after payment by AstraZeneca of a mid-single-digit royalty to Amgen. AstraZeneca continues to lead development and Amgen continues to lead manufacturing. All aspects of the collaboration are under the oversight of joint governing bodies. Under the amended agreement, Amgen and AstraZeneca will jointly commercialize Tezspire in North America. Amgen will record product sales in the U.S., with AZ recording its share of US profits as Collaboration Revenue. Outside of the U.S., AstraZeneca will record product sales, with Amgen recording profit share as Other/Collaboration revenue.
For more information, visit www.amgen.com and follow us on www.twitter.com/amgen.
View original content:https://www.prnewswire.com/news-releases/fda-approves-tezspire-tezepelumab-ekko-in-the-us-for-severe-asthma-301447689.html
SOURCE Amgen
Dec. 17, 2021 4:46 PM ET
By: Dulan Lokuwithana, SA News Editor2 Comments
December 17, 2021 6:45 am ET
Recommendation Based on Significant Disease-Free Survival Benefit Demonstrated With KEYTRUDA Versus Placebo in Phase 3 KEYNOTE-564 TrialKENILWORTH, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, today announced the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) has adopted a positive opinion recommending approval of KEYTRUDA, Merck’s anti-PD-1 therapy, as monotherapy for the adjuvant treatment of adults with renal cell carcinoma (RCC) at increased risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions.The positive opinion is based on results from the pivotal Phase 3 KEYNOTE-564 trial, in which KEYTRUDA demonstrated a statistically significant improvement in disease-free survival, reducing the risk of disease recurrence or death by 32% (HR=0.68 [95% CI, 0.53-0.87]; p=0.0010) compared to placebo, in patients at increased risk of recurrence (defined in the clinical trial protocol as intermediate-high or high-risk following nephrectomy and those with resected advanced disease). The CHMP’s recommendation will now be reviewed by the European Commission for marketing authorization in the European Union, and a final decision is expected in the first quarter of 2022.“Patients in Europe with earlier-stage renal cell carcinoma, who are at increased risk of recurrence following nephrectomy, have not had an approved treatment option in the adjuvant setting that can help reduce the risk of their cancer returning,” said Dr. Scot Ebbinghaus, vice president, clinical research, Merck Research Laboratories. “This positive CHMP opinion for KEYTRUDA is an important step forward in bringing adjuvant immunotherapy to these patients and demonstrates Merck’s progress in providing new options to treat earlier stages of cancer.”Merck has a broad clinical development program exploring KEYTRUDA, as monotherapy or in combination, as well as several other investigational and approved medicines across multiple settings and stages of RCC, including adjuvant and advanced or metastatic disease.
About KEYTRUDA® (pembrolizumab) Injection, 100 mgKEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.MelanomaKEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.KEYTRUDA is indicated for the adjuvant treatment of adult and pediatric (12 years and older) patients with stage IIB, IIC, or III melanoma following complete resection.Non-Small Cell Lung CancerKEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [Tumor Proportion Score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.Head and Neck Squamous Cell CancerKEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [Combined Positive Score (CPS) ≥1] as determined by an FDA-approved test.KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.Classical Hodgkin LymphomaKEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.Primary Mediastinal Large B-Cell LymphomaKEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy.KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.Urothelial CarcinomaKEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
Non-muscle Invasive Bladder CancerKEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.Microsatellite Instability-High or Mismatch Repair Deficient CancerKEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options.This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with MSI-H central nervous system cancers have not been established.Microsatellite Instability-High or Mismatch Repair Deficient Colorectal CancerKEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC).Gastric CancerKEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.Esophageal CancerKEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or GEJ (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical CancerKEYTRUDA, in combination with chemotherapy, with or without bevacizumab, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.Hepatocellular CarcinomaKEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.Merkel Cell CarcinomaKEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.Renal Cell CarcinomaKEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).KEYTRUDA is indicated for the adjuvant treatment of patients with RCC at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions.Tumor Mutational Burden-High CancerKEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.Cutaneous Squamous Cell CarcinomaKEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.Triple-Negative Breast CancerKEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
For more information about our oncology clinical trials, visit www.merck.com/clinicaltrials.
For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
https://www.keytruda.com/Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf . Source: Merck & Co., Inc.
Dec. 17, 2021 7:02 AM ET Merck & Co., Inc. (MRK) By: Mamta Mayani, SA News Editor
For further information please visit www.gsk.com/aboutus.
12/17/21 at 8:16 AM ESTPDF Version
LONDON and SAN FRANCISCO, Dec. 17, 2021 (GLOBE NEWSWIRE) -- GlaxoSmithKline plc (LSE/NYSE: GSK) and Vir Biotechnology, Inc. (Nasdaq: VIR) today announced that the European Commission (EC) has granted marketing authorization to Xevudy (sotrovimab) for the early treatment of COVID-19. Sotrovimab is now approved in the European Union (EU) for the treatment of adults and adolescents (aged 12 years and over and weighing at least 40kg) with COVID-19 who do not require supplemental oxygen and who are at increased risk of progressing to severe COVID-19.
The grant of the marketing authorization in the EU is a result of the positive opinion issued on December 16 by the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP).
In July 2021, GSK and Vir announced a Joint Procurement Agreement (JPA) with the EC to supply up to 220,000 doses of sotrovimab. Following the grant of the marketing authorization in the EU, Member States participating in the JPA can now order sotrovimab to support their pandemic responses.
Dr Hal Barron, Chief Scientific Officer and President R&D, GSK, said: “Since the start of the pandemic we have seen an unprecedented effort by governments, academia and industry to find solutions to help as many people as quickly as possible. COVID-19 therapeutics are an important part of the solution. We have already been working to lay the foundation for more patients across Europe to access sotrovimab through the Joint Procurement Agreement with the European Commission. With today’s marketing authorization we are now able to expand access, and we are discussing with governments how we can bring sotrovimab to more patients.”
George Scangos,Ph.D., Chief Executive Officerof Vir, said: “The grant of the marketing authorization in the European Union for sotrovimab marks yet another important milestone in our efforts to combat COVID-19, as it allows us to expand access across multiple countries working to address this challenge. Given recent preclinical data from our own labs, as well as that of other independent labs, demonstrating that sotrovimab retains activity against the rapidly spreading Omicron variant and all other currently tested variants of concern and interest, we remain confident in the critical role of sotrovimab and look forward to further contributing to the fight against this pandemic.”
The grant of the marketing authorization in the EU is based on data from the COMET-ICE Phase 3 trial, demonstrating that intravenous treatment with sotrovimab resulted in a 79% reduction (adjusted relative risk reduction) (p<0.001) in all-cause hospitalizations for more than 24 hours or death due to any cause by Day 29 compared to placebo, meeting the primary endpoint of the trial. In absolute numbers, 30 (6%) of the 529 patients in the placebo arm progressed, compared to six (1%) of the 528 patients receiving sotrovimab. In clinical trials conducted to date, sotrovimab has been well-tolerated. The most common adverse reactions are hypersensitivity and infusion-related reactions, seen in approximately 2% and 1% of cases, respectively.
GSK and Vir are committed to the ongoing evaluation of sotrovimab as the COVID-19 landscape continues to evolve at different rates across the globe and new variants of concern and interest emerge. Updated in vitro data, published in bioRxiv, demonstrate that sotrovimab retains activity against all tested variants of concern and interest of the SARS-CoV-2 virus as defined by the World Health Organization, including, but not limited to, Omicron (B.1.1.529), Delta (B.1.617.2), Delta Plus (AY.1 or AY.2) and Mu (B.1.621).
About Xevudy (Sotrovimab)
Xevudy (sotrovimab) is an investigational SARS-CoV-2 neutralizing monoclonal antibody. The antibody binds to an epitope on SARS-CoV-2 shared with SARS-CoV-1 (the virus that causes SARS), indicating that the epitope is highly conserved, which may make it more difficult for resistance to develop. Sotrovimab, which incorporates Xencor, Inc.’s Xtend™ technology, has also been designed to achieve high concentration in the lungs to ensure optimal penetration into airway tissues affected by SARS-CoV-2 and to have an extended half-life.
Authorized Use
The US FDA has issued an EUA to permit the emergency use of the unapproved product sotrovimab for the treatment of mild-to-moderate coronavirus disease 2019 (COVID-19) in adults and pediatric patients (12 years of age and older weighing at least 40kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death.
Please see the Food and Drug Administration (FDA) Letter of Authorization, full Fact Sheet for Healthcare Providers and full Fact Sheet for Patients, Parents, and Caregivers.
About the GSK and Vir Collaboration
In April 2020, GSK and Vir entered into a collaboration to research and develop solutions for coronaviruses, including SARS-CoV-2, the virus that causes COVID-19. The collaboration uses Vir’s proprietary monoclonal antibody platform technology to accelerate existing and identify new anti-viral antibodies that could be used as therapeutic or preventive options to help address the current COVID-19 pandemic and future outbreaks. The companies will leverage GSK’s expertise in functional genomics and combine their capabilities in CRISPR screening and artificial intelligence to identify anti-coronavirus compounds that target cellular host genes. They will also apply their combined expertise to research SARS-CoV-2 and other coronavirus vaccines.
For further information please visit www.gsk.com/aboutus.
For more information, please visit www.vir.bio.
Dec. 17, 2021 8:58 AM ET GlaxoSmithKline plc (GSK), VIR
By: Dulan Lokuwithana, SA News Editor
December 16, 2021
-- Kite and Daiichi Sankyo to Expand YESCARTA® Collaboration in Japan --
SANTA MONICA, Calif. & TOKYO--(BUSINESS WIRE)-- Kite, a Gilead Company, and Daiichi Sankyo Company, Limited (hereafter, Daiichi Sankyo) today announced that YESCARTA® (axicabtagene ciloleucel), a chimeric antigen receptor (CAR) T-cell therapy, will be available to patients with relapsed or refractory large B-cell lymphomas in Japan through the first treatment center now authorized by Daiichi Sankyo. Kite and Daiichi Sankyo will also build on the exclusive licensing deal for commercialization rights for axicabtagene ciloleucel in Japan, formalized in January 2017. Both partners are pleased to agree on a broadening of their business collaboration in Japan.
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20211216006041/en/
“We are pleased to bring the benefits of axicabtagene ciloleucel to eligible patients in Japan, in collaboration with Daiichi Sankyo,” said Warner Biddle, Kite’s Global Head of Commercial. “Japan has the second-largest number of people diagnosed with non-Hodgkin lymphoma globally1 and we remain committed to bringing our innovative CAR T-cell therapies to additional new markets.”
“We are pleased to be able to deliver axicabtagene ciloleucel, Daiichi Sankyo's first cell therapy product, to patients in Japan,” said Akio Sakurai, Daiichi Sankyo Corporate Officer, Head of Sales Division. “By strengthening our collaboration with Kite, the originator of axicabtagene ciloleucel and a world leader in cell therapy, we will strive to bring this innovative therapy to as many patients as possible.”
CAR T-cell therapy is a complex immunotherapy, and all hospitals must complete a rigorous training process before administering axicabtagene ciloleucel to patients. These hospitals receive specific training in handling and risk minimization procedures in order to ensure that patient safety remains a priority.
Several factors are considered when qualifying a hospital, including their specialist skills and services, geographic coverage and experience in managing other complex procedures, such as stem cell transplantation and a co-located intensive care unit.
Axicabtagene ciloleucel has been approved in Japan for treatment of patients with relapsed or refractory diffuse large B-cell lymphoma, primary mediastinal B-cell lymphoma, transformed follicular lymphoma or high-grade B-cell lymphoma. The use of axicabtagene ciloleucel is limited to patients not previously treated with a CD-19 CAR-positive T-cell infusion; patients previously treated with two or more lines of treatment including chemotherapy or an autologous stem cell transplant; and, patients ineligible for an autologous stem cell transplant. In January 2017, Daiichi Sankyo received exclusive development, manufacturing and commercialization rights for axicabtagene ciloleucel in Japan from California-based Kite, a Gilead Company.
The approval of axicabtagene ciloleucel in Japan is based on data from the global pivotal trial conducted by Kite (ZUMA-1)2 and results of a Phase 2 study conducted by Daiichi Sankyo in Japan. In the Japanese Phase 2, open-label, single-arm study, the same dose (2.0 x 106 cells/kg) of axicabtagene ciloleucel as used in the ZUMA-1 study was administered to assess efficacy and safety in 16 Japanese patients with relapsed or refractory large B-cell lymphoma, including diffuse large B-cell lymphoma, primary mediastinal B-cell lymphoma, transformed follicular lymphoma or high-grade B-cell lymphoma. The study reached its primary endpoint, demonstrating an objective response rate (ORR) of 86.7% (95% CI: 59.5 – 98.3%).
The overall safety and tolerability profile of axicabtagene ciloleucel in the Japan trial was consistent with that observed in ZUMA-1. Dose limiting toxicity was not observed. Grade ≥3 treatment emergent adverse event occurred in all patients; most commonly neutropenia (81.3%), lymphopenia (81.3%) and thrombocytopenia (62.5%). Cytokine release syndrome (CRS), a typical CAR T-cell therapy-emergent adverse event, occurred in 13 patients (81.3%, all Grade), with Grade ≥3 CRS in one patient (6.3%). No neurological events, another CAR T-cell therapy-emergent adverse event, were observed.
About YESCARTA ®
YESCARTA® (axicabtagene ciloleucel) is a CAR T-cell therapy directed against CD19 (a cell membrane protein), which harnesses a patient’s own immune system to fight cancer. Axicabtagene ciloleucel is made by removing a patient’s T cells and engineering them in the lab to express chimeric antigen receptors so that they can recognize and destroy cancer cells. The CAR T therapy is manufactured specifically for each patient and administered only once.3
Axicabtagene ciloleucel received
Orphan Drug Designation from the Japan MHLW in 2018 for the treatment of diffuse large B-cell lymphoma, primary mediastinal B-cell lymphoma, transformed follicular lymphoma and high-grade B-cell lymphoma.
YESCARTA® is approved in the U.S. and Europe for patients with certain types of relapsed or refractory B-cell lymphoma, where it is developed, manufactured and commercialized by Kite.
Please see full U.S. Prescribing Information, including BOXED WARNING and Medication Guide.
Yescarta® is a CD19-directed genetically modified autologous T cell immunotherapy indicated for the treatment of:
Limitations of Use: Yescarta is not indicated for the treatment of patients with primary central nervous system lymphoma.
For more information, please visit: www.daiichisankyo.com.
For more information on Kite, please visit www.kitepharma.com.
U.S. Prescribing Information for YESCARTA® including BOXED WARNING, is available at www.kitepharma.com and www.gilead.com.
Kite, the Kite logo, YESCARTA, and GILEAD are trademarks of Gilead Sciences, Inc. or its related companies.
For more information on Kite, please visit the company’s website at www.kitepharma.com Follow Kite on social media on Twitter (@KitePharma) and LinkedIn.
_________
View source version on businesswire.com: https://www.businesswire.com/news/home/20211216006041/en/
Source: Gilead Sciences, Inc.
Dec. 17, 2021 5:55 AM ET
By: Mamta Mayani, SA News Editor
December 15 2021
Sanofi and GSK announce positive preliminary booster data for their COVID-19 vaccine candidate and continuation of Phase 3 trial per independent Monitoring Board recommendation
PARIS – December 15, 2021 - Sanofi and GSK announced today that a single booster dose of their recombinant adjuvanted COVID-19 vaccine candidate delivered consistently strong immune responses. Preliminary results from the VAT0002 clinical trial investigating the safety and immunogenicity of the booster showed neutralizing antibodies increased 9- to 43-fold regardless of the primary vaccine received (AstraZeneca, Johnson & Johnson, Moderna, Pfizer/BioNTech) and for all age groups tested. The booster was well tolerated, with a safety profile similar to currently approved COVID-19 vaccines. This is the most comprehensive booster trial to date to explore boosting across different vaccine technologies used for primary vaccination.
The ongoing global Phase 3 trial, VAT0008, includes regular reviews by an independent Data Safety Monitoring Board (DSMB). During its last review, the DSMB identified no safety concerns and recommended the trial to continue into early 2022 to accrue more data.
Regulatory authorities require Phase 3 efficacy to be demonstrated in “naive” populations, i.e. participants who have never been infected by the COVID-19 virus (seronegative). The Phase 3 trial recruited most participants in Q3 2021, coinciding with a significant increase in the number of people infected by the COVID-19 virus globally due to the Delta variant. To provide the necessary data to regulatory authorities for the booster vaccine submission, the trial will continue to accrue the number of events needed for analysis, with results expected in Q1, 2022.
“These preliminary data show we have a strong booster candidate, whatever primary vaccine you have received.” said Thomas Triomphe, Executive Vice President, Sanofi Pasteur. “This is consistent with our efforts to provide relevant responses to evolving public health needs. While pursuing a phase 3 trial is a challenge in a quickly shifting pandemic environment, we look forward to seeing the results to support submissions of our booster vaccine as quickly as possible.”
Roger Connor, President of GSK Vaccines, added: “As the pandemic threat continues with the current dominant Delta variant and Omicron rapidly gaining ground, booster vaccines will continue to be needed to help protect people over time. The initial booster data are promising, and we await the phase III results to determine the next steps on making protein-based adjuvanted COVID-19 vaccines available."
In parallel, Sanofi continues its contribution to global public health needs with the manufacturing of up to half a billion doses from BioNTech/Pfizer, Moderna, and Johnson & Johnson vaccines.
About the booster trial (VAT0002)
The VAT0002 extension trial is the most comprehensive heterologous booster trial conducted to date. In the first cohort of this trial, the four most-widely approved COVID-19 primary vaccines using mRNA and adenovirus vector technologies were boosted with the Sanofi/GSK protein-based adjuvanted vaccine candidate after full primary vaccination to assess its safety profile and immunogenicity.
Participants in the first cohort (n=521) had previously been vaccinated with the approved dosing schedule of an authorized COVID-19 mRNA vaccine (Moderna, Pfizer/BioNTech,) or adenovirus vector vaccine (AstraZeneca, Johnson & Johnson,). This preliminary analysis includes data from trial participants who received one 5µg booster dose of the adjuvanted recombinant protein vaccine targeting the D614 parent virus, between four and ten months after a complete primary vaccination schedule.
The trial is ongoing across sites in multiple countries, including the U.S., France, and the UK. To address the emergence of COVID-19 variants of concern, additional trial cohorts are assessing the boosting potential of monovalent and bivalent vaccine formulations also containing the Beta (B.1.351) variant. More data from this trial are expected during the first half of 2022. The Omicron variant was not circulating during the trial. Using sera from booster trial participants, testing is underway to establish the ability of the vaccine candidate to cross-neutralize against Omicron.
About the Phase 3 efficacy trial (VAT0008)
The primary endpoint of this ongoing Phase 3, randomized, double-blind, placebo-controlled trial is the prevention of symptomatic COVID-19 in SARS-CoV-2 naïve adults, with secondary endpoints of preventing severe COVID-19 disease and infection. Stage one of the trial is assessing the efficacy of a vaccine formulation containing the spike protein against the original D614 (parent) virus in more than 10,000 participants >18 years of age, randomized to receive two doses of 10µg vaccine or placebo at day 1 and day 22 across sites in the US, Asia, Africa and Latin America. A second stage in the trial is evaluating a second bivalent formulation, adding the spike protein of the B.1.351 (Beta) variant.
These efforts are supported by federal funds from the Biomedical Advanced Research and Development Authority, part of the office of the Assistant Secretary for Preparedness and Response at the U.S. Department of Health and Human Services in collaboration with the U.S. Department of Defense Joint Program Executive Office for Chemical, Biological, Radiological and Nuclear Defense under Contract # W15QKN-16-9-1002.
About the Sanofi and GSK collaboration
In the collaboration between the two companies, Sanofi provides its recombinant antigen and GSK contributes its pandemic adjuvant, both established vaccine platforms that have proven successful against influenza.
For further information please visit www.gsk.com.
Dec. 15, 2021 1:52 AM ET Sanofi (SNY), GSK By: Mamta Mayani, SA News Editor1 Comment
Thursday, December 16, 2021 - 10:30am
NEW YORK--(BUSINESS WIRE)-- Pfizer Inc. (NYSE: PFE) announced today that the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) issued advice on the use of PAXLOVID™ (nirmatrelvir [PF-07321332] tablets and ritonavir tablets), stating that PAXLOVID can be used to treat adults with COVID-19 who do not require supplemental oxygen and who are at increased risk of progressing to severe disease. The CHMP also recommend that PAXLOVID should be administered as soon as possible after diagnosis of COVID-19 and within five days of the start of symptoms. The EMA issued this advice under Article 5(3) of Regulation 726/2004 to support authorities of European Union (EU) Member States who may decide to allow the supply and use of PAXLOVID, for example in emergency use settings, prior to EU conditional marketing authorization. PAXLOVID is currently not authorized for use in the EU.
“The CHMP’s advice signifies the strength of our data for PAXLOVID in the treatment of high-risk adults diagnosed with COVID-19,” said Albert Bourla, Chairman and Chief Executive Officer, Pfizer. “COVID-19 continues to take lives at an unprecedented pace globally and exacts a devastating toll on health care systems. If authorized, PAXLOVID has the potential to help save lives and reduce hospitalizations. We look forward to working with the EMA and other regulatory agencies worldwide to bring this potential treatment to patients as quickly as possible.”
The CHMP based their advice on positive results from the Phase 2/3 EPIC-HR (Evaluation of Protease Inhibition for COVID-19 in High-Risk Patients) interim analysis, which enrolled non-hospitalized adults with confirmed COVID-19 who are at increased risk of progressing to severe illness. The data demonstrated an 89% reduction in risk of COVID-19-related hospitalization or death from any cause in patients treated with PAXLOVID compared to placebo within three days of symptom onset, with no deaths in the treatment group. Similar results were seen with those treated within five days of symptom onset. Treatment-emergent adverse events were comparable between PAXLOVID (19%) and placebo (21%), most of which were mild in intensity. Pfizer recently announced that results from the final analysis of the primary endpoint from all patients enrolled in EPIC-HR were consistent with the interim analysis, confirming robust efficacy and a similar safety profile.
Pfizer has also initiated rolling submission with the EMA for potential EU conditional marketing authorization of PAXLOVID. If authorized, PAXLOVID could be prescribed as an at-home treatment to high-risk patients at the first sign of infection, potentially helping patients avoid severe illness which can lead to hospitalization and death. PAXLOVID is also being studied in adults at standard risk of progressing to severe illness, as well as in adults who have been exposed to the virus through household contacts.
About PAXLOVID™ (nirmatrelvir [PF-07321332] tablets and ritonavir tablets)
PAXLOVID is an investigational SARS-CoV-2 protease inhibitor antiviral therapy. It was developed to be administered orally so that, if authorized or approved, it can be prescribed at the first sign of infection or at first awareness of an exposure – potentially helping patients avoid severe illness (which can lead to hospitalization and death) or disease development following contact – subject to the successful completion of the remainder of the EPIC clinical development program. Nirmatrelvir [PF-07321332], which originated in Pfizer’s laboratories, is designed to block the activity of the SARS-CoV-2-3CL protease, an enzyme that the coronavirus needs to replicate. Co-administration with a low dose of ritonavir helps slow the metabolism, or breakdown, of nirmatrelvir in order for it to remain active in the body for longer periods of time at higher concentrations to help combat the virus.
Nirmatrelvir is designed to inhibit viral replication at a stage known as proteolysis, which occurs before viral RNA replication. In preclinical studies, nirmatrelvir did not demonstrate evidence of mutagenic DNA interactions.
Current variants of concern can be resistant to treatments that are focused on the spike protein expressed on the surface of the SARS-CoV-2 virus, due to the mutations in this region. PAXLOVID, however, works intracellularly on the protease of the SARS-CoV-2 virus by inhibiting viral replication. Nirmatrelvir has shown consistent in vitro antiviral activity against the previously identified variants of concerns (i.e., alpha, beta, delta, gamma, lambda, and mu). In addition, nirmatrelvir potently inhibited the 3CL protease associated with Omicron in an in vitro biochemical assay. This indicates nirmatrelvir’s potential to maintain robust antiviral activity against Omicron. Additional in vitro antiviral studies with this variant are underway.
If authorized or approved, PAXLOVID will be administered at a dose of 300 mg (two 150 mg tablets) of nirmatrelvir with one 100 mg tablet of ritonavir, given twice-daily for five days. One box contains five blister packs of PAXLOVID, as co-packaged nirmatrelvir tablets with ritonavir tablets, providing all required doses for a full five-day treatment course.
For more information on the EPIC Phase 2/3 clinical trials for PAXLOVID, visit clinicaltrials.gov.
We routinely post information that may be important to investors on our website at www.Pfizer.com.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211215006145/en/
Source: Pfizer Inc.
Dec. 16, 2021 11:46 AM ET Pfizer Inc. (PFE)
By: Dulan Lokuwithana, SA News Editor6 Comments
PUBLISHED16 December 2021
AstraZeneca’s Evusheld (tixagevimab co-packaged with cilgavimab), a long-acting antibody combination for the prevention of COVID-19, retained neutralising activity against the Omicron SARS-CoV-2 variant (B.1.1.529), according to new preclinical data.
In this study, Evusheld’s Inhibitory Concentration 50 (IC50), a measure of neutralising potency of an antibody, was 171 ng/ml and 277 ng/ml in two confirmatory tests, which is within the range of neutralising titres found in someone who has been previously infected with COVID-19. Evusheld’s IC50 for the original strain of SARS-CoV-2, previously referred to as the Wuhan strain, was approximately 1.3 ng/ml and 1.5 ng/ml, respectively.
The early data, generated by pseudovirus testing of the full Omicron variant spike against the combination of tixagevimab with cilgavimab, the antibodies that comprise Evusheld, add to the growing body of preclinical evidence demonstrating that Evusheld retains activity against all tested variants of concern to date.1
The study was performed independently by investigators at the US Food and Drug Administration (FDA), Center for Biologics Evaluation and Research. The work was supported by US government research funds.
Mene Pangalos, Executive Vice President, BioPharmaceuticals R&D, AstraZeneca, said: “This study shows Evusheld retains neutralisation activity against the Omicron variant. By combining two potent antibodies with different and complementary activities against the virus, Evusheld was designed to evade potential resistance with the emergence of new SARS-CoV-2 variants. Evusheld is the first long-acting antibody to receive emergency use authorisation in the US for pre-exposure prophylaxis of COVID-19, in addition to authorisations in other countries, and we are working with regulators on applications for the use of Evusheld in treating COVID-19.”
The Omicron variant was not in circulation during the Evusheld clinical trials. The Company is continuing to collect further data to better understand the implications of this observation in clinical practice. Additional analyses to evaluate Evusheld against the Omicron variant are being conducted by AstraZeneca and third-party laboratories, with data anticipated very soon.
Evusheld received Emergency Use Authorization (EUA) in the US in December 2021 for pre-exposure prophylaxis (prevention) of COVID-19 in people with moderate to severe immune compromise due to a medical condition or immunosuppressive medications and who may not mount an adequate immune response to COVID-19 vaccination, as well as those individuals for whom COVID-19 vaccination is not recommended. The first doses are expected to become available within days.
About 2% of the global population is considered at increased risk of an inadequate response to a COVID-19 vaccine.3,4 Emerging evidence indicates that protecting vulnerable populations from getting COVID-19 could help prevent viral evolution that is an important factor in the emergence of variants.5
Additionally, the TACKLE Phase III outpatient treatment trial of Evusheld showed it reduced the risk of developing severe COVID-19 or death (from any cause) by 50% compared to placebo in non-hospitalised patients with mild to moderate COVID-19 who had been symptomatic for seven days or less.6
Notes
Evusheld
Evusheld, formerly known as AZD7442 is a combination of two LAABs - tixagevimab (AZD8895) and cilgavimab (AZD1061) - derived from B-cells donated by convalescent patients after SARS-CoV-2 virus. Discovered by Vanderbilt University Medical Center and licensed to AstraZeneca in June 2020, the human monoclonal antibodies bind to distinct sites on the SARS-CoV-2 spike protein7 and were optimised by AstraZeneca with half-life extension and reduced Fc receptor and complement C1q binding. The half-life extension more than triples the durability of its action compared to conventional antibodies and could afford up to 12 months of protection from COVID-19 following a single administration;8-10 data from the Phase III PROVENT trial show protection lasting at least six months.11 The reduced Fc receptor binding aims to minimise the risk of antibody-dependent enhancement of disease - a phenomenon in which virus-specific antibodies promote, rather than inhibit, infection and/or disease.12
In December 2021, the U.S. Food and Drug Administration issued an Emergency Use Authorisation (EUA) for the use of Evusheld (tixagevimab co-packaged with cilgavimab) for the pre-exposure prophylaxis (prevention) of COVID-19. It is the only antibody authorised in the US to prevent COVID-19 symptoms before virus exposure. Evusheld is also authorised for emergency use for prevention of COVID-19 in several other countries.
In August 2021, AstraZeneca announced that Evusheld demonstrated a statistically significant reduction in the risk of developing symptomatic COVID-19 in the PROVENT trial; efficacy was 83% compared to placebo in a six-month analysis announced on 18 November 2021. In October 2021, AstraZeneca announced positive high-level results from the Evusheld TACKLE Phase III outpatient treatment trial.
Evusheld is also being studied as a potential treatment for hospitalised COVID-19 patients as part of the National Institute of Health’s ACTIV-3 trial and in an additional collaborator hospitalisation treatment trial.
Evusheld is being developed with support from the US government, including federal funds from the Department of Health and Human Services; Office of the Assistant Secretary for Preparedness and Response; Biomedical Advanced Research and Development Authority in partnership with the Department of Defense; Joint Program Executive Office for Chemical, Biological, Radiological and Nuclear Defense, under Contract No. W911QY-21-9-0001.
Under the terms of the licensing agreement with Vanderbilt, AstraZeneca will pay single-digit royalties on future net sales.
Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Dec. 16, 2021 12:11 PM ET
By: Dulan Lokuwithana, SA News Editor
Tuesday, December 14, 2021 - 06:45am
NEW YORK--(BUSINESS WIRE)-- Pfizer Inc. (NYSE: PFE) today announced final results from an analysis of all 2,246 adults enrolled in its Phase 2/3 EPIC-HR (Evaluation of Protease Inhibition for COVID-19 in High-Risk Patients) trial of its novel COVID-19 oral antiviral candidate PAXLOVID™ (nirmatrelvir [PF-07321332] tablets and ritonavir tablets). These results were consistent with the interim analysis announced in November 2021, showing PAXLOVID significantly reduced the risk of hospitalization or death for any cause by 89% compared to placebo in non-hospitalized, high-risk adult patients with COVID-19 treated within three days of symptom onset. In a secondary endpoint, PAXLOVID reduced the risk of hospitalization or death for any cause by 88% compared to placebo in patients treated within five days of symptom onset, an increase from the 85% observed in the interim analysis. The EPIC-HR data have been shared with the U.S. Food and Drug Administration (FDA) as part of an ongoing rolling submission for Emergency Use Authorization (EUA).
“This news provides further corroboration that our oral antiviral candidate, if authorized or approved, could have a meaningful impact on the lives of many, as the data further support the efficacy of PAXLOVID in reducing hospitalization and death and show a substantial decrease in viral load. This underscores the treatment candidate’s potential to save the lives of patients around the world,” said Albert Bourla, Chairman and Chief Executive Officer, Pfizer. “Emerging variants of concern, like Omicron, have exacerbated the need for accessible treatment options for those who contract the virus, and we are confident that, if authorized or approved, this potential treatment could be a critical tool to help quell the pandemic.”
EPIC-HR Final Results
In the final analysis of the primary endpoint from all patients enrolled in EPIC-HR, an 89% reduction in COVID-19-related hospitalization or death from any cause compared to placebo in patients treated within three days of symptom onset was observed, consistent with the interim analysis. In addition, a consistent safety profile was observed.
0.7% of patients who received PAXLOVID were hospitalized through Day 28 following randomization (5/697 hospitalized with no deaths), compared to 6.5% of patients who received placebo and were hospitalized or died (44/682 hospitalized with 9 subsequent deaths). The statistical significance of these results was high (p<0.0001). In a secondary endpoint, PAXLOVID reduced the risk of hospitalization or death for any cause by 88% compared to placebo in patients treated within five days of symptom onset; 0.8% of patients who received PAXLOVID were hospitalized or died through Day 28 following randomization (8/1039 hospitalized with no deaths), compared to 6.3% of patients who received placebo (66/1046 hospitalized with 12 subsequent deaths), with high statistical significance (p<0.0001). Relative risk reduction was 94% in patients 65 years of age or older, one of the populations at highest risk for hospitalization or death; 1.1% of patients who received PAXLOVID were hospitalized through Day 28 (1/94 hospitalized with no deaths), compared to 16.3% of patients who received placebo (16/98 hospitalized with 6 deaths), with high statistical significance (p<0.0001). In the overall study population through Day 28, no deaths were reported in patients who received PAXLOVID as compared to 12 (1.2%) deaths in patients who received placebo.
In the EPIC-HR trial, in a secondary endpoint, SARS-CoV-2 viral load at baseline and Day 5 have been evaluated for 499 patients. After accounting for baseline viral load, geographic region, and serology status, PAXLOVID reduced viral load by approximately 10-fold, or 0.93 log10 copies/mL, relative to placebo, indicating robust activity against SARS-CoV-2 and representing the strongest viral load reduction reported to date for an oral COVID-19 agent.
Treatment-emergent adverse events were comparable between PAXLOVID (23%) and placebo (24%), most of which were mild in intensity. Fewer serious adverse events (1.6% vs. 6.6%) and discontinuation of study drug due to adverse events (2.1% vs. 4.2%) were observed in patients dosed with PAXLOVID, compared to placebo, respectively.
All other secondary endpoints for this study, which are available on clinicaltrials.gov (NCT04960202), were not yet available for this review. Full study data are expected to be released later this month and submitted to a peer-reviewed publication.
EPIC-SR Interim Results
Interim analyses of the EPIC-SR (Evaluation of Protease Inhibition for COVID-19 in Standard-Risk Patients) Phase 2/3 study, which included unvaccinated adults who were at standard risk (i.e., low risk of hospitalization or death) as well as vaccinated adults who had one or more risk factors for progressing to severe illness, showed that the novel primary endpoint of self-reported, sustained alleviation of all symptoms for four consecutive days, as compared to placebo, was not met.
The key secondary endpoint showed a 70% reduction in hospitalization and no deaths in the treated population for any cause compared to placebo. Additionally, there was approximately a 10-fold, or 1 log10 copies/mL, decrease in viral load compared to placebo, consistent with results from the Phase 2/3 EPIC-HR study.
The data were reviewed by an independent Data Monitoring Committee (DMC) and, based on the totality of the data available, the DMC recommended that the trial continue.
At the EPIC-SR interim analysis, which included 45% of the trial’s planned enrollment, 0.6% of those who received PAXLOVID were hospitalized following randomization (2/333 hospitalized with no deaths), compared to 2.4% of patients who received placebo and were hospitalized or died (8/329 hospitalized with no deaths). A follow-on analysis at 80% of enrolled patients was consistent with these findings. In this analysis, 0.7% of those who received PAXLOVID were hospitalized following randomization (3/428 hospitalized with no deaths), compared to 2.4% of patients who received placebo and were hospitalized or died (10/426 hospitalized with no deaths); p=0.051.
Treatment-emergent adverse events were comparable between PAXLOVID (22%) and placebo (21%), most of which were mild in intensity. Rates of serious adverse events (1.4% vs. 1.9%) and discontinuation of study drug due to adverse events (2.1% vs. 1.2%) were also comparable between PAXLOVID and placebo.
All other secondary endpoints for this study, which are available on clinicaltrials.gov (NCT05011513), were not yet available for this review. The study is now fully enrolled, and further data will be released upon analysis of the full study data expected later this month.
About PAXLOVID™ (nirmatrelvir [PF-07321332] tablets and ritonavir tablets)
PAXLOVID is an investigational SARS-CoV-2 protease inhibitor antiviral therapy. It was developed to be administered orally so that, if authorized or approved, it can be prescribed at the first sign of infection or at first awareness of an exposure – potentially helping patients avoid severe illness (which can lead to hospitalization and death) or avoid disease development following contact with a household member who contracts COVID-19 – subject to the clinical success of the rest of the EPIC development program. Nirmatrelvir [PF-07321332], which originated in Pfizer laboratories, is designed to block the activity of the SARS-CoV-2-3CL protease, an enzyme that the coronavirus needs to replicate. Co-administration with a low dose of ritonavir helps slow the metabolism, or breakdown, of nirmatrelvir in order for it to remain active in the body for longer periods of time at higher concentrations to help combat the virus.
Nirmatrelvir is designed to inhibit viral replication at a stage known as proteolysis, which occurs before viral RNA replication. In preclinical studies, nirmatrelvir did not demonstrate evidence of mutagenic DNA interactions.
Current variants of concern can be resistant to treatments that are focused on the spike protein expressed on the surface of the SARS-CoV-2 virus, due to the mutations in this region. PAXLOVID, however, works intracellularly on the protease of the SARS-CoV-2 virus by inhibiting viral replication. Nirmatrelvir has shown consistent in vitro antiviral activity against the previously identified variants of concerns (i.e., alpha, beta, delta, gamma, lambda, and mu). In addition, nirmatrelvir potently inhibited the 3CL protease associated with Omicron in an in vitro biochemical assay. This indicates nirmatrelvir’s potential to maintain robust antiviral activity against Omicron. Additional in vitro antiviral studies with this variant are underway.
If authorized or approved, PAXLOVID will be administered at a dose of 300 mg (two 150 mg tablets) of nirmatrelvir with one 100 mg tablet of ritonavir, given twice-daily for five days. One box contains five blister packs of PAXLOVID, as co-packaged nirmatrelvir tablets with ritonavir tablets, providing all required doses for a full five-day treatment course.
About the Phase 2/3 EPIC-HR Study Top-Line Results
The final analysis of the primary endpoint evaluated data from 2,246 adults who were enrolled by November 4, 2021. At the time of the decision to stop recruiting patients, enrollment was at 75% of the 3,000 planned patients from clinical trial sites across North and South America, Europe, Africa, and Asia, with 41% of patients located in the United States. Enrolled individuals had a laboratory-confirmed diagnosis of mild to moderate SARS-CoV-2 infection within a five-day period and were required to have at least one characteristic or underlying medical condition associated with an increased risk of developing severe illness from COVID-19. Each patient was randomized (1:1) to receive PAXLOVID or placebo orally every 12 hours for five days.
About the Phase 2/3 EPIC-SR Study Interim Analyses
The primary analysis of the interim data, consisting of the first 45% of patients enrolled in the study, included 673 adults, of whom 338 received PAXLOVID and 335 received placebo. At the time of the interim analyses, EPIC-SR had reached its planned enrollment of more than 1,140 adults from clinical trial sites across North and South America, Europe, Africa, and Asia, and the United States. Enrolled individuals had a laboratory-confirmed diagnosis of mild to moderate SARS-CoV-2 infection within a five-day period and were either unvaccinated adults who were at standard risk (i.e., low risk of hospitalization or death) or vaccinated adults who had one or more risk factors for progressing to severe illness from COVID-19. Each patient was randomized (1:1) to receive PAXLOVID or placebo orally every 12 hours for five days.
For more information on the EPIC Phase 2/3 clinical trials for PAXLOVID, visit clinicaltrials.gov.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211214005548/en/
Source: Pfizer Inc.
Dec. 14, 2021 7:04 AM ET Pfizer Inc. (PFE) By: Dulan Lokuwithana, SA News Editor13 Comments
December 16, 2021 at 4:43 AM ESTPDF Version
NUZYRA demonstrated efficacy against common pathogens in three Phase 3 studies, including pathogens resistant to other antibiotic classes as a potential best-in-class tetracycline
NUZYRA is Zai Lab’s fourth new product approval and the first outside oncology
SHANGHAI, SAN FRANCISCO, and CAMBRIDGE, Mass., Dec. 16, 2021 (GLOBE NEWSWIRE) -- Zai Lab Limited (NASDAQ: ZLAB; HKEX: 9688), a patient-focused, innovative, commercial-stage, global biopharmaceutical company, today announced that the China National Medical Products Administration (NMPA) has approved its New Drug Application (NDA) for NUZYRA® (omadacycline), a novel antibiotic with both oral and intravenous (IV) formulations, for the treatment of community-acquired bacterial pneumonia (CABP) and acute bacterial skin and skin structure infections (ABSSSI). NUZYRA was approved as a Category 1 innovative drug by the NMPA and is locally manufactured in China. It is the fourth Zai Lab product approved over the last 24 months.
“In the face of ever-increasing antibiotic resistance, the NMPA’s approval of NUZYRA brings an important new treatment option for CABP and ABSSSI to millions of patients in China,” said Dr. Harald Reinhart, Chief Medical Officer for Autoimmune and Infectious Diseases at Zai Lab. “We believe NUZYRA is particularly well positioned due to its broad activity against a wide spectrum of pathogens, including multi-drug-resistant (MDR) bacteria, associated with these serious infections. In addition, NUZYRA offers clinicians the ability to treat patients in the hospital with the intravenous formulation and transition them to complete treatment at home with the oral formulation. This flexible treatment regimen potentially helps reduce exposure to hospital pathogens and the costs associated with hospital stays.”
“CABP is a common secondary infection associated with respiratory viruses like influenza,” said Professor Haihui Huang, Chief Physician, Fudan University Affiliated Huashan Hospital, Deputy Director of Antibiotic Research Institute. “We believe omadacycline is a potential best-in-class tetracycline, with demonstrated efficacy comparable to moxifloxacin in CABP and to linezolid in ABSSSI.”
“Omadacycline is one of the most potent antibiotics with intravenous and oral formulations in these indications,” said Professor Jing Zhang, Chief of Pharmacy, Fudan University Affiliated Huashan Hospital, Deputy Director of Drug Clinical Trial Center. “Importantly, it also has a favorable safety and tolerability profile, particularly regarding GI side effects, which can be a serious liability with other tetracyclines.”
NUZYRA was approved by the U.S. Food and Drug Administration (FDA) for both CABP and ABSSSI based on comprehensive clinical trial programs involving more than 2,000 patients and, since 2019, it has been marketed in the United States by Paratek Pharmaceuticals, Inc. In 2017, while NUZYRA was still in its clinical stage, Zai Lab in-licensed the rights to NUZYRA for the Greater China region (mainland China, Hong Kong, Macau, and Taiwan). Since then, Zai Lab conducted three clinical trials involving Chinese patients in support of NUZYRA’s registration in mainland China.
About CABP and ABSSSI
CABP is the most common type of pneumonia that is acquired outside the hospital. It is one of the most common infectious diseases and is an important cause of mortality and morbidity worldwide. ABSSSI are bacterial infections of skin and associated soft tissues, such as loose connective tissue and mucous membranes. ABSSSI are common and encompass a variety of disease presentations and degrees of severity. In 2015, the estimated incidences of ABSSSI and CABP were 2.8 million patients and 16.5 million patients, respectively, in China alone. Linezolid and moxifloxacin are the current standards of care for ABSSSI and CABP, respectively. There are significant unmet needs for broad-spectrum antibiotics addressing MDR infections with a favorable safety profile.
About NUZYRA
NUZYRA® (omadacycline), a novel tetracycline-class antibacterial with both once-daily oral and IV formulations, is specifically designed to overcome tetracycline resistance and to improve activity across a broad spectrum of bacterial infections, such as those caused by Gram-positive, Gram-negative, atypical, and many other pathogens. NUZYRA was launched in the United States in February 2019 as a once-daily oral and intravenous antibiotic for the treatment of adults with CABP and ABSSSI. It was approved in June 2021 by the FDA as an oral-only dosing regimen for the treatment of adults with CABP.
For additional information about the company, please visit www.zailaboratory.com or follow us at www.twitter.com/ZaiLab_Global.
For more investor-related information about Zai Lab, please go to www.SEC.gov or visit www.zailaboratory.com.
https://www.nuzyra.com/nuzyra-pi.pdf
Dec. 16, 2021 4:57 AM ETZai Lab Limited (ZLAB)By: Mamta Mayani, SA News Editor
Paratek has global rights with the exception of the greater China region where Paratek has entered into a collaboration agreement with Zai Lab (Shanghai) Co., Ld.
https://www.paratekpharma.com/pipelineandproducts
December 14, 2021 DownloadPDF Format (opens in new window)
WILMINGTON, Del.--(BUSINESS WIRE)-- Incyte (Nasdaq:INCY) today announced that the U.S. Food and Drug Administration (FDA) has accepted for Priority Review the supplemental New Drug Application (sNDA) for ruxolitinib cream 1.5% (Opzelura™) a topical JAK inhibitor, as a potential treatment for adolescents and adults (age ≥12 years) with vitiligo. The FDA grants Priority Review to medicines that may offer a major advance in treatment where none currently exists. The Prescription Drug User Fee Act (PDUFA) target action date is April 18, 2022.
“Vitiligo is a chronic autoimmune disease that can have a profound impact on people’s lives,” said Jim Lee, M.D., Ph.D., Group Vice President, Inflammation & Autoimmunity, Incyte. “Currently, there are no FDA-approved drug therapies for repigmentation in people with vitiligo. The FDA’s acceptance of our sNDA for ruxolitinib cream brings us one step closer to offering patients with vitiligo an additional treatment option.”
The sNDA is supported by data from the Phase 3 TRuE-V clinical trial program evaluating the safety and efficacy of ruxolitinib cream in more than 600 people with non-segmental vitiligo, age 12 and older. Results from the Phase 3 program were recently presented at the 30th European Academy of Dermatology and Venereology (EADV) congress during a late-breaking research session. The data showed that at Week 24, 29.9% of patients applying ruxolitinib cream achieved ≥75% improvement from baseline in the facial Vitiligo Area Scoring Index (F-VASI75), the primary endpoint.
In September 2021, Opzelura™ (ruxolitinib) cream was approved by the FDA for the topical short-term and non-continuous chronic treatment of mild to moderate atopic dermatitis (AD) in non-immunocompromised patients 12 years of age and older whose disease is not adequately controlled with topical prescription therapies, or when those therapies are not advisable. Use of Opzelura™ in combination with therapeutic biologics, other JAK inhibitors or potent immunosuppressants such as azathioprine or cyclosporine is not recommended.
About Vitiligo
Vitiligo is a chronic autoimmune disease characterized by depigmentation of skin that results from the loss of pigment-producing cells known as melanocytes. Over-activity of the JAK signaling pathway is believed to drive inflammation involved in the pathogenesis and progression of vitiligo. It affects approximately 1.5 million+ people in the U.S.1 and there are no U.S. Food and Drug Administration (FDA)-approved drug therapies for repigmentation in vitiligo. It can occur at any age, although many patients with vitiligo will experience initial symptoms before the age of 20.2
About TRuE-V
The TRuE-V clinical trial program includes two Phase 3 studies, TRuE-V1 (NCT04052425) and TRuE-V2 (NCT04057573), evaluating the safety and efficacy of ruxolitinib cream in patients with vitiligo.
The studies each enrolled approximately 300 patients (age ≥12 years) who have been diagnosed with non-segmental vitiligo and have depigmented areas including at least 0.5% of the body surface area (BSA) on the face, ≥0.5 facial Vitiligo Area Scoring Index [F-VASI] score, at least 3% BSA on nonfacial areas, ≥3 total body Vitiligo Area Scoring Index [T-VASI] score and total BSA involvement (facial and nonfacial) of up to 10%. Participants were randomized into two arms: 1.5% ruxolitinib cream twice daily (BID) and vehicle control for the 24-week double-blind period. Patients who successfully completed baseline and Week 24 assessments, including those that received vehicle control during the double-blind phase, were offered treatment extension with 1.5% ruxolitinib cream BID for an additional 28 weeks.
The primary endpoint of both studies in the TRuE-V program is the proportion of patients achieving F-VASI75, defined as at least a 75% improvement from baseline in the F-VASI score at Week 24. Key secondary endpoints include: the proportion of patients achieving F-VASI50 (at least 50% improvement from baseline in the F-VASI), F-VASI90 (at least 90% improvement from baseline in the F-VASI) and T-VASI50 (at least 50% improvement from baseline in the T-VASI) at Week 24, the proportion of patients achieving a Vitiligo Noticeability Scale (VNS) score of 4 (a lot less noticeable) or 5 (no longer noticeable) at Week 24, and the percentage change from baseline in facial BSA (F-BSA) at Week 24. The studies also track the frequency, duration and severity of adverse events associated with the use of ruxolitinib cream.
For more information on the TRuE-V studies, please visit https://clinicaltrials.gov/ct2/show/NCT04052425 and https://clinicaltrials.gov/ct2/show/NCT04057573.
About Ruxolitinib Cream (Opzelura™)
Ruxolitinib cream (Opzelura) a novel cream formulation of Incyte’s selective JAK1/JAK2 inhibitor ruxolitinib, is the first and only topical JAK inhibitor approved for use in the United States for the topical short-term and non-continuous chronic treatment of mild to moderate atopic dermatitis (AD) in non-immunocompromised patients 12 years of age and older whose disease is not adequately controlled with topical prescription therapies, or when those therapies are not advisable. Use of Opzelura in combination with therapeutic biologics, other JAK inhibitors, or potent immunosuppressants, such as azathioprine or cyclosporine, is not recommended.
Incyte has worldwide rights for the development and commercialization of ruxolitinib cream, marketed in the United States as Opzelura. On October 28, 2021, Incyte announced the validation of the European Marketing Authorization Application (MAA) for ruxolitinib cream as a potential treatment for adolescents and adults (age >12 years) with non-segmental vitiligo with facial involvement.
Opzelura is a trademark of Incyte.
To learn more, visit the Dermatology section of Incyte.com.
For additional information on Incyte, please visit Incyte.com and follow @Incyte.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211214006227/en/
Source: Incyte
Dec. 14, 2021 5:01 PM ET
By: Jonathan M Block, SA News Editor
December 15, 2021 6:45 am ET
KENILWORTH, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, announced today the European Commission (EC) has approved VAXNEUVANCE™ (Pneumococcal 15-valent Conjugate Vaccine) for active immunization for the prevention of invasive disease and pneumonia caused by Streptococcus pneumoniae in individuals 18 years of age and older. The approval allows marketing of VAXNEUVANCE in all 27 European Union (EU) Member States plus Iceland, Norway and Lichtenstein. The use of VAXNEUVANCE in the EU should be in accordance with official recommendations.
The EC’s decision follows a positive opinion from the European Medicines Agency’s Committee for Medicinal Products on Human Use (CHMP), which reviewed data from seven randomized, double-blind clinical studies evaluating VAXNEUVANCE in 7,438 individuals from a variety of adult populations and clinical circumstances. These included healthy adults ages 50 years and older, adults ages 18 to 49 with risk factors for pneumococcal disease, and immunocompromised adults living with HIV. In the pivotal, double blind, active-comparator controlled study in 1,205 immunocompetent pneumococcal vaccine-naïve adults ages 50 and older, immune responses elicited by VAXNEUVANCE were non-inferior to the currently available 13-valent pneumococcal conjugate vaccine (PCV13) for the 13 shared serotypes, as assessed by opsonophagocytic activity (OPA) Geometric Mean Titers (GMTs) at 30 days post-vaccination. Additionally, immune responses for VAXNEUVANCE were superior to PCV13 for shared serotype 3 and for the two serotypes unique to VAXNEUVANCE, 22F and 33F. Randomized controlled trials assessing the clinical efficacy of VAXNEUVANCE compared to PCV13 have not been conducted.
“At Merck, we are committed to helping protect more people from invasive pneumococcal disease (IPD), as well as from pneumococcal pneumonia, the most common form of pneumococcal disease in adults,” said Dr. Roy Baynes, senior vice president and head of global clinical development, chief medical officer, Merck Research Laboratories. “With VAXNEUVANCE, we developed a conjugate vaccine that elicits a strong immune response to pneumococcal serotypes that contribute substantially to the burden of disease, including serotype 3, a leading cause of IPD in the EU. This approval provides physicians and patients in the European Union with a new option that can help protect against pneumococcal serotypes responsible for around 40 percent of IPD cases in adults over 65 in the largest EU member countries.”
In July 2021, VAXNEUVANCE received approval from the U.S. Food and Drug Administration (FDA) for active immunization for the prevention of invasive disease caused by sand 33F in adults 18 years and older.
Select Safety Information for VAXNEUVANCE (Pneumococcal 15-valent Conjugate Vaccine) in Adults 18 Years of Age and Older in the U.S.
Do not administer VAXNEUVANCE to individuals with a severe allergic reaction (e.g., anaphylaxis) to any component of VAXNEUVANCE or to diphtheria toxoid.
Some individuals with altered immunocompetence, including those receiving immunosuppressive therapy, may have a reduced immune response to VAXNEUVANCE.
The most commonly reported solicited adverse reactions in individuals 18 through 49 years of age were: injection site pain (75.8%), fatigue (34.3%), myalgia (28.8%), headache (26.5%), injection site swelling (21.7%), injection site erythema (15.1%) and arthralgia (12.7%).
The most commonly reported solicited adverse reactions in individuals 50 years of age and older were: injection site pain (66.8%), myalgia (26.9%), fatigue (21.5%), headache (18.9%), injection site swelling (15.4%), injection site erythema (10.9%) and arthralgia (7.7%).
Vaccination with VAXNEUVANCE may not protect all vaccine recipients.
To learn more about Merck’s infectious diseases pipeline, visit www.merck.com.
For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Please see U.S. Prescribing Information for VAXNEUVANCE (Pneumococcal 15-valent Conjugate Vaccine) at https://www.merck.com/product/usa/pi_circulars/v/vaxneuvance/vaxneuvance_pi.pdf and Patient Information at https://www.merck.com/product/usa/pi_circulars/v/vaxneuvance/vaxneuvance_ppi.pdf.
Dec. 15, 2021 7:37 AM ET
By: Mamta Mayani, SA News Editor
December 14, 2021Download PDFSignificantly more patients treated with mirikizumab maintenance dosing achieved the primary endpoint of clinical remission at one year (52 weeks), and all key secondary endpoints were met Mirikizumab is the first and only anti-IL23p19 to demonstrate maintenance of clinical remission in a Phase 3 study in UC
INDIANAPOLIS, Dec. 14, 2021 /PRNewswire/ -- Eli Lilly and Company (NYSE: LLY) announced today that mirikizumab met the primary endpoint of clinical remission and all key secondary endpoints at one year in LUCENT-2, a Phase 3 maintenance study evaluating the efficacy and safety of mirikizumab for the treatment of patients with moderately-to-severely active ulcerative colitis (UC). Patients in this study were previously enrolled in a 12-week induction study, LUCENT-1. These results build on the positive outcomes from LUCENT-1.
In LUCENT-2, for patients who achieved clinical response with mirikizumab in the 12-week induction study and were re-randomized to mirikizumab maintenance dosing, a statistically higher proportion met the primary endpoint of clinical remission at one year compared to patients who were re-randomized to placebo (p<0.001). Clinical remission is reached when inflammation of the colon is controlled or resolved, leading to normalization or near-normalization of symptoms such as frequent and bloody stools. All key secondary endpoints were also met (p<0.001), including significantly higher proportions of patients treated with mirikizumab achieving endoscopic remission, corticosteroid-free remission, resolution or near-resolution of bowel urgency, improvement in endoscopic histologic intestinal inflammation and maintenance of remission, and greater reduction from baseline in bowel urgency symptoms at one year compared to placebo.
"In this maintenance study, treatment with mirikizumab demonstrated clinically meaningful and statistically significant improvements in clinical, endoscopic and histologic measures, including reduction of bowel urgency – a novel endpoint in the LUCENT program," said Bruce E. Sands, M.D., M.S., Dr. Burrill B. Crohn Professor of Medicine, Chief of the Dr. Henry D. Janowitz Division of Gastroenterology at the Icahn School of Medicine at Mount Sinai. "Bowel urgency is one of the most bothersome and disruptive symptoms people living with ulcerative colitis experience, and the LUCENT program leveraged an innovative and systematic patient-centric approach to assess patients' symptoms."
In the placebo-controlled maintenance cohort, the frequency of serious adverse events among patients treated with mirikizumab was numerically lower compared to placebo, and the overall safety profile was consistent with that of the previous mirikizumab studies in UC and other studies within the anti-IL-23p19 antibody class. The most common treatment emergent adverse events reported among patients treated with mirikizumab were nasopharyngitis, arthralgia and exacerbation of ulcerative colitis. Additional adverse events of interest reported among patients treated with mirikizumab included hypersensitivity, injection site reaction, depression, liver enzyme elevation, herpes zoster and oral candidiasis.
"Existing therapies aren't fully meeting the needs of people with ulcerative colitis who still have unresolved symptoms that impact their health and quality of life," said Lotus Mallbris, M.D., Ph.D., vice president of global immunology development and U.S. and global medical affairs at Lilly. "These positive long-term results provide evidence that mirikizumab has the potential to be an effective treatment option and become the first medicine of its kind for people with ulcerative colitis, including those who suffer from bowel urgency."
With these data, Lilly plans to submit a Biologics License Application (BLA) to the FDA for mirikizumab in UC, followed by submissions to other regulatory agencies around the world in the first half of 2022.
"The results announced today are encouraging for those who live with ulcerative colitis," said Michael Osso, President and CEO for Crohn's & Colitis Foundation. "We're excited about potential new options in the inflammatory bowel disease treatment space that may be able to help people living with ulcerative colitis successfully control their disease symptoms and achieve remission."
Topline results from the Phase 3 induction study, LUCENT-1, were announced in March 2021. Data from the Phase 3 LUCENT program, including results from LUCENT-1 and LUCENT-2, will be disclosed at upcoming congresses and in publications in 2022. Additional Phase 3 clinical trials are ongoing for mirikizumab in Crohn's disease.
About Mirikizumab
Mirikizumab is a humanized IgG4 monoclonal antibody that binds to the p19 subunit of interleukin 23. Mirikizumab is being studied for the treatment of immune-mediated diseases, including ulcerative colitis and Crohn's disease.
About the LUCENT Clinical Trial Program
The LUCENT Phase 3 clinical development program for mirikizumab includes LUCENT-1, LUCENT-2 and LUCENT-3. LUCENT-1 (NCT03518086) is a multicenter, randomized, double-blind, placebo-controlled induction study of mirikizumab in patients with moderately-to-severely active ulcerative colitis who have previously failed conventional and/or biologic therapies and/or JAK inhibitors. LUCENT-2 (NCT03524092) is a multicenter, randomized, double-blind, placebo-controlled, Phase 3 maintenance study in patients who completed LUCENT-1. LUCENT-3 (NCT03519945) is an open label extension study for eligible patients who have participated in mirikizumab UC trials.
The program began in 2018, with full results from the induction and maintenance studies anticipated in early 2022.
To learn more about Lilly, please visit us at lilly.com and lilly.com/newsroom. P-LLY
View original content to download multimedia:https://www.prnewswire.com/news-releases/mirikizumab-demonstrates-superiority-over-placebo-in-phase-3-maintenance-study-in-ulcerative-colitis-supporting-regulatory-submissions-in-2022-301444707.html
SOURCE Eli Lilly and Company
Dec. 14, 2021 5:36 PM ET
By: Jonathan M Block, SA News Editor
12/14/21 at 4:39 PM ESTPDF Version
– Data demonstrate sotrovimab, developed by Vir in conjunction with GlaxoSmithKline, continues to retain in vitro neutralizing activity against all tested variants of concern, including Omicron –
SAN FRANCISCO, Dec. 14, 2021 (GLOBE NEWSWIRE) -- Vir Biotechnology, Inc. (Nasdaq: VIR) today announced new preclinical data, published to the preprint server bioRxiv, demonstrating the impact of the significant antigenic shift of the new SARS-CoV-2 Omicron variant (B.1.1.529). A significant reduction in plasma neutralizing activity was observed against Omicron in sera from vaccinated and convalescent individuals. Researchers also tested the in vitro neutralizing activity of 44 monoclonal antibodies (mAbs) (eight of which are currently authorized or approved). Data demonstrate that sotrovimab and five other preclinical mAbs, developed by Vir in conjunction with GlaxoSmithKline, retained their in vitro neutralizing activity against Omicron.
These preclinical results were generated through pseudo-virus testing, and support recent pre-published findings from Vir researchers1, as well as other independent laboratories and institutions2. These data confirm Omicron’s immune evasion and its impact on the neutralizing activity of the majority of tested monoclonal antibody therapies that target the receptor binding motif and other areas of the spike protein that are more prone to mutate.
“While we work to better understand the potential impact of the significant number of mutations in the Omicron variant and its anticipated trajectory, it is encouraging to see such a high level of consistency across a rapidly growing body of preclinical data generated from both industry and academia,” said Herbert “Skip” Virgin, M.D., Ph.D., executive vice president of research and chief scientific officer for Vir Biotechnology. “These results, together with new data from external sources, continue to validate our approach of targeting a highly conserved region of the spike protein. We believe this strategy is responsible for sotrovimab’s ability to maintain activity against all tested variants of concern and interest, including Omicron. We look forward to applying these learnings to our ongoing efforts to address both current and future pandemics.”
About sotrovimab
Sotrovimab is an investigational SARS-CoV-2 neutralizing monoclonal antibody. The antibody binds to an epitope on SARS-CoV-2 shared with SARS-CoV-1 (the virus that causes SARS), indicating that the epitope is highly conserved, which may make it more difficult for resistance to develop. Sotrovimab, which incorporates Xencor, Inc.’s Xtend™ technology, has also been designed to achieve high concentration in the lungs to ensure optimal penetration into airway tissues affected by SARS-CoV-2 and to have an extended half-life.
Preclinical data, published in bioRxiv, demonstrate that sotrovimab retains activity against all currently tested variants of concern and interest of the SARS-CoV-2 virus as defined by WHO, plus others, including but not limited to Delta (B.1.617.2), Delta Plus (AY.1 or AY.2), Mu (B.1.621) and Omicron (B.1.1.529).
About the sotrovimab clinical development program
About global access to sotrovimab
Sotrovimab is authorized for emergency use in the United States. Xevudy (sotrovimab) received a positive scientific opinion under Article 5(3) of Regulation 726/2004 from the Committee for Human Medicinal Products in the EU, conditional marketing authorization in Great Britain, provisional marketing authorization in Australia and conditional marketing authorization in Saudi Arabia. It has been approved via the Special Approval for Emergency Pathway in Japan. Temporary authorizations for sotrovimab have been granted in a dozen countries.
GSK and Vir also recently submitted the Marketing Authorization Application to the European Medicines Agency for Xevudy for the treatment of adults and adolescents (aged 12 years and over and weighing at least 40kg) with coronavirus disease 2019 (COVID-19) who do not require oxygen supplementation and who are at risk of progressing to severe COVID-19.
Sotrovimab is supplied in several countries worldwide, including through national agreements in the United States, United Kingdom, Japan, Australia, Canada, Singapore, Switzerland and the United Arab Emirates. The companies have also signed a Joint Procurement Agreement with the European Commission to supply doses of sotrovimab to participating Member States of the EU. Additional agreements are yet to be announced due to confidentiality or regulatory requirements.
Sotrovimab in the United States
The following is a summary of information for sotrovimab. Healthcare providers in the US should review the Fact Sheets for information about the authorized use of sotrovimab and mandatory requirements of the EUA. Please see the Food and Drug Administration (FDA) Letter of Authorization, full Fact Sheet for Healthcare Providers and full Fact Sheet for Patients, Parents, and Caregivers.
Sotrovimab has been authorized by the US FDA for the emergency use described below. Sotrovimab is not FDA-approved for this use.
Sotrovimab is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of sotrovimab under section 564(b)(1) of the Act, 21 USC § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.
Authorized Use
The US FDA has issued an Emergency Use Authorization (EUA) to permit the emergency use of the unapproved product sotrovimab for the treatment of mild-to-moderate coronavirus disease 2019 (COVID-19) in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death.
Limitations of Authorized Use
For more information, please visit www.vir.bio.
Dec. 15, 2021 9:21 AM ET
Vir Biotechnology, Inc. (VIR) GSK
By: Jonathan Block, SA News Editor
PUBLISHED12 December 2021
Updated results from the ASCEND Phase III trial showed AstraZeneca’s Calquence (acalabrutinib) maintained a statistically significant progression-free survival (PFS) benefit at three years compared to investigator’s choice of rituximab combined with either idelalisib (IdR) or bendamustine (BR) in adults with relapsed or refractory chronic lymphocytic leukaemia (CLL), the most common type of leukaemia in adults.1,2
These data, presented at the 63rd American Society of Hematology (ASH) Annual Meeting & Exposition, demonstrated Calquence reduced the risk of disease progression or death by 71% versus IdR/BR as assessed by investigators at three years (based on a hazard ratio [HR] of 0.29; 95% confidence interval [CI]: 0.21-0.41; p<0.0001). Similar clinical benefits were observed in an exploratory analysis comparing each regimen with Calquence. Safety and tolerability of Calquence were consistent with earlier findings, with no new safety signals identified.1
Additional safety analyses from the ELEVATE-RR Phase III trial were also presented at ASH to further characterise adverse events (AEs) related to treatment with Bruton’s tyrosine kinase (BTK) inhibitors Calquence and ibrutinib. Overall, patients on ibrutinib experienced a 37% higher burden of AEs of any grade versus patients on Calquence.3
For any grade atrial fibrillation/flutter, a key secondary endpoint in the ELEVATE-RR trial, median time to onset was longer for Calquence versus ibrutinib (28.8 versus 16.0 months), and cumulative incidence was lower at all timepoints from six months through two years.3
Additionally, the ELEVATE-RR Phase III trial showed incidence of all-grade atrial fibrillation/flutter was lower for Calquence across subgroups of age, prior line of therapy and among patients without prior history of heart complications.3 Atrial fibrillation is an irregular heart rate that can increase the risk of stroke, heart failure and other heart-related complications.4
John F. Seymour, MBBS PhD, Peter MacCallum Centre and the Royal Melbourne Hospital, and a lead investigator on the ELEVATE-RR trial, said: “Patients with relapsed or refractory chronic lymphocytic leukaemia face limited options to successfully manage their disease, as they are often older and dealing with significant comorbidities. The risk of cardiac adverse events is an important consideration, especially for treatment with Bruton’s tyrosine kinase inhibitors because they can produce significant morbidity in some cases and also lead patients to discontinue treatment. The ELEVATE-RR data provide compelling evidence that acalabrutinib is a more tolerable option with reduced cardiovascular toxicity, giving clinicians further reassurance when prescribing this medicine that fewer patients will need to cease treatment due to adverse events, thus maintaining ongoing control of their disease, even in this complex setting.”
Anas Younes, Senior Vice President, Haematology R&D, AstraZeneca, said: “These impressive new long-term data support Calquence as the preferred therapy for the most common type of leukaemia in adults, with favourable safety compared to the current standards of care. The totality of the ASCEND and ELEVATE-RR data, in addition to data introducing a new tablet formulation for patients who need alternative methods of taking Calquence, continues to reinforce the positive experience that this medicine can deliver for patients with chronic lymphocytic leukaemia.”
ELEVATE-RR: Additional safety analyses of Calquence versus ibrutinib in relapsed or refractory CLL (abstract #3721)
Results from the ELEVATE-RR Phase III trial were first presented on 7 June 2021 at the American Society of Clinical Oncology (ASCO) Annual Meeting and published in the Journal of Clinical Oncology on 26 July 2021.
Additional safety data were used to characterise BTK inhibitor-related AEs, using measures of frequency, duration and drug exposure (versus incidence alone) to measure AE burden. Median treatment exposures were 38.3 months in the Calquence arm and 35.5 months in the ibrutinib arm.3
For any-grade hypertension, median time to onset was similar for Calquence and ibrutinib (8.1 months versus 7.0), but cumulative incidence was lower for Calquence at 6 months (5% versus 12%), 12 months (6% versus 16%), 18 months (8% versus 20%) and 24 months (8% versus 23%).
Hypertension also occurred less frequently with Calquence versus ibrutinib in subgroups of age, prior line of therapy and among patients without prior history.3
Among cardiovascular AEs of clinical interest, incidences of any-grade atrial fibrillation/flutter, hypertension and bleeding were statistically higher with ibrutinib versus Calquence, with higher exposure-adjusted incidence (2.0-, 2.8-, and 1.6-fold, respectively) and exposure-adjusted time with event (2.8-, 3.7-, and 1.8-fold).3
ASCEND
ASCEND (ACE-CL-309) is a global, randomised, multicentre, open-label Phase III trial evaluating the efficacy of Calquence in patients with relapsed or refractory CLL.5,9
In the trial, 310 patients were randomised (1:1) into two treatment arms. Patients in the first arm received Calquence monotherapy (100mg twice-daily until disease progression or unacceptable toxicity). Patients in the second arm received physician’s choice of either rituximab, a CD20 monoclonal antibody, in combination with idelalisib, a PI3-kinase inhibitor, or rituximab in combination with bendamustine, a chemotherapy.9
The primary endpoint at the interim analysis was PFS assessed by an Independent Review Committee (IRC), and key secondary endpoints included investigator-assessed PFS, IRC- and investigator-assessed overall response rate and duration of response, as well as overall survival, patient-reported outcomes and time to next treatment.
ASCEND is the first randomised Phase III trial to directly compare a BTK inhibitor as monotherapy to these combinations in relapsed or refractory CLL.5,9
ELEVATE-RR
ELEVATE-RR (ACE-CL-006) is a randomised, multicentre, open-label Phase III non-inferiority trial of Calquence versus ibrutinib in patients with relapsed or refractory CLL after at least one prior therapy, and at least one of the following prognostic factors: presence of 17p deletion, or presence of 11q deletion.10,11
In the trial, 533 patients were randomised (1:1) into two arms. Patients in the first arm received Calquence (100mg orally twice-daily until disease progression or unacceptable toxicity). Patients in the second arm received ibrutinib (420mg orally once-daily until disease progression or unacceptable toxicity).11
The primary endpoint for the trial was IRC-assessed PFS (non-inferiority; tested after 250 events, upper margin of 95% CI for HR<1.429). Secondary endpoints included incidence of atrial fibrillation, incidence of Grade 3 or higher infections, incidence of Richter’s transformation (a condition in which CLL changes into an aggressive form of lymphoma12) and OS.11
ELEVATE-RR is the first randomised Phase III trial to directly compare two BTK inhibitors as monotherapy in relapsed or refractory CLL.
Calquence
Calquence (acalabrutinib) is a next-generation, selective inhibitor of BTK. Calquence binds covalently to BTK, thereby inhibiting its activity.13,14 In B cells, BTK signalling results in activation of pathways necessary for B-cell proliferation, trafficking, chemotaxis and adhesion.13
Calquence is approved for the treatment of CLL and small lymphocytic lymphoma (SLL) in the US, approved for CLL in the EU and several other countries worldwide and approved in Japan for relapsed or refractory CLL and SLL. A Phase I trial is currently underway in Japan for the treatment of front-line CLL.
In the US and several other countries, Calquence is also approved for the treatment of adult patients with mantle cell lymphoma (MCL) who have received at least one prior therapy. The US MCL indication is approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials. Calquence is not currently approved for the treatment of MCL in Europe or Japan.
As part of an extensive clinical development programme, AstraZeneca and Acerta Pharma are currently evaluating Calquence in more than 20 company-sponsored clinical trials. Calquence is being evaluated for the treatment of multiple B-cell blood cancers including CLL, MCL, diffuse large B-cell lymphoma, Waldenström’s macroglobulinaemia, follicular lymphoma and other haematologic malignancies.
Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Dec. 13, 2021 3:29 AM ET AstraZeneca PLC (AZN) By: Mamta Mayani, SA News Editor
Sunday, Dec 12, 2021
South San Francisco, CA -- December 12, 2021 --
Genentech, a member of the Roche Group (SIX: RO, ROG; OTCQX: RHHBY), today announced results from an interim analysis of the Phase III HAVEN 6 study, which show Hemlibra® (emicizumab-kxwh) demonstrated a favorable safety profile and effective bleed control in people with moderate or mild hemophilia A without factor VIII inhibitors. The data were presented at the 63rd American Society of Hematology (ASH) Annual Meeting and Exposition as an oral presentation on December 12, 2021.
While the treatment and management of severe hemophilia A are well established, there is less information and treatment guidance on moderate and mild hemophilia A, which can lead to delayed or missed diagnoses of bleeding episodes. Considering this population may not use preventative treatments, these patients may experience worsened clinical burden, with less than 30% of people with moderate or mild hemophilia A living a bleed-free life.
“We are pleased to see that Hemlibra continues to show benefit in additional hemophilia A populations, regardless of severity,” said Levi Garraway, M.D., Ph.D., chief medical officer and head of Global Product Development. “The clinical evidence for Hemlibra derives from one of the largest pivotal clinical trial programs in hemophilia A, with and without factor VIII inhibitors. We remain committed to working together with the hemophilia community as we further explore the efficacy and safety of Hemlibra in broader populations.”
HAVEN 6 is a Phase III study evaluating the safety, efficacy, pharmacokinetics and pharmacodynamics of Hemlibra in people with moderate or mild hemophilia A without factor VIII inhibitors. This interim analysis included data from 71 participants (69 men and two women), 20 of whom had mild hemophilia A without factor VIII inhibitors and 51 of whom had moderate hemophilia A without factor VIII inhibitors. Thirty-seven participants were on factor VIII prophylaxis at baseline.
This interim analysis was conducted after 50 participants with moderate hemophilia A completed at least 24 weeks in the study or withdrew. Data cutoff was on April 16, 2021. These data show Hemlibra demonstrated a favorable safety profile and effective bleed control in the HAVEN 6 study, with 80.3% of participants experiencing no bleeding episodes that required treatment and 90.1% experiencing no joint bleeds that required treatment. Annualized bleeding rates (ABR) remained low, consistent with previously reported observations from the HAVEN 1-4 studies. In addition, of the 50 participants aged 12 years or older who responded to the EmiPref questionnaire, 48 (96.0%) preferred Hemlibra to their previous treatment, one preferred their old treatment, and one expressed no preference.
The most common adverse events (AEs) occurring in 10% or more people in the HAVEN 6 study were headache (14.1%) and local injection site reactions (ISRs) (12.7%). Eleven people (15.5%) reported a Hemlibra-related AE, with ISRs being the most common (12.7%). There were no deaths, or cases of thrombotic microangiopathy (TMA) or serious thrombotic events (TEs) in the study as of the data cutoff, reinforcing Hemlibra’s favorable safety profile.
A separate analysis of TE and TMA events in people taking Hemlibra, including real-world data, will also be presented as a poster at ASH. These results showed that the evaluation of reported events without concomitant activated prothrombin complex concentrate (aPCC) remains similar to previous analyses as exposure increases, and the benefit/risk profile of Hemlibra remains unchanged. These data further confirm the favorable safety profile of Hemlibra, consistent with results from previous HAVEN and STASEY studies.
Hemlibra is approved to treat people with hemophilia A with factor VIII inhibitors in more than 100 countries worldwide and people without factor VIII inhibitors in more than 90 countries worldwide, including the U.S., EU and Japan. Hemlibra has been studied in one of the largest clinical trial programs in people with hemophilia A with and without factor VIII inhibitors, including eight Phase III studies.
About Hemlibra
Hemlibra is a bispecific factor IXa- and factor X-directed antibody. It is designed to bring together factor IXa and factor X, proteins required to activate the natural coagulation cascade and restore the blood clotting process for hemophilia A patients. Hemlibra is a prophylactic (preventative) treatment that can be administered by an injection of a ready-to-use solution under the skin (subcutaneously) once weekly, every two weeks or every four weeks. Hemlibra was created by Chugai Pharmaceutical Co., Ltd. and is being co-developed globally by Chugai, Roche and Genentech.
Hemlibra U.S. Indication
Hemlibra is a prescription medicine used for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children, ages newborn and older, with hemophilia A with or without factor VIII inhibitors.
Please see the Hemlibra full Prescribing Information and Medication Guide for more important safety information including Serious Side Effects.
For more information visit http://www.gene.com/hemophilia.
For additional information about the company, please visit http://www.gene.com.
Dec. 13, 2021 1:54 AM ET
Roche Holding AG (RHHBY), CHGCYCHGCF
By: Mamta Mayani, SA News Editor
TARRYTOWN, N.Y. and PARIS, Dec. 13, 2021 /PRNewswire/ --
Dupixent significantly improved skin clearance and reduced overall disease severity and itch in pivotal trial that met all primary and secondary endpoints
Global regulatory filings planned in the coming months, starting with the U.S. by the end of 2021
Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) and Sanofi today announced detailed positive Phase 3 results that showed adding Dupixent® (dupilumab) to standard-of-care topical corticosteroids (TCS) significantly improved skin clearance and reduced overall disease severity and itch in infants and children aged 6 months to 5 years with uncontrolled moderate-to-severe atopic dermatitis. These data will be presented today in a late-breaking session at the 2021 Revolutionizing Atopic Dermatitis Conference (RAD 2021).
"One of most challenging aspects of my job as a physician is having limited treatment options to help babies and young children suffering from moderate-to-severe atopic dermatitis, which can disrupt their ability to fully thrive in these early years of life," said Amy S. Paller, M.D, Walter J. Hamlin Professor and Chair of Dermatology and Professor of Pediatrics at Northwestern University Feinberg School of Medicine, and principal investigator of the trial. "These results show dupilumab can significantly improve the signs and overall severity of atopic dermatitis in children as young as 6 months. Safety is of paramount importance when treating children at such a young age. We are encouraged that these data show a safety profile consistent with what has been seen in other age groups. We will continue to follow these patients for up to 5 years in an open-label trial."
Eighty-five to 90% of patients with atopic dermatitis develop symptoms before the age of 5, which can often continue through adulthood. Symptoms include intense, persistent itch and skin lesions that cover much of the body (58% on average for the patients in this trial at baseline), resulting in skin dryness, cracking, redness or darkening, and crusting and oozing, along with increased risk of skin infections. Moderate-to-severe atopic dermatitis may also significantly impact the quality of life of a young child, their parents and caregivers. In addition, the underlying type 2 inflammation involved in atopic dermatitis can contribute to the development of other diseases, like asthma and certain allergies, that may also appear throughout a person's life.
Topline results from the randomized, placebo-controlled pivotal trial, which met all primary and secondary endpoints, were announced in August 2021. Data presented at RAD 2021 showed that at 16 weeks, patients who added Dupixent to low-potency TCS experienced the following, compared to low-potency TCS alone (placebo):
The safety profile observed in the randomized, placebo-controlled trial was consistent with the well-established safety profile of Dupixent in adults, adolescents and children 6 years and older with moderate-to-severe atopic dermatitis. Overall rates of adverse events (AEs) were 64% for Dupixent and 74% for placebo. Most common AEs and AEs of special interest included nasopharyngitis (8% Dupixent, 9% placebo), upper respiratory tract infection (6% Dupixent, 8% placebo), conjunctivitis (5% Dupixent, 0% placebo) and herpes viral infections (6% Dupixent, 5% placebo).
These results will form the basis of global regulatory submissions for this age group, beginning with the U.S. in 2021 and European Union in the first half of 2022.
Additionally, long-term data from the Phase 3 trial in patients aged 6 to 11 years with moderate-to-severe atopic dermatitis are also being presented in a late-breaking session. Efficacy and safety results at one year were consistent with the known profile of Dupixent in atopic dermatitis.
The data from these trials add to the extensive LIBERTY AD clinical program – the largest Phase 3 clinical trial program in atopic dermatitis, involving approximately 3,500 infants, children, adolescents and adults to date.
Dupixent is the first biologic medicine to demonstrate positive results in this young patient population. The efficacy and safety of Dupixent in children below the age of 6 years have not been fully evaluated by any regulatory authority.
About the Dupixent Trial
LIBERTY AD PRESCHOOL is a two-part Phase 2/3 trial. The Phase 3 randomized, double-blind, placebo-controlled part of the trial (Part B) evaluated the efficacy and safety of Dupixent added to standard-of-care low-potency TCS compared to low-potency TCS alone (placebo) in 162 children aged 6 months to 5 years with uncontrolled moderate-to-severe atopic dermatitis.
The primary endpoints assessed the proportion of patients achieving an Investigator's Global Assessment (IGA) score of 0 (clear) or 1 (almost clear) and 75% improvement in Eczema Area and Severity Index (EASI-75) at week 16. EASI measures extent and severity of the disease. Itch was assessed using a caregiver-reported 0 to 10 Numerical Rating Scale. Patients treated with Dupixent received either 200 mg (for children weighing ≥5 to <15 kg) or 300 mg (for children weighing ≥15 to <30 kg) every four weeks.
In total, there were 162 patients in the trial (83 Dupixent, 79 placebo), the average age was 3.8 years and 61% were male. Approximately 12% of patients were Latino/Hispanic and 19% were Black/African American. At the start of the trial, 77% of patients had severe disease and 29% had previously used systemic immunosuppressants for their atopic dermatitis, and on average, patients had atopic dermatitis covering 58% of their body. Furthermore, 81% of these patients had at least one concurrent type 2 inflammatory and/or allergic condition such as allergic rhinitis and asthma.
Part B of the Phase 3 trial was informed by Part A, which was an open-label, single-ascending-dose, sequential cohort Phase 2 trial designed to assess the pharmacokinetics and safety of Dupixent in children aged 6 months to 5 years with uncontrolled severe atopic dermatitis.
Children who completed Part A or Part B of the trial were eligible to enroll in an open-label extension trial to assess the safety and efficacy of long-term treatment with Dupixent in this age group for an additional five years.
About Dupixent
Dupixent, which was invented using Regeneron's proprietary VelocImmune® technology, is a fully human monoclonal antibody that inhibits the signaling of the interleukin-4 (IL-4) and interleukin-13 (IL-13) pathways. Dupixent is not an immunosuppressant and does not require lab monitoring. IL-4 and IL-13 are key and central drivers of the type 2 inflammation that plays a major role in atopic dermatitis, asthma and chronic rhinosinusitis with nasal polyposis (CRSwNP).
Dupixent is currently approved in the U.S., Europe, Japan and other countries around the world for use in specific patients with moderate-to-severe atopic dermatitis, as well as certain patients with asthma or CRSwNP in different age populations. Dupixent is also approved in one or more of these indications in more than 60 countries around the world and more than 300,000 patients have been treated globally.
U.S. Indications
DUPIXENT is a prescription medicine used:
Please see accompanying full Prescribing Information including Patient Information.
For additional information about the company, please visit www.regeneron.com or follow @Regeneron on Twitter.
SOURCE Regeneron Pharmaceuticals, Inc.
Dec. 13, 2021 1:32 AM ET Regeneron Pharmaceuticals, Inc. (REGN), SNY
By: Mamta Mayani, SA News Editor1 Comment
12/08/2021
– Exploratory Analyses from the Pivotal HER2CLIMB Trial Show Patients with Stable and Active Brain Metastases Treated with a TUKYSA Regimen Maintained a Survival Benefit After Additional 15.6 Months of Follow-Up –
BOTHELL, Wash.--(BUSINESS WIRE)-- Seagen Inc. (Nasdaq:SGEN) today announced the presentation of new data from exploratory analyses from the pivotal HER2CLIMB trial showing that improvement in overall survival (OS) was maintained after an additional 15.6 months of follow-up when TUKYSA® (tucatinib) was combined with trastuzumab and capecitabine in patients with HER2-positive metastatic breast cancer (MBC) who had stable or active brain metastases. The data were featured today in a spotlight poster (Abstract #PD4-04) at the 2021 San Antonio Breast Cancer Symposium (SABCS).
“The risk of breast cancer spreading to the brain is more pronounced for patients with aggressive subtypes of breast cancer, including HER2-positive breast cancer,” said Nancy U. Lin, M.D., Director of the Metastatic Breast Cancer Program in the Susan F. Smith Center for Women’s Cancers at Dana-Farber in Boston, MA. “These analyses provide a hopeful outcome for patients with HER2-positive metastatic breast cancer when cancer has spread to the brain as they show that the TUKYSA regimen not only helped patients live longer but also slowed disease progression in the brain.”
“After more than two years of follow up, these exploratory analyses show the impact that TUKYSA can have on HER2-positive metastatic breast cancer patients and underscore its importance as a treatment option,” said Roger D. Dansey, M.D., Chief Medical Officer of Seagen.
After a median follow-up of 29.6 months, the TUKYSA regimen improved OS for patients with brain metastases by 9.1 months compared to trastuzumab and capecitabine alone (21.6 months vs. 12.5 months) (HR: 0.60; [95% CI: 0.44, 0.81]). The benefit extended to patients with active or stable brain metastases.
About HER2CLIMB
HER2CLIMB is a multinational randomized (2:1), double-blind, placebo-controlled, active comparator, pivotal clinical trial comparing TUKYSA in combination with trastuzumab and capecitabine compared with trastuzumab and capecitabine alone in patients with locally advanced unresectable or metastatic HER2-positive breast cancer who were previously treated with trastuzumab, pertuzumab and T-DM1. The primary endpoint of the trial was PFS per Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 as determined by blinded independent central review (BICR) in the first 480 patients enrolled in the trial. HER2CLIMB enrolled a total of 612 patients to support the analyses of key secondary endpoints, including overall survival, PFS per BICR in patients with brain metastases at baseline and confirmed objective response rate. Safety data were evaluated throughout the study.1
About TUKYSA (tucatinib)
TUKYSA is an oral medicine that is a tyrosine kinase inhibitor of the HER2 protein. In vitro (in lab studies), TUKYSA inhibited phosphorylation of HER2 and HER3, resulting in inhibition of downstream MAPK and AKT signaling and cell growth (proliferation), and showed anti-tumor activity in HER2-expressing tumor cells. In vivo (in living organisms), TUKYSA inhibited the growth of HER2-expressing tumors. The combination of TUKYSA and the anti-HER2 antibody trastuzumab showed increased anti-tumor activity in vitro and in vivo compared to either medicine alone.
TUKYSA is approved in 36 countries. It was approved by the U.S. FDA in April 2020 and by the European Medicines Agency and the UK Medicines and Healthcare Products Regulatory Agency in February 2021. Merck, known as MSD outside the U.S. and Canada, has exclusive rights to commercialize TUKYSA in Asia, the Middle East and Latin America and other regions outside of the U.S., Canada and Europe.
U.S. Indication and Important Safety Information
TUKYSA is indicated in combination with trastuzumab and capecitabine for treatment of adult patients with advanced unresectable or metastatic HER2-positive breast cancer, including patients with brain metastases, who have received one or more prior anti-HER2-based regimens in the metastatic setting.
For more information on the company’s marketed products and robust pipeline, visit www.seagen.com and follow @SeagenGlobal on Twitter.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211208005218/en/
Source: Seagen Inc.
Dec. 09, 2021 4:48 AM ETSeagen Inc. (SGEN)By: Mamta Mayani, SA News Editor
Dec 11, 2021
Basel, December 11, 2021 — Novartis today announced new 48-week data from the Phase III ASCEMBL trial of Scemblix® (asciminib) demonstrating that the results observed in the primary analysis (24 weeks) vs. Bosulif®* (bosutinib) were maintained in longer-term follow up for patients with Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase (Ph+ CML-CP) previously treated with two or more tyrosine kinase inhibitors (TKIs)1-4.
In this analysis, presented at the 63rd American Society of Hematology Annual Meeting & Exposition (ASH), the major molecular response (MMR) rate at 48 weeks was 29.3% for patients treated with Scemblix vs. 13.2% for patients in the Bosulif arm, which is consistent with a doubling of the efficacy at 24 weeks (25% vs. 13% [P=0.029])1-4. The proportion of patients treated with Scemblix who experienced adverse reactions leading to discontinuation was more than three times lower than those in the Bosulif arm (7.1% vs. 25%)1.
Scemblix is the first FDA-approved CML treatment that works by binding to the ABL myristoyl pocket2. This novel mechanism of action, also known in scientific literature as a STAMP inhibitor, can help address resistance to TKI therapy in patients with CML and overcome mutations at the defective BCR-ABL1 gene, which is associated with the over-production of leukemic cells2-4. Scemblix continues to be studied across multiple lines of treatment for CML-CP3-12.
“We often see that sequential use of TKI treatments can be associated with increased failure rates and greater concerns regarding potential treatment side effects as patients move to later lines. Scemblix offers an increasingly proven option for patients living with CML who have previously tried two or more TKIs, and takes a different approach to targeted inhibition to better manage CML,” said Dr. Michael J. Mauro**, Hematologist and Myeloproliferative Neoplasms Program Leader at Memorial Sloan Kettering Cancer Center (MSK).
In this updated analysis, responses were also durable, with 60 out of 62 patients on Scemblix maintaining MMR at time of their last assessment1. Scemblix continued to deliver more favorable deep molecular responses (MRs) with MR4 and MR4.5 rates at 48 weeks of 10.8% and 7.6%, compared to 3.9% and 1.3% in patients treated with Bosulif, respectively1. Additionally, the cumulative proportion of patients achieving a level of BCR-ABL1IS ≤1% at 48 weeks – a predictor of better long-term outcomes in this heavily pretreated patient population – was higher in the Scemblix arm than in the Bosulif arm (50.8% vs 33.7%)1.
The most common reason for treatment discontinuation was lack of efficacy in 37 (23.6%) patients treated with Scemblix and 27 (35.5%) patients treated with Bosulif1. Median duration of exposure was 15.4 months (range, 0.0–37.3 months) for Scemblix and 6.8 months (range, 0.2–34.3 months) for Bosulif1. With a longer duration of exposure, the safety and tolerability profile remains consistent with the primary analysis of the ASCEMBL trial1-4. The most common (incidence ≥ 20%) adverse reactions reported in this analysis were thrombocytopenia (29.5%) and neutropenia (23.1%) in the Scemblix arm; and diarrhea (71.1%), nausea (46.1%), increased ALT (28.9%), vomiting (26.3%), rash (23.7%), increased AST (21.1%) and neutropenia (21.1%) in the Bosulif arm1.
“We are excited to see the continued benefit with Scemblix for this long-underserved patient population,” said Jeff Legos, Executive Vice President, Global Head of Oncology & Hematology Development at Novartis. “These data are encouraging as we continue to challenge the current standard of care in CML by exploring if and how Scemblix can help more patients living with this disease.”
Scemblix received FDA approval in October 2021 and is currently available for physicians to prescribe to appropriate patients in the US2. Scemblix is also being evaluated in studies across multiple treatment lines and indications for CML-CP, including the ASC4FIRST Phase III study for newly diagnosed adult patients, as well as in a Phase Ib/II dose assessment study in pediatric patients with Ph+ CML-CP. Trial-in-progress posters for both are being presented at ASH13-22.
To learn more about our long-standing commitment to transforming the lives of patients with CML with bold science, the latest information from Novartis and access to our ASH 2021 scientific presentations, visit the Novartis Oncology Congress Hub at https://www.hcp.novartis.com/virtual-congress/ash-2021/.
About Scemblix® (asciminib)
Scemblix (asciminib) is indicated for the treatment of adult patients with Ph+ CML-CP pre-treated with two or more TKIs, as well as adult patients with Ph+ CML-CP with the T315I mutation. The first indication is approved under the US FDA Accelerated Approval Program based on MMR rate at 24 weeks; continued approval for the first indication may be contingent upon verification and description of clinical benefit from confirmatory evidence2.
Scemblix is the first FDA-approved CML treatment that binds to the ABL myristoyl pocket2. This novel mechanism of action, also known in scientific literature as a STAMP inhibitor, can help address resistance in patients with CML previously treated with two or more TKIs and overcome mutations at the defective BCR-ABL1 gene, which is associated with the over-production of leukemic cells3-12.
Novartis has initiated regulatory filings for Scemblix in multiple countries and regions across the globe.
Scemblix represents an important development for patients who experience resistance and/or intolerance to currently available TKI therapies, and it is being studied across multiple treatment lines for CML-CP3-20. Specifically, the ASC4FIRST Phase III study (NCT04971226) evaluates Scemblix as a first-line treatment and is in the recruitment phase14,21.
About Novartis Commitment to CML
Novartis has a long-standing scientific commitment to patients living with CML. For more than 20 years, our bold science has helped transform CML into a chronic disease for many patients. Despite these advancements, we’re not standing still. We continue to research ways to target the disease, seeking to address the challenges with treatment resistance and/or intolerance that many patients face. Novartis also continues to reimagine CML care through its commitment to sustainable access for patients and collaboration with the global CML community.
Indication
SCEMBLIX® (asciminib) tablets is a prescription medicine used to treat adults with Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase (CP), previously treated with 2 or more tyrosine kinase inhibitor (TKI) medicines. The effectiveness of SCEMBLIX in these patients is based on a study that measured major molecular response (MMR) rates. No clinical information is available to show if these patients treated with SCEMBLIX live longer or if their symptoms improve. Ongoing studies exist to find out how SCEMBLIX works over a longer period of time.
SCEMBLIX is also approved for use in adults with Ph+ CML in CP with the T315I mutation.
It is not known if SCEMBLIX is safe and effective in children.
Please see full Prescribing Information for SCEMBLIX, available at https://www.novartis.us/sites/www.novartis.us/files/scemblix.pdf.
Find out more at https://www.novartis.com.
Dec. 13, 2021 6:11 AM ET Novartis AG (NVS)
By: Mamta Mayani, SA News Editor
Dec 11, 2021
Basel, December 11, 2021 — Novartis announced Kymriah® (tisagenlecleucel) demonstrated strong efficacy in high-risk patients with relapsed or refractory (r/r) follicular lymphoma (FL) based on a subgroup analysis from an approximately 17-month median follow-up of the pivotal Phase II ELARA study1. These results were presented in an oral session at the 63rd American Society of Hematology Annual Meeting & Exposition (ASH) (Abstract #131).
In the subgroup analysis, results showed high rates of durable responses were induced by Kymriah in patients for the majority of high-risk disease subgroups, who typically have a poor prognosis. Complete response rate (CRR), overall response rate (ORR), and durability of response (DOR) were maintained in most patients in the high-risk subgroups, with the exception of those in three of the nine subgroups analyzed: those with progression-of-disease within two years (POD24), high total metabolic tumor volume (TMTV) and patients who had received five or more prior lines of therapy1.
“It’s truly exciting to see that after treatment with Kymriah in patients with difficult-to-treat, high-risk follicular lymphoma, patients are experiencing long-lasting responses with a low risk of severe adverse events,” said Catherine Thieblemont, MD, PhD, Professor of Hematology in the Paris VII- University, France and Head of the Hemato-Oncology Unit of St-Louis Hospital in Paris.
High and durable responses were seen in the overall population of the ELARA study in which 94 patients were evaluable for efficacy with a median follow-up of approximately 17 months. The CRR was 69% (95% CI, 60-78), ORR was 86% (95% CI, 78-92), 12-month progression-free survival (PFS) was 67% (95% CI, 56-76) and nine-month DOR was 76% (95% CI, 65-84). For patients who had a complete response (CR), 12-month PFS was 86% (95% CI, 74-92) and the estimated DOR rate was 87% (95% CI, 75-93). In the safety analysis (n=97), the safety profile of Kymriah continued to reflect the remarkable results seen in earlier ELARA analyses. Within eight weeks of infusion, 48% of patients experienced cytokine release syndrome (CRS), with no patients experiencing CRS of grade 3 or higher as defined by the Lee scale, 37% had neurological events (3% were greater than or equal to grade 3) and there were no treatment-related deaths1.
A separate analysis of hospitalization and intensive care unit patterns for patients treated in the inpatient and outpatient settings in the ELARA trial suggest Kymriah may reduce healthcare resource utilization for patients with r/r FL treated in the outpatient setting (Abstract #3533). Among patients treated in the outpatient setting (n=17), 35% did not require hospitalization during the first two months of the post-infusion period; those who did had a lower median average length of stay than the patients infused in an inpatient setting (4 days [n=17] vs 12 days [n=80]). Additionally, the mean hospitalization costs in the post-infusion period were substantially lower in the outpatient versus inpatient setting2.
“The ability to administer Kymriah, a potentially definitive treatment, in the outpatient setting may reduce the burden of therapy for patients and their care teams,” said Jeff Legos, Executive Vice President, Global Head of Oncology & Hematology Development, Novartis. “The breadth of follicular lymphoma data presented at this year’s ASH demonstrate the potential for Kymriah to provide transformative results and a positive impact on health systems overall.”
Novartis is committed to bringing the benefits of Kymriah to more patients with advanced blood cancers worldwide, with regulatory submissions for follicular lymphoma in the US and EU complete in October 2021. If approved in this indication, Novartis will look to confirm these results of the ELARA trial and related analyses in the real-world setting.
About the ELARA trial
ELARA is a Phase II, single-arm, multicenter, open-label trial investigating the efficacy and safety of Kymriah in adult patients with r/r FL after at least two prior therapies. This international trial has enrolled patients from over 30 sites in 12 countries worldwide. The primary endpoint is complete response rate (CRR) based on best response by central review (Lugano 2014 criteria). Patients evaluable for efficacy had measurable disease at infusion and more than six months of follow-up from infusion or discontinued early. After infusion, disease assessments were performed every three months. In a high-risk subgroup analysis, patients evaluated included those with prior hematopoietic stem cell transplant, double-refractory disease, high Follicular Lymphoma International Prognostic Index at study entry, high lactate dehydrogenase at baseline, high C-reactive protein prior to infusion, radiological bulky disease, progression-of-disease within two years (POD24), high total metabolic tumor volume (TMTV), and patients who had received five or more prior lines of therapy1. Secondary endpoints include overall response rate, duration of response, progression-free survival, overall survival and safety. Primary analysis data announced at ASCO 2021 showed Kymriah led to responses for the majority of patients treated, with 66% achieving a complete response (95% CI, 56-75). The overall response rate was 86% (95% CI, 78-92)9. Importantly, no patients in ELARA trial experienced grade 3 or higher cytokine release syndrome related to Kymriah within the first 8 weeks following infusion, the most common side effect associated with CAR-T therapy9.
US FDA approved indications for Kymriah
Kymriah® (tisagenlecleucel) is a CD19-directed genetically modified autologous T cell immunotherapy, which is indicated for:
Please see the full Prescribing Information for KYMRIAH, including Boxed WARNING, and Medication Guide at www.KYMRIAH.com
Find out more at https://www.novartis.com.
Dec. 13, 2021 6:21 AM ET
By: Mamta Mayani, SA News Editor
12/11/2021CATEGORY:
Phase 3 TRANSFORM study shows Breyanzi significantlyimproved event-free survival (EFS) vs. chemotherapy plus autologous stem cell transplant, with a 65% reduction in risk of EFS events in first disclosure of results presented at ASH 2021
Breyanzi showed a manageable safety profile and no new safety signals observed, with low rates of severe cytokine release syndrome and neurologic events
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) today announced the first disclosure of results from a prespecified interim analysis of the pivotal TRANSFORM study, a global, randomized, multicenter, Phase 3 study evaluating Breyanzi (lisocabtagene maraleucel; liso-cel), a CD19-directed chimeric antigen receptor (CAR) T cell therapy, as a second-line treatment in adults with relapsed or refractory large B-cell lymphoma (LBCL) compared to the standard of care consisting of salvage chemotherapy followed by high-dose chemotherapy plus autologous hematopoietic stem cell transplant (HSCT). Results show, at a median follow up of 6.2 months, Breyanzi significantly improved event-free survival (EFS) compared to standard of care, the study’s primary endpoint, with a median EFS of 10.1 months (95% CI: 6.1-NR) for Breyanzi and 2.3 months (95% CI: 2.2-4.3) for standard of care (HR: 0.349; p<0.0001), representing a 65% reduction in risk of EFS events with Breyanzi. The data will be presented in an oral session during the 63rd American Society of Hematology (ASH) Annual Meeting and Exposition (Abstract #91) and has been selected for inclusion in the ASH Annual Meeting Press Program.
“For more than 20 years, salvage chemotherapy followed by high-dose chemotherapy and stem cell transplant have been the mainstay of care for patients with second-line relapsed or refractory LBCL, but only a small portion of patients experience long-term benefit with this approach,” said Manali Kamdar, M.D., lead investigator and Associate Professor, Clinical Director of Lymphoma Services, Division of Hematology, Hematologic Malignancies and Stem Cell Transplantation, University of Colorado Cancer Center. “With liso-cel outperforming the current standard of care for patients with hard-to-treat disease in the TRANSFORM study, these results may pave the way for a practice-changing treatment approach where patients whose disease relapses or is refractory to frontline therapy can be treated with a personalized CAR T cell therapy to increase the potential for improved outcomes.”
In the TRANSFORM study, 184 patients with primary refractory LBCL or relapsed disease within ≤12 months after first-line therapy who were eligible for autologous HSCT were randomized to receive Breyanzi (n=92) or salvage chemotherapy followed by high-dose chemotherapy and autologous HSCT (n=92), which is considered the current standard of care for these patients. In the trial, which allowed for crossover, 50 patients switched from the standard of care arm to receive Breyanzi following failure to achieve a response by nine weeks post-randomization (after three cycles of salvage chemotherapy) or after disease progression at any time.
The majority of patients (86%) treated with Breyanzi achieved a complete or partial response, with 66% of patients achieving a complete response. In comparison, less than half (48%) of patients who received the standard of care achieved a response, and only 39% of these patients achieved a complete response (p<0.0001). Median progression-free survival was significantly longer with Breyanzi compared to standard of care (14.8 months vs. 5.7 months [HR: 0.406; p=0.0001]). Although overall survival data were not yet mature, the prespecified interim analysis showed a trend favoring Breyanzi compared with the standard of care (HR: 0.509, 95% CI: 0.258-1.004, p=0.0257).
Breyanzi exhibited a manageable safety profile with very low rates of severe cytokine release syndrome (CRS) and neurologic events, and no new safety signals were observedin this second-line setting. In the trial, no Grade 4/5 CRS or neurologic events were reported. Any-grade CRS was reported in 49% of patients, with Grade 3 CRS reported in only one patient. Any-grade neurologic events were reported in 12% of patients treated with Breyanzi, with Grade 3 neurologic events reported in four patients (4%).
Results from the long-term follow-up of treatment with Breyanzi in the TRANSCEND NHL 001 study, the largest pivotal trial in third-line plus relapsed or refractory LBCL, reinforcing durable remissions demonstrated with Breyanzi, will also be presented at the meeting during a poster presentation on Sunday, December 12 (Abstract #2840).
“Breyanzi, a differentiated CD-19 directed CAR T cell therapy,has the potential to transform the treatment paradigm for relapsed or refractory LBCL across lines of therapy, with a proven significant clinical benefit and a consistent safety profile in the TRANSFORM and TRANSCEND NHL 001 trials presented at this year’s ASH Annual Meeting,” said Anne Kerber, senior vice president, Cell Therapy Development, Bristol Myers Squibb. “We designed a patient-centric clinical trial program for Breyanzi with the strategic intent to improve outcomes for patients with some of the most aggressive blood cancers, aligned with our commitment to advancing a leading cell therapy portfolio for patients in need.”
Breyanzi is approved by the U.S. Food and Drug Administration for the treatment of adult patients with relapsed or refractory LBCL after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified (including DLBCL arising from indolent lymphoma), high-grade B-cell lymphoma, primary mediastinal large B-cell lymphoma, and follicular lymphoma grade 3B. Breyanzi is not indicated for the treatment of patients with primary central nervous system lymphoma. The U.S. Prescribing Information for Breyanzi has a BOXED WARNING for the risks of CRS and neurologic toxicities. Breyanzi is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the BREYANZI REMS. The use of Breyanzi in primary refractory or relapsed LBCL is investigational and not approved in any geography.
About TRANSFORM
TRANSFORM (NCT03575351) is a pivotal, global, randomized, multicenter Phase 3 trial evaluating Breyanzi compared to current standard of care regimens in adults with high-risk, transplant-eligible, relapsed and refractory large B-cell lymphoma (LBCL). All enrolled patients have LBCL and were relapsed or refractory within ≤12 months from CD20 antibody and anthracycline containing first-line therapy. Patients were randomized to receive Breyanzi or standard of care salvage therapy, including rituximab plus dexamethasone, high-dose cytarabine, and cisplatin (R-DHAP), rituximab plus ifosfamide, carboplatin and etoposide (R-ICE), or rituximab plus gemcitabine, dexamethasone and cisplatin (R-GDP) per the investigators’ choice before proceeding to high-dose chemotherapy and hematopoietic stem cell transplant. The primary endpoint of the study is event-free survival, defined as time from randomization to death from any cause, progressive disease, failure to achieve complete response or partial response, or start of new antineoplastic therapy due to efficacy concerns, whichever occurs first. Complete response rate is a key secondary endpoint. Other efficacy endpoints include progression-free survival, overall survival, overall response rate and duration of response.
About TRANSCEND NHL 001
TRANSCEND NHL 001 is an open-label, multicenter, pivotal Phase 1 study to determine the safety, pharmacokinetics, and antitumor activity of Breyanzi in patients with relapsed/refractory B-cell non-Hodgkin lymphoma, including diffuse large B-cell lymphoma, high-grade B-cell lymphoma, primary mediastinal B-cell lymphoma, follicular lymphoma Grade 3B and mantle cell lymphoma. The primary outcome measures were treatment-related adverse events, dose-limiting toxicities and objective response rate. Secondary outcome measures included complete response rate, duration of response and progression-free survival.
About Breyanzi
Breyanzi is a CD-19 directed chimeric antigen receptor (CAR) T cell therapy with a defined composition and 4-1BB costimulatory domain. Breyanzi is administered as a defined composition to reduce variability of the CD8 and CD4 component dose. The 4-1BB signaling domain enhances the expansion and persistence of the CAR T cells. Breyanzi is approved by the U.S. Food and Drug Administration for the treatment of adult patients with relapsed or refractory LBCL after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified (including DLBCL arising from indolent lymphoma), high-grade B-cell lymphoma, primary mediastinal large B-cell lymphoma, and follicular lymphoma grade 3B. Breyanzi is not indicated for the treatment of patients with primary central nervous system lymphoma.
Breyanzi is also approved in Japan for third-line plus relapsed and refractory LBCL, and Marketing Authorization Applications for Breyanzi for this indicationare currently under review in the European Union, Switzerland and Canada. Bristol Myers Squibb’s clinical development program for Breyanzi includes clinical studies in earlier lines of treatment for patients with relapsed or refractory LBCL and other types of lymphoma. For more information, visit clinicaltrials.gov.
Please see full Prescribing Information, including Boxed WARNINGS and Medication Guide.
About Bristol Myers Squibb
Bristol Myers Squibb is a global biopharmaceutical company whose mission is to discover, develop and deliver innovative medicines that help patients prevail over serious diseases. For more information about Bristol Myers Squibb, visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
Celgene and Juno Therapeutics are wholly owned subsidiaries of Bristol-Myers Squibb Company. In certain countries outside the U.S., due to local laws, Celgene and Juno Therapeutics are referred to as, Celgene, a Bristol Myers Squibb company and Juno Therapeutics, a Bristol Myers Squibb company.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211211005007/en/
Source: Bristol Myers Squibb
https://www.nasdaq.com/market-activity/stocks/bmy/dividend-history
https://seekingalpha.com/symbol/BMY
Friday, December 10, 2021 - 09:27am
Cibinqo is a once-daily oral treatment with proven efficacy demonstrated in a large-scale clinical trial program
NEW YORK--(BUSINESS WIRE)-- Pfizer Inc. (NYSE: PFE) today announced that the European Commission (EC) has approved the 100 mg and 200 mg doses of Cibinqo® (abrocitinib), an oral, once-daily, Janus kinase 1 (JAK1) inhibitor, for the treatment of moderate-to-severe atopic dermatitis (AD) in adults who are candidates for systemic therapy. Additionally, a 50 mg dose was approved to treat moderate-to-severe AD specifically in patients with moderate and severe renal impairment (kidney failure) or certain patients receiving treatment with inhibitors of cytochrome P450 (CYP) 2C19.
“For adults living with moderate-to-severe atopic dermatitis, Cibinqo could help provide relief from the hallmark symptom of intense itch and has demonstrated rapid improvements in skin clearance, extent, and severity of disease, versus placebo,” said Dr. Stephan Weidinger, Professor of Dermatology at Christian-Albrechts University Kiel and Vice Head of the Department of Dermatology at the University Hospital Schleswig-Holstein, Kiel, Germany. “The approval of Cibinqo in the European Union makes me hopeful for many patients who will have this additional option to help manage the often painful and disruptive symptoms of moderate-to-severe atopic dermatitis.”
The approval of Cibinqo was based on the results of five clinical studies of more than 2,800 patients including four Phase 3 studies and an ongoing long-term open label extension study. Cibinqo demonstrated meaningful improvements across measures of symptom relief and disease control versus placebo. In one trial including an active control arm with dupilumab, which evaluated patients on background topical medicated therapy, Cibinqo 200 mg was associated with a greater improvement in itch relief after two weeks than dupilumab. Cibinqo also demonstrated a consistent safety profile across trials, including in a long-term extension study, showing a favorable benefit-risk profile.
“There have been few treatment innovations over the last decade for those in the European Union suffering with the daily discomfort, distress, and pain caused by moderate-to-severe atopic dermatitis,” said Mike Gladstone, Global President of Pfizer Inflammation & Immunology. “The safety and efficacy established through a rigorous clinical trial program, designed to evaluate measures of symptom relief most important to patients, gives us great confidence in the positive impact Cibinqo could have on those living with this debilitating immuno-inflammatory condition.”
The most common adverse events reported with Cibinqo in ≥5% of patients were nausea (15.1%) and headache (7.9%). The most frequent serious adverse reactions were infections (0.3%).
Additional Details on the Cibinqo Clinical Trial Program
Findings from the following five studies in the Cibinqo JAK1 Atopic Dermatitis Efficacy and Safety (JADE) global development program were included in the submission to support this approval. The trials evaluated measures of improvements for AD including the Investigator Global Assessment (IGA), Eczema Area and Severity Index (EASI), and Peak Pruritus Numerical Rating Scale (PP-NRS):
Select findings for Cibinqo 100 mg, 200 mg, and placebo follow. P-value differences versus placebo across endpoints in JADE MONO-1, JADE MONO-2, and JADE COMPARE were *p<0.01 or **p<0.001. Treatment effects in subgroups, such as age or weight, were consistent with the results in the overall study populations.
About Cibinqo® (abrocitinib)
Cibinqo is an oral small molecule that selectively inhibits Janus kinase (JAK) 1. Inhibition of JAK1 is thought to modulate multiple cytokines involved in pathophysiology of atopic dermatitis, including interleukin IL-4, IL-13, IL-31, IL-22, and thymic stromal lymphopoietin (TSLP).
Cibinqo received marketing authorization from the UK Medicines and Healthcare products Regulatory Agency (MHRA), the Japanese Ministry of Health, Labour and Welfare (MHLW) and Korea’s Ministry of Food and Drug Safety (MFDS) earlier this year.
We routinely post information that may be important to investors on our website at www.Pfizer.com.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211210005319/en/
Source: Pfizer Inc.
Dec. 10, 2021 9:46 AM ET Pfizer Inc. (PFE)
By: Dulan Lokuwithana, SA News Editor
Dec. 10, 2021 8:02 AM ETNovartis AG (NVS)
PR Newswire
EAST HANOVER, N.J., Dec. 10, 2021 /PRNewswire/ -- Novartis today announced new Piqray® (alpelisib) data indicating benefit across a broad range of patient and disease characteristics as seen in analyses from all three cohorts of BYLieve. BYLieve is an ongoing Phase II, open-label, 3-cohort non-comparative study evaluating Piqray with endocrine therapy including men and pre- and postmenopausal women with hormone-receptor positive, human epidermal growth factor receptor-2 negative (HR+/HER2-) advanced or metastatic breast cancer (mBC) who have progressed on or after prior therapies, including CDK4/6 inhibitor plus endocrine therapy1-5. These data will be presented at the 2021 San Antonio Breast Cancer Symposium (SABCS) from December 7-10.
"The data from all three cohorts of the BYLieve study have value for the medical community and for the patients we care for with mBC, because these cohorts show a benefit from alpelisib in the post-CDK4/6i setting for patients with HR+/HER2- PIK3CA-mutated cancer," said Dr. Hope S. Rugo, Director, Breast Oncology and Clinical Trials Education, University of California San Francisco (UCSF) Helen Diller Family Comprehensive Cancer Center. "Beyond illustrating the efficacy and safety of alpelisib, regardless of the duration of prior CDK4/6i treatment, the data provide meaningful insights into how alpelisib may benefit different subgroups of patients."
Highlights from the BYLieve data presented at SABCS
About Piqray® (alpelisib)
Piqray is a kinase inhibitor developed for use in combination with fulvestrant for the treatment of postmenopausal women, and men, with HR+/HER2-, PIK3CA-mutated, advanced or metastatic breast cancer following progression on or after endocrine-based regimen7. Piqray is approved in 64 countries, including the US and European member states12.
Novartis is continuing to reimagine cancer with additional trials of Piqray. EPIK-B5 will be a large Phase III clinical trial of Piqray in combination with fulvestrant to complement the SOLAR-1 study13. Novartis is also studying the potential of Piqray in triple negative breast cancer (TNBC) in the EPIK-B3 Phase III clinical trial, in advanced HER2+ breast cancer in the EPIK-B2 Phase III clinical trial and in ovarian cancer in the EPIK-O Phase III clinical trial14-16.
Indication
PIQRAY® (alpelisib) tablets is a prescription medicine used in combination with the medicine fulvestrant to treat women who have gone through menopause and men who have hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced breast cancer or breast cancer that has spread to other parts of the body (metastatic), with an abnormal phosphatidylinositol-3-kinase catalytic subunit alpha (PIK3CA) gene, and whose disease has progressed on or after endocrine therapy. Your health care provider will test your cancer for an abnormal "PIK3CA" gene to make sure that PIQRAY is right for you. It is not known if PIQRAY is safe and effective in children.
Important Safety Information
Patients should not take PIQRAY if they have had a severe allergic reaction to PIQRAY or are allergic to any of the ingredients in PIQRAY.
PIQRAY may cause serious side effects. PIQRAY can cause severe allergic reactions. Patients should tell their health care provider or get medical help right away if they have trouble breathing, flushing, rash, fever, or fast heart rate during treatment with PIQRAY. PIQRAY can cause severe skin reactions. Patients should tell their health care provider or get medical help right away if they get severe rash or rash that keeps getting worse, reddened skin, flu-like symptoms, blistering of the lips, eyes or mouth, blisters on the skin or skin peeling, with or without fever. PIQRAY can cause high blood sugar levels (hyperglycemia). Hyperglycemia is common with PIQRAY and its complications can be severe. Health care providers will monitor patients' blood sugar levels before they start and during treatment with PIQRAY. Health care providers may monitor patients' blood sugar levels more often if they have a history of type 2 diabetes. Patients should tell their health care provider right away if they develop symptoms of hyperglycemia or its complications, including excessive thirst, dry mouth, urinating more often than usual or having a higher amount of urine than normal, increased appetite with weight loss, confusion, nausea, vomiting, fruity odor on breath, difficulty breathing, or dry or flushed skin. PIQRAY can cause lung problems (pneumonitis). Patients should tell their health care provider right away if they develop new or worsening symptoms of lung problems, including shortness of breath or trouble breathing, cough, or chest pain. Diarrhea is common with PIQRAY and can be severe. Severe diarrhea can lead to the loss of too much body water (dehydration) and kidney problems. Patients who develop diarrhea during treatment with PIQRAY should tell their health care provider right away.
Before taking PIQRAY, patients should tell their health care provider if they have a history of diabetes, skin rash, redness of skin, blistering of the lips, eyes or mouth, or skin peeling, are pregnant, or plan to become pregnant as PIQRAY can harm their unborn baby. Females who are able to become pregnant should use effective birth control during treatment with PIQRAY and for 1 week after the last dose. Do not breastfeed during treatment with PIQRAY and for 1 week after the last dose. Males with female partners who are able to become pregnant should use condoms and effective birth control during treatment with PIQRAY and for 1 week after the last dose. Patients should also read the full Prescribing Information of fulvestrant for important pregnancy, contraception, infertility, and lactation information.
Patients should tell their health care provider all of the medicines they take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. PIQRAY and other medicines may affect each other causing side effects. Know the medicines you take. Keep a list of them to show your health care provider or pharmacist when you get a new medicine.
The most common side effects of PIQRAY when used with fulvestrant are rash, nausea, tiredness and weakness, decreased appetite, mouth sores, vomiting, weight loss, hair loss, and changes in certain blood tests.
Please see full Prescribing Information for PIQRAY, available at www.piqray.com.
For more information, please visit https://www.novartis.us.
https://seekingalpha.com/symbol/NVS
Dec 10, 2021 3:00 AM
In RATIONALE 309, tislelizumab in combination with chemotherapy significantly prolonged progression-free survival for patients, with survival benefit observed across patient subgroups
The safety profile of the combination was consistent with known risks of each treatment agent
Following the positive topline at an interim analysis, a supplemental biologics license application in this indication is currently under review in China
CAMBRIDGE, Mass. & BEIJING--(BUSINESS WIRE)-- BeiGene (NASDAQ: BGNE; HKEX: 06160), a global, science-driven biotechnology company focused on developing innovative and affordable medicines to improve treatment outcomes and access for patients worldwide, today announced results from the RATIONALE 309 trial of tislelizumab versus placebo in combination with chemotherapy as a first-line treatment for patients with recurrent or metastatic nasopharyngeal cancer (RM-NPC) at the European Society for Medical Oncology Immuno-Oncology (ESMO I-O) Congress 2021, taking place on December 8-11, 2021.
“We are pleased that tislelizumab in combination with chemotherapy demonstrated a statistically significant progression-free survival benefit for patients with RM-NPC over chemotherapy,” commented Yong (Ben) Ben, M.D., Chief Medical Officer, Immuno-Oncology at BeiGene. “A filing based on these results is currently under review in China, where NPC as an endemic disease remains a significant unmet medical need. We look forward to continued discussions with the health authority and are working to bring this important immunotherapy to patients in China as soon as we can.”
In August 2021, the Center for Drug Evaluation (CDE) of the China National Medical Products Administration (NMPA) accepted a supplement Biologics License Application (sBLA) for tislelizumab in combination with chemotherapy as a first-line treatment for patients with RM-NPC based on results from the interim analysis of the RATIONALE 309 trial.
Results from RATIONALE 309: Tislelizumab vs. Placebo in Combination with Chemotherapy in First-Line RM-NPC
Proffered Paper: 121O
RATIONALE 309 is a multicenter, randomized, double-blind, placebo-controlled Phase 3 clinical trial (NCT03924986) designed to evaluate the efficacy and safety of tislelizumab combined with gemcitabine and cisplatin (Arm A) versus placebo combined with gemcitabine and cisplatin (Arm B) as a first-line treatment for patients with RM-NPC. The primary endpoint of the trial is progression-free survival (PFS) in the intent-to-treat (ITT) population as assessed by an independent review committee (IRC) per RECIST v1.1 criteria; secondary endpoints include IRC-assessed overall response rate (ORR), IRC-assessed duration of response (DoR), overall survival (OS), investigator-assessed PFS, time to second objective disease progression (PFS2), and safety. A total of 263 patients were enrolled in the trial, with 131 and 132 randomized to Arm A and Arm B, respectively, with balanced baseline characteristics between both arms.
“In the RATIONALE 309 trial, the addition of tislelizumab to chemotherapy significantly prolonged PFS for previously untreated patients with RM-NPC, an aggressive head and neck cancer prevalent in Asia, with consistent survival benefit across patient subgroups. Safety results in both arms remained similar to known risks and no new safety signals were identified. The promising results support the potential of tislelizumab in combination with chemotherapy as a new standard of care in China for the first-line treatment of RM-NPC,” commented Yunpeng Yang, M.D., Professor at Sun Yat-sen University Cancer Center and principal investigator of the study.
As of March 26, 2021, with a median follow-up time of 10.0 months, RATIONALE 309 achieved the primary endpoint at the interim analysis, with the combination of tislelizumab and chemotherapy demonstrating a statistically significant improvement in PFS, compared to the combination of placebo and chemotherapy, per IRC assessment. Efficacy results included:
The safety profile of tislelizumab and chemotherapy combination was manageable, consistent with known risks of each treatment agent. Safety results included:
About Tislelizumab
Tislelizumab (BGB-A317) is a humanized IgG4 anti-PD-1 monoclonal antibody specifically designed to minimize binding to FcγR on macrophages. In pre-clinical studies, binding to FcγR on macrophages has been shown to compromise the anti-tumor activity of PD-1 antibodies through activation of antibody-dependent macrophage-mediated killing of T effector cells. Tislelizumab is the first drug from BeiGene’s immuno-oncology biologics program and is being developed internationally as a monotherapy and in combination with other therapies for the treatment of a broad array of both solid tumor and hematologic cancers.
The China National Medical Products Administration (NMPA) has approved tislelizumab in five indications, including full approval for first-line treatment of patients with advanced squamous non-small cell lung cancer (NSCLC) in combination with chemotherapy and for first-line treatment of patients with advanced non-squamous NSCLC in combination with chemotherapy. NMPA also granted conditional approval for the treatment of patients with classical Hodgkin’s lymphoma (cHL) who received at least two prior therapies, for the treatment of patients with locally advanced or metastatic urothelial carcinoma (UC) with PD-L1 high expression whose disease progressed during or following platinum-containing chemotherapy or within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy, and for the treatment of patients with hepatocellular carcinoma (HCC) who have received at least one systemic therapy. Full approval for these indications is contingent upon results from ongoing randomized, controlled confirmatory clinical trials.
In addition, four supplemental Biologics License Applications for tislelizumab are under review by the Center for Drug Evaluation (CDE) of the NMPA, including as second- or third-line treatment of patients with locally advanced or metastatic NSCLC who progressed on prior platinum-based chemotherapy, for patients with previously treated, locally advanced unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair-deficient (dMMR) solid tumors, for the treatment of patients with locally advanced or metastatic esophageal squamous cell carcinoma (ESCC) who have disease progression following or are intolerant to first-line standard chemotherapy, and for first-line treatment of patients with recurrent or metastatic nasopharyngeal cancer (NPC).
In the U.S., a Biologics License Application for tislelizumab as a treatment for patients with unresectable recurrent locally advanced or metastatic ESCC after prior systemic therapy is currently under review by the U.S. Food and Drug Administration with a PDUFA target action date of July 12, 2022.
BeiGene has initiated or completed 17 potentially registration-enabling clinical trials in China and globally, including 13 Phase 3 trials and four pivotal Phase 2 trials.
In January 2021, BeiGene and Novartis entered into a collaboration and license agreement granting Novartis rights to develop, manufacture, and commercialize tislelizumab in North America, Europe, and Japan.
Tislelizumab is not approved for use outside of China.
To learn more about BeiGene,
please visit www.beigene.com and follow us on Twitter at @BeiGeneGlobal.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211210005022/en/
BeiGene
Dec. 10, 2021 4:28 AM ET BeiGene, Ltd. (BGNE)
By: Mamta Mayani, SA News Editor
Dec 09, 2021
Basel, December 9, 2021 — Novartis today announced the first interpretable results from year two (week 100) of the Phase III KESTREL study. KESTREL assessed the safety and efficacy of Beovu® (brolucizumab) 6 mg in patients with visual impairment due to diabetic macular edema (DME). Results from year two confirmed the visual acuity gains, fluid reduction findings and safety profile from year one, while addressing the burden of frequent treatments for DME patients1,2.
Results from year two of KESTREL were consistent with those seen at year one, including maintenance of best-corrected visual acuity (BCVA) and sustained reductions in central subfield thickness (CSFT)1,2. Additionally, numerically fewer Beovu patients had intraocular fluid and/or sub-retinal fluid (IRF/SRF) versus patients treated with aflibercept1. CSFT is a key indicator of fluid in the retina, and fluid is a key marker of disease activity3,4.
More than 40% of Beovu patients were maintained on 12-week dosing intervals, and 70% of patients who completed the first 12-week cycle after loading remained on 12-week dosing through year two, showing the potential for Beovu to offer fluid resolution in more DME patients with fewer injections versus aflibercept1.
“With an average age at diagnosis of 48 years, DME primarily affects working-age adults, which means managing their vision, in addition to multiple comorbidities related to diabetes, may result in loss of work productivity and employment instability5,6,” said Dr. David M Brown MD, Director of Research, Retina Consultants of Texas. “The extended dosing and fluid resolution observed in year two of the KESTREL clinical trial suggest Beovu has the potential to help appropriate patients more conveniently and effectively manage their disease with dosing intervals every 12 weeks after an initial loading phase.”
Further details of year-two findings from the KESTREL trial, along with findings from KITE*, another pivotal Phase III trial of Beovu in DME, will be presented at upcoming medical congresses.
About the KESTREL year two safety results
In KESTREL (NCT03481634), rates of intraocular inflammation (IOI) were 4.2% for Beovu 6 mg, 5.3% for Beovu 3 mg and 1.1% for aflibercept; retinal vasculitis (RV) rates were 0.5% for Beovu 6 mg, 1.6% for Beovu 3 mg and 0% for aflibercept1. Rates of retinal vascular occlusion (RO) were 1.6% for both Beovu 6 mg and 3 mg versus 0.5% for aflibercept1. The majority of IOI events were manageable and resolved without any clinical complications1. No new RV events were reported during year two of KESTREL1. Of the four new RO events reported during year two (two in Beovu 6 mg, one in Beovu 3 mg and one in aflibercept), none were associated with IOI or RV1.
Brolucizumab 6 mg is the commercialized dose of Beovu in wet age-related macular degeneration (AMD)7. Novartis is committed to bringing Beovu 6 mg to DME patients and has submitted data from KESTREL and KITE (NCT03481660), to global health authorities in H2 2021.
About the KESTREL and KITE clinical trials
KESTREL and KITE are global, randomized, double-masked, Phase III, two-year studies comparing the safety and efficacy of Beovu and aflibercept in the treatment of patients with visual impairment due to DME8,9.
KESTREL and KITE involved 926 total patients in 36 countries8,9. In the loading phase of both trials, patients in the Beovu arms were treated every six weeks for a total of five doses; patients in the aflibercept arms were treated every four weeks for a total of five doses, in line with its label at the start of the studies8,9. Following the loading phase, patients in the Beovu arms were subsequently treated every 12 weeks, with those demonstrating disease activity moved to dosing every eight weeks for the remainder of the study8,9. At week 72 of KITE, Beovu patients dosed every 12 weeks could be extended to dosing every 16 weeks, and patients dosed every eight weeks could be extended to every 12 weeks9.
About Beovu (brolucizumab) 6 mg
Beovu (brolucizumab, also known as RTH258) 6 mg is approved for the treatment of wet age-related macular degeneration (AMD) in more than 70 countries, including in the US, EU, UK, Japan, Canada and Australia7,13-16. Additional trials, which study the effects of brolucizumab in patients with wet AMD, diabetic macular edema (DME), and proliferative diabetic retinopathy (PDR), are currently ongoing.
Find out more at https://www.novartis.com.
https://www.nasdaq.com/market-activity/stocks/nvs/dividend-history
https://seekingalpha.com/symbol/NVS
December 9, 2021
- Presbyopia, or age-related blurry near vision, is a common, progressive condition that reduces the eye's ability to focus on near objects and affects nearly half of the U.S. adult population, usually over age 40
- VUITY is a once-daily prescription eye drop that improves near and intermediate vision without impacting distance vision for adults with age-related blurry near vision
NORTH CHICAGO, Ill., Dec. 9, 2021 /PRNewswire/ -- Allergan, an AbbVie (NYSE: ABBV) company, today announced that VUITY™ (pilocarpine HCl ophthalmic solution) 1.25%, the first and only eye drop approved by the U.S. Food and Drug Administration (FDA) to treat presbyopia, is now available by prescription in pharmacies nationwide. Presbyopia, or age-related blurry near vision, can be diagnosed through a basic eye exam by an eye doctor (optometrist or ophthalmologist) and is a common and progressive eye condition that affects 128 million Americans, or nearly half of the U.S. adult population.
Experience the interactive Multichannel News Release here: https://www.multivu.com/players/English/8969851-allergan-abbvie-vuity-now-available/.
"We are pleased to be able to bring this first-of-its-kind treatment to market sooner than expected for the millions of Americans with presbyopia who may benefit from it," said Jag Dosanjh, senior vice president medical therapeutics, Allergan, an AbbVie company. "This significant innovation in age-related eye health reflects our commitment to advance vision care and expands our leading portfolio of treatments for eye care providers and their patients."
"Many Americans deal with presbyopia, which typically begins around age 40, by relying on reading glasses or resorting to work-arounds like zooming in on their digital devices to see up close. As an optometrist who also has presbyopia, I'm personally and professionally excited to try VUITY for myself, as well as offer it to my patients with age-related blurry near vision," said optometrist Dr. Selina McGee, Fellow of the American Academy of Optometry. "With VUITY now available, it is a good time for those who experience age-related blurry near vision to visit their eye doctor for an exam and to discuss their options to manage this common condition."
VUITY is an optimized formulation of pilocarpine, an established eye care therapeutic, specifically designed to treat age-related blurry near vision. It is delivered with proprietary pHast™ technology, which allows VUITY to rapidly adjust to the physiologic pH of the tear film. This was studied in simulated tear film, and the clinical significance is unknown. VUITY uses the eye's own ability to reduce pupil size, improving near and intermediate vision while maintaining distance vision.
"As I've gotten older, my vision has changed, and it has become almost impossible to see clearly up close unless I wear my readers. Realizing that I needed to start using readers showed me how important it was to address this condition," said Toni Wright, clinical trial participant. "It was great to have the opportunity to participate in the clinical study investigating a new potential treatment option. I'm so excited the investigational treatment, which has been identified as VUITY, is now approved and available as a treatment to manage age-related blurry near vision."
For product information, visit www.VUITY.com and talk to an eye care professional.
About the VUITY Clinical Development Program
The FDA approval of VUITY in October 2021 was based on data from two pivotal phase 3 clinical studies, GEMINI 1 and GEMINI 2, which evaluated the efficacy, safety and tolerability of VUITY for the treatment of presbyopia.
About Presbyopia
Presbyopia, known as age-related blurry near vision, is a common and progressive eye condition that reduces the eye's ability to focus on near objects and usually impacts people after age 40. In a non-presbyopic eye, the clear lens behind the iris can change shape and focus light to the retina, making it easier to see things up close. In a presbyopic eye, the clear lens hardens and does not change shape as easily, making it difficult to focus on near objects. Presbyopia can be diagnosed by an eye doctor (ophthalmologist/optometrist).
Approved Use and Important Safety Information
USE
VUITY™ (pilocarpine hydrochloride ophthalmic solution) 1.25% is a prescription eye drop used to treat age-related blurry near vision (presbyopia) in adults.
Please see full Prescribing Information at www.VUITY.com.
About Allergan Eye Care
As a leader in eye care, Allergan has discovered, developed, and delivered some of the most innovative products in the industry for more than 70 years. Allergan has launched over 125 eye care products and invested billions of dollars in treatments for the most prevalent eye conditions including glaucoma, ocular surface disease, and retinal diseases such as diabetic macular edema and retinal vein occlusion.
About AbbVie
AbbVie's mission is to discover and deliver innovative medicines that solve serious health issues today and address the medical challenges of tomorrow. We strive to have a remarkable impact on people's lives across several key therapeutic areas: immunology, oncology, neuroscience, eye care, virology, women's health and gastroenterology, in addition to products and services across its Allergan Aesthetics portfolio. For more information about AbbVie, please visit us at www.abbvie.com. Follow @abbvie on Twitter, Facebook, Instagram, YouTube and LinkedIn.
https://seekingalpha.com/symbol/ABBV
PUBLISHED9 December 2021
New DESTINY-Breast03 data presented at SABCS 2021 showed AstraZeneca and Daiichi Sankyo’s ENHERTU demonstrated a similar benefit in patient subgroups, including in patients with stable brain metastases, versus T-DM1
New results from the DESTINY-Breast03 Phase III trial showed that ENHERTU® (fam-trastuzumab deruxtecan-nxki) demonstrated a higher progression-free survival (PFS) and objective response rate (ORR) in pre-specified patient subgroups compared to trastuzumab emtansine (T-DM1) in patients with HER2 positive unresectable and/or metastatic breast cancer previously treated with trastuzumab and a taxane.
ENHERTU is a HER2-directed antibody drug conjugate (ADC) being jointly developed by AstraZeneca and Daiichi Sankyo Company, Limited (hereafter, Daiichi Sankyo).
A similar PFS and ORR benefit was observed in exploratory analyses in patients defined by stable brain metastases, hormone receptor status, number of prior lines of therapy, prior pertuzumab treatment, or status of visceral metastasis. Results were presented as an oral presentation at the 2021 San Antonio Breast Cancer Symposium (SABCS).
In patients with stable brain metastases at baseline, treatment with ENHERTU resulted in higher PFS compared to T-DM1 (PFS by blinded independent central review (BICR) hazard ratio [HR] = 0.25; 95% confidence interval [CI]: 0.13-0.45). Additionally, in this subgroup, ENHERTU improved PFS to a median of 15 months versus 3 months for T-DM1.
Sarah Hurvitz, MD, MD, FACP, medical oncologist, professor of medicine, and director of the Breast Cancer Clinical Trials Program in the division of hematology-oncology at the David Geffen School of Medicine at UCLA, and medical director for the Clinical Research Unit at the UCLA Jonsson Comprehensive Cancer Center in Santa Monica, CA, said: “The main goals in the treatment of HER2-positive metastatic breast cancer, including those with stable brain metastases, are to improve symptoms, stabilize or reduce tumor size and improve overall survival. The higher progression-free survival seen in DESTINY-Breast03 in the subgroup of patients with stable brain metastases are encouraging, and underscores the excitement around another potential treatment option for patients who have experienced disease progression on currently available therapies.”
Susan Galbraith, Executive Vice President, Oncology R&D, AstraZeneca, said: “More treatment options are needed to delay progression and extend survival for patients with HER2-positive metastatic breast cancer who develop brain metastases. These additional analyses from DESTINY-Breast03 reinforce the potential of ENHERTU with similar benefits in the different subgroups.”
Ken Takeshita, Global Head, R&D, Daiichi Sankyo, said: “These additional analyses from DESTINY-Breast03 continue to demonstrate the benefit of ENHERTU compared to T-DM1 in patient subgroups, including 15-month progression-free survival in those with stable brain metastases, illustrating the potential of this treatment to become the new standard of care in patients with previously treated HER2-positive metastatic breast cancer. These data will support our ongoing conversations with global health authorities to realize our commitment to bring ENHERTU to patients with previously treated HER2-positive metastatic breast cancer earlier in the metastatic setting.”
Between 30 to 50% of patients with HER2-positive metastatic breast cancer will develop brain metastases, and while increased availability of HER2 therapies has improved systemic disease control, prognosis following the development of brain metastases remains poor.1-5
Confirmed ORR for patients with stable brain metastases at baseline was 67.4% with ENHERTU versus 20.5% with T-DM1. A retrospective, non-prespecified evaluation of intracranial response among patients with stable brain metastases who received scans at baseline provided preliminary evidence that treatment with ENHERTU is associated with intracranial tumor response and reduction in Central Nervous System disease with 10 (27.8%) complete responses (CR) and 13 (36.1%) partial responses (PR) compared to one (2.8%) CR and 11 (30.6%) PRs in those treated with T-DM1.
Indications
ENHERTU is a HER2-directed antibody and topoisomerase inhibitor conjugate indicated for the treatment of adult patients with:
Please see accompanying full Prescribing Information, including Boxed WARNINGS, and Medication Guide.
ENHERTU
ENHERTU (fam-trastuzumab deruxtecan-nxki) is a HER2-directed ADC. Designed using Daiichi Sankyo’s proprietary DXd ADC technology, ENHERTU is the lead ADC in the oncology portfolio of Daiichi Sankyo and the most advanced program in AstraZeneca’s ADC scientific platform. ENHERTU consists of a HER2 monoclonal antibody attached to a topoisomerase I inhibitor payload, an exatecan derivative, via a stable tetrapeptide-based cleavable linker.
ENHERTU (5.4mg/kg) is approved in Canada, the EU, Israel, Japan, the UK and the US for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received two or more prior anti-HER2-based regimens in the metastatic setting based on the results from the DESTINY-Breast01 trial.
ENHERTU (6.4mg/kg) is also approved in Israel, Japan and the US for the treatment of adult patients with locally advanced or metastatic HER2-positive gastric or gastroesophageal junction adenocarcinoma who have received a prior trastuzumab-based regimen based on the results from the DESTINY-Gastric01 trial.
Daiichi Sankyo collaboration
Daiichi Sankyo and AstraZeneca entered into a global collaboration to jointly develop and commercialize ENHERTU (a HER2-directed ADC) in March 2019, and datopotamab deruxtecan (DS-1062; a TROP2-directed ADC) in July 2020, except in Japan where Daiichi Sankyo maintains exclusive rights. Daiichi Sankyo is responsible for manufacturing and supply of ENHERTU and datopotamab deruxtecan.
For more information, please visit www.astrazeneca-us.com and follow us on Twitter @AstraZenecaUS.
Dec. 09, 2021 12:03 PM ET Daiichi Sankyo Company, Limited (DSKYF), AZN By: Jonathan M Block, SA News Editor
PUBLISHED8 December 2021
AstraZeneca's Evusheld (tixagevimab co-packaged with cilgavimab), a long-acting antibody (LAAB) combination, has received emergency use authorisation (EUA) in the US for the pre-exposure prophylaxis (prevention) of COVID-19, with first doses expected to become available very soon.
The Food and Drug Administration (FDA) granted the EUA for Evusheld for pre-exposure prophylaxis of COVID-19 in adults and adolescents (aged 12 and older who weigh 40kg or more) with moderate to severe immune compromise due to a medical condition or immunosuppressive medications and who may not mount an adequate immune response to COVID-19 vaccination, as well as those individuals for whom COVID-19 vaccination is not recommended. Recipients should not be currently infected with or had recent known exposure to a person infected with SARS-CoV-2.
Myron J. Levin, MD, Professor of Pediatrics and Medicine, University of Colorado School of Medicine, US, and principal investigator on the PROVENT trial, said: “Millions of people in the US and around the world remain at serious risk for COVID-19 because their immune systems do not generate a sufficient immune response, even after receiving all recommended doses of vaccine. I am excited to offer my patients Evusheld as an easily-administered new option that provides long-lasting protection that could help them return to their everyday lives.”
Mene Pangalos, Executive Vice President, BioPharmaceuticals R&D, AstraZeneca, said: “We are proud to play a leading role in fighting the COVID-19 pandemic and, with Evusheld, we now have the first antibody therapy authorised in the US to prevent COVID-19 symptoms before virus exposure, while also providing long lasting protection with a single dose. Evusheld neutralises all previous SARs-CoV-2 variants to date, and we are working quickly to establish its efficacy against the new Omicron variant. We thank our clinical trial participants, the investigators, scientists, and government agencies and our colleagues at AstraZeneca who have all contributed to the development of Evusheld.”
Brian Koffman, MDCM (retired), MS Ed, Co-Founder, Executive Vice President and Chief Medical Officer of the CLL (Chronic Lymphocytic Leukemia) Society, US, said: “One of the primary questions I keep getting asked by patients is ‘When can I hug my grandchildren again?’ As a physician and person with a weakened immune system, l am filled with hope now that Evusheld will soon be available to those who can’t count on vaccination alone to provide the protection they need.”
Evusheld is a combination of two long-acting monoclonal antibodies and is the only antibody therapy authorised in the US for COVID-19 pre-exposure prophylaxis and the only COVID-19 antibody delivered as an intramuscular dose (150mg tixagevimab and 150mg cilgavimab).
About 2% of the global population is considered at increased risk of an inadequate response to a COVID-19 vaccine.1,2 About seven million people in the US are immunocompromised and may benefit from Evusheld for pre-exposure prophylaxis of COVID-19.1,3,4 This includes people with blood cancers or other cancers being treated with chemotherapy, and those taking medications after an organ transplant or who are taking immunosuppressive drugs for conditions including multiple sclerosis and rheumatoid arthritis.5-9
The primary data supporting the Evusheld EUA are from the ongoing PROVENT Phase III pre-exposure prevention trial, which showed a statistically significant reduction (77% at primary analysis, 83% at median six-month analysis) in the risk of developing symptomatic COVID-19 compared to placebo, with protection from the virus continuing for at least six months. More follow-up is needed to establish the full duration of protection provided by Evusheld. Data from the Phase III STORM CHASER post-exposure trial and the Evusheld Phase I trial also supported the EUA. Evusheld was well-tolerated in the trials.
Evusheld and SARS-CoV-2 variants
Studies are underway to provide information on the impact of the new Omicron variant (B.1.1.529) on Evusheld.10,11 Of the Omicron binding site substitutions relevant to Evusheld that have been tested to date in preclinical assays, none have been associated with escape from Evusheld neutralisation.10,11 In vitro findings demonstrate Evusheld neutralises other recent emergent SARS-CoV-2 viral variants, including the Delta and Mu variants.10
Evusheld is being developed with support from the US government, including federal funds from the Department of Health and Human Services; Office of the Assistant Secretary for Preparedness and Response; Biomedical Advanced Research and Development Authority in partnership with the Department of Defense; Joint Program Executive Office for Chemical, Biological, Radiological and Nuclear Defense, under Contract No. W911QY-21-9-0001.
AstraZeneca has agreed to supply the US government with 700,000 doses of Evusheld. The U.S. government has indicated that it plans to distribute these doses to states and territories at no cost and on a pro rata basis.
AstraZeneca is progressing with filings around the globe for potential emergency use authorisation or conditional approval of Evusheld in both COVID-19 prophylaxis and treatment.
Notes
Evusheld
Evusheld, formerly known as AZD7442 is a combination of two LAABs - tixagevimab (AZD8895) and cilgavimab (AZD1061) - derived from B-cells donated by convalescent patients after SARS-CoV-2 virus. Discovered by Vanderbilt University Medical Center and licensed to AstraZeneca in June 2020, the human monoclonal antibodies bind to distinct sites on the SARS-CoV-2 spike protein13 and were optimised by AstraZeneca with half-life extension and reduced Fc receptor and complement C1q binding. The half-life extension more than triples the durability of its action compared to conventional antibodies and could afford up to 12 months of protection from COVID-19 following a single administration;14-16 data from the Phase III PROVENT trial show protection lasting at least six months.17 The reduced Fc receptor binding aims to minimise the risk of antibody-dependent enhancement of disease - a phenomenon in which virus-specific antibodies promote, rather than inhibit, infection and/or disease.18 Evusheld is delivered as an IM dose of 150mg tixagevimab and 150mg cilgavimab administered in two separate, consecutive injections.
In August 2021, AstraZeneca announced that Evusheld demonstrated a statistically significant reduction in the risk of developing symptomatic COVID-19 in the PROVENT trial; efficacy was 83% compared to placebo in a six-month analysis announced on 18 November 2021. In October 2021, AstraZeneca announced positive high-level results from the Evusheld TACKLE Phase III outpatient treatment trial. Evusheld is also being studied as a potential treatment for hospitalised COVID-19 patients as part of the National Institute of Health’s ACTIV-3 trial and in an additional collaborator hospitalisation treatment trial.
Under the terms of the licensing agreement with Vanderbilt, AstraZeneca will pay single-digit royalties on future net sales.
Dec. 08, 2021 4:02 PM ET
By: Dulan Lokuwithana, SA News Editor
DOWNLOAD AS PDFDECEMBER 09, 2021
- Study demonstrated a favorable risk/benefit profile over a three-year period, with safety comparable to AURORA 1, and sustained efficacy -
- Topline results show sustained meaningful reductions in proteinuria compared to mycophenolate mofetil (MMF) and low-dose oral corticosteroids alone, the active study control -
- Similar to the active control, the voclosporin-treated group maintained stable renal function at the end of the study -
- Aurinia to host conference call today, December 9, at 8:30 a.m. EST -
VICTORIA, British Columbia--(BUSINESS WIRE)-- Aurinia Pharmaceuticals Inc. (NASDAQ:AUPH) (Aurinia or the Company), a biopharmaceutical company committed to delivering therapeutics that change the course of autoimmune disease, today announced positive topline results from the AURORA 2 continuation study evaluating the long-term safety and tolerability of LUPKYNIS™ (voclosporin) for the treatment of adults with active lupus nephritis (LN), a serious complication in patients with systemic lupus erythematosus (SLE). In combination with background immunosuppressive therapy, LUPKYNIS is the first and only FDA-approved medicine with three years of pivotal trial results, including long-term safety data, within LN.
“Up to half of patients with systemic lupus erythematosus will develop lupus nephritis, that can result in severe and permanent damage to the kidneys and, in some cases, renal failure,” said Amit Saxena, M.D., assistant professor, department of medicine at NYU Langone Medical Center and an investigator in the AURORA 2 study. “These highly anticipated long-term results, of voclosporin in adult patients with LN, show consistent safety with the phase 3 AURORA 1 study and a benign impact on eGFR even after up to three years of treatment while maintaining the impressive reductions in proteinuria seen in AURORA 1.”
“We are pleased by the final results of the AURORA 2 continuation study evaluating LUPKYNIS for the treatment of lupus nephritis, which supports the long-term safety and tolerability for up to three years,” said Neil Solomons, M.D., Chief Medical Officer of Aurinia. “Furthermore, we observed that efficacy was maintained in combination with MMF and low-dose steroids.”
Highlights of topline results from AURORA 2:
“Aurinia’s LN clinical program, including AURA-LV and both the AURORA trials, represents the largest successful LN program to date. The latest results, featuring consistent outcomes over three years, serve to reinforce LUPKYNIS as a safe and effective, important treatment option for patients living with lupus nephritis,” said Peter Greenleaf, President and CEO of Aurinia. “On behalf of Aurinia, I extend our deepest gratitude to the participants, their families, and the health care providers involved in these studies.”
AURORA 2 Study Design
AURORA 2 (NCT03597464) is a Phase 3 randomized, double-blind, placebo-controlled clinical trial to assess the long-term safety and tolerability of voclosporin, in addition to MMF/steroids. Patients who completed 12 months of treatment in the Phase 3 AURORA 1 study were eligible to enroll in the AURORA 2 continuation study with the same randomized treatment of voclosporin at 23.7 mg twice daily or placebo, in combination with MMF at 1 g twice daily with low-dose oral steroids, for up to an additional 24 months. A total of 216 LN patients out of 357 who were enrolled in the AURORA 1 study continued into AURORA 2, with 116 patients in the voclosporin-treated group and 100 patients in the active control group. 90 and 78 patients, respectively, received 36 months of total treatment at the completion of the study. Results from the completed Phase 3 randomized, double-blind, placebo-controlled, multicenter AURORA 1 study (NCT03021499) were recently published in The Lancet.
About LUPKYNIS
LUPKYNIS is the first FDA-approved oral medicine for the treatment of adult patients with active LN. LUPKYNIS is a novel, structurally modified calcineurin inhibitor (CNI) with a dual mechanism of action, acting as an immunosuppressant through inhibition of T-cell activation and cytokine production and promoting podocyte stability in the kidney. The recommended starting dose of LUPKYNIS is three capsules twice daily with no requirement for serum drug monitoring. Dose modifications can be made based on Aurinia’s proprietary personalized eGFR-based dosing protocol. Boxed Warning, warnings and precautions for LUPKYNIS are consistent with those of other CNI-immunosuppressive treatments.
INDICATIONS
LUPKYNIS is indicated in combination with a background immunosuppressive therapy regimen for the treatment of adult patients with active LN. Limitations of Use: Safety and efficacy of LUPKYNIS have not been established in combination with cyclophosphamide. Use of LUPKYNIS is not recommended in this situation.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211209005485/en/
Source: Aurinia Pharmaceuticals Inc.
Released December 9, 2021
Dec. 09, 2021 6:30 AM ET Aurinia Pharmaceuticals Inc. (AUPH)
By: Mamta Mayani, SA News Editor6 Comments
12/07/21 at 3:48 AM ESTPDF Version
– New preclinical findings generated through in vitro testing of sotrovimab against
the complete pseudo-virus updated to bioRxiv
– Data build on promising signal published last week, underscoring the importance of
sotrovimab for early treatment of COVID-19 –
– Sotrovimab is authorized and available for the treatment of early COVID-19 in the US
and multiple countries around the world –
LONDON and SAN FRANCISCO, Dec. 07, 2021 (GLOBE NEWSWIRE) -- GlaxoSmithKline plc (LSE/NYSE: GSK) and Vir Biotechnology, Inc. (Nasdaq: VIR) today announced an update to preclinical data on bioRxiv1, a preprint server, demonstrating that sotrovimab, an investigational monoclonal antibody, retains in vitro activity against the full known Omicron spike protein, the new SARS-CoV-2 variant (B.1.1.529). The preclinical data was generated through pseudo-virus testing of the combined known mutations of the Omicron variant, which included the maximum number of changes (37 mutations) identified to date in the spike protein. These findings build on the initial preclinical data generated through pseudo-virus testing, provided last week, showing sotrovimab retained in vitro activity against key individual mutations of the Omicron variant, including those found in the binding site of sotrovimab. These data add to the growing body of preclinical evidence demonstrating that sotrovimab retains activity against all tested variants of concern.
George Scangos, Ph.D., Chief Executive Officer of Vir, said: “Sotrovimab is the first monoclonal antibody to report preclinical data demonstrating activity against all tested SARS-CoV-2 variants of concern and interest to date, including Omicron, as well as the still prevalent and highly contagious Delta variant. Given the less than three-fold neutralization shift demonstrated in the pre-clinical pseudo-virus assay, we are confident that sotrovimab will continue to provide significant benefit for the early treatment of patients hoping to avoid the most severe consequences of COVID-19.”
Dr. Hal Barron, Chief Scientific Officer and President R&D, GSK, said: “From the outset of our collaboration with Vir we hypothesized that sotrovimab would have a high barrier to resistance and thus could deliver best-in-class potential for the early treatment of patients with COVID-19. These pre-clinical data demonstrate the potential for our monoclonal antibody to be effective against the latest variant, Omicron, plus all other variants of concern defined to date by the WHO, and we look forward to discussing these results with regulatory authorities around the world.”
About sotrovimab
Sotrovimab is an investigational SARS-CoV-2 neutralizing monoclonal antibody. The antibody binds to an epitope on SARS-CoV-2 shared with SARS-CoV-1 (the virus that causes SARS), indicating that the epitope is highly conserved, which may make it more difficult for resistance to develop. Sotrovimab, which incorporates Xencor, Inc.’s Xtend™ technology, has also been designed to achieve high concentration in the lungs to ensure optimal penetration into airway tissues affected by SARS-CoV-2 and to have an extended half-life.
Pre-clinical data, published in bioRxiv, demonstrate that sotrovimab retains activity against all currently tested variants of concern and interest of the SARS-CoV-2 virus as defined by WHO, plus others, including but not limited to Delta (B.1.617.2), Delta Plus (AY.1 or AY.2), Mu (B.1.621) and Omicron (B.1.1.529).
About the sotrovimab clinical development program
Authorized Use
The U.S. FDA has issued an Emergency Use Authorization (EUA) to permit the emergency use of the unapproved product sotrovimab for the treatment of mild-to-moderate coronavirus disease 2019 (COVID-19) in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death.
About the Vir and GSK Collaboration
In April 2020, Vir and GSK entered into a collaboration to research and develop solutions for coronaviruses, including SARS-CoV-2, the virus that causes COVID-19. The collaboration uses Vir’s proprietary monoclonal antibody platform technology to accelerate existing and identify new anti-viral antibodies that could be used as therapeutic or preventive options to help address the current COVID-19 pandemic and future outbreaks. The companies will leverage GSK’s expertise in functional genomics and combine their capabilities in CRISPR screening and artificial intelligence to identify anti-coronavirus compounds that target cellular host genes. They will also apply their combined expertise to research SARS-CoV-2 and other coronavirus vaccines.
For further information please visit www.gsk.com/aboutus.
For more information, please visit www.vir.bio.
Dec. 07, 2021 4:24 AM ET GlaxoSmithKline plc (GSK), VIR By: Mamta Mayani, SA News Editor1 Comment
Injection, 500 mg, for intravenous use
12/07/21PDF Version- Highly Statistically Significant Reduction in Chorea Movements (p < 0.0001) as Measured by the Unified Huntington's Disease Rating Scale (UHDRS®) Total Maximal Chorea (TMC) Score
- Placebo-Adjusted Mean Reduction in TMC Score of 3.2 Units in Valbenazine-Treated Patients
- Company Plans to Submit Supplemental New Drug Application to U.S. Food and Drug Administration in 2022
SAN DIEGO, Dec. 7, 2021 /PRNewswire/ -- Neurocrine Biosciences (Nasdaq: NBIX) today announced positive top-line data from its Phase 3 KINECT-HD study evaluating the efficacy, safety and tolerability of valbenazine, a selective vesicular monoamine transporter 2 (VMAT2) inhibitor being investigated as a once-daily treatment in adults with chorea associated with Huntington disease (HD). The study met the primary endpoint of reduction in severity of chorea, the cardinal motor feature in Huntington disease, as measured by change in the Unified Huntington's Disease Rating Scale (UHDRS®) Total Maximal Chorea (TMC) score from baseline to the average score at weeks 10 and 12.
"The positive results of the KINECT-HD study move us closer to bringing valbenazine as a potential treatment option to patients in the U.S. living with chorea, one of the most common symptoms of Huntington disease," said Eiry W. Roberts, M.D., Chief Medical Officer at Neurocrine Biosciences. "We are immensely grateful to our partners at the Huntington Study Group and the Clinical Trials Coordination Center at the University of Rochester, New York, who were instrumental in completing this study, as well as the study participants and the families and caregivers who supported them. We will review the complete data and begin preparing a supplemental new drug application (sNDA) for submission to the U.S. Food and Drug Administration next year. In the meantime, we will continue dosing in the KINECT-HD2 study, which is evaluating the long-term safety and tolerability of valbenazine in this same patient population."
KINECT-HD2 is an open-label study to evaluate the long-term safety and tolerability of valbenazine for the treatment of chorea in Huntington disease.
About the KINECT-HD Study
KINECT-HD is a Phase 3, randomized, double-blind, placebo-controlled study designed to: evaluate the efficacy of valbenazine as a once-daily treatment to reduce chorea associated with Huntington disease (HD) and evaluate the safety and tolerability of valbenazine in patients with HD. The study enrolled 128 adults 18 to 75 years of age who have been diagnosed with motor manifest HD and who have sufficient chorea symptoms to meet study protocol criteria. For more information on this KINECT-HD study, please visit www.huntingtonstudygroup.org.
About KINECT-HD2
KINECT-HD2 is an open-label study to evaluate the long-term safety and tolerability of valbenazine in patients with chorea associated with Huntington disease (HD). The 112-week study will enroll up to 150 adults 18 to 75 years of age who have been diagnosed with motor manifest HD and who have sufficient chorea symptoms to meet study protocol criteria. For more information on the KINECT-HD2 study, please visit www.huntingtonstudygroup.org. or clinicaltrials.gov.
For more information, visit neurocrine.com, and follow the company on LinkedIn.
View original content to download multimedia:https://www.prnewswire.com/news-releases/neurocrine-biosciences-announces-positive-phase-3-data-for-kinect-hd-study-evaluating-valbenazine-for-chorea-associated-with-huntington-disease-301439605.html
SOURCE Neurocrine Biosciences, Inc.
Dec. 07, 2021 5:36 PM ET
Neurocrine Biosciences, Inc. (NBIX)
By: Jonathan M Block,SA News Editor
Basel, 06 December 2021 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) has recommended extending the marketing authorisation for Actemra®/RoActemra® (tocilizumab) to include the treatment of COVID-19 in adults who are receiving systemic corticosteroids and require supplemental oxygen or mechanical ventilation. A final decision regarding the approval of Actemra/RoActemra is expected from the European Commission in the near future.
“As COVID-19 cases in Europe rise and with pressure on hospitals likely to increase, the need for effective treatments for those suffering most severely with COVID-19 could intensify,” said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “We are proud that the CHMP has recognised the potential of Actemra/RoActemra as we continue our efforts to bring treatment options to those most in need.”
In August 2021, the EMA’s CHMP began an accelerated assessment of Actemra/RoActemra – this rapid analysis is reserved for medicines that may offer significant benefit to public health. The assessment reviewed results from four studies in over 5,500 patients with severe or critical COVID-19. These include the Roche-led phase III COVACTA, EMPACTA and REMDACTA trials, and the University of Oxford’s Randomised Evaluation of COVID-19 Therapy (RECOVERY) study, which was supported by Roche. The totality of this clinical evidence demonstrated that Actemra/RoActemra reduced the risk of death in patients with severe or critical COVID-19.1
Actemra/RoActemra has been provisionally approved in Australia, authorised for emergency use in the United States and Ghana, and recommended by the World Health Organization (WHO) for the treatment of COVID-19. 1,2,3,4 Roche is working closely with regulatory bodies and other partners around the world on the next steps to bring this medicine to as many people as possible.
Following the recent emergence of the new SARS-CoV-2 variant of concern, Omicron (B.1.1.529), WHO has reported that interleukin 6 receptor blockers, such as Actemra/RoActemra, are expected to still be effective for managing patients with severe COVID-19.5
In these exceptional times, Roche stands together with society, governments, healthcare providers and all those working towards the common goal of overcoming the COVID-19 pandemic.
About Actemra®/RoActemra® (tocilizumab) in COVID-19 clinical trials
Roche has evaluated Actemra/RoActemra in COVID-19 in three phase III randomised studies: COVACTA, EMPACTA and REMDACTA.
COVACTA was a global, randomised, double-blind, placebo-controlled phase III study (COVACTA, NCT04320615), which evaluated the safety and efficacy of intravenous Actemra/RoActemra plus standard of care in adult patients hospitalised with severe COVID-19 pneumonia compared to placebo plus standard of care. The primary and secondary endpoints included clinical status, mortality, mechanical ventilation and intensive care unit (ICU) variables. Patients were followed for 60 days post-randomisation.
EMPACTA (Evaluating Minority Patients with Actemra) was a phase III, randomised, double-blind, placebo-controlled multicentre study (EMPACTA, NCT04372186) which evaluated the efficacy and safety of Actemra/RoActemra in the treatment of COVID-19 pneumonia among hospitalised patients that are often underrepresented in clinical trials. The primary endpoint was the cumulative proportion of participants dying or requiring mechanical ventilation by Day 28. Secondary endpoints included: time to clinical failure (defined as the time to death), mechanical ventilation, ICU admission, or withdrawal (whichever occured first); mortality rate by Day 28; and time to hospital discharge or “ready for discharge.”
REMDACTA was a two-armed global phase III, randomised, double-blind, multicentre study (REMDACTA, NCT04409262) to evaluate the efficacy and safety of Actemra/RoActemra plus Veklury® (remdesivir), versus placebo plus Veklury in hospitalised patients with severe COVID-19 pneumonia receiving standard of care. Veklury is an antiviral medicine that works to stop replication of SARS-CoV-2, the virus that causes COVID-19. The REMDACTA trial was conducted in collaboration with Gilead Sciences, Inc. The primary endpoint was improvement in time to hospital discharge by Day 28. Key secondary endpoints included likelihood of death, likelihood of progression to mechanical ventilation or death, and clinical status. Clinical status was measured by the 7-category ordinal scale, which tracks patients’ clinical status based on the need for intensive care and/or ventilator use, as well as supplemental oxygen requirements. Patients were followed for 60 days post-randomisation.
There have also been a number of clinical trials with an external third party as the sponsor exploring the efficacy and safety of Actemra/RoActemra for the treatment of patients hospitalised with COVID-19, including the University of Oxford’s RECOVERY study, which was supported by Roche. RECOVERY was a phase III, randomised trial (NCT04381936), which evaluated whether multiple potential treatments, including Actemra/RoActemra, prevent death in hospitalised adult patients with severe COVID-19.
Results of a prospective meta-analysis of almost 11,000 patients across 27 clinical trials, published by researchers from the World Health Organization in The Journal of the American Medical Association, found that treatment of hospitalised patients with severe or critical COVID-19 with IL-6 receptor blockers, including Actemra/RoActemra, was associated with improved mortality and reduced progression to invasive mechanical ventilation or death compared with usual care or placebo. The prospective meta-analysis included data on Actemra/RoActemra in COVID-19 from COVACTA, EMPACTA and REMDACTA, along with 16 additional third-party studies.
About Actemra®/RoActemra® (tocilizumab)
Actemra/RoActemra was the first approved anti-IL-6 receptor biologic and is available in both intravenous (IV) and subcutaneous (SC) formulations for the treatment of adult patients with moderate-to-severe active rheumatoid arthritis (RA). Actemra/RoActemra can be used alone or with methotrexate (MTX) in adult RA patients who are intolerant to, or have failed to respond to, other disease-modifying anti-rheumatic drugs (DMARDs). In Europe, RoActemra IV and SC are also approved for use in adult patients with severe, active and progressive RA who previously have not been treated with MTX. Actemra/RoActemra IV and SC are also approved globally for polyarticular juvenile idiopathic arthritis (pJIA) and systemic juvenile idiopathic arthritis (sJIA) in children two years of age and older. Actemra/RoActemra SC is approved globally for giant cell arteritis (GCA), and Actemra/RoActemra IV is approved for the treatment of chimeric antigen receptor (CAR) T-cell-induced severe or life-threatening cytokine release syndrome (CRS) in people two years of age and older. Actemra/RoActemra was the first approved treatment for sJIA, GCA and CRS. Actemra SC is now approved in the United States for slowing the rate of decline in pulmonary function in adult patients with systemic sclerosis-associated interstitial lung disease (SSc-ILD). In addition to the above-mentioned indications, in Japan Actemra IV is also approved for the treatment of Castleman’s disease and adult Still's disease, and the Actemra SC formulation is approved for Takayasu arteritis.
Actemra/RoActemra is part of a co-development agreement with Chugai Pharmaceutical Co., Ltd and has been approved in Japan since April 2005. Actemra/RoActemra is approved in more than 110 countries worldwide.
For more information on how Roche is responding to the global COVID-19 pandemic, please visit our COVID-19 response page.
For more information, please visit www.roche.com.
Basel, 07 December 2021 - Roche (SIX: RO, ROG; OTCQX: RHHBY)
https://www.roche.com/media/releases/med-cor-2021-12-07.htm
Dec. 06, 2021 1:17 PM ET Roche Holding AG (RHHBY) By: Jonathan M Block, SA News Editor
December 8, 2021
Patients receiving zuranolone 50 mg in the WATERFALL Study demonstrated rapid improvements in depressive and anxiety symptoms, as early as the first measured timepoint (Day 3 for HAMD-17 and Day 8 for HAM-A), with average improvements maintained through the end of the study (Day 42)
Safety data from the WATERFALL Study was consistent with the zuranolone safety profile seen to date across the LANDSCAPE program
Zuranolone was generally well-tolerated in a subgroup of patients aged 65 years and older in the SHORELINE Study, showing similar efficacy and safety results in the datasets analyzed to that observed in the general study population
CAMBRIDGE, Mass.--(BUSINESS WIRE)--Dec. 8, 2021-- Sage Therapeutics, Inc. (Nasdaq: SAGE) and Biogen Inc. (Nasdaq: BIIB) today announced new data from the LANDSCAPE clinical program evaluating the efficacy and safety of zuranolone for the treatment of major depressive disorder (MDD) presented at the American College of Neuropsychopharmacology (ACNP) Congress taking place December 5-8 in San Juan, Puerto Rico. Data from the SHORELINE and WATERFALL Studies in the LANDSCAPE clinical program further the understanding of the potential efficacy and safety profile of zuranolone for the treatment of MDD. Across the studies, zuranolone treatment led to improvements in depressive symptoms as well as in symptoms of elevated anxiety as assessed by multiple scales (HAMD-17, MADRS and HAM-A, respectively). In the WATERFALL Study, a rapid onset of effect in HAMD-17 was observed compared to placebo as early as Day 3, reaching statistical significance, followed by a stabilization of depressive symptoms through the follow-up period.
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20211208005327/en/
Zuranolone has demonstrated a consistent safety profile in the totality of clinical data to date, with no increased incidence of adverse events (AEs) of weight gain, sexual dysfunction, or sleep disruption relative to placebo, symptoms that are often the cause of standard of care antidepressant discontinuation. In an analysis of a subgroup of patients (N=96) over the age of 65 in the SHORELINE Study, zuranolone efficacy and safety results for the initial 2-week dose treatment course were similar to that of the general study population. At the time of the analysis, retreatment data were only available for the zuranolone 30 mg cohort of the SHORELINE Study. In a subgroup of patients 65 years and older who responded to the initial 2-week treatment course of zuranolone 30 mg, and were followed for up to one year in the SHORELINE Study, more than half did not receive a second course of treatment during their time in the study.
December 8, 2021
Patients receiving zuranolone 50 mg in the WATERFALL Study demonstrated rapid improvements in depressive and anxiety symptoms, as early as the first measured timepoint (Day 3 for HAMD-17 and Day 8 for HAM-A), with average improvements maintained through the end of the study (Day 42)
Safety data from the WATERFALL Study was consistent with the zuranolone safety profile seen to date across the LANDSCAPE program
Zuranolone was generally well-tolerated in a subgroup of patients aged 65 years and older in the SHORELINE Study, showing similar efficacy and safety results in the datasets analyzed to that observed in the general study population
CAMBRIDGE, Mass.--(BUSINESS WIRE)--Dec. 8, 2021-- Sage Therapeutics, Inc. (Nasdaq: SAGE) and Biogen Inc. (Nasdaq: BIIB) today announced new data from the LANDSCAPE clinical program evaluating the efficacy and safety of zuranolone for the treatment of major depressive disorder (MDD) presented at the American College of Neuropsychopharmacology (ACNP) Congress taking place December 5-8 in San Juan, Puerto Rico. Data from the SHORELINE and WATERFALL Studies in the LANDSCAPE clinical program further the understanding of the potential efficacy and safety profile of zuranolone for the treatment of MDD. Across the studies, zuranolone treatment led to improvements in depressive symptoms as well as in symptoms of elevated anxiety as assessed by multiple scales (HAMD-17, MADRS and HAM-A, respectively). In the WATERFALL Study, a rapid onset of effect in HAMD-17 was observed compared to placebo as early as Day 3, reaching statistical significance, followed by a stabilization of depressive symptoms through the follow-up period.
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20211208005327/en/
Zuranolone has demonstrated a consistent safety profile in the totality of clinical data to date, with no increased incidence of adverse events (AEs) of weight gain, sexual dysfunction, or sleep disruption relative to placebo, symptoms that are often the cause of standard of care antidepressant discontinuation. In an analysis of a subgroup of patients (N=96) over the age of 65 in the SHORELINE Study, zuranolone efficacy and safety results for the initial 2-week dose treatment course were similar to that of the general study population. At the time of the analysis, retreatment data were only available for the zuranolone 30 mg cohort of the SHORELINE Study. In a subgroup of patients 65 years and older who responded to the initial 2-week treatment course of zuranolone 30 mg, and were followed for up to one year in the SHORELINE Study, more than half did not receive a second course of treatment during their time in the study.
“The data shared at ACNP continue to provide insight to help us better understand how zuranolone could impact the treatment of depression and potentially differentiate further from current antidepressants, if approved,” said Barry Greene, Chief Executive Officer at Sage Therapeutics. “The analysis conducted evaluating zuranolone’s effects on measures of anxiety in patients with MDD is critical. Symptoms of anxiety are highly present in patients with depression, which can pose unique challenges to care. We are also pleased with the data for those 65 and older, who can struggle with current therapies to treat their depression. Zuranolone has consistently demonstrated rapid and sustained improvements in depressive symptoms and a well-tolerated safety profile in our clinical trials to date without the adverse events that are often associated with discontinuation of standard of care antidepressants.”
ACNP presentations included data from the WATERFALL Study, a Phase 3 placebo-controlled trial that evaluated the efficacy and safety of zuranolone 50 mg in adults 18 to 64 years old with MDD as well as the ongoing open-label SHORELINE Study in MDD. In an oral session, an analysis from the WATERFALL Study assessing zuranolone (50 mg) on symptoms of anxiety as measured by the Hamilton Anxiety Rating Scale (HAM-A) showed improvements in symptoms of anxiety compared with placebo at Days 8 and 15. Additional data presented provided an efficacy analysis from the WATERFALL Study as measured by the Montgomery-Åsberg Depression Rating Scale (MADRS) total score, which addresses core mood symptoms such as sadness, lack of energy, and suicidal thoughts. The group of patients receiving zuranolone (50 mg) showed rapid improvements in depressive symptoms and anxiety symptoms, as early as the first measured timepoint (Day 3 for HAMD-17 and Day 8 for HAM-A). Similar results have been observed across the LANDSCAPE program. Additionally, data from the SHORELINE Study support the potential of zuranolone as an oral, as-needed treatment for patients with MDD, including those age 65 and older
“The new data shared at ACNP further our confidence in the potential of zuranolone to help rapidly mitigate various symptoms associated with depression, including elevated anxiety,” said Priya Singhal, M.D., M.P.H., Head of Global Safety and Regulatory Sciences and Interim Head of Research and Development at Biogen. “As we continue to evaluate the totality of the data from the LANDSCAPE clinical develop program – which includes the Phase 3 WATERFALL and SHORELINE Studies – we aim to gain a more comprehensive understanding of how zuranolone may one day be a valuable option for people worldwide who seek a new way to treat depression.”
Data presented at ACNP:
About Major Depressive Disorder (MDD)
Major depressive disorder (MDD) is a common but serious mood disorder in which people experience depressive symptoms that impair their social, occupational, educational, or other important functioning, such as a depressed mood or loss of interest or pleasure in daily activities, consistently for at least a two-week period. It is estimated that approximately 19 million people in the U.S. and more than 250 million people worldwide suffer from MDD each year. While antidepressants are widely used to treat MDD, large-scale studies have demonstrated the need for additional therapies with a differentiated profile.
About Zuranolone
Zuranolone (SAGE-217/BIIB125) is a once-daily, two-week, investigational drug in development for the treatment of major depressive disorder (MDD) and postpartum depression (PPD). Zuranolone is an investigational oral neuroactive steroid (NAS) GABA-A receptor positive allosteric modulator (PAM). The GABA system is the major inhibitory signaling pathway of the brain and central nervous system and contributes to regulating brain function. Zuranolone has been granted Breakthrough Therapy Designation by the U.S. Food & Drug Administration.
Zuranolone is being evaluated in the NEST and LANDSCAPE clinical trial programs. The two development programs include multiple studies examining use of zuranolone in several thousand patients with a variety of dosing, clinical endpoints, and treatment paradigms. The LANDSCAPE program includes five studies of zuranolone in patients with MDD (MDD-201B, MOUNTAIN, SHORELINE, WATERFALL, and CORAL Studies). The NEST program includes two placebo-controlled studies of zuranolone in patients with PPD (ROBIN and SKYLARK Studies). Additionally, Shionogi recently completed a Phase 2 study of zuranolone in MDD in Japan.
For more information, please visit www.sagerx.com.
We routinely post information that may be important to investors on our website at www.biogen.com.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211208005327/en/
Source: Sage Therapeutics, Inc.
Dec. 08, 2021 7:16 AM ET Biogen Inc. (BIIB), SAGE
By: Mamta Mayani,SA News Editor
Latest Phase 3 Data for First-in-Class TREMFYA® (guselkumab) Demonstrates Significant and Durable Improvement in Signs and Symptoms of Active Psoriatic Arthritis while Maintaining its Safety Profile in Patients with Inadequate Response to Tumor Necrosis Factor Inhibition (TNFi-IR)The COSMOS study met its primary endpoint, with a significantly higher proportion of TREMFYA-treated patients (44.4 percent) versus placebo-treated patients (19.8 percent) achieving ACR20 response at week 24; the TREMFYA treatment effect in these TNFi-IR patients was seen by week four
Response rates continued to improve at one year (week 48) with 57.7 percent of TREMFYA patients achieving ACR20 with a similar safety profile (week 56)
TREMFYA is the first and only selective interleukin (IL)-23 inhibitor therapy approved in the U.S. for moderate to severe plaque psoriasis and active psoriatic arthritis irrespective of prior TNFi exposure
SPRING HOUSE, PENNSYLVANIA, December 3, 2021 – The Janssen Pharmaceutical Companies of Johnson & Johnson today announced new TREMFYA® (guselkumab) efficacy and safety data from the Phase 3b COSMOS trial published in Annals of the Rheumatic Diseases (ARD), evaluating this selective interleukin (IL)-23 inhibitor in adults with active psoriatic arthritis (PsA) who demonstrated inadequate efficacy or intolerance to tumor necrosis factor inhibition (TNFi).1 Results showed significantly higher proportions of patients treated with TREMFYA had improvement in joint signs and symptoms and complete skin clearance versus placebo at week 24 in this documented TNFi-IRa patient population, which is often more difficult to treat.1 Furthermore, improvements in signs and symptoms of PsA were maintained or numerically increased through one year (week 48) among TREMFYA-randomized patients.1
“Active psoriatic arthritis is a heterogenous disease and a significant number of patients do not respond adequately to TNF inhibition,” said lead author Laura Coates, M.D., Ph.D., Associate Professor, University of Oxford, UK.b “The COSMOS data demonstrate that TREMFYA significantly improved signs and symptoms of active psoriatic arthritis across multiple clinical disease domains, including patient reported outcomes, with treatment effects observed by week four. These findings reinforce the utility of this alternative mechanism of action as a therapeutic option for adults with active psoriatic arthritis who have not responded to one or more therapies.”
Results show:
Joint Symptom Improvement:
Improvements in Physical Function, Health-Related Quality of Life (HRQoL) and Fatigue:
About COSMOS (NCT03796858)2,18
COSMOS was a Phase 3b, multicenter, randomized, double-blind, placebo-controlled study to evaluate the safety and efficacy of TREMFYA in 285 patients with active PsA and IR to TNFi therapy. The primary endpoint was ACR20 response at week 24. Participants were randomized (2:1) to receive TREMFYA 100 mg at weeks 0, 4, and every 8 weeks thereafter, or placebo. The study included two periods: a 24-week double-blind, placebo-controlled period for the primary analysis of the efficacy and safety of TREMFYA, compared with placebo and a 32-week active-treatment and safety follow-up period for additional analysis of the efficacy and safety of TREMFYA. Through week 48, NRI rules were used for missing data (after the application of treatment failure rules [TFR]). Safety was monitored throughout the study to week 56.
About TREMFYA® (guselkumab)18
Developed by Janssen, TREMFYA is the first approved fully human monoclonal antibody that selectively binds to the p19 subunit of IL-23 and inhibits its interaction with the IL-23 receptor. IL-23 is an important driver of the pathogenesis of inflammatory diseases such as moderate to severe plaque PsO and active PsA.19 TREMFYA is approved in the U.S., Canada, Japan, and a number of other countries worldwide for the treatment of adults with moderate to severe plaque PsO who are candidates for injections or pills (systemic therapy) or phototherapy (treatment using ultraviolet light), and for the treatment of adult patients with active PsA. It is also approved in the EU for the treatment of moderate to severe plaque PsO in adults who are candidates for systemic therapy and for the treatment of active PsA in adult patients who have had an inadequate response or who have been intolerant to a prior disease-modifying antirheumatic drug therapy.
The Janssen Pharmaceutical Companies of Johnson & Johnson maintain exclusive worldwide marketing rights to TREMFYA®.
Infections. TREMFYA® may lower the ability of your immune system to fight infections and may increase your risk of infections. Your healthcare provider should check you for infections and tuberculosis (TB) before starting treatment with TREMFYA® and may treat you for TB before you begin treatment with TREMFYA® if you have a history of TB or have active TB. Your healthcare provider should watch you closely for signs and symptoms of TB during and after treatment with TREMFYA®.
Tell your healthcare provider right away if you have an infection or have symptoms of an infection, including:
Dec. 03, 2021 8:33 AM ET Johnson & Johnson (JNJ) By: Jonathan M Block, SA News Editor
https://www.nasdaq.com/market-activity/stocks/jnj/dividend-history
December 6, 2021
- In U-EXCEED, a significantly higher proportion of patients with moderate to severe Crohn's disease treated with upadacitinib (45 mg once daily for induction) achieved both primary endpoints of clinical remission[a,b] and endoscopic response[c] compared to placebo at week 12[1]
- The study showed that a significantly higher proportion of upadacitinib-treated patients achieved steroid-free clinical remission[d] at week 12 compared to placebo[1]
- The safety results in this study were consistent with the known profile of upadacitinib, with no new safety risks observed[1-6]
- Upadacitinib, a selective and reversible JAK inhibitor discovered and developed by AbbVie, is being studied as an oral therapy for moderate to severe Crohn's disease and several other immune-mediated inflammatory diseases[1,6-14]
NORTH CHICAGO, Ill., Dec. 6, 2021 /PRNewswire/ -- AbbVie (NYSE: ABBV) today announced positive top-line results from U-EXCEED, a Phase 3 induction study, showing upadacitinib (45 mg once daily) achieved both primary endpoints of clinical remissiona,b and endoscopic responsec at week 12.1 The U-EXCEED study enrolled patients with moderate to severe Crohn's disease who had an inadequate response or were intolerant to biologic therapy, with over 60 percent having previously failed two or more biologics.1 U-EXCEED is the first of two Phase 3 induction studies to evaluate the safety and efficacy of upadacitinib in adults with moderate to severe Crohn's disease.1
"The data from this first Phase 3 induction study in Crohn's disease suggest upadacitinib may help address the needs of patients suffering from this disease, as demonstrated in stringent endpoints such as endoscopic response," said Michael Severino, M.D., vice chairman and president, AbbVie. "We continue to leverage our expertise in IBD by driving research and development that help shape the IBD landscape and elevate standards of care for patients."
In U-EXCEED, clinical remission was measured by the Crohn's Disease Activity Index (CDAI) and by the patient-reported symptoms of stool frequency/abdominal pain (SF/AP).1 A significantly greater proportion of patients treated with a 12-week induction regimen of upadacitinib 45 mg daily achieved clinical remission per CDAI at week 12 compared to placebo (39 percent, versus 21 percent; p<0.0001).1 Similar results were seen with clinical remission per SF/AP (40 percent in upadacitinib-treated patients versus 14 percent in placebo-treated patients; p<0.0001).1 In this study, all patients were also evaluated for improvement in the intestinal mucosa by endoscopy.1 At week 12, a significantly greater proportion of patients treated with upadacitinib 45 mg achieved endoscopic response compared to the placebo group (35 percent versus 4 percent; p<0.0001).1
Among patients taking corticosteroids at baseline, a significantly higher proportion of patients receiving upadacitinib 45 mg achieved steroid-free clinical remissiond per CDAI and per SF/AP compared to placebo at week 12.1 A significantly higher proportion of patients receiving upadacitinib compared to placebo also achieved early symptom improvement measured by CR-100 (defined as reduction of CDAI ≥100 points from baseline) at week 2 as well as clinical remission at week 4.1
"I am thrilled to see the results of this first Phase 3 induction study of upadacitinib, particularly in this difficult-to-treat refractory patient population," said Jean-Frederic Colombel, M.D., professor of medicine and director of Inflammatory Bowel Disease Center, Icahn School of Medicine, Mount Sinai, and U-EXCEED study investigator. "These results demonstrate upadacitinib's potential to achieve endoscopic response and clinical remission, including steroid-free clinical remission, at 12 weeks in patients living with Crohn's disease."
* Co-primary endpoints were clinical remission (per CDAI for the U.S. FDA and per SF/AP for the EU EMA) and endoscopic response at week 12. All primary endpoints achieved statistical significance with p-values of <0.0001 versus placebo.
a Clinical remission (per CDAI) is defined as CDAI <150.
b Clinical remission per SF (stool frequency)/AP (abdominal pain) (also referred to as PRO-2) is defined as average daily very soft or liquid stool frequency ≤2.8 AND average daily abdominal pain score ≤1.0, and both not greater than baseline.
c Endoscopic response is defined as a decrease in simple endoscopic score for Crohn's disease (SES-CD) of >50 percent from baseline (or at least a 2-point reduction from baseline for subjects with a baseline SES-CD of 4), as scored by central reviewer.
During the 12-week, double-blind, placebo-controlled period, the safety profile of upadacitinib 45 mg was consistent with the safety profile observed in previous studies across indications, with no new safety risks observed.1 The most common adverse event was nasopharyngitis for upadacitinib and exacerbation of Crohn's disease for placebo.1 Serious adverse events occurred in 9.3 percent of patients in the upadacitinib 45 mg group compared to 9.9 percent of patients in the placebo group.1 Rates of serious infections were 2.8 percent in those treated with upadacitinib 45 mg and 1.8 percent in patients who received placebo.1 All events of herpes zoster (1.5 percent of patients) were nonserious and reported in the upadacitinib group only.1 There was one case of adjudicated gastrointestinal perforation in the upadacitinib group.1
An additional 207 patients at week 12 received an upadacitinib 45 mg daily induction regimen (including non-responders to placebo or from an open label arm).1 Among these patients, there were two additional cases of adjudicated gastrointestinal perforation reported.1 Overall, the safety results in these patients were consistent with what was observed in the upadacitinib 45 mg group during the placebo-controlled period.1
In the study, no treatment-emergent cases of adjudicated cardiovascular event, malignancy, thromboembolic event or death were reported across treatment groups.1
Full results from the U-EXCEED study will be presented at upcoming medical conferences and published in a peer-reviewed medical journal. Use of upadacitinib in Crohn's disease is not approved and its safety and efficacy have not been evaluated by regulatory authorities.
dSteroid-free clinical remission is defined as clinical remission (per CDAI <150, or per SF/AP with average daily SF ≤2.8 and not worse than baseline and average daily AP score ≤1 and not worse than baseline) and discontinuation of corticosteroid use among patients taking corticosteroids at baseline.
About Upadacitinib (RINVOQ®)
Discovered and developed by AbbVie scientists, RINVOQ is a selective and reversible JAK inhibitor that is being studied in several immune-mediated inflammatory diseases.6-14,21 Based on enzymatic and cellular assays, RINVOQ demonstrated greater inhibitory potency for JAK-1 vs JAK-2, JAK-3, and TYK-2.21 The relevance of inhibition of specific JAK enzymes to therapeutic effectiveness is not currently known. RINVOQ 15 mg is approved by the U.S. Food and Drug Administration (FDA) for adults with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more TNF blockers. RINVOQ is also approved by the European Commission for adults (15 mg and 30 mg) and adolescents (15 mg) with moderate to severe atopic dermatitis. RINVOQ 15 mg is approved by the European Commission for adults with moderate to severe active rheumatoid arthritis, adults with active psoriatic arthritis and adults with active ankylosing spondylitis. Phase 3 trials of RINVOQ in rheumatoid arthritis, atopic dermatitis, psoriatic arthritis, axial spondyloarthritis, Crohn's disease, ulcerative colitis, giant cell arteritis and Takayasu arteritis are ongoing.7-14 The use of upadacitinib in Crohn's disease is not approved and its safety and efficacy have not been evaluated by regulatory authorities.
Please click here for the Full Prescribing Information and Medication Guide.
Dec. 06, 2021 9:06 AM ET
By: Jonathan M Block, SA News Editor3 Comments
December 3, 2021 6:15 pm ET
KEYTRUDA Is the First Anti-PD-1/L1 Therapy to Show Recurrence-Free Survival Benefit in the Adjuvant Setting for Stage IIB and IIC Melanoma
KEYTRUDA Is Now Approved as Adjuvant Treatment for Patients (≥12 Years of Age) With Completely Resected Melanoma Across Stage IIB, Stage IIC and Stage III Disease
KENILWORTH, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, today announced that the U.S. Food and Drug Administration (FDA) has approved KEYTRUDA, Merck’s anti-PD-1 therapy, for the adjuvant treatment of adult and pediatric (12 years and older) patients with stage IIB or IIC melanoma following complete resection. Additionally, the FDA expanded the indication for KEYTRUDA as adjuvant treatment for stage III melanoma following complete resection to include pediatric patients (12 years and older).
The approval in stage IIB and IIC melanoma is based on data from the first interim analysis of the Phase 3 KEYNOTE-716 trial, in which KEYTRUDA showed a statistically significant improvement in recurrence-free survival (RFS), reducing the risk of disease recurrence or death by 35% (HR=0.65 [95% CI, 0.46-0.92]; p=0.0132) compared to placebo. Median RFS was not reached for either group. After a median follow-up of 14.4 months, 11% (n=54/487) of patients who received KEYTRUDA had recurrence or died compared to 17% (n=82/489) of patients who received placebo. Efficacy in pediatric patients (12 years and older) with stage IIB, IIC and III melanoma is supported by extrapolation of efficacy data from adults, given similar biology, pharmacology of drug effect, as well as similar exposure-response for efficacy and safety.
Immune-mediated adverse reactions, which may be severe or fatal, can occur in any organ system or tissue and can affect more than one body system simultaneously. Immune-mediated adverse reactions can occur at any time during or after treatment with KEYTRUDA, including pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, dermatologic reactions, solid organ transplant rejection, and complications of allogeneic hematopoietic stem cell transplantation. Important immune-mediated adverse reactions listed here may not include all possible severe and fatal immune-mediated adverse reactions. Early identification and management of immune-mediated adverse reactions are essential to ensure safe use of KEYTRUDA. Based on the severity of the adverse reaction, KEYTRUDA should be withheld or permanently discontinued and corticosteroids administered if appropriate. KEYTRUDA can also cause severe or life-threatening infusion-related reactions. Based on its mechanism of action, KEYTRUDA can cause fetal harm when administered to a pregnant woman. For more information, see “Selected Important Safety Information” below.
“The standard of care for patients with resected stage IIB and IIC melanoma has been observation, despite the fact that for these patients, the risk of recurrence is nearly the same as for patients with later-stage disease for whom treatment is recommended,” said Dr. Jason Luke, director, Cancer Immunotherapeutics Center at UPMC Hillman Cancer Center. “Today’s approval of pembrolizumab for the adjuvant treatment of patients 12 years and older with stage IIB and IIC melanoma following complete resection is an important advance that provides these patients with a new option that can help reduce the risk of their cancer returning.”
“KEYTRUDA was the first anti-PD-1 therapy to be approved in metastatic melanoma in the U.S. seven years ago. Since then, we have built on this foundation in melanoma and have expanded the use of KEYTRUDA into earlier stages of this disease,” said Dr. Scot Ebbinghaus, vice president, clinical research, Merck Research Laboratories. “With today’s approval, we can now offer healthcare providers and patients 12 years and older the opportunity to help prevent melanoma recurrence with KEYTRUDA across resected stage IIB, stage IIC and stage III melanoma.”
Study Design and Additional Data From KEYNOTE-716
KEYNOTE-716 (ClinicalTrials.gov, NCT03553836) is a multicenter, randomized (1:1), double-blind, placebo-controlled Phase 3 trial that enrolled 976 patients with completely resected stage IIB or IIC melanoma. Patients were randomized to KEYTRUDA 200 mg or the pediatric (≥12 years old) dose of KEYTRUDA 2 mg/kg intravenously (up to a maximum of 200 mg) every three weeks or placebo for up to one year until disease recurrence or unacceptable toxicity. Randomization was stratified by AJCC eighth edition T Stage (>2.0-4.0 mm with ulceration vs. >4.0 mm without ulceration vs. >4.0 mm with ulceration). Patients must not have been previously treated for melanoma beyond complete surgical resection for their melanoma prior to study entry. The main efficacy outcome measure was investigator-assessed RFS (defined as the time between the date of randomization and the date of first recurrence [local, in-transit or regional lymph nodes or distant recurrence] or death, whichever occurred first). New primary melanomas were excluded from the definition of RFS. Patients underwent imaging every six months for one year from randomization, every six months from years two to four, and then once in year five from randomization or until recurrence, whichever came first.
Adverse reactions occurring in patients with stage IIB or IIC melanoma were similar to those occurring in 1,011 patients with stage III melanoma from KEYNOTE-054 or the 2,799 patients with melanoma or NSCLC treated with KEYTRUDA as a single agent. For more information, see “Selected Important Safety Information” below.
About KEYTRUDA® (pembrolizumab) Injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of adult and pediatric (12 years and older) patients with stage IIB, IIC, or III melanoma following complete resection.
Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [tumor proportion score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [combined positive score (CPS) ≥1] as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy. KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Carcinoma
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
Non-muscle Invasive Bladder Cancer
KEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with MSI-H central nervous system cancers have not been established.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC).
Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or GEJ (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
KEYTRUDA, in combination with chemotherapy, with or without bevacizumab, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
KEYTRUDA is indicated for the adjuvant treatment of patients with RCC at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions.
Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf.
Dec. 03, 2021 4:55 PM ET
By: Jonathan M Block, SA News Editor1 Comment
12/06/2021
- Intranasal zavegepant 10 mg met the study's co-primary endpoints and demonstrated statistically significant superiority versus placebo on a total of 15 consecutive, prespecified primary and secondary outcome measures in the acute treatment of migraine- Zavegepant showed ultra-rapid pain relief at the earliest measured time point of 15 minutes and sustained efficacy through 48 hours after a single intranasal dose- Biohaven plans to file a New Drug Application for zavegepant with the U.S. Food and Drug Administration in 1Q2022
NEW HAVEN, Conn., Dec. 6, 2021 /PRNewswire/ -- Biohaven Pharmaceutical Holding Company Ltd. (NYSE: BHVN) today announced positive topline results from the second pivotal clinical trial (NCT04571060) evaluating the safety and efficacy of its investigational therapy, intranasal zavegepant, for the acute treatment of migraine in adults. The Phase 3 study achieved its co-primary regulatory endpoints of pain freedom and freedom from most bothersome symptom at 2 hours and showed broad efficacy by demonstrating statistically significant superiority to placebo across a total of 15 prespecified primary and secondary outcome measures. Based upon these results, combined with the prior positive Phase 2/3 trial, Biohaven is moving forward with plans for regulatory submissions in the United States and other countries. Full results from this Phase 3 trial will be presented at upcoming medical conferences and/or published in peer-reviewed journals.
Richard B. Lipton, M.D., Professor and Vice Chair of Neurology at the Albert Einstein College of Medicine and Director of the Montefiore Headache Center, said "Patients with migraine rate speed of onset as one of the most important aspects of an effective therapy. The data from this trial shows that intranasal zavegepant delivered impressive performance on this metric by demonstrating statistically significant pain relief within 15 minutes and return to normal function within 30 minutes. Additionally, non-oral treatments offer additional benefits for patients who experience nausea, vomiting or gastroparesis (with slow absorption). Intranasal zavegepant will be an important new treatment option for patients who require a rapid and non-oral option for acute treatment of their migraine attacks."
Zavegepant was statistically superior to placebo on the co-primary endpoints of pain freedom (24% vs 15%, p < 0.0001) and freedom from most bothersome symptom (40% vs 31%, p = 0.0012) at 2 hours. Zavegepant was superior to placebo demonstrating pain relief as early as 15 minutes (see Figure 1).
Patients achieved return to normal function as early as 30 minutes after dosing (p < 0.006). The efficacy benefits of zavegepant were durable, including superiority versus placebo (p < 0.05) on: sustained pain freedom 2 to 24 hours; sustained pain freedom 2 to 48 hours; sustained pain relief 2 to 24 hours; and sustained pain relief 2 to 48 hours.
Vlad Coric, M.D., Chief Executive Officer at Biohaven stated, "Intranasal zavegepant was designed to provide ultra-rapid pain relief and expand our CGRP receptor-antagonist franchise by providing patients with another important tool to combat migraine. The trial results clearly show that the performance of this formulation exceeded expectations by demonstrating superiority over placebo on pain relief at 15 minutes and return to normal function by 30 minutes. The impressive efficacy, safety and tolerability profile shown in this trial highlights the potential of zavegepant to usher in a new era of non-oral CGRP targeting migraine therapies that may transcend the traditional boundaries of older legacy intranasal migraine approaches. Biohaven is committed to delivering on its promise to provide new treatment options for the millions of people living with this debilitating disease and these data represent a major milestone in that endeavor," (see Figure 2).
The Phase 3 pivotal study is a randomized, double-blind, placebo-controlled clinical trial that randomized 1,405 adults with at least a one-year history of migraine (with or without aura) and migraine attacks lasting, on average, 4 to 72 hours if untreated. Conducted at 94 sites in the United States, the study evaluated the safety and efficacy of zavegepant intranasal spray taken as needed in a single dose compared to placebo for the acute treatment of a moderate to severe migraine attack.
Zavegepant showed a favorable safety and tolerability profile among study participants that was consistent with prior clinical trial experience. The most common individual adverse event in the pivotal study reported with a frequency ≥ 5% in the zavegepant treatment arm and greater than placebo was abnormal taste (21% vs 5%). The majority of AEs were mild in intensity.
Biohaven plans to file a New Drug Application (NDA) for zavegepant with the U.S. Food and Drug Administration (FDA) in 1Q 2022 and other countries thereafter. If ultimately approved, zavegepant would be the first intranasal calcitonin gene-related peptide (CGRP) receptor antagonist for the acute treatment of migraine. Zavegepant is the second clinical candidate for Biohaven after FDA-approved Nurtec® ODT (rimegepant) for the acute treatment of migraine and preventive treatment of episodic migraine in adults.
About Zavegepant
Zavegepant is a third generation, high affinity, selective and structurally unique, small molecule CGRP receptor antagonist from Biohaven's NOJECTION® Migraine Platform and the only CGRP receptor antagonist in clinical development with both intranasal and oral formulations. Previously the efficacy and safety of intranasal zavegepant was shown in a randomized controlled Phase 2/3 dose-ranging trial with a total of over 1000 patients treated. In this study, zavegepant showed statistical superiority to placebo on the coprimary endpoints of 2 hour freedom from pain and freedom from patients' most bothersome symptom (either nausea, photophobia or phonophobia). The present announcement represents the second zavegepant pivotal clinical trial to meet these coprimary endpoints. For more information, visit https://www.biohavenpharma.com.
More information about Biohaven is available at www.biohavenpharma.com.
View original content to download multimedia:https://www.prnewswire.com/news-releases/biohaven-reports-positive-topline-results-from-pivotal-migraine-trial-of-intranasal-zavegepant-demonstrating-ultra-rapid-pain-relief-by-15-minutes-prepares-for-submission-of-new-drug-application-301437767.html
SOURCE Biohaven Pharmaceutical Holding Company Ltd.
Dec. 06, 2021 9:33 AM ET
Biohaven Pharmaceutical Holding Company Ltd. (BHVN)
By: Dulan Lokuwithana, SA News Editor
Dec 02, 2021 11:00 PM
CAMBRIDGE, Mass. & BEIJING & HEMEL HEMPSTEAD, England--(BUSINESS WIRE)-- BeiGene, Ltd. (NASDAQ: BGNE; HKEX: 06160) and EUSA Pharma (UK), Ltd. today announced that the China National Medical Products Administration (NMPA) has approved SYLVANT® (siltuximab for injection) for the treatment of adult patients with multicentric Castleman disease (MCD) who are human immunodeficiency virus (HIV) negative and human herpes virus-8 (HHV-8) negative, also known as idiopathic MCD (iMCD). This disease is a rare, life-threatening, and debilitating condition of the lymph nodes and related tissues. Siltuximab is a monoclonal antibody approved in the United States, European Union, and other countries and regions around the world.
“Today’s approval provides a new treatment for patients in China with this rare systemic disorder,” commented Xiaobin Wu, Ph.D., President, Chief Operating Officer, and General Manager of China at BeiGene. “This approval is our second product in our collaboration with EUSA, highlighting the combined expertise of our companies as we work together on behalf of patients. We are excited to have reached this important milestone and we look forward to launching siltuximab in 2022 and to helping patients with iMCD in China.”
“We are delighted that siltuximab has been approved in China and that this innovative treatment will soon be made available to patients suffering from this devastating disease,” said Carsten Thiel, Ph.D., Chief Executive Officer of EUSA Pharma.
The approval of siltuximab for the treatment of iMCD was supported by clinical results from a multinational, randomized, double blind, placebo-controlled Phase 2 trial (NCT01024036) conducted in 79 patients from 19 countries and regions, including 16 patients from China.
The primary endpoint of the study was durable tumor and symptomatic response, defined as tumor response assessed by independent review and complete resolution or stabilization of prospectively collected iMCD symptoms, for at least 18 weeks without treatment failure. A statistically significant difference in independently reviewed durable tumor and symptomatic response rate in the siltuximab arm compared with the placebo arm (34% vs. 0%, respectively; 95% CI: 11.1, 54.8; p = 0.0012) was observed.
Data from all patients treated with siltuximab monotherapy (n = 370) form the overall basis of the safety evaluation. Infections (including upper respiratory tract infections), pruritus, rash, arthralgia, and diarrhea were the most common adverse reactions, occurring in > 20% of siltuximab-treated patients in iMCD clinical studies. The most serious adverse reaction associated with the use of siltuximab was anaphylactic reaction.
In the study, a total of 34 Asian patients were enrolled, including 16 from China; 24 of 34 Asian patients were treated in the siltuximab arm and the remaining 10 patients were treated in the placebo arm. The subgroup analyses showed that there was no significant difference in demographic and baseline disease characteristics between Asian patients and the overall patient population; the efficacy data on primary and secondary study endpoints of Asian patients was consistent with those of the overall patient population, and the same for safety data; no other safety signals were found.
An open label, long-term extension Phase 2 trial (NCT01400503) was also conducted in patients with iMCD treated in prior trials. The median duration of siltuximab treatment was 5.52 years (range: 0.8 to 10.8 years); more than 50% of patients received siltuximab treatment for ≥5 years. The rate of serious or Grade ≥3 adverse events did not increase over time as a function of cumulative exposure.
About SYLVANT® (siltuximab for injection)
Siltuximab is a monoclonal antibody that directly neutralises IL-6, an inflammatory cytokine detected at elevated levels in multiple inflammatory conditions. SYLVANT® is approved in a number of countries and regions and is indicated for the treatment of patients with multicentric Castleman disease (MCD) who are human immunodeficiency virus (HIV) negative and human herpesvirus-8 (HHV-8) negative. iMCD is a rare, life-threatening and debilitating lymphoproliferative disorder, which causes abnormal overgrowth of immune cells and shares many symptomatic and histological features with lymphoma. The Approved EU and U.S. Indications and Usage information are available for additional information at: EMA Summary of Product Characteristics (SmPC) and FDA Prescribing Information.
For more information please visit www.eusapharma.com.
To learn more about BeiGene, please visit www.beigene.com and follow us on Twitter at @BeiGeneGlobal.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211202006046/en/
Source: BeiGene, Ltd.
Dec. 03, 2021 1:20 AM ET
By: Mamta Mayani, SA News Editor
December 3, 2021Download PDF
Expanded EUA includes both treatment of patients with COVID-19 and post-exposure prophylaxis (PEP) in high-risk pediatric and infant patients
INDIANAPOLIS, Dec. 3, 2021 /PRNewswire/ -- The U.S. Food and Drug Administration (FDA) has expanded the Emergency Use Authorization (EUA) for bamlanivimab and etesevimab administered together to include certain high-risk pediatric patients from birth to <12 years old, Eli Lilly and Company (NYSE: LLY) announced today. This expansion allows for bamlanivimab and etesevimab to be administered together in high-risk pediatric patients for the treatment of mild to moderate COVID-19 as well as post-exposure prophylaxis.
"Our mission since the start of the pandemic has been to offer crucial support by developing therapeutic options that could prevent hospitalization and death for as many people as possible," said Daniel Skovronsky, M.D., Ph.D., Lilly's chief scientific and medical officer, and president of Lilly Research Laboratories. "With the FDA's decision to allow use of bamlanivimab with etesevimab in children and infants, Lilly can now offer treatment and prevention options to high-risk individuals of any age."
The expanded authorization is based on safety and efficacy data of pediatric and infant patients in BLAZE-1, a phase 2/3 clinical trial studying bamlanivimab and etesevimab administered together for the treatment of mild to moderate COVID-19 and who are at high risk for severe disease progression. The median time to complete symptom resolution as recorded in a trial specific daily symptom diary was 7 days for subjects treated with bamlanivimab 700 mg and etesevimab 1,400 mg and 5 days for subjects treated with weight-based dosing of bamlanivimab and etesevimab. No pediatric subject died or required hospitalization due to COVID-19.
Bamlanivimab and etesevimab, when administered together, retain neutralization activity against the Delta variant, which is the predominant variant of concern within the U.S. Lilly has performed pseudovirus and authentic virus studies to confirm that bamlanivimab with etesevimab retain neutralization activity against the Delta variant of concern. Lilly is working quickly to understand neutralization activity of our therapies on the Omicron variant of concern.
To date, over 700,000 patients have been treated with bamlanivimab or bamlanivimab and etesevimab, potentially preventing more than 35,000 hospitalizations and at least 14,000 deaths during the worst of the pandemic.
For more information about the use of bamlanivimab and etesevimab together for the treatment of mild to moderate COVID-19 and prevention of COVID-19 in high risk pediatric and infant patients under the FDA's emergency use authorization, please see the Fact Sheet for Healthcare Providers, and Fact Sheet for Patients, Parents and Caregivers (English) (Spanish), click here or contact Lilly's 24-hour support line at 1-855-LillyC19 (1-855-545-5921). Patients and physicians can visit the NICA Infusion Center Locator or the HHS Therapeutics Distribution locator to find a potential therapy location.
For media resources, including product images and fact sheets, please click here.
Important Information about bamlanivimab and etesevimab together
Bamlanivimab and etesevimab together have not been approved by the FDA for any use. It is not known if bamlanivimab and etesevimab together are safe and effective for the treatment or post-exposure prophylaxis of COVID-19.
Bamlanivimab and etesevimab together are authorized under Emergency Use Authorization only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use under Section 564(b)(1) of the Act, 21 U.S.C § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.
Healthcare providers should review the Fact Sheet for information on the authorized use of bamlanivimab and etesevimab together and mandatory requirements of the EUA. Please see the FDA Letter of Authorization, Fact Sheet for Healthcare Providers, and Fact Sheet for Patients, Parents and Caregivers (English) (Spanish) for bamlanivimab and etesevimab together.
Authorized Use and Important Safety Information
TREATMENT
The U.S. Food and Drug Administration (FDA) has issued an Emergency Use Authorization (EUA) to permit the emergency use of the unapproved products bamlanivimab and etesevimab administered together for the treatment of mild to moderate coronavirus disease 2019 (COVID-19) in adults and pediatric patients, including neonates, with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death.
About bamlanivimab and etesevimab
Bamlanivimab is a recombinant, neutralizing human IgG1 monoclonal antibody (mAb) directed against the spike protein of SARS-CoV-2. It was designed to block viral attachment and entry into human cells, thus neutralizing the virus. Bamlanivimab emerged from the collaboration between Lilly and AbCellera to create antibody therapies for the prevention and treatment of COVID-19. Lilly scientists rapidly developed the antibody in less than three months after it was discovered by AbCellera and the scientists at the National Institute of Allergy and Infectious Diseases (NIAID) Vaccine Research Center. Bamlanivimab was identified from a blood sample taken from one of the first U.S. patients who recovered from COVID-19.
Etesevimab (LY-CoV016, also known as JS016) is a recombinant fully human monoclonal neutralizing antibody, which specifically binds to the SARS-CoV-2 surface spike protein receptor binding domain with high affinity and can block the binding of the virus to the ACE2 host cell surface receptor. Point mutations were introduced into the native human IgG1 antibody to mitigate effector function. Lilly licensed etesevimab from Junshi Biosciences after it was jointly developed by Junshi Biosciences and the Institute of Microbiology, Chinese Academy of Science (IMCAS). Junshi Biosciences leads development in Greater China, while Lilly leads development in the rest of the world.
Results from a Phase 2/3 study in people recently diagnosed with COVID-19 in the ambulatory setting (BLAZE-1, NCT04427501) were published in the New England Journal of Medicine. Results from a Phase 3 study of bamlanivimab in residents and staff at long-term care facilities (BLAZE-2, NCT04497987) were published in the Journal of American Medical Association (JAMA). A Phase 2 study assessing the efficacy and safety of bamlanivimab alone, and bamlanivimab with other neutralizing antibodies versus placebo for the treatment of symptomatic low-risk COVID-19 in the outpatient setting (BLAZE-4. NCT04634409) has completed enrollment.
To learn more about Lilly, please visit us at www.lilly.com and www.lilly.com/news. P-LLY
View original content to download multimedia:https://www.prnewswire.com/news-releases/lillys-bamlanivimab-with-etesevimab-authorized-as-the-first-and-only-neutralizing-antibody-therapy-for-emergency-use-in-covid-19-patients-under-the-age-of-12-301437329.html
SOURCE Eli Lilly and Company
Dec. 03, 2021 2:59 PM ET Eli Lilly and Company (LLY), ABCL By: Dulan Lokuwithana, SA News Editor2 Comments
https://www.nasdaq.com/market-activity/stocks/lly/dividend-history
Dec 02, 2021 11:00 PM
BEIJING & CAMBRIDGE, Mass.--(BUSINESS WIRE)-- BeiGene (NASDAQ:BGNE; HKEX:06160), a global science-driven biotechnology company focused on developing innovative and affordable medicines to improve treatment outcomes and access for patients worldwide, today announced that three of its medicines have been added to the most recent National Reimbursement Drug List (NRDL) in China by the National Healthcare Security Administration (NHSA). BeiGene-discovered medicines in the updated NRDL include: anti-PD-1 antibody tislelizumab in three new indications, including in lung and liver cancers; BTK inhibitor BRUKINSA® (zanubrutinib) in one new indication; and the initial listing for PARP inhibitor pamiparib. The changes to the NRDL will be effective on January 1, 2022.
“The inclusion of our three internally-discovered innovative medicines in the latest NRDL will help expand access to these high-quality oncology treatments across China at affordable prices and reduce the financial burden for patients and their families,” commented Xiaobin Wu, Ph.D., President of BeiGene, Chief Operating Officer, and General Manager of China. “Since its establishment, the NHSA has accelerated the frequency of adjustment to the NRDL, forming a dynamic mechanism for annual updates. Through the establishment of a comprehensive healthcare system, life-saving innovative oncology medicines are now more quickly included in the NRDL at affordable prices, covering different types of medical care and providing benefits for people living with cancer. As an innovative company with strong R&D capabilities and global reach, BeiGene is working to change the status quo in the field of treatment and fill the gap in clinical treatment options. We look forward to working with the NHSA to fulfill the demand for these treatments across China as soon as possible.”
The following indications have been included in the updated NRDL:
About BRUKINSA® (zanubrutinib)
BRUKINSA® (zanubrutinib) is a small molecule inhibitor of Bruton’s tyrosine kinase (BTK) discovered by BeiGene scientists that is currently being evaluated globally in a broad clinical program as a monotherapy and in combination with other therapies to treat various B-cell malignancies. Because new BTK is continuously synthesized, BRUKINSA® was specifically designed to deliver complete and sustained inhibition of the BTK protein by optimizing bioavailability, half-life, and selectivity. With differentiated pharmacokinetics compared to other approved BTK inhibitors, BRUKINSA® has been demonstrated to inhibit the proliferation of malignant B cells within a number of disease relevant tissues.
BRUKINSA is approved in one or more indications in a total of 40 countries and regions, including the United States, China, the European Union, Australia and Canada. To date, more than 20 marketing authorization applications have been submitted for BRUKINSA for various indications.
About Pamiparib
Pamiparib is an inhibitor of PARP1 and PARP2 which has demonstrated pharmacological properties such as brain penetration and PARP-DNA complex trapping in preclinical models. Discovered by BeiGene scientists, pamiparib was the first PARP inhibitor approved in both platinum-sensitive and platinum-resistant relapsed ovarian cancer in China. Pamiparib is currently being evaluated globally as a monotherapy or in combination with other agents for a variety of solid tumor malignancies.
In China, pamiparib received conditional approval for the treatment of patients with germline BRCA (gBRCA) mutation-associated recurrent advanced ovarian, fallopian tube, or primary peritoneal cancer who have been treated with two or more lines of chemotherapy in May 2021. Full approval for this indication is contingent upon results from ongoing corroborative trials confirming the clinical benefit of pamiparib in this population.
To learn more about BeiGene, please visit www.beigene.com.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211202006057/en/
Source: BeiGene
https://seekingalpha.com/symbol/BGNE
December 3, 2021
WHIPPANY, N.J.--(BUSINESS WIRE)-- The Phase III ARASENS trial investigating the use of the oral androgen receptor inhibitor (ARi) NUBEQA® (darolutamide) in metastatic hormone-sensitive prostate cancer (mHSPC) has met its primary endpoint. In the ARASENS trial, NUBEQA in combination with docetaxel and androgen deprivation therapy (ADT) significantly increased overall survival (OS) compared to docetaxel and ADT. The overall incidence of reported adverse events was similar between treatment arms. Detailed results of the study are planned to be presented at an upcoming scientific congress. NUBEQA is currently indicated for the treatment of patients with non-metastatic castration-resistant prostate cancer (nmCRPC).
The ARASENS trial investigating NUBEQA is the only Phase III randomized, multi-center, double-blind trial, which was prospectively designed to evaluate the efficacy and safety of a combination of an ARi with docetaxel and ADT compared to docetaxel and ADT in patients with mHSPC.1
“For patients with mHSPC, there remains a significant need for new therapeutic approaches that improve treatment outcomes. ARASENS was prospectively designed to investigate whether combining NUBEQA with docetaxel and ADT could lead to an increase in overall survival for men with mHSPC,” said Scott Z. Fields, M.D., Senior Vice President and Head of Oncology Development at Bayer's Pharmaceutical Division. “We are especially grateful to the patients and investigators for participating in this important trial and look forward to presenting the full results at an upcoming meeting.”
Bayer plans to discuss the data from ARASENS with health authorities worldwide regarding the submission of NUBEQA for marketing authorization in this indication.
About the ARASENS Trial1
The ARASENS trial (NCT02799602) is a randomized, Phase III, multi-center, double-blind, placebo-controlled trial, which was prospectively designed to investigate the safety and efficacy of oral NUBEQA, an androgen receptor inhibitor (ARi), in combination with the chemotherapy docetaxel and androgen deprivation therapy (ADT) in patients with metastatic hormone-sensitive prostate cancer (mHSPC). 1,306 newly diagnosed patients were randomized in a 1:1 ratio to receive 600 mg of NUBEQA twice a day or matching placebo, in addition to docetaxel and standard ADT.
The primary endpoint of this trial is overall survival (OS). Secondary endpoints include time to castration-resistant prostate cancer (CRPC), time to initiation of subsequent anticancer therapy, time to first symptomatic skeletal event (SSE), time to pain progression, all measured at 12‐week intervals, as well as adverse events as a measure of safety and tolerability.
About the ARANOTE Trial
The ARANOTE trial (NCT04736199) is a randomized, double-blind, placebo-controlled Phase III study of NUBEQA in addition to androgen deprivation therapy (ADT) versus placebo plus ADT in men with metastatic hormone-sensitive prostate cancer (mHSPC). The primary endpoint of this study is radiological progression-free survival (rPFS), as measured as the time from the date of randomization to the date of first documentation of radiological progressive disease or death due to any cause, whichever occurs first.
About NUBEQA® (darolutamide) 2
NUBEQA is an androgen receptor inhibitor (ARi) with a distinct chemical structure that competitively inhibits androgen binding, AR nuclear translocation, and AR-mediated transcription.2
On July 30, 2019, the FDA approved NUBEQA® (darolutamide) based on the ARAMIS trial, a randomized, double-blind, placebo-controlled, multi-center Phase III study, which evaluated the safety and efficacy of oral NUBEQA in patients with non-metastatic castration-resistant prostate cancer (nmCRPC) who were receiving a concomitant gonadotropin-releasing hormone (GnRH) analog or had a bilateral orchiectomy. In the clinical study, 1,509 patients were randomized in a 2:1 ratio to receive 600 mg of NUBEQA orally twice daily or androgen deprivation therapy (ADT) alone. The primary efficacy endpoint was metastasis-free survival (MFS). NUBEQA is also being investigated in further studies across various stages of prostate cancer, including another Phase III trial in metastatic hormone-sensitive prostate cancer (mHSPC) (ARANOTE) as well as a Phase III trial evaluating NUBEQA as an adjuvant treatment for localized prostate cancer with very high risk of recurrence (DASL-HiCaP). Information about these trials can be found at www.clinicaltrials.gov.
Developed jointly by Bayer and Orion Corporation, a globally operating Finnish pharmaceutical company, NUBEQA is currently indicated for the treatment of men with nmCRPC.2 The approvals of NUBEQA in the U.S., European Union (EU), and other global markets have been based on the pivotal Phase III ARAMIS trial data evaluating the efficacy and safety of NUBEQA plus ADT compared to ADT alone.2 Filings in other regions are underway or planned.
INDICATION FOR NUBEQA® (darolutamide)
NUBEQA® (darolutamide) is an androgen receptor inhibitor indicated for the treatment of patients with non-metastatic castration-resistant prostate cancer.
IMPORTANT SAFETY INFORMATION FOR NUBEQA® (darolutamide)
Embryo-Fetal Toxicity: Safety and efficacy of NUBEQA have not been established in females. NUBEQA can cause fetal harm and loss of pregnancy. Advise males with female partners of reproductive potential to use effective contraception during treatment with NUBEQA and for 1 week after the last dose.
For more information, go to www.bayer.com.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211202006066/en/
Source: Bayer
Dec. 03, 2021 7:12 AM ET Bayer Aktiengesellschaft (BAYRY), BAYZF By: Dulan Lokuwithana, SA News Editor
12/03/2021CATEGORY:
Submission based on results from Phase 2 BEYOND study of Reblozyl plus best supportive care in adults with NTD beta thalassemia
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) today announced that the U.S. Food and Drug Administration (FDA) has accepted for priority review the supplemental Biologics License Application (sBLA) for Reblozyl® (luspatercept-aamt), a first-in-class erythroid maturation agent, for the treatment of anemia in adults with non-transfusion dependent (NTD) beta thalassemia. The FDA has set a Prescription Drug User Fee Act (PDUFA) goal date of March 27, 2022. In addition, the European Medicines Agency has validated the Type II variation for Reblozyl inNTD beta thalassemia. Reblozyl is being co-developed and co-commercialized with Merck & Co., Inc., known as MSD outside the United States and Canada, following Merck’s recent acquisition of Acceleron Pharma, Inc.
These applications were based on safety and efficacy results from the pivotal Phase 2 BEYOND study evaluating Reblozyl plus best supportive care in patients with NTD beta thalassemia.
“Patients with non-transfusion dependent beta thalassemia may not require lifelong blood transfusions for survival, but their need for effective treatment options is significant as they face a range of clinical complications due to chronic anemia and iron overload,” said Noah Berkowitz, M.D., Ph.D., senior vice president, Hematology Development, Bristol Myers Squibb. “Reblozyl is an important therapy approved for anemia associated with beta thalassemia and lower-risk myelodysplastic syndromes in multiple countries, including the United States and within the European Union. Along with our partners at Merck, we are committed to continuing to advance our clinical program for Reblozyl and look forward to working with the FDA during its review of our application for this underserved patient population.”
Updated analyses from the BEYOND study will be presented at the 63rd American Society of Hematology Annual Meeting and Exposition from December 11-14.
About BEYOND
BEYOND (NCT03342404) is a Phase 2, double-blind, randomized, placebo-controlled, multicenter study to determine the efficacy and safety of luspatercept-aamt (ACE-536) versus placebo in adults with non-transfusion dependent beta thalassemia. The study is divided into the Screening Period, Double-blind Treatment Period (DBTP) and Post-Treatment Follow-up Period (PTFP) and randomized 145 subjects at a 2:1 ratio of Reblozyl versus placebo. The primary endpoint of the study is the proportion of subjects who have an increase from baseline ≥1.0 g/dL in mean of hemoglobin values over a continuous 12-week interval from Week 13 to Week 24 of treatment in the absence of transfusions. Key secondary endpoints include mean change in non-transfusion dependent beta thalassemia-patient reported outcome (NTDT-PRO) Tiredness and Weakness (TW) domain score and baseline hemoglobin (Hb).
About Reblozyl®
Reblozyl, the first and only erythroid maturation agent, promotes late-stage red blood cell maturation in animal models.1 Bristol Myers Squibb and Merck & Co., Inc., through Merck’s acquisition of Acceleron, are jointly developing Reblozyl as part of a global collaboration. Reblozyl is currently approved in the U.S. for the treatment of:
Reblozyl is not indicated for use as a substitute for red blood cell transfusions in patients who require immediate correction of anemia.
Please see full Prescribing Information for REBLOZYL
For more information about Bristol Myers Squibb, visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211203005095/en/
Source: Bristol Myers Squibb
Dec. 03, 2021 7:12 AM ET
Bristol-Myers Squibb Company (BMY)
By: Mamta Mayani, SA News Editor
12/02/21 at 2:07 AM ESTPDF Version
– Preclinical data demonstrate sotrovimab, authorized in multiple countries around the world, retains activity against all tested variants of concern, including key mutations of Omicron –
– Data to be confirmed by further in vitro pseudo-virus testing –
LONDON and SAN FRANCISCO, Dec. 02, 2021 (GLOBE NEWSWIRE) -- GlaxoSmithKline plc (LSE/NYSE: GSK) and Vir Biotechnology, Inc. (Nasdaq: VIR) today announced an update to bioRxiv, a preprint server, with preclinical data demonstrating that sotrovimab, an investigational monoclonal antibody, retains activity against key mutations1 of the new Omicron SARS-CoV-2 variant (B.1.1.529), including those found in the binding site of sotrovimab. These data were generated through pseudo-virus testing of specific individual mutations found in Omicron. To date, sotrovimab has demonstrated ongoing activity against all tested variants of concern and interest defined by the World Health Organization (WHO). The companies are now completing in vitro pseudo-virus testing to confirm the neutralizing activity of sotrovimab against the combination of all the Omicron mutations with the intent to provide an update by the end of 2021.
George Scangos, Ph.D., Chief Executive Officer of Vir, said: “Sotrovimab was deliberately designed with a mutating virus in mind. By targeting a highly conserved region of the spike protein that is less likely to mutate, we hoped to address both the current SARS-CoV-2 virus and future variants that we expected would be inevitable. This hypothesis has borne out again and again – with its ongoing ability to maintain activity against all tested variants of concern and interest to date, including key mutations found in Omicron, as demonstrated by pre-clinical data. We have every expectation that this positive trend will continue and are working rapidly to confirm its activity against the full combination sequence of Omicron.”
Dr. Hal Barron, Chief Scientific Officer and President R&D, GSK, said: “Since the beginning of the pandemic, we have been working with Vir to combine our scientific expertise and technologies to deliver an enduring treatment option for patients with COVID-19. Though early, these pre-clinical data support our long-held view on the potential for sotrovimab to maintain its activity as the virus continues to mutate. We are pleased that this treatment option is available to patients in the US and many other countries, and are working to expand access worldwide.”
About Sotrovimab
Sotrovimab is an investigational SARS-CoV-2 neutralizing monoclonal antibody. The antibody binds to an epitope on SARS-CoV-2 shared with SARS-CoV-1 (the virus that causes SARS), indicating that the epitope is highly conserved, which may make it more difficult for resistance to develop. Sotrovimab, which incorporates Xencor’s Xtend™ technology, has also been designed to achieve high concentration in the lungs to ensure optimal penetration into airway tissues affected by SARS-CoV-2 and to have an extended half-life.
Updated in vitro data, published in bioRxiv, demonstrate that sotrovimab retains activity against all current tested variants of concern and interest of the SARS-CoV-2 virus as defined by WHO, plus others, including but not limited to Delta (B.1.617.2), Delta Plus (AY.1 or AY.2), Mu (B.1.621) and key mutations of Omicron (B.1.1.529).
About the sotrovimab clinical development program
Sotrovimab in the United States
The following is a summary of information for sotrovimab. Healthcare providers in the U.S. should review the Fact Sheets for information about the authorized use of sotrovimab and mandatory requirements of the EUA. Please see the Food and Drug Administration (FDA) Letter of Authorization, full Fact Sheet for Healthcare Providers and full Fact Sheet for Patients, Parents, and Caregivers.
Sotrovimab has been authorized by the US FDA for the emergency use described below. Sotrovimab is not FDA-approved for this use.
Sotrovimab is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of sotrovimab under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.
Authorized Use
The U.S. FDA has issued an Emergency Use Authorization (EUA) to permit the emergency use of the unapproved product sotrovimab for the treatment of mild-to-moderate coronavirus disease 2019 (COVID-19) in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death.
Limitations of Authorized Use
Sotrovimab is not authorized for use in patients:
Benefit of treatment with sotrovimab has not been observed in patients hospitalized due to COVID‑19. SARS-CoV-2 monoclonal antibodies may be associated with worse clinical outcomes when administered to hospitalized patients with COVID‑19 requiring high flow oxygen or mechanical ventilation.
About the Vir and GSK Collaboration
In April 2020, Vir and GSK entered into a collaboration to research and develop solutions for coronaviruses, including SARS-CoV-2, the virus that causes COVID-19. The collaboration uses Vir’s proprietary monoclonal antibody platform technology to accelerate existing and identify new anti-viral antibodies that could be used as therapeutic or preventive options to help address the current COVID-19 pandemic and future outbreaks. The companies will leverage GSK’s expertise in functional genomics and combine their capabilities in CRISPR screening and artificial intelligence to identify anti-coronavirus compounds that target cellular host genes. They will also apply their combined expertise to research SARS-CoV-2 and other coronavirus vaccines.
For further information please visit www.gsk.com/aboutus.
For more information, please visit www.vir.bio.
Dec. 02, 2021 3:07 AM ET
GlaxoSmithKline plc (GSK), VIR
By: Mamta Mayani, SA News Editor1 Comment
December 2, 2021Download PDF
SAN FRANCISCO, INDIANAPOLIS, and SUZHOU, China, Dec. 2, 2021 /PRNewswire/ -- Innovent Biologics, Inc. ("Innovent") (HKEX: 01801), a world-class biopharmaceutical company that develops, manufactures and commercializes high quality medicines for the treatment of oncology, metabolic, autoimmune and other major diseases, and Eli Lilly and Company ("Lilly") (NYSE: LLY), announce that the innovative PD-1 inhibitor sintilimab has been successfully included in the updated National Reimbursement Drug List ("NRDL") for all approved indications, according to the latest announcement from the China National Healthcare Security Administration ("NHSA"). The updated NRDL will officially take effect on January 1, 2022.
A total of four approved indications for sintilimab are now included in the updated NRDL:
Dr. Michael Yu, Founder, Chairman and CEO of Innovent, stated, "Two years ago, sintilimab was the first and only PD-1 inhibitor included in the NRDL. This year, three additional first-line indications for sintilimab have been successfully included in the NRDL, further enhancing the accessibility of this anti-cancer drug and alleviating financial burden for Chinese patients and their families. We have witnessed the profound reform and rapid development of pharmaceutical industry in China, driven by the government's commitment to continuously support innovation and emphasize a healthier and better life for the people of China. Innovent is honored to be a part of the Chinese government's initiative to improve health, and are devoted to the deepening of the national health care reform. With our company's mission 'to develop and commercialize high quality biopharmaceuticals that are affordable to ordinary people,' we hope to continue to work together with all relevant parties to improve drug affordability and accessibility, and contribute to the 'Healthy China 2030' initiative."
Julio Gay-Ger, President and General Manager, Lilly China, stated, "In recent years, China has continued to intensify medical insurance reform, giving strategic priority to safeguarding people's health. As a multinational pharmaceutical company tied with China for over 100 years, Lilly always adheres to the philosophy of 'In China, For China', and actively participates in China's health reform, especially in the drug supply system. The indication expansion of sintilimab in the National Reimbursement Drugs List (NRDL) can further reduce the burden of healthcare, enabling the patients to afford innovative drugs and have a higher quality of life through persistent treatment. Lilly will continue to keep a close eye on the major healthcare challenges in China in the future, and play an important role in the country's 'all-round and full-cycle health' ecosystem, to support the accelerated implementation of the 'Healthy China 2030' initiatives."
Mr. Min Liu, Chief Commercial Officer of Innovent, stated, "Sintilimab is the only PD-1 inhibitor in China with four major indications (1L nsq NSCLC, 1L sq NSCLC, 1L HCC and cHL) approved and included in China's NRDL. Particularly, lung cancer and liver cancer are two of the most prevalent tumor types in China, accounting for the first and third largest numbers of new cases each year – representing a large unmet medical need. We will proactively support the work of the government departments at all levels, cooperate with the implementation of medical insurance policies in all regions, and help relieve patients' economic burden to a further extent, to allow this high-quality immunotherapy product to benefit more lives of Chinese patients and their families."
About Sintilimab
Sintilimab, marketed as TYVYT® (sintilimab injection) in China, is an innovative PD-1 inhibitor with global quality standards jointly developed by Innovent and Eli Lilly and Company. Sintilimab is an immunoglobulin G4 monoclonal antibody, which binds to PD-1 molecules on the surface of T-cells, blocks the PD-1 / PD-Ligand 1 (PD-L1) pathway, and reactivates T-cells to kill cancer cells. Innovent is currently conducting more than 20 clinical studies of sintilimab worldwide, to evaluate its safety and efficacy in a wide variety of cancer indications, including more than 10 registrational or pivotal clinical trials.
In China, sintilimab has been approved and included in the National Reimbursement Drug List (NRDL) for four indications, including:
Additionally, Innovent currently has two regulatory submissions under review in China for sintilimab, for the first-line treatment of esophageal squamous cell carcinoma, and the first-line treatment of unresectable, locally advanced, recurrent or metastatic gastric or gastroesophageal junction adenocarcinoma.
Additionally, three clinical studies of sintilimab have met their primary endpoints:
In May 2021, the U.S. FDA accepted for review the Biologics License Application (BLA) for sintilimab in combination with pemetrexed and platinum chemotherapy for the first-line treatment of nonsquamous non-small cell lung cancer.
For more information, please visit: www.innoventbio.com. and www.linkedin.com/company/innovent-biologics/.
To learn more about Lilly, please visit lilly.com and lilly.com/newsroom.
About Innovent Biologics' strategic collaboration with Eli Lilly and Company
Innovent entered into a strategic collaboration with Lilly focused on biological medicine in March 2015 – a groundbreaking partnership between a Chinese pharmaceutical company and a multinational pharmaceutical company. Under the agreement, Innovent and Lilly are co-developing and commercializing oncology medicines, including sintilimab, in China. In October 2015, the two companies announced the extension of their existing collaboration to include co-development of three additional antibodies targeting oncology indications. In August 2019, Innovent further entered into a licensing agreement with Lilly to develop and commercialize a potentially global best-in-class diabetes medicine in China. The collaboration with Lilly is an example of how Innovent has established a comprehensive level of cooperation between China's innovative pharmaceuticals sector and the international pharmaceuticals sector in fields such as R&D, CMC, clinical development and commercialization. In August 2020,Lilly and Innovent announced a global expansion of their strategic alliance for sintilimab, whereby Lilly obtained an exclusive license for sintilimab for geographies outside of China and plans to pursue registration of sintilimab in the U.S. and other geographies outside of China.
Note:
TYVYT® (sintilimab injection) is not an approved product in the United States.
BYVASDA® (bevacizumab biosimilar injection), SULINNO®, and HALPRYZA® (rituximab biosimilar injection) are not approved products in the United States.
TYVYT® (sintilimab injection, Innovent)
BYVASDA® (bevacizumab biosimilar injection, Innovent)
HALPRYZA® (rituximab biosimilar injection, Innovent)
SULINNO® (adalimumab biosimilar injection, Innovent)
Pemazyre® (pemigatinib oral inhibitor, Incyte Corporation). Pemazyre® was discovered by Incyte Corporation and licensed to Innovent for development and commercialization in Mainland China, Hong Kong, Macau and Taiwan.
Dec. 03, 2021 8:21 AM ET
Innovent Biologics, Inc. (IVBIY), LLY
By: Dulan Lokuwithana, SA News Editor
Novavax Statement on Omicron Variant ResponseDec 2, 2021
Novavax is rapidly responding to the emergence of the latest potential threat of the SARS-CoV-2 Omicron (B.1.1.529) variant of concern (VoC). The company is executing a two-pronged variant strategy.
First, Novavax is evaluating its vaccine against the Omicron variant, as the company has done for previous variants including Alpha, Beta and Delta. Second, Novavax has initiated development of an Omicron-specific vaccine construct.
Novavax is conducting ongoing studies to evaluate multiple variants, and we are encouraged by our current and ongoing data, which shows efficacy and safety against variants of interest (VoIs) and VoCs (see clinical data below). Based on this, we will evaluate whether immune responses induced by NVX-CoV2373, the company’s recombinant nanoparticle protein-based COVID-19 vaccine, will offer similar cross-protection against Omicron as seen with other variants.
Novavax will begin testing whether antibodies from previously vaccinated individuals can neutralize the Omicron variant, with lab-based data expected in the coming weeks. Clinical samples from participants in Novavax’ clinical trials will be evaluated as samples and viral reagents become available, and we will work diligently to expedite this analysis. Additionally, we plan on testing whether vaccinees’ antibodies can block Omicron-hACE2 receptor binding (a step that is required in the viral invasion process).
In parallel, Novavax has initiated development of an Omicron-specific construct of its SARS-CoV-2 Spike protein (rS) antigen, currently in use in NVX-CoV2373. The initial steps required to manufacture an Omicron-specific spike are underway and GMP manufacturing in a commercial facility is anticipated in January 2022. Lab-based assessment of a new strain-matched nanoparticle vaccine will begin within a few weeks.
Clinical Trial Data Supporting Novavax’ Nanoparticle Vaccine Technology with Matrix-M™ Adjuvant:
Following the emergence of other VoCs (Alpha, Beta, Delta), antibody responses were analyzed in Novavax’ ongoing Phase 1/2 study in the US and Australia. Patients who received a third (6-month booster) dose of NVX-CoV2373 were evaluated for the production of antibodies against known VoCs in August 2021. The responses were encouraging for the protection against new variants:
In PREVENT-19, Novavax’ Phase 3 trial of NVX-CoV2373 in the U.S. and Mexico, we observed 93% efficacy against predominantly circulating VoI/VoCs, which represents 79% of cases for which sequence is available. Further, a post-hoc analysis showed 94% efficacy against the Alpha variant. We also observed 100% efficacy against variants not considered VoI/VoC.
Novavax’ Phase 3 trial of NanoFlu™, our quadrivalent nanoparticle vaccine candidate against seasonal influenza, demonstrated the strong role our proprietary Matrix-M™ adjuvant can play in addressing rapid viral genetic drift. Matrix-M adjuvant was shown to increase and broaden immune response, with a demonstrated ability to stimulate high levels of neutralizing antibodies and T-cell responses.
Cumulatively, these clinical experiences are encouraging for insights into the performance of and our confidence in NVX-CoV2373 during an evolving COVID-19 pandemic.
Dec. 02, 2021 10:08 AM ETNovavax, Inc. (NVAX)By: Dulan Lokuwithana, SA News Editor27 Comments

December 2, 2021
CAMBRIDGE, Massachusetts, December 2, 2021– Takeda Pharmaceutical Company Limited (TSE:4502/NYSE:TAK) (“Takeda”) today announced the United States commercial availability of LIVTENCITY™ (maribavir), the first and only treatment for adults and pediatric patients (12 years of age and older and weighing at least 35 kg) with post-transplant cytomegalovirus (CMV) infection/disease that is refractory to treatment (with or without genotypic resistance) with ganciclovir, valganciclovir, cidofovir or foscarnet.1 LIVTENCITY, an orally bioavailable anti-CMV compound, became available for prescription on December 2, 2021, just over a week after the U.S. Food and Drug Administration (FDA) approval which took place on November 23, 2021.
“Those undergoing a life-saving transplant often have an incredibly complex medical journey to overcome, so when faced with the subsequent impact of a difficult-to-manage infection/disease such as CMV, it was a priority for our teams to expedite access to this additional treatment option,” said Cheryl Schwartz, Senior Vice President and U.S. Business Unit Lead, Rare Diseases, Takeda.
LIVTENCITY is now available to healthcare providers through a network of specialty pharmacies and distributors. For appropriate patients, physicians can submit a prescription and initiate access to treatment through a specialty pharmacy by visiting www.takedapatientsupport.com or by calling Takeda Patient Support at 1-855-268-1825. Requests for acquisition through a distributor for inpatient administration can be fulfilled by emailing customerservice@takeda.com.
“Treatment of post-transplant CMV has historically been a challenge for clinicians, given that conventional antivirals have been the only treatment option,” said Emily Blumberg Director, Transplant Infectious Diseases, Penn Medicine. “The availability of LIVTENCITY, the first and only FDA-approved oral treatment for post-transplant resistant/refractory CMV, represents a significant step forward for the transplant community in addressing this difficult to treat infection/disease.”
LIVTENCITY is a new molecular entity which targets CMV at UL97, resulting in inhibition of viral DNA replication, encapsidation, and nuclear egress.1,2,3,4,5,6 Though a rare disease overall, CMV is one of the most common infections experienced by transplant recipients, with an estimated incidence rate of around 16%–56% in solid organ transplant (SOT) recipients7 and 30%–70% in hematopoietic stem cell (HSCT) transplant recipients.8 CMV can be acquired or reactivated following transplant leading to serious consequences—including loss of the transplanted organ and failure of the graft—or loss of life. In patients with compromised immunity, CMV causes clinically challenging complications that can be fatal.9,10,11
Takeda Patient Support is available to help patients prescribed LIVTENCITY gain access to their medication, find educational resources, and understand financial assistance options. A team of experts is available Monday through Friday, 8:00am to 8:00pm ET. For additional information visit www.takedapatientsupport.com or call 1-855-268-1825.
LIVTENCITY (maribavir), an orally bioavailable anti-CMV compound, is the first and only antiviral agent that targets and inhibits the pUL97 protein kinase and its natural substrates.1 It is approved in the U.S. for the treatment of adults and pediatric patients (12 years of age or older and weighing at least 35 kg) with post-transplant cytomegalovirus (CMV) infection/disease that is refractory to treatment (with or without genotypic resistance) with ganciclovir, valganciclovir, cidofovir or foscarnet. For more information on LIVTENCITY, visit LIVTENCITY.com.1
LIVTENCITY is indicated for the treatment of adults and pediatric patients (12 years of age and older and weighing at least 35 kg) with post-transplant cytomegalovirus (CMV) infection/disease that is refractory to treatment (with or without genotypic resistance) with ganciclovir, valganciclovir, cidofovir or foscarnet.1
For more information, visit https://www.takeda.com.
Takeda Pharmaceutical Company Limited (TSE: 4502/NYSE: TAK)
Dec. 02, 2021 11:48 AM ET
Takeda Pharmaceutical Company Limited (TAK)
By: Jonathan M Block, SA News Editor
Soligenix Announces Successful Protection using a Bivalent Thermostabilized Filovirus VaccineThermostabilized single vial vaccine targeting Sudan and Marburg filoviruses protects 100% of non-human primates against lethal Sudan ebolavirus challenge
PRINCETON, N.J., Dec. 2, 2021 /PRNewswire/ -- Soligenix, Inc. (Nasdaq: SNGX) (Soligenix or the Company), a late-stage biopharmaceutical company focused on developing and commercializing products to treat rare diseases where there is an unmet medical need, announced today 100% protection of non-human primates (NHPs) against lethal Sudan ebolavirus (SUDV) challenge using a bivalent, thermostabilized vaccine formulated in a single vial, reconstituted only with water immediately prior to use. This milestone is part of an ongoing collaboration with the University of Hawaiʻi at Mānoa (UHM), demonstrating the successful presentation of one or more antigen(s) within the same formulation while maintaining full potency and thermostability. It further demonstrates the broad applicability of the vaccine platform, and its potential role in the US government's initiative for pandemic preparedness. This same heat stable vaccine platform has also been applied to the development of a COVID-19 vaccine, CiVax™, which the Company anticipates testing against the new omicron variant, having shown immunogenicity against previous variants including delta.
The antigens and adjuvants used in this study have been previously shown to protect NHPs from subsequent infection, and represent the only recombinant subunit vaccines that have demonstrated efficacy against Zaire ebolavirus and other filoviruses in NHPs. Lyophilization of the antigens within monovalent vaccine formulations has also been demonstrated to thermostabilize the antigens at temperatures as high as 40 degrees Celsius (104 degrees Fahrenheit) for up to 12 weeks, enabling storage at ambient temperature. No currently approved lyophilized vaccine that contains adjuvant is presented in a single vial format and there are few reports of successfully using nano-emulsions in lyophilized formulations. Previous work has demonstrated the use of a single vial platform to co-lyophilize antigen(s) and a nano-emulsion adjuvant, CoVaccine HT™, maintaining key adjuvant stability characteristics including particle size and colloidal stability, as well as maintaining immunogenicity. This most recent milestone confirms that in the context of lethal challenge with Sudan ebolavirus, complete protection is maintained with the thermostabilized formulation.
"Filoviruses such as Zaire ebolavirus, Sudan ebolavirus and Marburg marburgvirus are some of the most lethal viruses known, and they are endemic in areas of the world where the power supply and distribution network can be uncertain. There are no vaccines for either Sudan ebolavirus or Marburg marburgvirus, while vaccines for Zaire ebolavirus are constrained by cold chain logistics. A heat stable vaccine in a single vial format would significantly enhance any public health preparedness or response to a new outbreak, at its source," stated Axel Lehrer, PhD, Associate Professor, Department of Tropical Medicine, Medical Microbiology and Pharmacology, John A. Burns School of Medicine (JABSOM), UHM. "Our work to date has demonstrated the feasibility of rapid and efficient manufacturing, as well as the ability to thermostabilize multiple antigens that can then be stored at temperatures exceeding 100 degrees Fahrenheit. Having a vaccine platform available such as this has the potential to accelerate worldwide vaccination campaigns addressing future health emergencies, including another global pandemic like the one we are unfortunately experiencing with COVID-19."
"Our next generation combined vaccine platform includes three major components: a robust protein manufacturing process that has been demonstrated on multiple protein antigens, a novel nano-emulsion adjuvant which induces broad immunity and a formulation procedure which enables thermostabilization of the combination of adjuvant and antigen in a single vial," stated Oreola Donini, PhD, Senior Vice President and Chief Scientific Officer of Soligenix. "Elements of this vaccine platform have been utilized in our ricin toxin, filovirus and COVID-19 vaccine candidates, indicating its broad applicability. The ability to package more than one vaccine candidate in a single vial platform further adds to their developability, whether as a multivalent or individual monovalent vaccine, particularly against Marburg marburgvirus and Sudan ebolavirus where there are currently no available vaccines."
Under the Company's Public Health Solutions business segment, ongoing collaborations with Dr. Lehrer, as well as work conducted by Theodore Randolph, PhD, Professor, Center for Pharmaceutical Biotechnology, Department of Chemical and Biological Engineering at the University of Colorado, Boulder have demonstrated the feasibility of developing thermally-stable subunit protein vaccine formulations for filoviruses. The thermostabilized filovirus vaccine program is continuing to advance with the support of a National Institute of Health (NIH) grant (R01-AI132323; awarded to the University of Hawaii) and a Small Business Innovation Research grant (#1R44AI157593-01; awarded to Soligenix). Work to date has demonstrated the compatibility of lyophilizing both antigen and adjuvant in the same vial, with reconstitution with sterile water for injection immediately prior to use. This simple delivery format, as well as the compatibility with ambient storage, enables vaccines that significantly reduce the logistical hurdles that have been required for addressing the current pandemic or deployment of other Ebola virus vaccines in recent outbreaks in Central and West Africa.
For further information regarding Soligenix, Inc., please visit the Company's website at https://www.soligenix.com and follow us on LinkedIn and Twitter at @Soligenix_Inc.
Dec. 02, 2021 8:01 AM ET
By: Mamta Mayani, SA News Editor
November 30, 2021 6:45 am ET
First Anti-PD-1/L1-Based Regimen Approved in Japan for First-Line Treatment of Advanced Esophageal Cancer
KEYTRUDA Is Now Approved for 16 Indications in Japan
KENILWORTH, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, today announced that KEYTRUDA, Merck’s anti-PD-1 therapy, has been approved by the Japan Pharmaceuticals and Medical Devices Agency (PMDA) for the first-line treatment of patients with radically unresectable, advanced or recurrent esophageal carcinoma in combination with chemotherapy (5-fluorouracil [5-FU] plus cisplatin) based on data from the Phase 3 KEYNOTE-590 trial.
“In Japan, patients living with advanced esophageal cancer face a poor prognosis with current chemotherapy regimens,” said Dr. Scot Ebbinghaus, vice president, clinical research, Merck Research Laboratories. “This approval offers a new treatment option with KEYTRUDA, which has been shown to improve overall survival and progression-free survival when combined with chemotherapy compared to standard of care chemotherapy as a first-line treatment for patients with radically unresectable, advanced or recurrent esophageal carcinoma.”
“The burden of esophageal cancer is high in Japan, where cases are increasing,” said Kyle Tattle, president, MSD Japan. “It is encouraging that now appropriate patients with esophageal cancer have an immunotherapy regimen option with KEYTRUDA earlier in the treatment course that can potentially extend their lives. We remain committed to addressing the most challenging cancers affecting Japanese patients and working with the government to provide access to patients in Japan.”
In the KEYNOTE-590 trial, KEYTRUDA in combination with chemotherapy (5-FU plus cisplatin) demonstrated statistically significant improvements in overall survival and progression-free survival versus chemotherapy alone (5-FU plus cisplatin) in chemotherapy-naïve patients with radically unresectable, advanced or recurrent esophageal squamous cell carcinoma or esophageal adenocarcinoma or adenocarcinoma of the esophagogastric junction (Siewert type 1), regardless of histology or PD-L1 expression status. KEYTRUDA plus chemotherapy reduced the risk of death by 27% (HR=0.73 [95% CI, 0.62-0.86]; p<0.0001) and reduced the risk of disease progression or death by 35% (HR=0.65 [95% CI, 0.55-0.76]; p<0.0001) versus chemotherapy alone.
The Japanese package insert notes that in KEYNOTE-590, adverse reactions were observed in 364 patients (98.4%) out of the safety analysis set of 370 patients (including 73 out of 74 Japanese patients) receiving KEYTRUDA at a dose of 200 mg every three weeks in combination with 5-FU and cisplatin. The most commonly observed adverse reactions (≥20%) were nausea in 233 patients (63.0%), decreased appetite in 145 patients (39.2%), anemia in 143 patients (38.6%), fatigue in 135 patients (36.5%), neutrophil count decreased in 135 patients (36.5%), vomiting in 110 patients (29.7%), diarrhea in 97 patients (26.2%), neutropenia and stomatitis in 96 patients (25.9% each) and white blood cell count decreased in 89 patients (24.1%).
This is the second indication for KEYTRUDA in esophageal cancer in Japan and is the 16th indication overall. In 2020, KEYTRUDA was approved in Japan for the second-line treatment of patients with radically unresectable, advanced or recurrent esophageal squamous cell carcinoma whose tumors express PD-L1.
Esophageal cancer begins in the inner layer (mucosa) of the esophagus and grows outward. Esophageal cancer is the eighth most commonly diagnosed cancer and the sixth leading cause of cancer death worldwide. In Japan, esophageal cancer is the tenth most commonly diagnosed cancer and the ninth leading cause of cancer death. It is estimated there were more than 26,000 new cases of esophageal cancer diagnosed and more than 12,000 deaths from the disease in 2020. Merck is studying KEYTRUDA across multiple settings and stages of gastrointestinal cancer through its broad clinical program, including studies in esophageal, gastric, hepatobiliary, pancreatic, colorectal and anal cancers.
About KEYTRUDA® (pembrolizumab) Injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of patients with melanoma with involvement of lymph node(s) following complete resection.
Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [tumor proportion score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [combined positive score (CPS) ≥1] as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy. KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Carcinoma
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
Non-muscle Invasive Bladder Cancer
KEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with MSI-H central nervous system cancers have not been established.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC).
Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or GEJ (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
KEYTRUDA, in combination with chemotherapy, with or without bevacizumab, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
KEYTRUDA is indicated for the adjuvant treatment of patients with RCC at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions.
Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
Selected Important Safety Information for KEYTRUDA
For more information about our oncology clinical trials, visit www.merck.com/clinicaltrials.
For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf.
Nov. 30, 2021 7:12 AM ET Merck & Co., Inc. (MRK) By: Mamta Mayani, SA News Editor
December 1, 2021 6:45 am ET
KENILWORTH, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, today announced the U.S. Food and Drug Administration (FDA) has accepted for Priority Review a supplemental Biologics License Application (sBLA) for VAXNEUVANCE (Pneumococcal 15-valent Conjugate Vaccine) for the prevention of invasive pneumococcal disease in children 6 weeks through 17 years of age. The FDA grants priority review to medicines and vaccines that, if approved, would provide a significant improvement in the safety or effectiveness of the treatment or prevention of a serious condition. The FDA set a Prescription Drug User Fee Act (PDUFA), or target action date, of April 1, 2022.
“VAXNEUVANCE has the potential to provide meaningful protection against invasive pneumococcal disease for children and infants by targeting pneumococcal strains, or serotypes, that contribute to substantial disease burden, including serotype 3, and broadening coverage to additional disease-causing serotypes, 22F and 33F, which are not included in the pneumococcal conjugate vaccine (PCV) currently available for this population,” said Dr. Roy Baynes, senior vice president and head of global clinical development, chief medical officer, Merck Research Laboratories. “We look forward to working with the U.S. FDA as it reviews what would be the first new option in pediatric pneumococcal vaccination in over a decade.”
The sBLA is supported by results from Phase 2 and Phase 3 clinical studies in pediatric populations including infants, children, and adolescents. These studies support the potential use of VAXNEUVANCE in a variety of clinical settings, including immunization of infants and toddlers as well as of special populations at increased risk for pneumococcal disease, such as children with HIV infection or sickle cell disease. The submission also includes data supporting the potential use of VAXNEUVANCE as part of a mixed dosing regimen following initiation of an infant vaccination schedule with PCV13 as well as in a catch-up setting for older children who are either pneumococcal vaccine-naïve or who previously received a partial or full regimen of a lower-valency pediatric PCV.
Invasive pneumococcal disease can cause serious and potentially life-threatening infections in infants and children. Children under the age of 2 are particularly vulnerable to pneumococcal infection, and incidence of invasive pneumococcal disease remains highest in the first year of life. There are 100 different types of pneumococcal bacteria, of which some continue to put children at risk, including serotypes 22F, 33F and 3, which represent more than a quarter of invasive pneumococcal disease in children under the age of 5.
About VAXNEUVANCE (Pneumococcal 15-valent Conjugate Vaccine)
VAXNEUVANCE, Merck’s 15-valent pneumococcal conjugate vaccine, consists of purified capsular polysaccharides from S. pneumoniae serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, 22F, 23F and 33F individually conjugated to CRM197 carrier protein. VAXNEUVANCE is indicated for active immunization of adults 18 years of age and older for the prevention of invasive disease caused by the S. pneumoniae serotypes contained in the vaccine. It is currently under investigation in the pediatric population. VAXNEUVANCE previously received Breakthrough Therapy designation from the FDA for the prevention of invasive pneumococcal disease in pediatric patients 6 weeks through 17 years of age.
For more information, visit www.merck.com and connect with us on Twitter, Facebook, Instagram, YouTube and LinkedIn.
Please see Prescribing Information for VAXNEUVANCE (Pneumococcal 15-valent Conjugate Vaccine) at https://www.merck.com/product/usa/pi_circulars/v/vaxneuvance/vaxneuvance_pi.pdf.
and Patient Information at https://www.merck.com/product/usa/pi_circulars/v/vaxneuvance/vaxneuvance_ppi.pdf.
Dec. 01, 2021 7:08 AM ET
By: Mamta Mayani, SA News Editor
12.01.21PDF VersionPrescription Drug User Fee Act (PDUFA) Target Action Date of March 25, 2022
EMERYVILLE, Calif., Dec. 01, 2021 (GLOBE NEWSWIRE) -- Zogenix (NASDAQ: ZGNX), a global biopharmaceutical company developing rare disease therapies today announced the U.S. Food and Drug Administration (FDA) has accepted for filing and granted Priority Review to the company’s supplemental New Drug Application (sNDA) for the use of FINTEPLA® for the treatment of seizures associated with Lennox-Gastaut syndrome (LGS). The FDA granted Priority Review, which means that the FDA has a goal to complete its review within six months from the date of receipt, with a Prescription Drug User Fee Act (PDUFA) target action date of March 25, 2022. FDA Priority Review is granted for investigational therapies that, if approved, may offer significant improvements in the treatment, prevention or diagnosis of a serious condition.
“This is a critically important milestone for our FINTEPLA development program in LGS and brings us one step closer to a potential new treatment option for this rare and difficult to treat childhood developmental and epileptic encephalopathy,” said Gail Farfel, Ph.D., Executive Vice President and Chief Development Officer of Zogenix. “We look forward to working closely with the FDA to potentially bring FINTEPLA for the treatment of seizures associated with LGS to market as quickly as possible.”
The sNDA submission is based on a positive, global, randomized, placebo-controlled Phase 3 clinical trial, Study 1601, in 263 patients (age 2-35 years) that demonstrated FINTEPLA at a dose of 0.7/mg/kg/day was superior to placebo in reducing the frequency of drop seizures (p=0.0012), as well as long-term safety and effectiveness data from Zogenix’s ongoing open-label extension trials.
In June 2020, FINTEPLA was approved by the FDA for the treatment of seizures associated with Dravet syndrome in patients two years of age and older.
About Lennox-Gastaut Syndrome
Lennox-Gastaut Syndrome (LGS) is a rare and devastating lifelong childhood-onset epilepsy that can arise from multiple different causes. LGS is characterized by many different seizure types, including many that result in frequent falls and injuries and that often don't respond to currently available seizure medications. The intellectual and behavioral problems associated with LGS, as well as around-the-clock care requirements, add to the complexity of life with this disease. There are an estimated 30,000 to 50,000 people who have LGS in the United States.1
In the U.S. and Europe, treatment with FINTEPLA is initiated and supervised by physicians with experience in the treatment of Dravet syndrome under a Risk Evaluation Mitigation Strategy (REMS) program (U.S.) or Controlled Access Program (EU). In the United States, please see important prescribing and safety information at www.Fintepla.com. In Europe, please see important prescribing and safety information at www.Fintepla.eu.
Dec. 01, 2021 8:21 AM ET Zogenix, Inc. (ZGNX)
By: Mamta Mayani, SA News Editor
PUBLISHED30 November 2021
AstraZeneca’s supplemental New Drug Application (sNDA) for Lynparza (olaparib) has been accepted and granted Priority Review in the US for the adjuvant treatment of patients with BRCA-mutated (BRCAm) HER2-negative high-risk early breast cancer who have already been treated with chemotherapy either before or after surgery.Lynparza is being jointly developed and commercialised by AstraZeneca and MSD.The Food and Drug Administration (FDA) grants Priority Review to applications for medicines that offer significant advantages over available options by demonstrating safety or efficacy improvements, preventing serious conditions, or enhancing patient compliance.1 The Prescription Drug User Fee Act date, the FDA action date for their regulatory decision, is during the first quarter of 2022.Breast cancer is now the most diagnosed cancer worldwide with an estimated 2.3 million patients diagnosed in 2020.2 Nearly 91% of all breast cancer patients are diagnosed at an early stage of disease and BRCA mutations are found in approximately 5% of patients.3,4,5The sNDA was based on results from the OlympiA Phase III trial presented during the 2021 American Society of Clinical Oncology Annual Meeting and simultaneously published in The New England Journal of Medicine.These results showed Lynparza demonstrated a statistically significant and clinically meaningful improvement in invasive disease-free survival (iDFS), reducing the risk of invasive breast cancer recurrence, second cancers or death by 42% versus placebo (based on a hazard ratio of 0.58; 99.5% confidence interval 0.41-0.82; p<0.0001). The safety and tolerability profile of Lynparza in this trial was in line with that observed in prior clinical trials.Lynparza is approved in the US, EU, Japan and several other countries for the treatment of patients with germline BRCAm, HER2-negative, metastatic breast cancer previously treated with chemotherapy based on results from the OlympiAD Phase III trial. In the EU, this indication also includes patients with locally advanced breast cancer.
OlympiAOlympiA is a Phase III, double-blind, parallel group, placebo-controlled, multicentre trial testing the efficacy and safety of Lynparza tablets versus placebo as adjuvant treatment in patients with germline BRCAm high-risk HER2-negative early breast cancer, who have completed definitive local treatment and neoadjuvant or adjuvant chemotherapy.11The primary endpoint of the trial is iDFS defined as time from randomisation to date of first loco-regional or distant recurrence or new cancer or death from any cause.11The OlympiA Phase III trial is led by the Breast International Group (BIG) in partnership with the Frontier Science & Technology Research Foundation (FSTRF), NRG Oncology, AstraZeneca and MSD.11
Lynparza Lynparza (olaparib) is a first-in-class PARP inhibitor and the first targeted treatment to block DNA damage response (DDR) in cells/tumours harbouring a deficiency in homologous recombination repair (HRR), such as those with mutations in BRCA1 and/or BRCA2, or those where deficiency is induced by other agents (such as new hormonal agents).Inhibition of PARP with Lynparza leads to the trapping of PARP bound to DNA single-strand breaks, stalling of replication forks, their collapse and the generation of DNA double-strand breaks and cancer cell death. Lynparza is being tested in a range of PARP-dependent tumour types with defects and dependencies in the DDR pathway.Lynparza is currently approved in a number of countries, including those in the EU, for the maintenance treatment of platinum-sensitive relapsed ovarian cancer. It is approved in the US, the EU, Japan, China, and several other countries as 1st-line maintenance treatment of BRCA-mutated advanced ovarian cancer following response to platinum-based chemotherapy.It is also approved in the US, EU and Japan as a 1st-line maintenance treatment with bevacizumab for patients with HRD-positive advanced ovarian cancer (BRCAm and/or genomic instability).Lynparza is approved in the US, Japan, and a number of other countries for germline BRCA-mutated, HER2-negative, metastatic breast cancer, previously treated with chemotherapy; in the EU, this includes locally advanced breast cancer.It is also approved in the US, the EU, Japan and several other countries for the treatment of germline BRCAm metastatic pancreatic cancer.Lynparza is approved in the US for HRR gene-mutated metastatic castration-resistant prostate cancer (BRCAm and other HRR gene mutations) and in the EU and Japan for BRCAm metastatic castration-resistant prostate cancer.Regulatory reviews are underway in several countries for ovarian, breast, pancreatic and prostate cancers. Lynparza, which is being jointly developed and commercialised by AstraZeneca and MSD, has been used to treat over 40,000 patients worldwide.Lynparza has the broadest and most advanced clinical trial development programme of any PARP inhibitor, and AstraZeneca and MSD are working together to understand how it may affect multiple PARP-dependent tumours as a monotherapy and in combination across multiple cancer types.Lynparza is the foundation of AstraZeneca's industry-leading portfolio of potential new medicines targeting DDR mechanisms in cancer cells.
The AstraZeneca and MSD strategic oncology collaborationIn July 2017, AstraZeneca and Merck & Co., Inc., Kenilworth, NJ, US (known as MSD outside the US and Canada) announced a global strategic oncology collaboration to co-develop and co-commercialise Lynparza, the world’s first PARP inhibitor, and Koselugo (selumetinib), a mitogen-activated protein kinase (MEK) inhibitor, for multiple cancer types.Working together, the companies will develop Lynparza and Koselugo in combination with other potential new medicines and as monotherapies. Independently, the companies will develop Lynparza and Koselugo in combination with their respective PD-L1 and PD-1 medicines.
Please visit astrazeneca.com and follow the Company on Twitter @AstraZeneca.
Nov. 30, 2021 6:39 AM ET AstraZeneca PLC (AZN), MRK By: Mamta Mayani, SA News Editor
November 30, 2021
- Submission supported by three pivotal Phase 3 studies in which risankizumab demonstrated significant improvements in clinical remission and endoscopic response as both induction and maintenance therapy[1,2]
- No new safety risks were observed compared to the known safety profile of risankizumab[1-6]
- If approved, Crohn's disease will mark the third indication for risankizumab in the European Union, following plaque psoriasis and psoriatic arthritis[7]
- Crohn's disease is a chronic, systemic disease that manifests as inflammation within the gastrointestinal (or digestive) tract, causing persistent diarrhea and abdominal pain that affects over 2 million people worldwide[8-11]
NORTH CHICAGO, Ill., Nov. 30, 2021 /PRNewswire/ -- AbbVie (NYSE:ABBV) today announced it has submitted an application to the European Medicines Agency (EMA) seeking approval for risankizumab (SKYRIZI®, 600 mg intravenous (IV) induction and 360 mg subcutaneous (SC) maintenance therapy), an interleukin-23 (IL-23) inhibitor, for the treatment of patients 16 years and older with moderate to severe active Crohn's disease who have had inadequate response, lost response or were intolerant to conventional or biologic therapy. The submission is supported by three pivotal Phase 3 studies, ADVANCE, MOTIVATE and FORTIFY.1,2
"Patients with moderate to severe Crohn's disease live with challenging symptoms, such as persistent diarrhea and abdominal pain, impacting their quality of life," said Tom Hudson, senior vice president of research and development, chief scientific officer, AbbVie. "We look forward to working with the regulatory authorities and hope to offer risankizumab as a potential first-in-class treatment option for patients living with this disease."
In the Phase 3 ADVANCE and MOTIVATE induction studies, a significantly greater proportion of patients with Crohn's disease treated with risankizumab IV induction therapy 600 mg achieved both primary endpoints demonstrating statistically significant results for clinical remission and endoscopic response at week 12 compared to placebo.1
In the Phase 3 FORTIFY maintenance study evaluating Crohn's disease patients with clinical response to risankizumab IV induction treatment, a significantly greater proportion of patients treated with risankizumab 360 mg SC achieved endoscopic response and clinical remission at one year (52 weeks) versus those who were withdrawn from risankizumab (control group).2
No new safety risks were observed in moderate to severe Crohn's disease in the ADVANCE, MOTIVATE and FORTIFY studies compared to the known safety profile of risankizumab.1-6
The use of risankizumab for Crohn's disease is not approved and its safety and efficacy have not been established by regulatory authorities.
Risankizumab (SKYRIZI) is part of a collaboration between Boehringer Ingelheim and AbbVie, with AbbVie leading development and commercialization globally.
About the ADVANCE and MOTIVATE Studies1,13,14
The ADVANCE and MOTIVATE studies are Phase 3, multicenter, randomized, double-blind, placebo-controlled induction studies designed to evaluate the efficacy and safety of two doses of risankizumab, 600 mg and 1200 mg, in adults with moderate to severe Crohn's disease, compared to placebo. Both studies included different sets of primary and secondary endpoints for outside U.S. (OUS) protocol and U.S. protocol. The primary endpoints were achievement of clinical remission (per PRO-2 for the OUS protocol, which was measured by daily stool frequency and abdominal pain score, and per CDAI for the U.S. protocol, which was measured by a CDAI score less than 150) and endoscopic response (for both protocols) at week 12. Endoscopic response is defined as a decrease in SES-CD of greater than 50 percent from baseline (or at least a greater than or equal to 50 percent decrease from baseline in patients with isolated ileal disease and a baseline SES-CD of 4), as scored by a central reviewer.
The ADVANCE study included a mixed population of patients who had responded inadequately or are intolerant to conventional and/or biologic therapy. The MOTIVATE study evaluated patients who had responded inadequately or were intolerant to biologic therapy. Topline results of the studies were shared in January 2021. More information can be found on www.clinicaltrials.gov (ADVANCE: NCT03105128; MOTIVATE: NCT03104413).
About the FORTIFY Study2,15
The FORTIFY study is a Phase 3, multicenter, randomized, double-blind, control group, 52-week maintenance study designed to evaluate the efficacy and safety of risankizumab 180 mg and 360 mg as maintenance therapy versus withdrawal in patients who responded to risankizumab induction treatment in the ADVANCE and MOTIVATE studies. This study included different sets of primary and secondary endpoints for the OUS analysis plan and U.S. analysis plan due to regulatory requirements in the different regions. The co-primary endpoints were achievement of endoscopic response and clinical remission at week 52. Endoscopic response is defined as a decrease in SES-CD of greater than 50 percent from baseline (or at least a greater than or equal to 50 percent decrease from baseline in patients with isolated ileal disease and a baseline SES-CD of 4), as scored by a central reviewer. Clinical remission is defined by SF/AP, which was measured by daily stool frequency and abdominal pain score, in the OUS analysis plan and defined by CDAI, which was measured by a CDAI score less than 150, in the U.S. analysis plan.
Topline results were announced in June 2021. An open label extension of FORTIFY will continue to assess the long-term safety of risankizumab in subjects who completed participation in FORTIFY. More information can be found on www.clinicaltrials.gov (NCT03105102).
About SKYRIZI® (risankizumab)
SKYRIZI is an interleukin-23 (IL-23) inhibitor that selectively blocks IL-23 by binding to its p19 subunit.7,16 IL-23, a cytokine involved in inflammatory processes, is thought to be linked to a number of chronic immune-mediated diseases, including Crohn's disease.16 The approved dose for SKYRIZI for moderate to severe plaque psoriasis and active psoriatic arthritis in the European Union is 150 mg (either as two 75 mg pre-filled syringe injections or one 150 mg prefilled pen or pre-filled injections) administered by subcutaneous injections at week 0 and 4 and every 12 weeks thereafter. The use of risankizumab in Crohn's disease is not approved and its safety and efficacy have not been established by regulatory authorities. Phase 3 trials of SKYRIZI in psoriasis, Crohn's disease, ulcerative colitis and psoriatic arthritis are ongoing.15,17-19
For more information about AbbVie, please visit us at www.abbvie.com. Follow @abbvie on Twitter, Facebook, LinkedIn or Instagram.
Nov. 30, 2021 8:11 AM ET
By: Mamta Mayani, SA News Editor2 Comments
November 30, 2021 at 7:30 AM ESTPDF Version
• PRIME study demonstrates that niraparib treatment had a clinically meaningful and statistically significant benefit in improving progression-free survival in the overall study population regardless of biomarker status when compared to placebo
• Treatment was tolerable in the population studied and showed a safety profile consistent with previous trials
• Zai Lab plans to present the PRIME study data at an upcoming medical conference
SHANGHAI, SAN FRANCISCO, and CAMBRIDGE, Mass., Nov. 30, 2021 (GLOBE NEWSWIRE) -- Zai Lab Limited (NASDAQ: ZLAB; HKEX: 9688), a patient-focused, innovative, commercial-stage, global biopharmaceutical company, today announced that the Phase 3 PRIME study of ZEJULA (niraparib) as maintenance therapy met its primary endpoint. ZEJULA demonstrated a statistically significant and clinically meaningful progression-free survival (PFS) benefit with a tolerable safety profile in Chinese patients with newly diagnosed advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer (collectively termed as ovarian cancer) following a response to platinum-based chemotherapy, regardless of biomarker status.
“I believe the data of the PRIME study will have a significant impact on the clinical practice in the first-line treatment of ovarian cancer in China and beyond, as the individualized starting dose regimen has demonstrated an improved safety profile,” said Dr. Lingying Wu, Director of the Department of Gynecologic Oncology, National Cancer Center / National Clinical Research Center for Cancer / Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College. “In addition, the PRIME study is the only study conducted in China that has demonstrated that a PARP inhibitor significantly improved PFS when given as monotherapy maintenance therapy in all Chinese patients with newly diagnosed ovarian cancer, regardless of biomarker status.”
“The PRIME clinical data in Chinese patients confirmed the clinical profile of ZEJULA and were consistent with the results seen in the global PRIMA study,” said Alan Sandler, M.D., President and Head of Global Development, Oncology, Zai Lab. “Importantly, the PRIME study further underscores the status of ZEJULA as the first and only PARP inhibitor approved globally, including in China, as monotherapy for all-comer patients in the first-line maintenance treatment settings.”
In September 2020, the China National Medical Products Administration (NMPA) approved ZEJULA’s supplemental New Drug Application (sNDA) as a maintenance treatment of adult patients with advanced ovarian cancer who are in a complete or partial response to first-line platinum-based chemotherapy. ZEJULA was also approved by the Hong Kong Department of Health as a maintenance treatment for adult patients with high-grade serous epithelial ovarian cancer who are in a complete response or partial response to first-line platinum-based chemotherapy.
ZEJULA is seeking National Reimbursement Drug List (NRDL) inclusion for a first-line ovarian cancer indication.
About PRIME Study
The fully powered Phase 3 PRIME study was evaluated in 384 advanced ovarian cancer patients who were in a complete or partial response to first-line platinum-based chemotherapy and who were randomized 2:1 to receive ZEJULA or placebo until disease progression. The study evaluated the efficacy of ZEJULA as a maintenance treatment, with the primary endpoint of PFS as assessed by blinded independent central review. The starting dose was individualized at 200 mg except for those patients with a baseline body weight ≥77kg and a platelet count ≥150K/μL, in which case the starting dose was 300 mg.
About ZEJULA (niraparib)
ZEJULA (niraparib) is an oral, once-daily poly (ADP-ribose) polymerase (PARP) inhibitor indicated as monotherapy for the maintenance treatment of adult patients with advanced and recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in response (complete or partial) to first- and second-line platinum-based chemotherapy.
Zai Lab has completed several studies in Chinese patients with ovarian cancer:
ZEJULA is also being evaluated in China in a Phase 1b/2 study in combination with tebotelimab (PD-1 x LAG-3 DART molecule) for advanced gastric cancer, triple negative breast cancer, biliary tract cancer, and endometrial cancer.
Zai Lab has a collaboration and license agreement with GlaxoSmithKline for the development and commercialization of ZEJULA in mainland China, Hong Kong, and Macau.
For additional information about the Company, please visit www.zailaboratory.com or follow us at www.twitter.com/ZaiLab_Global.
Nov. 30, 2021 7:39 AM ET Zai Lab Limited (ZLAB) By: Mamta Mayani, SA News Editor
November 29, 2021 6:50 am ET
First Combination of Immunotherapy With Tyrosine Kinase Inhibitor Approved in Europe for Adult Patients With Advanced or Recurrent Endometrial Carcinoma With Disease Progression on or Following Prior Treatment With a Platinum-Containing Therapy in Any Setting and Who Are Not Candidates for Curative Surgery or Radiation
Approval Based on KEYNOTE-775/Study 309 Results Demonstrating Statistically Significant Improvements in Overall Survival and Progression-Free Survival Compared With Chemotherapy
KENILWORTH, N.J. & TOKYO--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, and Eisai today announced that the European Commission has approved the combination of KEYTRUDA, Merck’s anti-PD-1 therapy, plus LENVIMA, the orally available multiple receptor tyrosine kinase inhibitor discovered by Eisai, for the treatment of advanced or recurrent endometrial carcinoma in adults who have disease progression on or following prior treatment with a platinum‑containing therapy in any setting and who are not candidates for curative surgery or radiation. This marks the first combination of an immunotherapy with a tyrosine kinase inhibitor approved in Europe for these patients with advanced or recurrent endometrial carcinoma.
The approval is based on results from the pivotal Phase 3 KEYNOTE-775/Study 309 trial, in which KEYTRUDA plus LENVIMA demonstrated statistically significant improvements in overall survival (OS), reducing the risk of death by 38% (HR=0.62 [95% CI, 0.51-0.75]; p<0.0001), and progression-free survival (PFS), reducing the risk of disease progression or death by 44% (HR=0.56 [95% CI, 0.47-0.66]; p<0.0001), versus chemotherapy (investigator’s choice of doxorubicin or paclitaxel). The median OS was 18.3 months for KEYTRUDA plus LENVIMA versus 11.4 months for chemotherapy. The median PFS was 7.2 months for KEYTRUDA plus LENVIMA versus 3.8 months for chemotherapy. The objective response rate (ORR) was 32% (95% CI, 27-37) for patients treated with KEYTRUDA plus LENVIMA versus 15% (95% CI, 11-18) for patients treated with chemotherapy (p<0.0001). Patients treated with KEYTRUDA plus LENVIMA achieved a complete response (CR) rate of 7% and partial response (PR) rate of 25% versus a CR rate of 3% and a PR rate of 12% for patients treated with chemotherapy.
“This approval is welcome news for patients in Europe and is based on the first Phase 3 study evaluating an immunotherapy and tyrosine kinase inhibitor combination that showed superior overall survival for patients with advanced or recurrent endometrial cancer compared to chemotherapy,” said Dr. Gregory Lubiniecki, Vice President, Clinical Research, Merck Research Laboratories. “Regardless of mismatch repair status, patients whose endometrial cancer progresses or returns after prior platinum-containing systemic therapies now have a combination treatment option in KEYTRUDA plus LENVIMA that demonstrated a 38% reduction in risk of death compared to chemotherapy alone.”
“Until recently, women in Europe with advanced or recurrent endometrial cancer have faced a difficult prognosis and had few treatment options,” said Corina Dutcus, M.D., Vice President, Clinical Research, Oncology Business Group at Eisai Inc. “The approval of KEYTRUDA plus LENVIMA in this setting reflects the progress that we have made in our collaboration with Merck in developing solutions for those diagnosed with difficult-to-treat cancers. We thank the patients, families and healthcare providers who made this milestone possible.”
The safety of KEYTRUDA in combination with LENVIMA was evaluated in 530 patients with advanced endometrial carcinoma. The most common adverse reactions were hypertension (63%), diarrhea (57%), hypothyroidism (56%), nausea (51%), decreased appetite (47%), vomiting (39%), fatigue (38%), decreased weight (35%), arthralgia (33%), proteinuria (29%), constipation, headache and urinary tract infection (27% each), dysphonia (25%), abdominal pain, asthenia, palmar-plantar erythrodysesthesia syndrome and stomatitis (23% each), anemia (22%) and hypomagnesaemia (20%).
This approval allows marketing of KEYTRUDA plus LENVIMA in all 27 EU member states plus Iceland, Liechtenstein, Norway and Northern Ireland. KEYTRUDA plus LENVIMA is now approved by the European Commission for two different types of cancer: for advanced or recurrent endometrial carcinoma in adults who have disease progression on or following prior treatment with a platinum-containing therapy in any setting and who are not candidates for curative surgery or radiation and for the first-line treatment of adult patients with advanced renal cell carcinoma.
About KEYNOTE-775/Study 309 Trial
The approval was based on data from KEYNOTE-775/Study 309 (ClinicalTrials.gov, NCT03517449), a Phase 3, multicenter, open-label, randomized, active-controlled study conducted in 827 patients with advanced endometrial carcinoma who had been previously treated with at least one prior platinum-based chemotherapy regimen in any setting, including in the neoadjuvant and adjuvant settings. Participants may have received up to two platinum-containing therapies in total, as long as one was given in the neoadjuvant or adjuvant treatment setting. The study excluded patients with endometrial sarcoma, carcinosarcoma, pre-existing Grade ≥3 fistula, uncontrolled BP (>150/90 mmHg), significant cardiovascular impairment or event within previous 12 months or patients who had active autoimmune disease or a medical condition that required immunosuppression. The primary efficacy outcome measures were OS, and PFS as assessed by blinded independent central review (BICR) according to RECIST v1.1. Secondary efficacy outcome measures included ORR as assessed by BICR.
Patients were randomized 1:1 to receive KEYTRUDA (200 mg intravenously every three weeks) plus LENVIMA (20 mg orally once daily) or investigator’s choice, consisting of either doxorubicin (60 mg/m2 every three weeks) or paclitaxel (80 mg/m2 given weekly, three weeks on/one week off). Treatment with KEYTRUDA plus LENVIMA continued until RECIST v1.1-defined progression of disease as verified by BICR, unacceptable toxicity, or for KEYTRUDA, a maximum of 24 months. Administration of KEYTRUDA plus LENVIMA was permitted beyond RECIST-defined disease progression if the treating investigator considered the patient to be deriving clinical benefit and the treatment was tolerated. A total of 121/411 (29%) of patients treated with KEYTRUDA plus LENVIMA received continued study therapy beyond RECIST-defined disease progression. The median duration of the post-progression therapy was 2.8 months. Assessment of tumor status was performed every eight weeks.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of patients with melanoma with involvement of lymph node(s) following complete resection.
Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [tumor proportion score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [combined positive score (CPS ≥1)] as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy. KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Carcinoma
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
Non-muscle Invasive Bladder Cancer
KEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin (BCG)-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ (CIS) with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with MSI-H central nervous system cancers have not been established.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC).
Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or GEJ (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
KEYTRUDA, in combination with chemotherapy, with or without bevacizumab, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
KEYTRUDA, in combination with LENVIMA, is indicated for the first-line treatment of adult patients with advanced RCC.
KEYTRUDA is indicated for the adjuvant treatment of patients with renal cell carcinoma (RCC) at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions.
Endometrial Carcinoma
KEYTRUDA, in combination with LENVIMA, is indicated for the treatment of patients with advanced endometrial carcinoma that is not MSI-H or dMMR, who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation.
Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf.
About LENVIMA® (lenvatinib); available as 10 mg and 4 mg capsules
LENVIMA, discovered and developed by Eisai, is a multiple receptor tyrosine kinase inhibitor that inhibits the kinase activities of vascular endothelial growth factor (VEGF) receptors VEGFR1 (FLT1), VEGFR2 (KDR), and VEGFR3 (FLT4). LENVIMA inhibits other kinases that have been implicated in pathogenic angiogenesis, tumor growth, and cancer progression in addition to their normal cellular functions, including fibroblast growth factor (FGF) receptors FGFR1-4, the platelet derived growth factor receptor alpha (PDGFRα), KIT, and RET. The combination of LENVIMA and everolimus showed increased anti-angiogenic and anti-tumor activity as demonstrated by decreased human endothelial cell proliferation, tube formation, and VEGF signaling in vitro and tumor volume in mouse xenograft models of human renal cell cancer greater than each drug alone. In syngeneic mouse tumor models, the combination of lenvatinib with an anti-PD-1 monoclonal antibody decreased tumor-associated macrophages, increased activated cytotoxic T cells, and demonstrated greater antitumor activity compared to either treatment alone.
LENVIMA® (lenvatinib) Indications in the U.S.
Nov. 29, 2021 7:56 AM ET Merck & Co., Inc. (MRK) By: Mamta Mayani, SA News Editor
November 29, 2021Download PDF
• Pivotal GEM-3 trial met its primary endpoint of complete wound healing at six-month timepoints,
and its secondary endpoint of complete wound healing at three-month timepoints
PITTSBURGH, Nov. 29, 2021 (GLOBE NEWSWIRE) -- Krystal Biotech, Inc., (“Krystal”) (NASDAQ: KRYS), the leader in redosable gene therapies for rare diseases, today announced positive topline results from the pivotal GEM-3 trial of investigational beremagene geperpavec (B-VEC), now known as VYJUVEKTM, for the treatment of dystrophic Epidermolysis Bullosa (dystrophic EB).
The primary endpoint of the trial evaluated complete wound healing of topical VYJUVEKTM compared to placebo at six-month timepoints and met statistical significance. VYJUVEKTM is the first non-invasive, topical and redosable gene therapy in development, and the only genetically corrective approach to treat dystrophic EB that has successfully completed a double blinded Phase 3 trial.
Highlights of Topline Results from the GEM-3 Trial
“Dystrophic Epidermolysis Bullosa is referred to as ‘the worst disease you’ve never heard of’ because of the incredibly devastating reality that patients with this genetic condition face, and we are thrilled to announce positive results from our pivotal GEM-3 trial of VYJUVEKTM which showed that this topical gene therapy led to durable wound healing in dystrophic EB wounds,” said Suma Krishnan, Founder and Chief Operating Officer of Krystal. “With these results in hand, we look forward to advancing discussions with regulatory authorities and will work quickly to bring this potential first-ever treatment to patients with dystrophic EB and their families who are in desperate need.”
“Today’s positive B-VEC results represent the culmination of years of study on the molecular basis and genetic correction of this disease. Finally, dystrophic EB patients may have an easily administered genetically targeted therapy which has been shown to promote durable wound healing in this clinical trial. This is a long overdue milestone for patients living with this disease, and one that has potential to drastically change the treatment paradigm,” said Dr. Peter Marinkovich, M.D., Bullous Disease Clinic Director and Associate Professor of Dermatology at Stanford University.
About the GEM-3 Trial
The GEM-3 trial (NCT04491604) was a randomized, double-blind, intra-patient placebo-controlled study designed to evaluate the efficacy and safety of VYJUVEKTM for the treatment of dystrophic EB. Thirty-one (31) patients were enrolled across three sites and ranged in ages from one (1) year to forty-four (44) years old.
In each patient, a primary wound pair was identified by the investigator; one wound was randomized to receive a weekly topical application of VYJUVEKTM and the other to receive placebo. Wounds were dosed once-weekly with either VYJUVEKTM or placebo until closure. Weekly application was resumed if wounds re-opened at any point in the study.
The primary outcome measure was complete wound healing determined by the Investigator in VYJUVEKTM treated wounds versus placebo treated at the six-month timepoints, meaning week 22 and Week 24 or Week 24 and Week 26. Secondary endpoints included investigator assessed complete wound healing at the three-month timepoints, meaning weeks 8 and 10 or 10 and 12 and mean change in pain severity using either a VAS or FLACC-R Scale at weeks 22, 24 and 26.
In addition to the primary target wound pair(s), additional wounds (secondary wounds) were selected and treated with VYJUVEKTM giving the treating physicians and patients flexibility to treat multiple wounds during the weekly application. For more information about the pivotal GEM-3 study, visit www.clinicaltrials.gov (NCT04491604).
Subjects returned to the clinical site 30 days following the last dosing visit (Week 26) for safety evaluation by the investigator and subsequently had the option to roll into the Open Label Extension (OLE) Study (NCT04917874). In addition, new participants who were unable to participate in the Phase 3 study but met all enrollment criteria are eligible to enroll in the OLE.
About VYJUVEKTM
VYJUVEKTM is an investigational non-invasive, topical gene therapy designed to deliver two copies of the COL7A1 gene when applied directly to DEB wounds. Unlike the current standard of care, VYJUVEKTM was designed to treat DEB at the molecular level by providing the patient’s skin cells the template to make normal COL7 protein, thereby addressing the fundamental disease-causing mechanism.
The FDA and the EMA have each granted VYJUVEKTM orphan drug designation for the treatment of DEB, and the FDA has granted VYJUVEKTM fast track designation and rare pediatric designation for the treatment of DEB. In addition, in 2019, the FDA granted Regenerative Medicine Advanced Therapy (“RMAT”) to VYJUVEKTM for the treatment of DEB and the EMA granted PRIority MEdicines ("PRIME"), eligibility for VYJUVEKTM to treat DEB.
For more information, please visit http://www.krystalbio.com
Nov. 29, 2021 7:34 AM ET Krystal Biotech, Inc. (KRYS)
By: Dulan Lokuwithana, SA News Editor1 Comment
Announcing topline data from the pivotal GEM-3 trial for its experimental therapy, VYJUVEK in dystrophic Epidermolysis Bullosa (dystrophic EB), Krystal Biotech (NASDAQ:KRYS) said the study met the primary endpoint with statistical significance.
November 23, 2021
OSAKA, Japan, and CAMBRIDGE, Mass., November 23, 2021– Takeda Pharmaceutical Company Limited (TSE:4502/NYSE:TAK) (“Takeda”) today announced that the U.S. Food and Drug Administration (FDA) has approved LIVTENCITY™ (maribavir) for the treatment of adults and pediatric patients (12 years of age or older and weighing at least 35 kg) with post-transplant cytomegalovirus (CMV) infection/disease that is refractory to treatment (with or without genotypic resistance) with ganciclovir, valganciclovir, cidofovir, or foscarnet.1 Overall, more than twice the proportion of adult transplant patients with refractory or resistant (R/R) CMV infection/disease achieved confirmed CMV DNA level <LLOQ* (lower limit of quantification, i.e. <137 IU/mL) at Week 8 (end of treatment phase), the study’s primary endpoint, with LIVTENCITY (56%; n=131/235), compared to those treated with conventional antiviral therapies (24%; n=28/117) (adjusted difference: 33%, 95% CI: 23–43; p<0.001).†‡§ LIVTENCITY is Takeda’s second new molecular entity to receive FDA approval in FY2021.
“Today’s announcement redefines the management of post-transplant CMV with the approval of the first and only treatment for transplant patients with CMV that is refractory with or without resistance, a significantly underserved and vulnerable patient community,” said Ramona Sequeira, President, U.S. Business Unit and Global Portfolio Commercialization, Takeda Pharmaceutical Company Limited. “People undergoing transplants have a lengthy and complex healthcare journey; with the approval of this treatment, we’re proud to offer these individuals a new oral antiviral to fight CMV infection and disease. We are grateful for the contributions of the patients and clinicians who participated in our clinical trials, as well as the dedication of our scientists and researchers.”
LIVTENCITY is a new molecular entity which targets CMV at pUL97, resulting in inhibition of viral DNA replication, encapsidation and nuclear egress.1,6,7,8,9,10 Though a rare disease overall, CMV is one of the most common infections experienced by transplant recipients, with an estimated incidence rate of around 16%–56% in solid organ transplant (SOT) recipients3 and 30%–70% in hematopoietic stem cell (HSCT) transplant patients.2 CMV can be acquired or reactivated following transplant leading to serious consequences—including loss of the transplanted organ and failure of the graft—or loss of life. In patients with compromised immunity, CMV causes clinically challenging complications that can be fatal.5,11,12
LIVTENCITY will be available in the coming days. For appropriate patients, physicians can submit a prescription to initiate access to treatment by contacting Takeda Patient Support at 1-855-268-1825.
“The FDA approval of LIVTENCITY marks a major step forward in the treatment of post-transplant CMV, bringing a new therapeutic option to those living with this potential life-threatening opportunistic infection,” said Roy F. Chemaly, M.D., M.P.H., FACP, FIDSA, Department of Infectious Diseases, Infection Control & Employee Health at The University of Texas MD Anderson Cancer Center in Houston, TX. “In clinical studies, we observed LIVTENCITY was statistically superior to conventional antiviral therapies in achieving the primary endpoint at Week 8.”
Prior to FDA approval, LIVTENCITY (maribavir) was granted Orphan Drug Designation by the FDA for treatment of clinically significant CMV viremia and disease in at-risk patients, as well as Breakthrough Therapy Designation as a treatment for CMV infection and disease in transplant patients resistant or refractory to prior therapy. Takeda is looking forward to continuing our discussions with regulatory agencies across the globe to potentially bring maribavir to patients worldwide. The company is also investigating maribavir as a first-line treatment of CMV in hematopoietic stem cell transplant recipients in an ongoing Phase 3 clinical trial.
LIVTENCITY was evaluated in the TAK-620-303 (SOLSTICE) trial, a global, multicenter, randomized, open-label, active-controlled superiority trial assessing the efficacy and safety of treatment with either maribavir or investigator-assigned treatment (IAT, conventional antiviral therapy) in 352 HSCT and SOT adult recipients with CMV infection refractory, with or without or resistance, to one or a combination of conventional antiviral therapies: ganciclovir, valganciclovir, foscarnet, or cidofovir. Participants were randomized 2:1 to receive maribavir (N=235) (400 mg, twice daily) or IAT (N=117) (as dosed by the investigator) for up to 8-weeks. After completion of the treatment period, subjects entered a 12-week follow-up phase.1 The primary efficacy endpoint was confirmed CMV DNA level <LLOQ* (lower limit of quantification, [i.e. <137 IU/mL] as assessed by COBAS® AmpliPrep/COBAS® TaqMan® CMV test at the end of Week 8).1
The most common adverse events occurring in all grades, >10% of patients receiving maribavir were taste disturbance,†† nausea, diarrhea, vomiting, and fatigue.1 A higher proportion of subjects in the IAT group discontinued study medication due to an adverse event compared to the LIVTENCITY group (32%, n=37/116 versus 13%, n=31/234, respectively).1,13 Taste disturbance events (46%, n=108/234) were generally mild, and rarely led to discontinuation of maribavir (1%).1,13 In 37% of patients, these events resolved while patients remained on therapy (median duration 43 days; range 7 to 59 days).1 For the patients with ongoing taste disturbance after drug discontinuation, resolution occurred in 89%.1 In patients with resolution of symptoms after drug discontinuation, the median duration of symptoms off treatment was 6 days (range 2 to 85 days).1 All-cause mortality was similar in each treatment group (LIVTENCITY 11%, n=27/235; IAT 11%, n=13/117).1
*Confirmed CMV DNA level < LLOQ at the end of Week 8 (2 consecutive samples separated by at least 5 days with DNA levels <LLOQ [ie, <137 IU/mL])
†The difference in proportion of responders between treatment groups was obtained using Cochran-Mantel-Haenszel (CMH) weighted average across all strata and tested using stratum-adjusted CMH method, with transplant type and baseline plasma CMV DNA concentration as two stratification factors
‡Refractory defined as documented failure to achieve >1 log10 decrease in CMV DNA level in whole blood or plasma after a 14 day or longer treatment period with IV ganciclovir/oral valganciclovir, IV foscarnet, or IV cidofovir
§Resistant defined as refractory CMV and documentation of >1 CMV genetic mutations associated with resistance to ganciclovir, valganciclovir, foscarnet, and/or cidofovir
††Taste disturbance is defined as including dysgeusia, ageusia, hypogeusia and taste disorder.
The TAK-620-303 (SOLSTICE) trial (NCT02931539) was a multicenter, randomized, open-label, active-controlled superiority trial to assess the efficacy and safety comparing treatment with either LIVTENCITY (maribavir) or investigator assigned treatment, IAT, (conventional antiviral therapy) in 352 hematopoietic stem cell transplant and solid organ transplant recipients with CMV infection refractory, with or without resistance, to one or a combination of the conventional antiviral therapies: ganciclovir, valganciclovir, foscarnet or cidofovir. Adult patients underwent a 2-week screening period, followed by randomization 2:1 to LIVTENCITY (maribavir) (n=235) (400 mg, twice daily) or IAT (n=117) (as dosed by the investigator) for up to 8-weeks. After completion of the treatment period, subjects entered a 12-week follow-up phase.
The trial’s primary endpoint was confirmed CMV DNA level <LLOQ (lower limit of quantification, [i.e. <137 IU/mL] in 2 consecutive samples separated by at least 5 days as assessed by COBAS® AmpliPrep/COBAS® TaqMan® CMV test at the end of Week 8). The key secondary endpoint was CMV DNA level <LLOQ and CMV infection symptom control at the end of Study Week 8 with maintenance of this treatment effect through Study Week 16.
LIVTENCITY (maribavir), an orally bioavailable anti-CMV compound, is the first and only antiviral agent that targets and inhibits the pUL97 protein kinase and its natural substrates.1 It is approved in the U.S. for the treatment of adults and pediatric patients (12 years of age or older and weighing at least 35 kg) with post-transplant cytomegalovirus (CMV) infection/disease that is refractory to treatment (with or without genotypic resistance) with ganciclovir, valganciclovir, cidofovir or foscarnet. For more information on LIVTENCITY, visit LIVTENCITY.com.1
LIVTENCITY is indicated for the treatment of adults and pediatric patients (12 years of age and older and weighing at least 35 kg) with post-transplant cytomegalovirus (CMV) infection/disease that is refractory to treatment (with or without genotypic resistance) with ganciclovir, valganciclovir, cidofovir or foscarnet.1
Please click for Full Prescribing Information.
For more information, visit https://www.takeda.com.
Nov. 23, 2021 5:01 PM ET Takeda Pharmaceutical Company Limited (TAK)
By: Jonathan M Block, SA News Editor
November 23, 2021
-- Marketing Authorization Based on Phase 3 ASCENT Study Showing Trodelvy Significantly Improved Overall Survival vs. Physician’s Choice of Chemotherapy in Metastatic Triple-Negative Breast Cancer --
-- Trodelvy Offers an Important New Treatment Option for People with this Aggressive Type of Metastatic Breast Cancer --
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the European Commission (EC) has granted marketing authorization for Trodelvy® (sacituzumab govitecan), a first-in-class Trop-2-directed antibody-drug conjugate, as a monotherapy indicated for the treatment of adult patients with unresectable or metastatic triple-negative breast cancer (TNBC) who have received two or more prior systemic therapies, at least one of them for advanced disease.
“The metastatic stage of TNBC is particularly challenging to treat and until now we have urgently needed new treatment options for people in Europe living with this condition,” said Dr Véronique Diéras, Senior Medical Oncologist Head, Breast Cancer Group, Department of Medical Oncology, Centre Eugène Marquis, Rennes, France. “Today’s approval including second-line metastatic TNBC is significant for the community as it’s another important step forward in helping women with this disease live longer.”
TNBC is the most aggressive type of breast cancer and accounts for approximately 15% of all breast cancers. It is more frequently diagnosed in younger and premenopausal women and is more prevalent in Black and Hispanic women. The five-year survival rate for this sub-type of breast cancer is 12%, compared with 28% for other breast cancer types, and these poor outcomes are often coupled with a significant decrease in quality of life, especially in relapsed/refractory disease.
“At Gilead, we push boundaries to deliver transformative science and novel treatment options that address urgent medical needs,” said Merdad Parsey, MD, PhD, Chief Medical Officer, Gilead Sciences. “We understand how difficult metastatic TNBC is to treat and we’re proud that Trodelvy can now offer a second-line treatment option with the potential to bring longer life to people living with this aggressive disease.”
The EC’s decision is supported by results from the Phase 3 ASCENT study, where Trodelvy reduced the risk of death by 49% and improved median overall survival to 11.8 months versus 6.9 months with physician’s choice of chemotherapy (HR: 0.51; 95% CI: 0.41-0.62; p<0.0001). These data also showed a statistically significant and clinically meaningful 57% reduction in the risk of death or disease worsening and improved median progression free survival (PFS) to 4.8 months from 1.7 months seen with physician’s choice of chemotherapy alone among all randomized patients, which included those with and without brain metastases (HR: 0.43; 95% CI: 0.35-0.54; p<0.0001). The most common Grade 3 or higher adverse reactions were neutropenia (49.5%), leukopenia (12.0%), diarrhea (10.7%), anemia (10.1%), febrile neutropenia (6.6%), fatigue (5.2%), hypophosphatemia (5.2%), nausea (4.1%) and vomiting (3.0%).8 The Trodelvy U.S. Prescribing Information has a BOXED WARNING for severe or life-threatening neutropenia and severe diarrhea; see below for Important Safety Information.
In addition to this approval, Trodelvy is approved in Australia, Canada, Great Britain, Switzerland, and the United States in metastatic TNBC. Regulatory review is also underway in Singapore and China with applications submitted by Everest Medicines. Trodelvy was also recently included in the updated ESMO Clinical Practice Guidelines as a preferred treatment option for metastatic TNBC after taxanes.
About the ASCENT Study
The ASCENT study is a global, open-label, randomized Phase 3 study that enrolled more than 500 patients across 230 study locations. The study evaluated the efficacy and safety of Trodelvy compared with a single-agent chemotherapy of the physician’s choice in patients with unresectable, locally advanced or metastatic TNBC who had received at least two prior systemic treatments. Patients were randomly allocated to receive either Trodelvy or a chemotherapy chosen by the patient’s treating physician. The primary endpoint was progression-free survival (PFS, as determined by blinded independent central review) in patients without brain metastases. Secondary endpoints included: PFS for full study population or intention-to-treat (ITT) population, overall survival in both the ITT population and in the subgroup without brain metastasis, independently determined objective response rate, duration of response, time to onset of response according to Response Evaluation Criteria in Solid Tumors (RECIST 1.1), quality of life and safety. More information about ASCENT is available at http://clinicaltrials.gov/show/NCT02574455.
About Trodelvy
Trodelvy is a first-in-class antibody and topoisomerase inhibitor conjugate directed to the Trop-2 receptor, a protein overexpressed in multiple types of epithelial tumors, including metastatic TNBC and metastatic urothelial cancer (UC), where high expression is associated with poor survival and relapse. Trodelvy is approved in second-line metastatic TNBC in multiple countries worldwide, including Australia, Canada, Great Britain, the European Union, Switzerland and the United States. Trodelvy is also approved for use in metastatic UC in the United States. Trodelvy continues to be developed for potential use in other TNBC and metastatic UC populations and is also being developed as an investigational treatment for hormone receptor-positive/human epidermal growth factor receptor 2-negative (HR+/HER2-) metastatic breast cancer and metastatic non-small cell lung cancer. Additional evaluation across multiple solid tumors is also underway.
Trodelvy, Gilead and the Gilead logo are trademarks of Gilead Sciences, Inc., or its related companies.
For more information about Gilead, please visit the company’s website at www.gilead.com, follow Gilead on Twitter (@GileadSciences) or call Gilead Public Affairs at 1-800-GILEAD-5 or 1-650-574-3000.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211123005779/en/
Source: Gilead Sciences, Inc.
Nov. 23, 2021 8:57 AM ET Gilead Sciences, Inc. (GILD) By: Mamta Mayani, SA News Editor2 Comments
Nov 23, 2021 5:00 PM
EU approval follows recent approvals for BRUKINSA including U.S., China, Brazil, and Canada
The approval is based on Phase 3 ASPEN head-to-head trial comparing BRUKINSA against ibrutinib
BASEL, Switzerland & CAMBRIDGE, Mass. & BEIJING--(BUSINESS WIRE)-- BeiGene (NASDAQ: BGNE; HKEX: 06160) announced today that the European Commission (EC) approved BRUKINSA® (zanubrutinib) for the treatment of adult patients with Waldenström’s macroglobulinemia (WM) who have received at least one prior therapy or for the first-line treatment of patients unsuitable for chemo-immunotherapy. The approval is applicable to all 27 European Union (EU) member states, plus Iceland and Norway. BeiGene is working to make this new treatment option available to WM patients in the EU as quickly as possible.
“BTK inhibition is an established mode of treatment for patients with WM, and the approval of BRUKINSA provides an important new option for patients with WM that may offer improved outcomes,” said Prof. Christian Buske, Medical Director at the University Hospital Ulm, Germany, and a trial investigator of the ASPEN study. “Patients and their physicians in the EU will soon have access to an innovative medicine that has potential to offer deep and durable responses and improved tolerability, as seen in the ASPEN trial.”
The EC approval for BRUKINSA follows a positive opinion granted in September by the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA), based on the results of the ASPEN trial. Although the primary endpoint of statistical superiority related to deep response, very good partial response (VGPR) or better, was not met, BRUKINSA demonstrated clinical benefit with advantages in safety compared to ibrutinib.1 Read more about the positive CHMP opinion and ASPEN trial results here.
“With BRUKINSA now approved in the EU, we continue to execute on our commitment of making this potentially best-in-class BTK inhibitor available for more patients around the world who may benefit,” said Jane Huang, M.D., Chief Medical Officer, Haematology at BeiGene. “BRUKINSA was designed to maximize BTK occupancy and minimize off-target effects and has demonstrated efficacy and advantages in safety and tolerability over ibrutinib in the ASPEN trial. We believe BRUKINSA will become the preferred treatment option among patients with WM and their physicians.”
“We have built a strong team in Europe that is committed to creating access to BRUKINSA for patients living with WM,” said Gerwin Winter, Senior Vice President, Head of Commercial, Europe at BeiGene. “This approval by the European Commission is a significant milestone for BeiGene’s expansion in the region, representing another step towards BeiGene’s goal of increasing access to innovative oncology medicines globally.”
About Waldenström’s Macroglobulinemia
Waldenström’s macroglobulinemia (WM) is a generally indolent and relatively rare B-cell malignancy characterized by bone marrow infiltration with monoclonal immunoglobulin M (IgM) secreting lymphoplasmacytic cells. WM represents approximately one percent of all non-Hodgkin’s lymphomas and typically progresses slowly after diagnosis.2 The disease usually affects older adults and is primarily found in the bone marrow, although lymph nodes and the spleen may be involved.3 Throughout Europe, the estimated incidence rate of WM is approximately seven out of every one million men and four out of every one million women.4
About the ASPEN trial
The Phase 3 randomized, open-label, multicentre ASPEN clinical trial (NCT03053440) evaluated BRUKINSA (zanubrutinib) versus ibrutinib in patients with relapsed or refractory (R/R) WM or treatment-naïve (TN) WM considered unsuitable for treatment with chemoimmunotherapy. The primary objective was to establish superiority of BRUKINSA compared to ibrutinib as demonstrated by the proportion of patients achieving complete response or very good partial response. Secondary endpoints included major response rate (MRR), duration of response (DoR) and progression-free survival (PFS), and safety, measured by incidence, timing and severity of treatment-emergent adverse events. The pre-specified analysis populations for the trial included the overall population (n=201), of which the majority were R/R patients (n=164). Exploratory endpoints included quality of life measures.
As assessed by an independent review committee (IRC) based on the modified Sixth International Workshop on Waldenström’s Macroglobulinemia (IWWM-6) response criteria (Treon 2015), the combined rate of complete response (CR) and VGPR in the overall intention-to-treat (ITT) population was 28% with BRUKINSA (95% CI: 20, 38), compared to 19% with ibrutinib (95% CI: 12, 28). While this difference was not statistically significant, BRUKINSA did achieve numerically higher VGPR rates and trends towards increased response quality.1
In the ASPEN trial, BRUKINSA demonstrated a more favorable safety profile compared to ibrutinib with lower frequency of adverse reactions that have raised concern with BTK inhibitors, including atrial fibrillation or flutter (2% vs. 15%), minor bleeding (49% vs. 59%) and major hemorrhage (6% vs. 9%). Despite higher rates of grade ≥3 neutropenia, patients on BRUKINSA did not demonstrate higher rates of infection as compared to those receiving ibrutinib. Of the 101 patients with WM treated with BRUKINSA, 4% of patients discontinued due to adverse events, and adverse events leading to dose reduction occurred in 14% of patients.1
The study includes three arms in two cohorts, a randomized cohort (Cohort 1, N=201) consisting of patients with a MYD88 mutation (MYD88MUT) and a non-randomized cohort (Cohort 2, N=28) in which patients with MYD88 wild-type (MYD88WT) received BRUKINSA because historic data indicated they were unlikely to benefit from ibrutinib. The randomized Cohort 1 enrolled 102 patients (including 83 R/R patients and 19 TN patients) in the BRUKINSA arm and 99 patients (including 81 R/R patients and 18 TN patients) in the ibrutinib arm. Patients in the BRUKINSA arm were assigned to receive BRUKINSA 160 mg twice daily (BID) and patients in the ibrutinib arm received 420 mg of ibrutinib once daily (QD).
About BRUKINSA
BRUKINSA (zanubrutinib) is a small molecule inhibitor of Bruton’s tyrosine kinase (BTK) discovered by BeiGene scientists that is currently being evaluated globally in a broad clinical program as a monotherapy and in combination with other therapies to treat various B-cell malignancies. Because new BTK is continuously synthesized, BRUKINSA was specifically designed to deliver complete and sustained inhibition of the BTK protein by optimizing bioavailability, half-life, and selectivity. With differentiated pharmacokinetics compared to other approved BTK inhibitors, BRUKINSA has been demonstrated to inhibit the proliferation of malignant B cells within a number of disease relevant tissues.
BRUKINSA is now approved in the United States, China, the European Union and nine other countries and regions. To date, more than 20 marketing authorization applications have been submitted for BRUKINSA for various indications.
To learn more about BeiGene, please visit www.beigene.com and follow us on Twitter at @BeiGeneGlobal.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211123006162/en/
Source: BeiGene
Nov. 24, 2021 4:17 AM ET BeiGene, Ltd. (BGNE) By: Mamta Mayani, SA News Editor
11/23/2021CATEGORY:
Zeposia brings a new way of treating this chronic immune-mediated disease, approved for adults with moderately to severely active ulcerative colitis (UC) who have had an inadequate response, lost response, or were intolerant to either conventional therapy or a biologic agent
Zeposia is the first and only oral sphingosine 1-phosphate (S1P) receptor modulator for UC, with this approval marking its second indication in the European Union
Zeposia approval is based on the Phase 3 True North trial, which demonstrated clinically meaningful improvements in key clinical, endoscopic and mucosal healing endpoints, with no new safety signals observed
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE:BMY) today announced the European Commission has granted a Marketing Authorization for Zeposia (ozanimod) for the treatment of adults with moderately to severely active ulcerative colitis (UC) who have had an inadequate response, lost response, or were intolerant to either conventional therapy or a biologic agent. Zeposia, an oral medication taken once daily,is a sphingosine 1-phosphate (S1P) receptor modulator that binds with high affinity selectively to S1P subtypes 1 (S1P1) and 5 (S1P5). Zeposia is the first and only oral S1P receptor modulator approved for UC, and represents a new way of treating this chronic immune-mediated disease.
“With today’s European Commission approval of Zeposia for ulcerative colitis, patients and physicians now have a once-daily oral treatment option to help address this debilitating disease, with a demonstrated efficacy and safety profile and a different mechanism of action than other available therapies,” said Jonathan Sadeh, M.D., MSc., senior vice president of Immunology and Fibrosis Development, Bristol Myers Squibb. “We are proud of our heritage in transformational science and innovative medicines that has brought us to this stage and look forward to offering appropriate patients in Europe a new therapy that provides significant symptom relief and lasting clinical remission.”
The approval was based on data from True North, a pivotal Phase 3 trial evaluating Zeposia as an induction and maintenance therapy versus placebo in adult patients with moderately to severely active UC. Key findings from the trial include:
“The findings from the True North trial show that Zeposia demonstrated significant, durable efficacy in patients with moderate to severe ulcerative colitis across multiple key endpoints such as clinical improvement, endoscopic and mucosal healing and clinical remission,” said Dr. Silvio Danese, M.D., Director, Gastroenterology and Endoscopy, IRCCS, San Raffaele Hospital and University Vita-Salute San Raffaele in Milan. “The results for endoscopic improvement and histologic remission are particularly meaningful because they can be very difficult to achieve, indicating that Zeposia has the potential to be an effective and safe oral treatment option for clinicians treating adults living with this serious, chronic disease.”
“In Europe, over 3 million people are affected by inflammatory bowel disease, which includes ulcerative colitis, a challenging and often debilitating form of the disease,” said Luisa Avedano, CEO, European Federation of Crohn's & Ulcerative Colitis Associations. “I’m thrilled that we now have a new treatment option for patients and their caregivers as they manage the symptoms of a disease that can have a such detrimental impact on quality of life.”
Zeposia is contraindicated in patients with hypersensitivity to the active substance or to any of the excipients, as listed in the Summary of Product Characteristics (SmPC); immunodeficient state; patients who in the last six months experienced myocardial infarction, unstable angina, stroke, transient ischemic attack, decompensated heart failure requiring hospitalization or New York Heart Association (NYHA) Class III/IV heart failure; patients with history or presence of second-degree atrioventricular (AV) block Type II or third-degree AV block or sick sinus syndrome unless the patient has a functioning pacemaker; severe active infections, active chronic infections such as hepatitis and tuberculosis; active malignancies; severe hepatic impairment (Child-Pugh class C); and during pregnancy and in women of childbearing potential not using effective contraception. The most commonly reported adverse reactions (>5%) in controlled periods of the adult multiple sclerosis (MS) and UC clinical studies are nasopharyngitis, alanine aminotransferase (ALT) increased, and gamma-glutamyl transferase (GGT) increased. The most common adverse reactions leading to discontinuation were related to liver enzyme elevations (1.1%) in the MS clinical studies. Liver enzyme elevations leading to discontinuation occurred in 0.4% of patients, in UC controlled clinical studies. The overall safety profile was similar for patients with MS and UC.
Bristol Myers Squibb thanks the patients and investigators involved in the True North clinical trial.
About True North
True North is a Phase 3, multicenter, randomized, double-blind, placebo-controlled clinical trial assessing the efficacy and safety of Zeposia 0.92 mg in patients with moderately to severely active ulcerative colitis (UC) who had an inadequate response or were intolerant to any of the following: oral aminosalicylates, corticosteroids, immunomodulators or a biologic. Patients were to be receiving treatment with oral aminosalicylates and/or corticosteroids prior to and during the induction period.A total of 30% of patients had previously failed or were intolerant to TNF blockers. Of these patients, 63% received at least two biologics including TNF blockers. At study entry, mean age was 42 years, 60% were male and mean disease duration was 7 years; patient characteristics were well-balanced across treatment groups.In the 10-week induction study (UC Study 1), a total of 645 patients were randomized 2:1 to receive Zeposia (n=429) or placebo (n=216), of whom 94% and 89%, respectively, completed the induction study. No new safety signals were observed in the induction phase.
In maintenance, UC Study 2, a total of 457 patients who received Zeposia in either UC Study 1 or in an open-label arm and achieved clinical response at Week 10 were re-randomized 1:1 and were treated with either Zeposia 0.92 mg (n=230) or placebo (n=227) for 42 weeks (UC Study 2), for a total of 52 weeks of treatment. Concomitant aminosalicylates were required to remain stable through week 52. Patients on concomitant corticosteroids were to taper their dose upon entering the maintenance study. Of these, 80% and 54.6% of patients who received Zeposia and placebo, respectively, completed the study. In the maintenance phase, the overall safety profile was consistent with the known safety profile for Zeposia and patients with moderate to severe UC. More information about the True North trial can be found on www.clinicaltrials.gov, NCT02435992.
The clinical findings from True North, entitled “Ozanimod as Induction and Maintenance Therapy for Ulcerative Colitis,” were published in the September 30th issue of The New England Journal of Medicine.
All eligible patients were rolled into an open-label extension trial, which is ongoing and designed to assess the longer-term profile of Zeposia for the treatment of moderately to severely active UC. Among patients who entered the trial clinical remission, clinical response, endoscopic improvement, and symptomatic remission were generally maintained through week 142. No new safety concerns were identified in this study extension in patients with UC. More information about the open-label extension trial can be found on www.clinicaltrials.gov, NCT02531126.
About Zeposia (ozanimod)
Zeposia (ozanimod) is an oral, sphingosine 1-phosphate (S1P) receptor modulator that binds with high affinity to S1P receptors 1 and 5. Zeposia reduces the capacity of lymphocytes to migrate from lymphoid tissue, reducing the number of circulating lymphocytes in peripheral blood. The mechanism by which Zeposia exerts therapeutic effects in UC is unknown but may involve the reduction of lymphocyte migration into the intestines.
Bristol Myers Squibb is continuing to evaluate Zeposia in an open-label extension trial, which is ongoing and designed to assess the longer-term profile of Zeposia for the treatment of moderately to severely active UC. The company is also investigating Zeposia for the treatment of moderately to severely active Crohn’s disease in the ongoing Phase 3 YELLOWSTONE clinical trial program.
The U.S. Food and Drug Administration (FDA) approved Zeposia for the treatment of adults with moderately to severely active UC on May 27, 2021, and for the treatment of adults with relapsing forms of multiple sclerosis (RMS) in March 2020. The European Commission approved Zeposia for the treatment of adult patients with relapsing remitting multiple sclerosis (RRMS) with active disease as defined by clinical or imaging features in May 2020.
U.S. FDA APPROVED INDICATIONS
ZEPOSIA (ozanimod) is indicated for the treatment of:
1. Relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults.
2. Moderately to severely active ulcerative colitis (UC) in adults.
For additional safety information, please see the full Prescribing InformationandMedication Guide.
For more information about Bristol Myers Squibb, visit us at BMS.com or follow us on LinkedIn, Twitter, YouTube, Facebook and Instagram.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211123005558/en/
Bristol Myers Squibb
Nov. 23, 2021 7:25 AM ET Bristol-Myers Squibb Company (BMY) By: Mamta Mayani, SA News Editor
https://www.nasdaq.com/market-activity/stocks/bmy/dividend-history
November 18, 2021 6:45 am ET
KEYTRUDA Is Now Approved for the Adjuvant Treatment of Patients With RCC at Intermediate-High or High Risk of Recurrence Following Nephrectomy, or Following Nephrectomy and Resection of Metastatic Lesions
KEYTRUDA Is the First Immunotherapy Approved for the Adjuvant Treatment of These Patients With RCC
KENILWORTH, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, today announced that the U.S. Food and Drug Administration (FDA) has approved KEYTRUDA, Merck’s anti-PD-1 therapy, for the adjuvant treatment of patients with renal cell carcinoma (RCC) at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions. The approval is based on data from the pivotal Phase 3 KEYNOTE-564 trial, in which KEYTRUDA demonstrated a statistically significant improvement in disease-free survival (DFS), reducing the risk of disease recurrence or death by 32% (HR=0.68 [95% CI, 0.53-0.87]; p=0.0010) compared to placebo. Median DFS has not been reached for either group.
“Despite decades of research, limited adjuvant treatment options have been available for earlier-stage renal cell carcinoma patients who are often at risk for recurrence. In KEYNOTE-564, pembrolizumab reduced the risk of disease recurrence or death by 32%, providing a promising new treatment option for certain patients at intermediate-high or high risk of recurrence,” said Dr. Toni K. Choueiri, director, Lank Center for Genitourinary Oncology, Dana-Farber Cancer Institute, and professor of medicine, Harvard Medical School. “With this FDA approval, pembrolizumab may address a critical unmet treatment need and has the potential to become a new standard of care in the adjuvant setting for appropriately selected patients.”
Immune-mediated adverse reactions, which may be severe or fatal, can occur in any organ system or tissue and can affect more than one body system simultaneously. Immune-mediated adverse reactions can occur at any time during or after treatment with KEYTRUDA, including pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, dermatologic reactions, solid organ transplant rejection, and complications of allogeneic hematopoietic stem cell transplantation. Important immune-mediated adverse reactions listed here may not include all possible severe and fatal immune-mediated adverse reactions. Early identification and management of immune-mediated adverse reactions are essential to ensure safe use of KEYTRUDA. Based on the severity of the adverse reaction, KEYTRUDA should be withheld or permanently discontinued and corticosteroids administered if appropriate. KEYTRUDA can also cause severe or life-threatening infusion-related reactions. Based on its mechanism of action, KEYTRUDA can cause fetal harm when administered to a pregnant woman. For more information, see “Selected Important Safety Information” below.
“KEYTRUDA is foundational for the treatment of patients with certain advanced cancers, and this approval marks the fourth indication for KEYTRUDA in earlier stages of cancer,” said Dr. Scot Ebbinghaus, vice president, clinical research, Merck Research Laboratories. “KEYTRUDA is now the first immunotherapy approved for the adjuvant treatment of certain patients with renal cell carcinoma. This milestone is a testament to our commitment to help more people living with cancer.”
In RCC, Merck has a broad clinical development program exploring KEYTRUDA, as monotherapy or in combination, as well as other investigational products across multiple settings and stages of RCC, including adjuvant and advanced or metastatic disease.
Data Supporting the Approval
KEYTRUDA demonstrated a statistically significant improvement in DFS in patients with RCC at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions compared with placebo (HR=0.68 [95% CI, 0.53-0.87]; p=0.0010). The trial will continue to assess overall survival (OS) as a secondary outcome measure.
In KEYNOTE-564, the median duration of exposure to KEYTRUDA was 11.1 months (range, 1 day to 14.3 months). Serious adverse reactions occurred in 20% of these patients receiving KEYTRUDA. Serious adverse reactions (≥1%) were acute kidney injury, adrenal insufficiency, pneumonia, colitis and diabetic ketoacidosis (1% each). Fatal adverse reactions occurred in 0.2% of those treated with KEYTRUDA, including one case of pneumonia. Adverse reactions leading to discontinuation occurred in 21% of patients receiving KEYTRUDA; the most common (≥1%) were increased alanine aminotransferase (1.6%), colitis and adrenal insufficiency (1% each). The most common adverse reactions (all grades ≥20%) in the KEYTRUDA arm were musculoskeletal pain (41%), fatigue (40%), rash (30%), diarrhea (27%), pruritus (23%) and hypothyroidism (21%).
About KEYNOTE-564
KEYNOTE-564 (ClinicalTrials.gov, NCT03142334) is a multicenter, randomized, double-blind, placebo-controlled Phase 3 trial evaluating KEYTRUDA as adjuvant therapy for RCC in 994 patients with intermediate-high or high risk of recurrence of RCC or M1 no evidence of disease (NED). Patients must have undergone a partial nephroprotective or radical complete nephrectomy (and complete resection of solid, isolated, soft tissue metastatic lesion[s] in M1 NED participants) with negative surgical margins for at least four weeks prior to the time of screening. Patients were excluded from the trial if they had received prior systemic therapy for advanced RCC. Patients with active autoimmune disease or a medical condition that required immunosuppression were also ineligible. The major efficacy outcome measure was investigator-assessed DFS, defined as time to recurrence, metastasis or death. An additional outcome measure was OS. Patients were randomized (1:1) to receive KEYTRUDA 200 mg administered intravenously every three weeks or placebo for up to one year until disease recurrence or unacceptable toxicity.
About KEYTRUDA® (pembrolizumab) Injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of patients with melanoma with involvement of lymph node(s) following complete resection.
Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [tumor proportion score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [combined positive score (CPS ≥1)] as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy. KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Carcinoma
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
Non-muscle Invasive Bladder Cancer
KEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with MSI-H central nervous system cancers have not been established.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC).
Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or GEJ (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
KEYTRUDA, in combination with chemotherapy, with or without bevacizumab, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
KEYTRUDA is indicated for the adjuvant treatment of patients with RCC at intermediate-high or high risk of recurrence following nephrectomy, or following nephrectomy and resection of metastatic lesions.
Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
More information is available by calling 855-257-3932 or visiting www.merckaccessprogram-keytruda.com.
For more information about our oncology clinical trials, visit www.merck.com/clinicaltrials.
For more information, visit www.merck.com
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf.
Source: Merck & Co., Inc.
Nov. 18, 2021 7:03 AM ET Merck & Co., Inc. (MRK) By: Dulan Lokuwithana, SA News Editor2 Comments
Pfizer’s XELJANZ® (Tofacitinib) Receives Marketing Authorization In The European Union For The Treatment Of Active Ankylosing Spondylitis
Thursday, November 18, 2021 - 01:30pm
NEW YORK--(BUSINESS WIRE)-- Pfizer Inc. (NYSE: PFE) announced today that the European Commission has approved XELJANZ® (tofacitinib) 5 mg twice daily for the treatment of adults with active ankylosing spondylitis (AS) who have responded inadequately to conventional therapy.
XELJANZ is the first and only oral Janus kinase (JAK) inhibitor approved for five indications in the European Union (EU), including in adults with moderate to severe active rheumatoid arthritis (RA), adults with active psoriatic arthritis (PsA), adults with moderately to severely active ulcerative colitis (UC), and patients 2 years of age or older with active polyarticular juvenile idiopathic arthritis (pcJIA) and juvenile psoriatic arthritis (PsA).
“Ankylosing spondylitis is a debilitating and chronic immuno-inflammatory disease that affects the lives of more than one million people in the EU who are in need of additional treatment options,” 1 said Ana Paula Carvalho, International Developed Markets Regional President, Inflammation & Immunology at Pfizer. “We are proud to make XELJANZ, a medicine that does not require an injection or an infusion, available to patients and their healthcare providers to help address this unmet medical need.”
The approval of XELJANZ for AS is based on data from a Phase 3, multicenter, randomized, double-blind, placebo-controlled study that evaluated the efficacy and safety of tofacitinib 5 mg twice daily versus placebo in 269 adult patients living with active AS. The study met its primary endpoint showing that at week 16, the percentage of patients achieving an Assessment in SpondyloArthritis International Society (ASAS) 20 response was significantly greater with tofacitinib (56.4%) versus placebo (29.4%) (p<0.0001). In addition, the percentage of ASAS40 response was significantly greater with tofacitinib (40.6%) versus placebo (12.5%) (p<0.0001), a key secondary endpoint of the study.2 ASAS20/40 are used for defining improvement or response to treatment.3 In general, the types of adverse drug reactions in patients with ankylosing spondylitis were consistent with the known safety profile of XELJANZ.
XELJANZ has been studied in more than 50 clinical trials worldwide, including more than 20 trials in RA patients, and prescribed to over 300,000 adult patients (the majority of whom were RA patients) since 2012.4,5,6
About XELJANZ® (tofacitinib)
XELJANZ (film-coated immediate release tablet, twice daily dosing) is the first and only oral JAK inhibitor approved in the European Union in five indications: adults with active ankylosing spondylitis (AS) who have responded inadequately to conventional therapy, adults with moderately to severely active rheumatoid arthritis (RA) after disease modifying antirheumatic drug (DMARD) failure or intolerance, adults with active PsA after DMARD failure or intolerance, adults with moderately to severely active ulcerative colitis (UC) who have had an inadequate response, lost response, or were intolerant to either conventional therapy or a biologic agent, and active polyarticular juvenile idiopathic arthritis (JIA) and juvenile psoriatic arthritis (PsA) in patients two years of age and older who have responded inadequately to previous therapy with DMARDs.
XELJANZ has been studied in more than 50 clinical trials worldwide, including more than 20 trials in RA patients, and prescribed to over 300,000 adult patients (the majority of whom were RA patients) worldwide since 2012.4,5,6 As the developer of tofacitinib, Pfizer is committed to advancing the science of JAK inhibition and enhancing understanding of tofacitinib through robust clinical development programs in the treatment of immune-mediated inflammatory conditions.
In June 2021, the Committee for Medicinal Products for Human (CHMP) Use of the European Medicines Agency adopted a recommendation from the Pharmacovigilance Risk Assessment Committee (PRAC) following the PRAC review of the co-primary endpoints data from ORAL Surveillance, which states that in patients over 65 years of age, patients who are current or past smokers, patients with other cardiovascular (CV) risk factors, and patients with other malignancy risk factors, XELJANZ should only be used if no suitable treatment alternatives are available. The updated summary of product characteristics reflecting the CHMP-endorsed PRAC recommendation is now effective and is applicable to all EU member states. PRAC’s review of the complete ORAL Surveillance data is ongoing.
In September 2021, the U.S. Food and Drug Administration (FDA) issued a Drug Safety Communication (DSC) related to XELJANZ/XELJANZ XR and two other arthritis medicines in the same drug class, based on its complete review of the ORAL Surveillance trial. More information is available on the DSC here. Pfizer is continuing to work with the FDA, EMA, and other regulatory agencies to update XELJANZ labeling across the world, in light of the ORAL Surveillance data.
U.S. FDA-APPROVED INDICATIONS
Rheumatoid Arthritis
Psoriatic Arthritis
Ulcerative Colitis
Polyarticular Course Juvenile Idiopathic Arthritis
In addition, to learn more, please visit us on www.Pfizer.com
View source version on businesswire.com: https://www.businesswire.com/news/home/20211118006167/en/
Source: Pfizer Inc.
Nov. 18, 2021 2:16 PM ET Pfizer Inc. (PFE) By: Dulan Lokuwithana, SA News Editor1 Comment
November 17, 2021
- Approval supported by data from two Phase 3 studies evaluating SKYRIZI in psoriatic arthritis patients, KEEPsAKE-1 and KEEPsAKE-2[1-3]
- These two Phase 3 studies evaluated SKYRIZI in adult patients with active psoriatic arthritis, and included patients who had responded inadequately or were intolerant to biologic therapy and/or non-biologic disease-modifying anti-rheumatic drugs (DMARDs)[1-6]
- In KEEPsAKE-1 and KEEPsAKE-2, statistical significance was achieved for the primary endpoint of ACR20 response for efficacy and multiple secondary endpoints, including physical function as measured by the Health Assessment Questionnaire Disability Index (HAQ-DI) and Minimal Disease Activity (MDA)[6]
- The safety profile of SKYRIZI in patients with psoriatic arthritis was consistent with the safety profile of SKYRIZI in plaque psoriasis patients[6]
- Psoriatic arthritis is a systemic inflammatory disease that impacts the skin and joints, affecting approximately 30 percent of patients with psoriasis[7-10]
NORTH CHICAGO, Ill., Nov. 17, 2021 /PRNewswire/ -- AbbVie (NYSE: ABBV) today announced that the European Commission (EC) has approved SKYRIZI® (risankizumab, 150 mg, subcutaneous injection at week 0, week 4 and every 12 weeks thereafter) alone or in combination with methotrexate (MTX), for the treatment of active psoriatic arthritis in adults who have had an inadequate response or who have been intolerant to one or more disease-modifying anti-rheumatic drugs (DMARDs). Marking the second indication for SKYRIZI, the Marketing Authorization will be valid in all member states of the European Union, as well as Iceland, Liechtenstein, Norway and Northern Ireland.
"People living with psoriatic arthritis struggle with psoriatic lesions and joint inflammation that causes swelling and pain. Reducing these symptoms may give people the ability to resume their daily activities and improve their quality of life," said Michael Severino, M.D., vice chairman and president, AbbVie. "We are excited by the EC approval of SKYRIZI for the treatment of adults with active psoriatic arthritis."
SKYRIZI received EC approval based on data from two Phase 3 clinical studies, KEEPsAKE-1 and KEEPsAKE-2.1-3,6 In these studies, SKYRIZI met the primary endpoint of ACR20 response at week 24 versus placebo, and ranked secondary endpoints including, but not limited to, improvements in several clinical manifestations of psoriatic arthritis such as physical function (as measured by the Health Assessment Questionnaire Disability Index [HAQ-DI]) and minimal disease activity (MDA) at week 24.1-3,6
Highlights from the pivotal Phase 3 program1-3,6
"Millions of people living with psoriatic arthritis are impacted by psoriatic lesions, joint pain, stiffness and fatigue," said Lars Erik Kristensen, M.D., Ph.D., consultant and head of science at the Parker Institute in Copenhagen Denmark, associate professor, Lund Sweden, SUS University Hospital. "As seen in this Phase 3 clinical trial program in psoriatic arthritis, SKYRIZI has the potential to be a valuable new treatment option, helping to improve the signs and symptoms of the disease."
The safety profile of SKYRIZI in psoriatic arthritis was consistent with the safety profile of SKYRIZI in plaque psoriasis, with no new safety risks observed.6 Through week 24, serious adverse events occurred in 2.5 percent and 4.0 percent of patients treated with SKYRIZI in KEEPsAKE-1 and KEEPsAKE-2, respectively, compared with 3.7 percent and 5.5 percent on placebo.1-3,6 Rates of serious infections were 1.0 and 0.9 percent in SKYRIZI-treated patients in KEEPsAKE-1 and KEEPsAKE-2, respectively, and 1.2 and 2.3 percent in patients who received placebo.1-3,6 The rates of adverse events leading to discontinuation of the study drug were 0.8 percent and 0.9 percent of patients treated with SKYRIZI in KEEPsAKE-1 and KEEPsAKE-2, respectively, compared with 0.8 percent and 2.3 percent on placebo.1-3,6 In KEEPsAKE-1, there was one death in the SKYRIZI group not related to the study drug per investigator.1,2,6 There were no deaths reported in KEEPsAKE-2.1,3,6
SKYRIZI (risankizumab) is part of a collaboration between Boehringer Ingelheim and AbbVie, with AbbVie leading development and commercialization globally.
About KEEPsAKE-1 and KEEPsAKE-21-6
KEEPsAKE-1 and KEEPsAKE-2 are both Phase 3, multicenter, randomized, double-blind, placebo-controlled studies designed to evaluate the safety and efficacy of SKYRIZI in adult patients with active psoriatic arthritis. KEEPsAKE-1 evaluated SKYRIZI in patients who had an inadequate response or intolerance to at least one DMARD. KEEPsAKE-2 evaluated SKYRIZI in patients who had an inadequate response or intolerance to biologic therapy and/or DMARDs. Patients were randomized to SKYRIZI 150 mg or placebo followed by SKYRIZI 150 mg at week 24. Patients randomized to SKYRIZI received four maintenance doses a year, following two initiation doses.
The primary endpoint for both studies was the achievement of ACR20 response at week 24. Ranked secondary endpoints included, but were not limited to, the achievement of MDA as well as the change from baseline in HAQ-DI at week 24. The studies are ongoing, and the long-term extension remains blinded to the original randomization and evaluates the long-term safety, tolerability and efficacy of SKYRIZI in patients who have completed the placebo-controlled period.
More information on these trials can be found at www.clinicaltrials.gov (KEEPsAKE-1: NCT03675308; KEEPsAKE-2: NCT03671148).
About SKYRIZI® (risankizumab)
SKYRIZI is an interleukin-23 (IL-23) inhibitor that selectively blocks IL-23 by binding to its p19 subunit.6,11 IL-23, a cytokine involved in inflammatory processes, is thought to be linked to a number of chronic immune-mediated diseases, including psoriasis.11 The approved dose for SKYRIZI is 150 mg (either as two 75 mg pre-filled syringe injections or one 150 mg pre-filled pen or pre-filled syringe injection), administered by subcutaneous injection at week 0 and 4, and every 12 weeks thereafter. The SKYRIZI 150 mg formulation was approved by the European Union in May 2021. Phase 3 trials of SKYRIZI in psoriasis, Crohn's disease, ulcerative colitis and psoriatic arthritis are ongoing.6,12-14
This is not a complete summary of all safety information. See SKYRIZI full summary of product characteristics (SmPC) at www.ema.europa.eu.
For more information about AbbVie, please visit us at www.abbvie.com
Nov. 17, 2021 2:50 AM ET AbbVie Inc. (ABBV)
By: Mamta Mayani, SA News Editor4 Comments
BioMarin Receives FDA Approval for VOXZOGO™ (vosoritide) for Injection, Indicated to Increase Linear Growth in Children with Achondroplasia Aged 5 and Up with Open Growth Plates
1st Therapeutic Treatment for Achondroplasia, Most Common Form of Disproportionate Short StatureInvestor Conference Call and Webinar to be Held on Friday, Nov. 19 at 11:30 AM EasternNov 19, 2021
SAN RAFAEL, Calif., Nov. 19, 2021 /PRNewswire/ -- BioMarin Pharmaceutical Inc. (NASDAQ: BMRN) today announced that the U.S. Food and Drug Administration (FDA) has granted accelerated approval to VOXZOGO™ (vosoritide) for Injection, indicated to increase linear growth in pediatric patients with achondroplasia five years of age and older with open epiphyses (growth plates). This indication is approved under accelerated approval based on an improvement in annualized growth velocity (AGV). Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory studies. To fulfill this post-marketing requirement, BioMarin intends to use the ongoing open-label extension studies compared to available natural history.
Voxzogo is the first FDA approved treatment for children with achondroplasia. In patients with achondroplasia, endochondral bone growth, an essential process by which bone tissue is created, is negatively regulated due to a gain of function mutation in fibroblast growth factor receptor 3 gene (FGFR3). Voxzogo, a C-type natriuretic peptide (CNP) analog, represents a new class of therapy, which acts as a positive regulator of the signaling pathway downstream of FGFR3 to promote endochondral bone growth.
"Voxzogo is a medical first that is rooted in BioMarin's focus on molecular genetics and targets the underlying cause of the condition. More than a decade of scientific research underpins the medical advance that Voxzogo represents. We thank the FDA for recognizing its value as the first therapeutic treatment option for children with achondroplasia," said Jean-Jacques Bienaimé, Chairman and Chief Executive Officer of BioMarin. "We extend our gratitude to the community, clinical investigators and the children and their families, who participated and continue to participate in our comprehensive clinical research program as we continue to investigate the full potential of vosoritide."
Vosoritide Safety
Safety and efficacy of Voxzogo in patients with achondroplasia were assessed in one 52–week, multi–center, randomized, double–blind, placebo–controlled, Phase 3 study. Transient decreases in blood pressure have been observed with Voxzogo. In the clinical study, 8 (13%) of 60 patients treated with Voxzogo had a total of 11 events of transient decreases in blood pressure compared to 3 (5%) of 61 patients on placebo, over a 52-week treatment period. Patients with significant cardiac or vascular disease or on anti-hypertensive medicine were excluded from the trial. To reduce the risk of a decrease in blood pressure and associated symptoms (dizziness, fatigue and/or nausea), patients should be well hydrated and have adequate food intake prior to administration.
The most common adverse reactions, occurring in greater than or equal to 5% of patients treated with Voxzogo and at a percentage greater than placebo in the Phase 3 study are injection site reactions (including redness, itching, swelling, bruising, rash, hives, pain), vomiting, joint pain, decreased blood pressure, gastroenteritis, diarrhea, dizziness, ear pain, influenza, fatigue, seasonal allergy, and dry skin. See Important Safety Information below and full Prescribing Information and Patient Prescribing Information for additional safety information.
Comprehensive Clinical Development Program
Voxzogo continues to be studied in a broad clinical development program in achondroplasia, and safety and efficacy are being further evaluated across different ages and over time. To date, 243 children with achondroplasia from eight countries have been enrolled in seven BioMarin clinical studies evaluating the safety and efficacy of vosoritide.
Patient Support for Accessing Voxzogo
To reach a BioMarin RareConnections® case manager, please call, toll-free, 1-866-906-6100 or e-mail support@biomarin-rareconnections.com. For more information about Voxzogo, please visit www.voxzogo.com. For additional information regarding this product, please contact BioMarin Medical Information at medinfo@bmrn.com.
For additional information, please visit www.biomarin.com.
What is VOXZOGO used for?
Nov. 19, 2021 11:01 AM ET BioMarin Pharmaceutical Inc. (BMRN)
By: Jonathan M Block, SA News Editor7 Comments
Basel, 19 November 2021 - Roche (SIX: RO, ROG; OTCQX: RHHBY) today announced that the European Commission (EC) has granted conditional marketing authorisation for Gavreto® (pralsetinib) as a monotherapy for the treatment of adults with rearranged during transfection (RET) fusion-positive advanced non-small cell lung cancer (NSCLC) not previously treated with a RET inhibitor. Gavreto is the first and only precision medicine approved in the European Union (EU) for the first-line treatment of people with RET fusion-positive advanced NSCLC.1
“Today’s approval represents an important step forward in delivering precision medicine to people with RET fusion-positive advanced non-small cell lung cancer, for whom treatment options have historically been limited," said Levi Garraway, M.D., Ph.D., Roche’s Chief Medical Officer and Head of Global Product Development. “By using cancer genomic profiling upfront, healthcare professionals may identify specific genetic alterations that predict clinical benefit of targeted treatment options like Gavreto in the first-line setting.”
The approval is based on results of the ongoing phase I/II ARROW study, in which Gavreto led to durable responses in people with advanced RET fusion-positive NSCLC.2 In 75 treatment-naïve patients, Gavreto demonstrated an overall response rate (ORR) of 72.0% (95% CI: 60.4%, 81.8%), and median duration of response (DOR) was not reached (NR) (95% CI: 9.0 months, NR).2 In 136 patients who had previously received platinum-based chemotherapy, Gavreto demonstrated an ORR of 58.8% (95% CI: 50.1%, 67.2%), and median DOR was 22.3 months (95% CI: 15.1 months, NR).2 Gavreto was also generally well-tolerated, with a low rate of treatment discontinuation; common grade 3-4 adverse reactions were neutropenia (reported in 20.1% of patients), anaemia (17.6%) and hypertension (16.1%).2
Approximately 37,500 people are diagnosed with RET fusion-positive NSCLC worldwide each year; the disease often affects people with minimal to no history of smoking, and who are typically younger than the average person diagnosed with lung cancer.3,4,5 Roche is committed to providing a tailored treatment option for every person with lung cancer, no matter how rare or difficult-to-treat their type of disease. Gavreto in RET fusion-positive advanced NSCLC, along with Alecensa® (alectinib) in ALK-positive advanced NSCLC and Rozlytrek® (entrectinib) in ROS1-positive advanced NSCLC, is part of Roche’s growing portfolio of precision medicines. Together, they offer personalised treatment options for almost one in ten people with advanced NSCLC, and biomarker testing is the most effective way to identify those people who may benefit.6
Beyond NSCLC, RET alterations are also key disease drivers in other cancer types, such as thyroid cancers. Gavreto has shown activity across multiple solid tumour types, reflecting tumour-agnostic potential.7 It is approved by the U.S. Food and Drug Administration (FDA) for the treatment of adults with metastatic RET fusion-positive NSCLC, and for the treatment of adult and paediatric patients 12 years of age and older with advanced RET-altered thyroid cancers. Gavreto is also approved in Canada, mainland China and Switzerland. In the EU, a submission for RET-altered thyroid cancers is planned. Regulatory submissions for advanced RET fusion-positive NSCLC and RET-altered thyroid cancers are also underway in multiple countries worldwide.
Blueprint Medicines and Roche are co-developing Gavreto globally, with the exception of certain territories in Asia, including China.* Blueprint Medicines and Genentech, a wholly owned member of the Roche Group, are commercialising Gavreto in the US and Roche has exclusive commercialisation rights for Gavreto outside of the US, with the exception of certain territories in Asia, including China.*
About the ARROW study8
ARROW is an ongoing phase I/II, open-label, first-in-human study designed to evaluate the safety, tolerability and efficacy of Gavreto, administered orally in people with rearranged during transfection (RET) fusion-positive non-small cell lung cancer (NSCLC), RET-mutant medullary thyroid cancer, RET fusion-positive thyroid cancer and other RET-altered solid tumours. ARROW is being conducted at multiple sites across the United States, Europe and Asia.
About Gavreto® (pralsetinib)
Gavreto is a once-daily, oral precision medicine designed to selectively target rearranged during transfection (RET) alterations, including fusions and mutations, regardless of the tissue of origin. Preclinical data have shown that Gavreto inhibits primary RET fusions and mutations that cause cancer in subsets of patients, as well as secondary RET mutations predicted to drive resistance to treatment. Blueprint Medicines and Roche are co-developing Gavreto for the treatment of people with various types of RET-altered cancers.
https://www.gavreto.com/index.html
For more information, please visit www.roche.com.
Blueprint Medicines, Gavreto and associated logos are trademarks of Blueprint Medicines Corporation.
Nov. 19, 2021 9:46 AM ET Roche Holding AG (RHHBY)
By: Mamta Mayani, SA News Editor
MAINZ, GERMANY, November 19, 2021 — BioNTech SE (Nasdaq: BNTX, “BioNTech” or “the Company”) today announced that the U.S. Food and Drug Administration (FDA) granted Fast Track Designation for BNT111, an investigational cancer immunotherapy for the potential treatment of advanced melanoma. BNT111 is the lead product candidate from BioNTech’s fully owned FixVac platform that utilizes a fixed combination of mRNA-encoded, tumor-associated antigens aiming to trigger a strong and precise immune response against cancer. The vaccine candidate is currently being investigated in a Phase 2 trial (EudraCT No.: 2020-002195-12; NCT04526899) in patients with anti-PD-1-refractory/relapsed unresectable Stage III or IV melanoma.
“The Fast Track Designation underlines the potential of our FixVac platform to address current treatment challenges of pre-treated and immune checkpoint blocker experienced melanoma with limited standard of care therapy options left. This is an important step to pave the way for this versatile new treatment approach in a high medical need setting,” said Özlem Türeci, M.D., Co-founder and Chief Medical Officer of BioNTech. “With the Fast Track status and support by the FDA, we aim to expedite the further development of the BNT111 program to provide a new therapeutic option for patients with life-threatening, hard-to-treat melanoma.”
Fast Track is a process designed to facilitate the development, and expedite the review, of new drugs and vaccines that are intended to treat or prevent serious conditions that have the potential to address an unmet medical need. The FDA’s decision is based on available preclinical and clinical data showing the potential of BNT111 to overcome current limitations in the treatment of inoperable therapy-resistant advanced-stage melanoma. With the Fast Track Designation, the development of BNT111 can benefit from more frequent engagement with the FDA, which will support the collection of appropriate data needed to accelerate BNT111’s development.
The ongoing randomized Phase 2 trial (BNT111-01) in patients with anti-PD1-refractory/relapsed unresectable Stage III or IV melanoma investigates BNT111 in combination with Libtayo® (cemiplimab), an anti-PD-1 monoclonal antibody being co-developed by Regeneron and Sanofi. The BNT111-01 trial which is conducted in collaboration with Regeneron is enrolling a total of 180 patients into three treatment arms in the United States, the United Kingdom, Australia, Spain, Germany, Italy and Poland. This trial seeks to support initial data reported from the ongoing Phase 1 Lipo-MERIT monotherapy dose escalation trial (EudraCT No. 2013-001646-33; NCT02410733; DOI: 10.1038/s41586-020-2537-9) that demonstrated a favorable safety profile and anti-tumor responses of BNT111 alone and in combination with immune checkpoint inhibitor therapy in patients with advanced melanoma.
About BNT111
BNT111 is an intravenous therapeutic cancer immunotherapy candidate encoding a fixed set of four cancer-specific antigens optimized for immunogenicity and delivered as RNA-lipoplex formulation. Based on current data from an exploratory interim analysis of the ongoing Phase 1 Lipo-MERIT monotherapy dose escalation trial, published in Nature, BNT111 induces novel antigen-specific anti-tumor immune responses and enhances pre-existing immune responses against the encoded melanoma-associated antigens (NY-ESO-1, MAGE-A3, tyrosinase, and TPTE), which are expressed in more than 90% of cutaneous melanomas. BNT111 is one of four clinical-stage FixVac product candidates within BioNTech’s development pipeline.
BNT111 is not yet authorized by any regulatory authority and the safety and efficacy has not yet been established.
For more information, please visit www.BioNTech.de.
Nov. 19, 2021 7:16 AM ET
By: Mamta Mayani, SA News Editor
NOVEMBER 16, 2021 • INVESTOR RELATIONS
CAMBRIDGE, Mass., Nov. 16, 2021 (GLOBE NEWSWIRE) -- Biogen Inc. (Nasdaq: BIIB) today announced that the European Commission (EC) has granted marketing authorization for VUMERITY® (diroximel fumarate) to treat adults with relapsing-remitting multiple sclerosis (MS). VUMERITY is a next-generation fumarate that offers the convenience of an oral medication with the established efficacy and well-characterized safety of TECFIDERA® (dimethyl fumarate). Globally, an estimated 2.8 million people live with MS, with more than 1 million people in Europe living with the disease.1,2
“The approval of VUMERITY provides a new oral treatment option with low gastrointestinal discontinuation rates that may help patients to start and adhere to treatment,” said Simon Faissner, M.D., PhD, Assistant Professor at the Department of Neurology, Ruhr-University Bochum. “This medication allows for MS patients in the EU to be treated without having to think about dietary restrictions or when to take a dose in relation to mealtimes which, when treating a chronic disease, may provide patients with additional flexibility in their daily lives.”
The EC’s approval of VUMERITY is based on data from pharmacokinetic bridging studies comparing VUMERITY and TECFIDERA to establish bioequivalent exposure of monomethyl fumarate, the active metabolite, and relied in part on the well-established long-term efficacy and safety profile of TECFIDERA. The approval was also based on findings from EVOLVE-MS-2, a large, randomized, double-blind, five-week, multi-center Phase 3 study to evaluate the gastrointestinal (GI) tolerability of VUMERITY compared to TECFIDERA in patients with relapsing-remitting MS. In EVOLVE-MS-2, the rate of overall treatment discontinuation was lower in participants treated with VUMERITY compared to those treated with TECFIDERA (1.6% compared to 6%, respectively). The difference in the discontinuation rates due to GI tolerability was 0.8% for VUMERITY compared to 4.8% for TECFIDERA. Additionally, flushing was reported in 32.8% of VUMERITY-treated patients and in 40.6% of TECFIDERA treated patients. There were no serious events of flushing or discontinuations due to flushing in the study.
“This approval is a significant step forward in improving treatment adherence for people living with relapsing MS, which can make a meaningful difference on treatment outcomes impacting their daily lives,” said Maha Radhakrishnan, M.D., Chief Medical Officer at Biogen. “The authorization of VUMERITY in the EU brings people with MS a new oral treatment option to meet their individual preferences and needs, with well-established efficacy and a positive GI tolerability profile that continues to be evaluated in the real-world setting.”
VUMERITY was first approved by the U.S. Food and Drug Administration in October 2019 and is also approved in Great Britain and Switzerland. Since its launch in the U.S., real-world data have reinforced the positive GI tolerability profile of VUMERITY and confirmed that the experience demonstrated in clinical trials is consistent with clinical practice.3 Biogen continues to file regulatory submissions in other countries.
About VUMERITY® (diroximel fumarate)
VUMERITY is an oral fumarate with a distinct chemical structure from TECFIDERA (dimethyl fumarate), approved in the U.S. for the treatment of relapsing forms of multiple sclerosis in adults, to include clinically isolated syndrome, relapsing-remitting disease and active secondary progressive disease. Once in the body, VUMERITY rapidly converts to monomethyl fumarate, the same active metabolite of dimethyl fumarate providing similar efficacy and safety profiles.
VUMERITY is contraindicated in patients with known hypersensitivity to diroximel fumarate, dimethyl fumarate or to any of the excipients of VUMERITY; and in patients taking dimethyl fumarate. Serious side effects for VUMERITY are based on data from dimethyl fumarate (which has the same active metabolite as VUMERITY) and include anaphylaxis and angioedema, progressive multifocal leukoencephalopathy, which is a rare opportunistic viral infection of the brain that has been associated with death or severe disability, a decrease in mean lymphocyte counts during the first year of treatment, herpes zoster and other serious infections, liver injury and flushing. The most common adverse events, obtained using data from dimethyl fumarate (which has the same active metabolite as VUMERITY), were flushing, abdominal pain, diarrhea and nausea.
Please click here for Important Safety Information and full Prescribing Information, including Patient Information for VUMERITY in the U.S., or visit your respective country’s product website.
About TECFIDERA® (dimethyl fumarate)
TECFIDERA, a treatment for relapsing forms of multiple sclerosis (MS) in adults, is the most prescribed oral medication for relapsing MS in the world and has been shown to reduce the rate of MS relapses, slow the progression of disability and impact the number of MS brain lesions, while demonstrating a well-characterized safety profile in people with relapsing forms of MS. TECFIDERA is approved in 69 countries, and more than 530,000 patients have been treated with it, representing more than 1,000,000 patient-years of exposure across clinical trial use and patients prescribed TECFIDERA.4
TECFIDERA is contraindicated in patients with a known hypersensitivity to dimethyl fumarate or any of the excipients of TECFIDERA. Serious side effects include anaphylaxis and angioedema, and cases of progressive multifocal leukoencephalopathy, a rare opportunistic viral infection of the brain which has been associated with death or severe disability, have been seen with TECFIDERA patients in the setting of prolonged lymphopenia although the role of lymphopenia in these cases is uncertain. Other serious side effects include a decrease in mean lymphocyte counts during the first year of treatment, herpes zoster and other serious infections, liver injury and flushing. In clinical trials, the most common adverse events associated with TECFIDERA were flushing, abdominal pain, diarrhea and nausea.
For information on TECFIDERA prescribing information in the EU, please visit: https://www.ema.europa.eu/en/medicines/human/EPAR/tecfidera. Please click here for Important Safety Information and full Prescribing Information, including Patient Information for TECFIDERA in the U.S., or visit your respective country’s product website.
To learn more, please visit www.biogen.com
Nov. 16, 2021 9:48 AM ETBiogen Inc. (BIIB)By: Dulan Lokuwithana, SA News Editor1 Comment
Nov. 17, 2021 10:03 AM ETAstraZeneca PLC (AZN)
Q3: 2021-11-12 Earnings Summary
TranscriptSlidesEPS of - misses by $0.91 | Revenue of $9.87B (49.98% Y/Y) beats by $286.00M
WILMINGTON, Del.--(BUSINESS WIRE)-- AstraZeneca (AZN) has been granted Fast Track Designation in the United States (US) for the development of LOKELMA® (sodium zirconium cyclosilicate) to reduce arrhythmia-related cardiovascular outcomes in patients on chronic hemodialysis with recurrent hyperkalemia (HK). The designation is based on the potential of LOKELMA to reduce serious adverse CV outcomes in this patient population, addressing a significant unmet medical need. This is being investigated in the ongoing Phase III DIALIZE-Outcomes trial.
HK is a prevalent condition in patients with chronic kidney disease (CKD) and heart failure (HF), affecting 24% to 48% of patients with moderate to advanced (stage 3-4) CKD and/or HF, and it remains a burden once patients are on chronic hemodialysis. In patients with end-stage renal disease (ESRD) receiving chronic hemodialysis, HK has been associated with an increased risk of all-cause and CV mortality, and hospitalizations.
The Food and Drug Administration’s (FDA) Fast Track program is designed to accelerate the development and review of new medicines for the treatment of serious conditions where there is an unmet treatment need.
Mene Pangalos, Executive Vice President, BioPharmaceuticals R&D, AstraZeneca said: “The DIALIZE-Outcomes trial is the first ever cardiovascular outcomes trial with a potassium binder in hemodialysis and has the potential to transform standard of care for these patients. The FDA decision demonstrates the importance of this trial, which will offer important information on LOKELMA’s ability to reduce potentially deadly cardiovascular complications associated with hyperkalemia for patients on chronic hemodialysis.”
The DIALIZE-Outcomes trial is part of the CRYSTALIZE evidence program, which is comprised of over 50 clinical and real-world evidence studies researching the potential benefit of LOKELMA in the management of HK across the cardiorenal spectrum. The DIALIZE-Outcomes trial is currently underway with results expected in 2024.
LOKELMA is a highly selective, oral potassium-removing agent currently approved in the US, EU, Canada, Hong Kong, China, Russia, Japan and other countries for the treatment of HK. In 2020, the FDA and the European Commission (EC) approved label updates in the US and EU, respectively, to include a dosing regimen specifically to treat HK in patients with ESRD on chronic hemodialysis.
INDICATION AND LIMITATION OF USE
LOKELMA® is indicated for the treatment of hyperkalemia in adults.
LOKELMA® should not be used as an emergency treatment for life-threatening hyperkalemia because of its delayed onset of action.
PLEASE READ FULL PRESCRIBING INFORMATION.
LOKELMA
LOKELMA® (sodium zirconium cyclosilicate) is an insoluble, non-absorbed sodium zirconium silicate, formulated as a powder for oral suspension, that acts as a highly selective potassium-removing medicine. It is administered orally, is odorless, tasteless and stable at room temperature. LOKELMA is indicated for the treatment of hyperkalemia in adults including patients on chronic hemodialysis.
For more information, please visit www.astrazeneca-us.com
View source version on businesswire.com: https://www.businesswire.com/news/home/20211117006015/en/
https://seekingalpha.com/symbol/AZN

Nov 15, 2021United States
6Milvexian demonstrated efficacy and no increase in bleeding across doses with no major bleeds in the milvexian arms, when compared with enoxaparin, for postoperative venous thromboembolism (VTE) prevention in patients undergoing elective total knee replacement (TKR) surgery
AXIOMATIC-TKR is the first of two studies to read out from the Phase 2 milvexian program, which together will inform the design and dose regimens of the Phase 3 program
Data simultaneously published in The New England Journal of Medicine and presented at the American Heart Association Scientific Sessions 2021
Milvexian is being developed by The Bristol Myers Squibb-Janssen Collaboration
RARITAN, NJ, November 15, 2021 – The Janssen Pharmaceutical Companies of Johnson & Johnson in collaboration with Bristol Myers Squibb today announced results from the Phase 2 AXIOMATIC-TKR study, which showed investigational oral milvexian reduced the risk of postoperative venous thromboembolism (VTE) in a dose dependent manner without increasing the risk of bleeding compared with enoxaparin in patients undergoing total knee replacement (TKR) surgery. These data were presented today at a Late-Breaking Science session at the American Heart Association (AHA) Scientific Sessions 2021 and simultaneously published in The New England Journal of Medicine (NEJM).
“This study establishes proof-of-principle for milvexian as a differentiated antithrombotic agent,” said Jeffrey Weitz, M.D., Professor of Medicine & Biochemistry and Biomedical Sciences at McMaster University and Executive Director of the Thrombosis and Atherosclerosis Research Institute.[i] “Furthermore, the consistently low rates of bleeding across a 16-fold range of milvexian doses suggest that it has a wide therapeutic window, which opens the opportunity to explore milvexian across a broad range of patients including those for whom factor Xa inhibitors are underutilized or not indicated.”
The trial met both of its pre-specified proof-of-principle requirements: the dose response for efficacy with twice-daily milvexian was significant (p<0.001), and the 12% rate of VTE with combined twice-daily doses of milvexian was significantly lower (p<0.0001) than the prespecified benchmark rate of 30%.
A total of 1,242 patients were randomized to receive one of seven regimens of oral milvexian given twice or once-daily or to receive 40 mg of subcutaneous enoxaparin once-daily. The assignment to milvexian or enoxaparin was open label, but the milvexian dose assignment was blinded. Treatment was given for 10-14 days. More information can be found on www.clinicaltrials.gov (NCT03891524).
About Milvexian*
Milvexian is a potential first-in-class oral factor XIa (FXIa) inhibitor (anti-thrombotic) for the prevention and treatment of major thrombotic conditions. Phase 2 TKR data, complementing human genetic, epidemiologic, and preclinical evidence, now provide further support for the hypothesis that inhibiting FXIa can reduce the risk of vascular events without increasing the risk of bleeding. The milvexian Phase 2 clinical trial program consists of two Phase 2 studies: AXIOMATIC-TKR (NCT03891524) evaluating milvexian in TKR surgery and AXIOMATIC-SSP (NCT03766581) evaluating milvexian for secondary stroke prevention (SSP).
*Milvexian is an investigational agent and has not been approved for use in any country, for any indication.
About The Bristol Myers Squibb-Janssen Collaboration
Bristol Myers Squibb and Janssen Pharmaceuticals, Inc., two leaders in thrombosis treatment and care, are collaborating to develop and commercialize milvexian, a potentially first-in-class oral factor XIa (FXIa) inhibitor, with the goal of improving upon the benefit-risk profile of existing anticoagulants. With extensive expertise and unparalleled leadership in cardiovascular treatments spanning decades, Bristol Myers Squibb and Janssen share a long-standing commitment to improving treatment options for patients with life-threatening cardiovascular conditions.
Learn more at https://www.janssen.com/.
https://clinicaltrials.gov/ct2/show/NCT04844424
Nov. 16, 2021 8:53 AM ET Bristol-Myers Squibb Company (BMY), JNJ
By: Jonathan M Block, SA News Editor
Data from a phase 2 study of Bristol-Myers Squibb's (NYSE:BMY) and Johnson & Johnson's (NYSE:JNJ) milvexian found the candidate reduced the risk of postoperative venous thromboembolism ("VTE") without increasing the risk of bleeding compared with enoxaparin in patients undergoing total knee replacement.
17 November 2021
Issued: London UK and San Francisco US
GlaxoSmithKline plc (LSE/NYSE: GSK) and Vir Biotechnology, Inc. (Nasdaq: VIR) today announced US government contracts totalling approximately $1 billion[1] (USD) to purchase sotrovimab, an investigational monoclonal antibody for the early treatment of COVID-19, which the US Food and Drug Administration (FDA) granted Emergency Use Authorization (EUA) in May 2021. GSK will supply these doses to the US government by December 17, 2021, enabling further expanded nationwide access to sotrovimab for patients.
In addition to the doses that will be supplied this year, the US government will have the option to purchase additional doses through March 2022.
Including the contracts announced today, GSK and Vir have received binding agreements for the sale of more than 750,000 doses of sotrovimab worldwide, with additional doses reserved through other agreements including the previously announced Joint Procurement Agreement with the European Commission.
Sotrovimab is an FDA EUA authorised investigational single-dose intravenous (IV) infusion SARS-CoV-2 monoclonal antibody. Under the EUA, sotrovimab can be used for the treatment of mild-to-moderate COVID-19 in adults and paediatric patients (12 years of age and older weighing at least 40 kg) with positive test results for COVID-19, and who are at high risk for progression to severe COVID-19, including hospitalisation or death.
About sotrovimab
Sotrovimab is an investigational SARS-CoV-2 neutralising monoclonal antibody. The antibody binds to an epitope on SARS-CoV-2 that is shared with SARS-CoV-1 (the virus that causes SARS), indicating that the epitope is highly conserved, which may make it more difficult for resistance to develop. Sotrovimab, which incorporates Xencor’s Xtend™ technology, has also been designed to achieve high concentration in the lungs to ensure optimal penetration into airway tissues affected by SARS-CoV-2 and to have an extended half-life.
About global access to sotrovimab
About the sotrovimab clinical development programme
Sotrovimab in the United States
The following is a summary of information for sotrovimab. Healthcare providers in the U.S. should review the Fact Sheets for information about the authorised use of sotrovimab and mandatory requirements of the EUA. Please see the FDA Letter of Authorization, full Fact Sheet for Healthcare Providers, and full Fact Sheet for Patients, Parents, and Caregivers.
Sotrovimab has been authorized by the U.S. FDA for the emergency use described below. Sotrovimab is not FDA-approved for this use.
Sotrovimab is authorized only for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of sotrovimab under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.
Authorized Use
The U.S. Food and Drug Administration (FDA) has issued an Emergency Use Authorization (EUA) to permit the emergency use of the unapproved product sotrovimab for the treatment of mild-to-moderate coronavirus disease 2019 (COVID-19) in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death.
Limitations of Authorized Use
Sotrovimab is not authorized for use in patients:
Benefit of treatment with sotrovimab has not been observed in patients hospitalized due to COVID‑19. SARS-CoV-2 monoclonal antibodies may be associated with worse clinical outcomes when administered to hospitalized patients with COVID‑19 requiring high flow oxygen or mechanical ventilation.
About the Vir and GSK Collaboration
In April 2020, Vir and GSK entered into a collaboration to research and develop solutions for coronaviruses, including SARS-CoV-2, the virus that causes COVID-19. The collaboration uses Vir’s proprietary monoclonal antibody platform technology to accelerate existing and identify new anti-viral antibodies that could be used as therapeutic or preventive options to help address the current COVID-19 pandemic and future outbreaks. The companies will leverage GSK’s expertise in functional genomics and combine their capabilities in CRISPR screening and artificial intelligence to identify anti-coronavirus compounds that target cellular host genes. They will also apply their combined expertise to research SARS-CoV-2 and other coronavirus vaccines.
For further information please visit www.gsk.com/about-us.
For more information, please visit www.vir.bio.
Nov. 17, 2021 6:12 AM ETGlaxoSmithKline plc (GSK), VIR
By: Mamta Mayani, SA News Editor1 Comment
17 November 2021
Issued: London UK
GlaxoSmithKline (GSK) plc today announced that the European Commission has approved Nucala (mepolizumab), a monoclonal antibody that targets interleukin-5 (IL-5), for use in three additional eosinophil-driven diseases. This authorisation follows positive opinions recommended by the Committee for Medicinal Products for Human Use and authorises mepolizumab for use as an add on treatment in hypereosinophilic syndrome (HES), eosinophilic granulomatosis with polyangiitis (EGPA) and chronic rhinosinusitis with nasal polyps (CRSwNP).
Eosinophil-driven diseases are inflammatory conditions associated with elevated levels of eosinophils, a type of white blood cell. CRSwNP is a condition in which patients develop soft tissue growths called nasal polyps which can cause chronic symptoms such as nasal obstruction, loss of smell and discharge. HES and EGPA are both potentially life-threatening rare diseases arising from inflammation in various tissues. The inflammation can cause a range of symptoms which are frequently severe. Mepolizumab is the first approved targeted treatment for EGPA and the first anti-IL-5 biologic treatment for patients with HES or CRSwNP in Europe. These approvals make mepolizumab the only treatment approved in Europe for use in four eosinophil-driven diseases as mepolizumab is already approved for use in Europe as an add-on treatment for patients aged six years and older with severe eosinophilic asthma (SEA).
About Nucala (mepolizumab)
First approved in 2015 for SEA, Nucala (mepolizumab) is the first-in-class monoclonal antibody that targets IL-5. It is believed to work by preventing IL-5 from binding to its receptor on the surface of eosinophils, reducing blood eosinophils and maintaining them within normal levels. The mechanism of action for mepolizumab has not been definitively established.
Nucala is available as a solution in a prefilled pen or syringe or as a powder that comes in a vial and is made up into an injection. The patient (adults and adolescents aged 12 years and older) or caregiver can use Nucala prefilled pen or syringe themselves if their healthcare professional determines that it is appropriate, and the patient or caregiver are trained in injection techniques, whereas the vial is only for use by a healthcare professional.
Mepolizumab has been developed for the treatment of diseases that are driven by inflammation caused by eosinophils. It has been studied in over 4,000 patients in 41 clinical trials across several eosinophilic indications and has been approved in the US, the EU and in over 25 other markets, as an add-on maintenance treatment for patients with SEA. Mepolizumab is approved in 17 markets, including the EU US, for paediatric use in SEA from ages six to 17 years of age, with approval in an additional seven markets for use in patients with SEA aged 12-17 years. The first approval for mepolizumab in CRSwNP was granted by the FDA in July 2021. Mepolizumab is approved for use in patients with EGPA in a total of 14 markets including the US, Japan and Canada. Mepolizumab was first approved for use in HES in the US in September 2020 and approvals have since then been granted in an additional 5 markets. Mepolizumab is currently in clinical development for chronic obstructive pulmonary disorder (COPD) and It is not currently approved for use in COPD anywhere in the world.
For further information please visit www.gsk.com/about-us.
Nov. 17, 2021 6:34 AM ETGlaxoSmithKline plc (GSK)By: Mamta Mayani, SA News Editor
PUBLISHED15 November 2021
Positive Phase IIa results from the EPICCURE trial demonstrated that AZD8601 met the primary endpoint of safety and tolerability in patients with heart failure.
In the trial, researchers injected mRNA encoding vascular endothelial growth factor (VEGF-A) – known as AZD8601 – directly into the myocardium of patients undergoing elective coronary artery bypass surgery (CABG) surgery. AZD8601 is being co-developed with Moderna.
In the study of 11 patients, seven were treated with AZD8601 and four received placebo injections. Trends were observed in the three exploratory efficacy endpoints: left ventricular ejection fraction (LVEF); NT-proBNP (a biomarker which measures the level of a hormone and is elevated in patients with heart failure); and functional patient reported outcomes, compared with placebo.
VEGF-A is a paracrine factor important for new blood vessel formation. In addition, it has been shown to stimulate progenitor cell division, descendants of stem cells that when stimulated differentiate to create specialised cell types that contribute to repair and regeneration of the heart.
Mene Pangalos, Executive Vice President, BioPharmaceuticals R&D, AstraZeneca, said: “Over one billion heart cells can be lost during a heart attack. These early results indicate the potential of mRNA therapeutics in stimulating VEGF-A production to provide reparative and disease-modifying options for patients with heart failure and other ischaemic vascular diseases.”
Stéphane Bancel, Chief Executive Officer, Moderna, said: “mRNA is a compelling therapeutic modality because of its ability to act locally and transiently, while driving dose-dependent protein expression. The results presented today are a result of pushing new boundaries in the treatment of cardiovascular and other ischaemic vascular diseases and address serious unmet needs with the goal of improving patients’ lives.”
Results from the EPICCURE trial were presented on 15 November 2021 at the American Heart Association’s Scientific Sessions 2021.
EPICCURE
EPICCURE is a Phase IIa randomised, double-blinded, multi-centre, placebo-controlled study in patients with stable coronary artery disease and moderately decreased LVEF who are undergoing CABG surgery. The primary endpoint was safety and tolerability of AZD8601. Seven patients received AZD8601 and four received placebo; all 11 were followed for six months to assess primary safety and exploratory efficacy endpoints. There were no deaths or treatment-related serious adverse events and no infections were observed related to the treatment of AZD8601. Exploratory efficacy endpoints included LVEF, NT-proBNP, and functional patient reported outcomes resulted in a positive trend for the treated group. Although the data is limited to show significant effect, it provides opportunity for AstraZeneca to move to further trials with AZD8601.
AZD8601
AZD8601 is a novel, investigational mRNA-based therapy, with mRNA formulated in a citrate saline buffer in the absence of lipid encapsulation, that encodes for VEGF-A for local administration in patients undergoing CABG.
mRNA is responsible for carrying genetic instructions transcribed from DNA, which cells then translate to produce proteins. Proteins are responsible for directing the body’s biological functions. Moderna’s pioneering mRNA therapeutics are designed to trigger the cellular machinery to produce specific proteins. In this application, AZD8601 may enable the delivery of mRNA instructions to spur the production of VEGF-A protein.
AstraZeneca and Moderna Therapeutics collaboration
AstraZeneca and Moderna Therapeutics have an exclusive agreement to discover, develop and commercialise pioneering messenger RNA therapeutics for the treatment of serious cardiovascular, metabolic and renal diseases as well as cancer. AstraZeneca brings unparalleled depth of understanding of cardiovascular disease biology and the discovery and development of medicines in this area, while Moderna brings advanced mRNA technology and the ability to engineer multiple different mRNAs into therapeutic proteins.
Please visit astrazeneca.com
November 15, 2021 at 8:35 AM ESTPDF Version
Phase 2a study met the primary endpoint of safety and tolerability; numerical trends observed in endpoints in the heart failure efficacy domains compared with placebo, including increase in LVEF and patient reported outcomes
Results support further investigation of AZD8601 for efficacy and safety in future larger studies
AZD8601 is being co-developed with AstraZeneca
CAMBRIDGE, Mass.--(BUSINESS WIRE)--Nov. 15, 2021-- Moderna, Inc. (Nasdaq: MRNA), a biotechnology company pioneering messenger RNA (mRNA) therapeutics and vaccines today announced positive data from the AstraZeneca-led Phase 2 (EPICCURE) study evaluating the use of an mRNA therapeutic that encodes for vascular endothelial growth factor-A (VEGF-A) (AZD8601) in patients undergoing coronary artery bypass grafting (CABG). These data were presented today at the American Heart Association’s Scientific Sessions 2021 annual meeting.
The Phase 2 study met the primary endpoint of safety and tolerability of AZD8601. In the study of 11 patients, seven were treated with AZD8601 VEGF-A mRNA and four received placebo injections. Numerical trends were observed in endpoints in the heart failure efficacy domains compared with placebo, including increase in left ventricular ejection fraction (LVEF) and patient reported outcomes. In addition, all seven patients treated with AZD8601 had NT-proBNP levels below heart failure (HF) limit at 6 months follow-up compared to one of four patients treated with placebo.
Nov. 15, 2021 10:03 AM ET Moderna, Inc. (MRNA) AZNBy: Mamta Mayani, SA News Editor
Press Release_EU decision UC_ENG_vFINAL for release
Mechelen, Belgium; 15 November 2021; 16.45 CET; Galapagos NV (Euronext & Nasdaq: GLPG) announced today that the European Commission has granted marketing authorization for Jyseleca® (filgotinib 200mg tablets) for the treatment of adult patients with moderately to severely active ulcerative colitis (UC).
The European Commission approved an additional indication for Jyseleca, an oral, once-daily, JAK1 preferential inhibitor, for adult patients with moderately to severely active UC who have had an inadequate response with, lost response to, or were intolerant to either conventional therapy or a biologic agent. The Commission based its decision on data from the Phase 2b/3 SELECTION program, which evaluated filgotinib as an induction and maintenance therapy in the patient population now included in the label. The SELECTION trial was published in The Lancet1.
“We are very pleased that the European Commission has approved Jyseleca as a treatment for people with UC. This decision further supports the efficacy and safety profile of Jyseleca, which was studied in over 1,250 UC patients. Our focus now is on making this treatment available as rapidly as possible to physicians and UC patients across the European Union,” said Onno van de Stolpe, CEO of Galapagos.
Professor Laurent Peyrin-Biroulet, Professor of Gastroenterology and Head of the Inflammatory Bowel Disease (IBD) group at the Gastroenterology Department, University Hospital of Nancy, France, and Principal Investigator for the SELECTION study, said: “Despite available treatments for managing UC, there is still a need for new and innovative therapies like Jyseleca. UC is an incurable and disabling disease; in severe cases we aim to keep patients out of hospital and reduce the need for surgical procedures such as colectomies. Overall, our goals are to manage the symptoms that have a significant, negative impact on a person’s overall well-being, to be able to stop the use of steroids and to improve the daily life of patients. In the SELECTION study we observed filgotinib’s tablet form was easily administered, provided significantly greater corticosteroid-free clinical remission from symptoms compared to placebo and was well tolerated by patients.”
UC is a life-long condition commonly starting in late adolescence or early adulthood and characterized by inflammation of the mucosal lining of the colon and rectum. As an increasingly prevalent disease, UC has a significant negative impact on the quality of life of more than 2 million2 people across Europe. Despite current treatments, many patients experience faecal urgency, incontinence, recurring bloody diarrhea, the need to empty their bowels frequently, abdominal pain, poor sleep, and fatigue. Patients with severe UC can be hospitalized and require life changing surgery. In addition to the physical symptoms, there is also a significant psychological impact associated with UC.
Luisa Avendano, CEO at the European Federation of Crohn’s and Ulcerative Colitis Associations (EFCCA) said: “The impact that living with UC can have on a person’s life cannot be underestimated. It is important for each individual to speak to their doctor about what approach will work best to help them manage their condition. Having new approved treatment choices is therefore very important, and at EFCCA we are pleased to see new options being made available.”
About filgotinib
Filgotinib is approved and marketed as Jyseleca (200mg and 100mg tablets) in the European Union, Great Britain, and Japan for the treatment of adults with moderate to severe active rheumatoid arthritis (RA) who have responded inadequately or are intolerant to one or more disease modifying anti-rheumatic drugs (DMARDs). Filgotinib may be used as monotherapy or in combination with methotrexate (MTX). Filgotinib is also approved and marketed as Jyseleca (200mg tablets) in the European Union for the treatment of adult patients with moderately to severely active UC who have had an inadequate response with, lost response to, or were intolerant to either conventional therapy or a biologic agent. The European Summary of Product Characteristics for filgotinib, which includes contraindications and special warnings and precautions, is available at www.ema.europa.eu. The interview form from the Japanese Ministry of Health, Labour and Welfare is available at www.info.pmda.go.jp. The Great Britain Summary of Product Characteristics for filgotinib can be found at www.medicines.org.uk/emc and the Northern Ireland Summary of Product Characteristics for filgotinib can be found at www.emcmedicines.com/en-GB/northernireland. Applications have been submitted to the UK’s Medicines and Healthcare products Regulatory Agency (MHRA), and Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) for the treatment of adults with moderately to severely active UC and are currently under review. A global Phase 3 program with filgotinib is ongoing in Crohn’s Disease. More information about clinical trials can be accessed at https://www.clinicaltrials.gov. Filgotinib is not approved in any other countries.
Jyseleca® is a trademark of Galapagos NV and Gilead Sciences, Inc. or its related companies.
https://www.glpg.com/filgotinib
About the filgotinib collaboration
Gilead and Galapagos NV are collaborative partners in the global development and commercialization of filgotinib. Galapagos will be responsible for the commercialization of filgotinib in Europe (transition anticipated to be completed by end of 2021), while Gilead will remain responsible for filgotinib outside of Europe, including in Japan, where filgotinib is co-marketed with Eisai.
More information at www.glpg.com.
Nov. 15, 2021 11:07 AM ET Galapagos NV (GLPG)
By: Jonathan M Block, SA News Editor
Nov 12, 2021
- If approved, more than 1,500 children would be eligible for a medicine that can treat the underlying cause of their disease for the first time -
LONDON--(BUSINESS WIRE)--Nov. 12, 2021-- Vertex Pharmaceuticals Incorporated (Nasdaq: VRTX) today announced that the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion for the label extension of KAFTRIO® (ivacaftor/tezacaftor/elexacaftor) in a combination regimen with ivacaftor, for the treatment of cystic fibrosis (CF) in patients ages 6 through 11 years old who have at least one F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene.
“Today marks an important milestone for the treatment of children with CF in Europe. If approved, KAFTRIO (ivacaftor/tezacaftor/elexacaftor) in a combination regimen with ivacaftor will offer physicians a new treatment option for these young patients to help combat this life-shortening condition as early as possible,” said Carmen Bozic, M.D., Executive Vice President, Global Medicines Development and Medical Affairs, and Chief Medical Officer at Vertex.
“The clinical data for ivacaftor/tezacaftor/elexacaftor plus ivacaftor in people with CF ages 6 through 11 with eligible CF genotypes demonstrated improvements in lung function, sweat chloride and respiratory symptoms and a safety and tolerability profile consistent with that observed in patients ages 12 years and older,” said Professor Marcus A. Mall, M.D., Head of the Department of Pediatric Respiratory Medicine, Immunology and Critical Care Medicine at Charité University Medical Center Berlin. “This medicine has already made a big impact on the lives of eligible people ages 12 years and above in Europe. The CF community is now looking forward to it being available for younger patients too, to enable treatment as early as possible in life.”
About KAFTRIO® (ivacaftor/tezacaftor/elexacaftor) in A Combination Regimen With Ivacaftor
In people with certain types of mutations in the CFTR gene, the CFTR protein is not processed or folded normally within the cell, and this can prevent the CFTR protein from reaching the cell surface and functioning properly. KAFTRIO® (ivacaftor/tezacaftor/elexacaftor) in combination with ivacaftor is an oral medicine designed to increase the quantity and function of the CFTR protein at the cell surface. Elexacaftor and tezacaftor work together to increase the amount of mature protein at the cell surface by binding to different sites on the CFTR protein. Ivacaftor, which is known as a CFTR potentiator, is designed to facilitate the ability of CFTR proteins to transport salt and water across the cell membrane. The combined actions of ivacaftor, tezacaftor and elexacaftor help hydrate and clear mucus from the airways.
KAFTRIO® (ivacaftor/tezacaftor/elexacaftor) in combination with ivacaftor is approved in the European Union for the treatment of cystic fibrosis (CF) in patients ages 12 years and older who have at least one copy of the F508del mutation in the CFTR gene.
For complete product information, please see the Summary of Product Characteristics that can be found on www.ema.europa.eu.
For company updates and to learn more about Vertex's history of innovation, visit https://global.vrtx.com/
View source version on businesswire.com: https://www.businesswire.com/news/home/20211112005560/en/
Source: Vertex Pharmaceuticals Incorporated
Nov. 12, 2021 8:14 AM ET Vertex Pharmaceuticals Incorporated (VRTX) By: Aakash Babu, SA News Editor
LATE BREAKING PRESENTATION AT AMERICAN HEART ASSOCIATION ANNUAL MEETING OF LANDMARK PHASE 3 TRIAL OF REXLEMESTROCEL-L IN CHRONIC HEART FAILURE Trial Results Highlighted Reduction in Cardiovascular Mortality, Heart Attacks and Strokes, with Greatest Effect Seen in Setting of Inflammation Melbourne, Australia; November 15, and New York, USA; November 14, 2021: Mesoblast Limited (ASX:MSB; Nasdaq:MESO), global leader in allogeneic cellular medicines for inflammatory diseases, today announced that results from the randomized, controlled Phase 3 trial of rexlemestrocel-L in 565 patients with New York Heart Association (NYHA) class II and class III chronic heart failure (CHF) with reduced ejection fraction (HFrEF) were presented as a late breaking presentation at the American Heart Association (AHA) annual Scientific Sessions during a featured program titled ‘Building on the Foundations of Treatment: Advances in Heart Failure Therapy’. The trial’s co-principal investigator Dr Emerson Perin, Medical Director of Texas Heart Institute, and Clinical Professor, Baylor College of Medicine, presented new results from the landmark study showing a significant relationship between presence of systemic inflammation as quantified by high-sensitivity C-reactive protein (hs-CRP) and treatment benefit with rexlemestrocel-L on risk of cardiovascular mortality, heart attacks or strokes. In a release by the American Heart Association, Dr Perin said: “Cell therapy has the potential to change how we treat heart failure. This study addresses the inflammatory aspects of heart failure, which go mostly untreated, despite significant pharmaceutical and device therapy development.” Key findings of pre-specified outcomes were: • A single dose of rexlemestrocel-L on top of standard of care reduced the incidence of heart attacks or strokes by 65% across all 537 NYHA class II or class III treated patients compared with standard of care alone, p=0.0011 • Across 301 NYHA class II and class III treated patients with high levels of inflammation (hsCRP ≥ 2mg/L), rexlemestrocel-L reduced the incidence of heart attacks or strokes by 79% compared with standard of care alone, p<0.0011 • In NYHA class II patients with high levels of inflammation (hs-CRP ≥ 2mg/L), a single dose of rexlemestrocel-L reduced the incidence of cardiovascular death by 80% compared with standard of care alone, p=0.0051 • Compared with standard of care alone, a single dose of rexlemestrocel-L on top of standard of care reduced the incidence of cardiovascular death, heart attacks or strokes by 33% across all 537 NYHA class II or class III patients, p=0.021, and by 45% in the 301 patients with high levels of inflammation (hs-CRP ≥ 2mg/L), p=0.0121 • Compared with standard of care alone, addition of rexlemestrocel-L did not further reduce the frequency of hospitalization for worsening HF symptoms as previously reported In a release from Texas Heart Institute, President & CEO Dr. Joseph G. Rogers said that the Texas Heart Institute prides itself upon consistently striving to uncover The Next First in cardiovascular discovery and treatment.
About Mesoblast Mesoblast is a world leader in developing allogeneic (off-the-shelf) cellular medicines for the treatment of severe and life-threatening inflammatory conditions. The Company has leveraged its proprietary mesenchymal lineage cell therapy technology platform to establish a broad portfolio of late-stage product candidates which respond to severe inflammation by releasing anti-inflammatory factors that counter and modulate multiple effector arms of the immune system, resulting in significant reduction of the damaging inflammatory process. Mesoblast has a strong and extensive global intellectual property portfolio with protection extending through to at least 2041 in all major markets. The Company’s proprietary manufacturing processes yield industrial-scale, cryopreserved, off-the-shelf, cellular medicines. These cell therapies, with defined pharmaceutical release criteria, are planned to be readily available to patients worldwide. Mesoblast has completed Phase 3 trials of rexlemestrocel-L for advanced chronic heart failure and chronic low back pain. Remestemcel-L is being developed for inflammatory diseases in children and adults including steroid refractory acute graft versus host disease and moderate to severe acute respiratory distress syndrome. Two products have been commercialized in Japan and Europe by Mesoblast’s licensees, and the Company has established commercial partnerships in Europe and China for certain Phase 3 assets. Mesoblast has locations in Australia, the United States and Singapore and is listed on the Australian Securities Exchange (MSB) and on the Nasdaq (MESO). For more information, please see www.mesoblast.com,
Nov. 15, 2021 3:10 AM ET Mesoblast Limited (MESO)
By: Mamta Mayani, SA News Editor1 Comment
11/12/21PDF Version
-- CHMP recommendation based on positive results from N-MOmentum, the largest clinical trial ever conducted in NMOSD to date --
-- In the trial, UPLIZNA was shown to reduce the risk of relapse by 77% compared to placebo in anti-aquaporin-4 antibody positive (AQP4-IgG+) adult patients --
-- Treatment is given via infusion every six months following an initial two treatments spaced two weeks apart --
-- UPLIZNA is under review by the European Medicines Agency (EMA) and was approved by the U.S. Food and Drug Administration (FDA) and the Japanese Ministry of Health, Labour and Welfare as a targeted CD19 B-cell depleting antibody for adults with AQP4-IgG+ NMOSD --
-- UPLIZNA received Breakthrough Therapy and Orphan Drug designation in the United States (U.S.) and Orphan Drug designation Japan --
DUBLIN--(BUSINESS WIRE)--Nov. 12, 2021-- Horizon Therapeutics plc (Nasdaq: HZNP) today announced that the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) has adopted a positive opinion recommending marketing authorization for UPLIZNA® (inebilizumab) as a monotherapy for the treatment of adult patients with neuromyelitis optica spectrum disorder (NMOSD) who are anti-aquaporin-4 immunoglobulin G seropositive (AQP4-IgG+). UPLIZNA has been granted orphan designation by the European Commission (EC). That designation is subject to additional review by the Committee for Orphan Medicinal Products.
UPLIZNA has not been approved for commercial use in the European Union (EU). Based on the CHMP recommendation, a decision by the EC, if granted, would authorize marketing approval in the EU. This centralized marketing authorization would be valid in all EU Member States, as well as in Iceland, Liechtenstein and Norway.
“Today’s positive CHMP opinion is a significant milestone in Horizon’s global growth and long-term commitment to address critical needs of those living with rare, autoimmune and severe inflammatory diseases,” said Tim Walbert, chairman, president and chief executive officer, Horizon. "In Europe, people living with NMOSD currently have very limited targeted treatment options and we look forward to advancing care for these patients who live daily with unpredictable attacks, pain and disability.”
UPLIZNA was approved by the FDA in June 2020 and by the Japanese Ministry of Health, Labour and Welfare in March 2021 as a targeted CD19 B-cell depleting antibody for adult patients with AQP4-IgG+ NMOSD, to reduce the risk of attacks. UPLIZNA is the only approved NMOSD therapy in the U.S. that has demonstrated a clinically relevant and durable effect on delaying worsening of disability, with a significant reduction in hospitalization. In Japan, Mitsubishi Tanabe Pharma Corporation has the rights for development and commercialization of UPLIZNA. Long-term UPLIZNA treatment has been shown to be well tolerated and provide a sustained reduction in NMOSD attack risk for four or more years.1
NMOSD is a rare, severe autoimmune disease where the body’s defense cells (B-cells) start to attack the optic nerve, spinal cord and brain stem.2,3 NMOSD is often misdiagnosed as multiple sclerosis (MS) and primarily damages the optic nerve(s) and spinal cord, causing permanent blindness, muscle weakness and paralysis.4 NMOSD is characterized by unpredictable attacks and severe disability that often occurs following the first attack, accumulating with each subsequent relapse.5 Preventing these attacks is the primary goal for disease management.6 UPLIZNA works by depleting B-cells in a targeted manner, and patients need only take it once every six months, via infusion, following an initial two treatments spaced two weeks apart.7
AQP4 is a membrane protein that is predominantly found in the central nervous system. It has a protective effect by regulating the water homeostasis, a self-regulating process, to maintain the stability of the physiological function in the body.8 A defining feature of NMOSD (seen in approximately 80% of patients) is the presence of serum autoantibodies which work against AQP4, leading to severe attacks in the central nervous system, resulting in substantial and often permanent disability.9
Globally, the prevalence of NMOSD is approximately 0.5–4/100,000 people.10, 11 Europe has an estimated disease population of at least 7,300 patients, 75-80% of which are AQP4-IgG+. Each year, approximately 370 new patients in Europe are diagnosed with NMOSD.12
The CHMP positive opinion was granted based on the data from the N-MOmentum clinical development program (NCT02200770), which found that UPLIZNA monotherapy reduced the risk of relapse by 77% compared to placebo in AQP4-IgG+NMOSD adult patients.7
About the N-MOmentum clinical program21
N-MOmentum was a multicenter, double-blind, randomized placebo-controlled Phase 2/3 clinical trial that was conducted in 25 countries. Participants were randomly assigned (3:1 with a ratio AQP4+: n=213 and AQP4-: n=17) to receive 300 mg of intravenous UPLIZNA or placebo. The study consisted of a 28-week randomized-controlled period (RCP), followed by an optional open-label period (OLP) of at least two years. The OLP lasted approximately four years, producing long-term data for a subset of patients (n=94 AQP4+ patients).
The trial Primary Endpoint was
The trial Secondary Endpoints were
For more information on how we go to incredible lengths to impact lives, please visit www.horizontherapeutics.com
View source version on businesswire.com: https://www.businesswire.com/news/home/20211112005564/en/
Source: Horizon Therapeutics plc
Nov. 12, 2021 7:58 AM ET
Horizon Therapeutics Public Limited Company (HZNP)
By: Aakash Babu, SA News Editor
November 12, 2021- Data presented further support Compugen's earlier findings that PVRIG plays a distinct role within the DNAM axis, with the potential to trigger robust immune responses in the tumor microenvironment
- Data demonstrate that PVRIG has a unique dominant expression in early memory (stem-like) T cells, in contrast to TIGIT and PD-1, and its ligand PVRL2 is highly expressed across dendritic cell subtypes, compared to PD-L1 and PVR, the ligand of TIGIT
- Translational data showing induction of activated dendritic cell markers in serum of two patients who clinically responded to treatment of COM701 in combination with nivolumab
- Data suggest that blockade of PVRIG/PVRL2 may enhance stem-like memory T cells - dendritic cells interaction potentially resulting in increased T cell expansion, differentiation, and infiltration in inflamed and in less 'inflamed' tumors
- Management will discuss the results as part of the Q3 earnings call, today at 8:30am ET
HOLON, Israel, Nov. 12, 2021 /PRNewswire/ -- Compugen Ltd. (Nasdaq: CGEN), a clinical-stage cancer immunotherapy company and a leader in predictive target discovery, today announced the presentation of new translational preliminary data detailing the differentiated profile of PVRIG compared to TIGIT and PD-1 as a novel checkpoint in the DNAM axis, supporting its potential role as a dominant checkpoint involved in stem-like memory T cells and dendritic cell (DCs) interaction at the 36th Annual Meeting of the Society for Immunotherapy of Cancer (SITC), being held on November 10-14, 2021.
"We believe evidence is growing to consistently demonstrate that PVRIG is a novel and differentiated checkpoint with a potential unique role in cancer immunotherapy" said Eran Ophir, Ph.D., Vice President of Research and Drug Discovery at Compugen. "For cancer immunotherapy to work, T cells are needed at the tumor site and recent studies suggest that early-memory (stem-like) T cells and DCs play an important role in this process. Here we show for the first-time preliminary data demonstrating greater induction of activated DC markers in the serum of two patients responding to our potentially first in class anti-PVRIG antibody, COM701, in combination with nivolumab compared to non-responders, potentially because of DC- T cell interaction. This preliminary data is in line with our recent scientific finding showing that PVRL2, the ligand of PVRIG, is abundantly expressed across DCs types, while PVRIG, measured by both gene and protein expression, is uniquely and dominantly expressed on early memory cells in contrast to TIGIT and PD-1."
Anat Cohen-Dayag, Ph.D., President and CEO of Compugen, added, "These new preliminary data further support our earlier findings that PVRIG plays a distinct role within the DNAM axis which we believe is important for triggering robust immune responses in the tumor microenvironment. PVRIG blockade may lead to key mechanistic differences as compared to other DNAM axis members, namely TIGIT and PD-1, with the potential to enhance T cell proliferation and tumor infiltration to address both inflamed and less inflamed tumor types where current checkpoint inhibitors have not shown success. With our potentially first-in-class anti- PVRIG antibody, COM701, we are uniquely positioned to target the DNAM axis in combination with TIGIT and PD-1/L1 inhibitors and look forward to continued translation of these scientific learnings across our ongoing clinical programs."
Key findings from the poster presentation titled, "Novel DNAM-1 axis member, PVRIG, is potentially a dominant checkpoint involved in stem-like memory T cells – dendritic cell interaction," presented by Zoya Alteber PhD, Associate Director, Research and Drug Discovery at Compugen include:
Nov. 12, 2021 8:18 AM ET Compugen Ltd. (CGEN)BMY
By: Dulan Lokuwithana, SA News Editor2 Comments
November 12, 2021 at 4:32 PM EST
ROCKVILLE, Md., Nov. 12, 2021 /PRNewswire/ --
REGENXBIO Inc. (Nasdaq: RGNX) today announced additional positive interim data from the ongoing Phase II AAVIATE® trial and the ongoing Phase II ALTITUDE™ trial of RGX-314 using in-office suprachoroidal delivery for the treatment of wet age-related macular degeneration (wet AMD) and diabetic retinopathy (DR) without center-involved diabetic macular edema (CI-DME), respectively. The results were presented at the American Academy of Ophthalmology (AAO) 2021 Annual Meeting by Robert L. Avery, M.D., Founder of California Retina Consultants and Research Foundation.
"We are pleased to share this initial data from Cohort 2 of the AAVIATE trial which provides encouraging evidence of the emerging clinical profile of RGX-314 for the treatment of wet AMD using suprachoroidal delivery. In the data reported today, RGX-314 was observed to be well tolerated in Cohort 2, with stable visual acuity and retinal thickness as well as a meaningful reduction in anti-VEGF treatment burden at six months," said Steve Pakola, M.D., Chief Medical Officer of REGENXBIO. "We look forward to providing additional updates from the program."
"These initial results from patients in Cohort 2 of the AAVIATE trial at six months after suprachoroidal administration of RGX-314 reinforce the potential impact that RGX-314 could have on the overall clinical management of patients with wet AMD," said Dr. Avery. "I am encouraged by the six-month data in Cohort 2 and look forward to reviewing further data from Cohorts 1-3 and from the higher dose level in Cohorts 4 and 5."
Study Design and Safety Update in Phase II AAVIATE Trial of RGX-314 for the Treatment of Wet AMD Using Suprachoroidal Delivery
AAVIATE is a multi-center, open-label, randomized, active-controlled, dose-escalation trial that will evaluate the efficacy, safety and tolerability of suprachoroidal delivery of RGX-314 using the SCS Microinjector®. Twenty patients in Cohort 1 were randomized to receive RGX-314 at a dose level of 2.5x1011 genomic copies per eye (GC/eye) versus monthly 0.5 mg ranibizumab intravitreal injection at a 3:1 ratio. Twenty patients in Cohort 2 were randomized to receive RGX-314 at a dose level of 5x1011 GC/eye through two injections versus monthly 0.5 mg ranibizumab intravitreal injection at a 3:1 ratio. Cohort 3 is evaluating RGX-314 at the same dose level as Cohort 2 in 20 patients who are neutralizing antibody (NAb) positive. Enrollment is ongoing in two additional cohorts (Cohorts 4 and 5) to evaluate RGX-314 at a third dose level of 1x1012 GC/eye. Cohort 4 will enroll 15 patients who will be dosed with RGX-314 and Cohort 5 will evaluate the same dose level of RGX-314 in 20 patients who are NAb positive. Patients do not receive prophylactic immune suppressive corticosteroid therapy before or after administration of RGX-314.
As of November 4, 2021, RGX-314 was reported to be well tolerated across 50 patients dosed in Cohorts 1-3. Four serious adverse events (SAEs) were reported in four patients, all of which were considered not related to RGX-314.1 For the total group of Cohorts 1 and 2, all common treatment emergent adverse events (TEAEs) through 6 months in the study eye were mild, and included conjunctival hemorrhage, worsening of wet AMD, dry eye, episcleritis, and conjunctival hyperemia. Mild intraocular inflammation observed on slit-lamp examination was reported at similar incidence across both dose levels in Cohorts 1 and 2, with four out of 15 patients in Cohort 1 and three out of 15 patients in Cohort 2. All cases of inflammation in both cohorts were resolved within days to weeks on topical corticosteroids.
Summary of Data for Cohort 2 in Phase II AAVIATE Trial of RGX-314 for the Treatment of Wet AMD at Six Months
In Cohort 2, patients dosed with RGX-314 demonstrated stable Best Corrected Visual Acuity (BCVA) and central retinal thickness (CRT) at 6 months. Fifteen patients in Cohort 2 dosed with RGX-314 had a mean BCVA change of -0.1 letters (95% Confidence Interval: -3.8, 3.6) when measured from Day 1 (at Screening) and +0.2 letters (-2.7, 3.1) when measured from Week 1 (prior to Randomization). These patients also demonstrated stable central retinal thickness (CRT), with a mean change of -33 µm (-71, 5) at six months from Day 1. Ten control patients receiving monthly injections of ranibizumab in Cohorts 1 and 2 had a mean BCVA change at six months of +4.0 letters (-0.5, 8.5) when measured from Day 1 and +1.3 letters (-2.2, 4.8) when measured from Week 1. Patients receiving monthly injections of ranibizumab had a mean change of CRT of -12 µm (-33, 8) at six months from Day 1.
There was a meaningful reduction in anti-vascular endothelial growth factor (anti-VEGF) treatment burden in patients following administration of RGX-314 compared to the mean annualized injection rate during the 12 months prior to administration. Patients in Cohort 2 received a mean of 1.3 injections over six months following administration of RGX-314, which represents a 71.8% reduction in anti-VEGF treatment burden. Six out of 15 patients (40%) in Cohort 2 received no anti-VEGF injections over six months following RGX-314 administration. In these patients, visual acuity and CRT was observed to be stable from Day 1 over six months, with a mean change of BCVA of +1.0 letters (-3.8, 5.8), and a mean change of CRT of +8 µm (-9.2, 24.2).
Summary of Visual Acuity Data for Cohort 1 in Phase II ALTITUDE Trial of RGX-314 for the Treatment of Diabetic Retinopathy at Three Months
REGENXBIO shared additional data from Cohort 1 of the ongoing ALTITUDE trial for the treatment of DR without CI-DME using in-office suprachoroidal delivery, supporting the positive initial data previously reported at the American Society of Retina Specialists (ASRS) Annual Meeting in October 2021. At three months, 15 patients dosed with 2.5x1011 GC/eye of RGX-314 demonstrated stable BCVA of +2.6 letters, while five patients in the observational control arm demonstrated stable BCVA of -0.4 letters.
Data presented today are available on the "Presentations and Publications" section of the REGENXBIO website at www.regenxbio.com.
About RGX-314
RGX-314 is being investigated as a potential one-time treatment for wet AMD, diabetic retinopathy, and other chronic retinal conditions. RGX-314 consists of the NAV AAV8 vector, which encodes an antibody fragment designed to inhibit vascular endothelial growth factor (VEGF). RGX-314 is believed to inhibit the VEGF pathway by which new, leaky blood vessels grow and contribute to the accumulation of fluid in the retina.
REGENXBIO is advancing research in two separate routes of administration of RGX-314 to the eye, through a standardized subretinal delivery procedure as well as delivery to the suprachoroidal space. REGENXBIO has licensed certain exclusive rights to the SCS Microinjector® from Clearside Biomedical, Inc. to deliver gene therapy treatments to the suprachoroidal space of the eye.
View original content to download multimedia:https://www.prnewswire.com/news-releases/regenxbio-presents-additional-positive-interim-data-from-trials-of-rgx-314-in-wet-amd-and-diabetic-retinopathy-using-suprachoroidal-delivery-at-aao-2021-301423126.html
SOURCE REGENXBIO Inc.
Nov. 12, 2021 5:13 PM ETREGENXBIO Inc. (RGNX), CLSDBy: Dulan Lokuwithana, SA News Editor2 Comments
November 10, 2021
-- The EVER-132-001 trial met its primary endpoint with an objective response rate of 38.8% --
FOSTER CITY, Calif., & SHANGHAI--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today reported that the Everest Medicines (HKEX 1952.HK) sponsored Phase 2b EVER-132-001 study of sacituzumab govitecan (marketed as Trodelvy® in the United States) met its primary endpoint of overall response rate (ORR) in metastatic triple-negative breast cancer (TNBC).
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20211110006163/en/
EVER-132-001 is a single-arm, multi-center Phase 2b registrational study evaluating sacituzumab govitecan in 80 patients enrolled in China for the treatment of adults with unresectable locally advanced or metastatic TNBC who have received two or more prior systemic therapies, at least one for metastatic disease. The results demonstrated an ORR of 38.8% (CI: 95%) as evaluated by an Independent Review Committee. The safety profile of sacituzumab govitecan was similar to that reported in prior studies, and no new safety signals were identified.
The primary endpoint measured ORR according to RECIST v1.1 by an Independent Review Committee. The results were consistent with results demonstrated in the global Phase 3 ASCENT study. Gilead and Everest Medicines are engaged in a joint partnership for the development and commercialization of sacituzumab govitecan in Asia.
In May 2020, the Center for Drug Evaluation (CDE) of the China National Medical Products Administration (NMPA) granted priority review to the Biologics License Application (BLA) for sacituzumab govitecan for adult patients with unresectable locally advanced or metastatic TNBC who have received two or more prior systemic therapies, at least one of them for metastatic disease.
“These topline results confirm that sacituzumab govitecan has the potential to help change the treatment outlook for people in China living with mTNBC,” said Yang Shi, Chief Medical Officer for Oncology at Everest Medicines. “These data, along with the benefit seen in the global ASCENT study, support its potential as a novel treatment for patients who currently have extremely limited options.”
Kerry Blanchard, MD, PhD, CEO of Everest Medicines added, “With the goal of delivering treatment to as many patients as quickly as possible, we are building the commercialization team for Trodelvy in preparation for product launch in China.”
“These data from the clinical trial of sacituzumab govitecan in China are extremely encouraging,” said Bill Grossman, MD, PhD, Senior Vice President, Oncology Clinical Research, Gilead Sciences. “We are confident in the potential of Trodelvy to help more women around the world.”
The Trodelvy U.S. Prescribing Information has a BOXED WARNING for severe or life-threatening neutropenia and severe diarrhea; see below for Important Safety Information.
About Sacituzumab Govitecan
Sacituzumab govitecan (Trodelvy) is a first-in-class antibody and topoisomerase inhibitor conjugate directed to the Trop-2 receptor, a protein overexpressed in multiple types of epithelial tumors, including metastatic TNBC and metastatic urothelial cancer (UC), where high expression is associated with poor survival and relapse. Trodelvy is approved in second-line metastatic TNBC in multiple countries worldwide, including Australia, Canada, Great Britain, Switzerland and the United States based on data submitted from the Phase 3 ASCENT study. Review is also underway in the European Union and Singapore and China through our partner Everest Medicines. Trodelvy is also approved for use in metastatic UC in the United States and continues to be developed for potential use in other TNBC and metastatic UC populations. It is also being developed as an investigational treatment for hormone receptor-positive/human epidermal growth factor receptor 2-negative (HR+/HER2-) metastatic breast cancer and metastatic non-small cell lung cancer. Additional evaluation across multiple solid tumors is also underway.
Under a licensing agreement with Gilead Sciences, Inc., Everest Medicines has exclusive rights to develop, register, and commercialize sacituzumab govitecan for all cancer indications in Greater China, South Korea, and certain Southeast Asian countries. In October 2020, sacituzumab govitecan was included in the updated 2020 China Guidelines for the Standardized Diagnosis and Treatment of Advanced Breast Cancer, compiled by the Breast Cancer Expert Committee of the National Cancer Control Center, the Breast Cancer Professional Committee of the Chinese Anti-Cancer Association, and the Cancer Drug Clinical Research Professional Committee of the Chinese Anti-Cancer Association.
The Ministry of Food and Drug Safety (MFDS) in South Korea has granted Fast Track Designation and Orphan Drug Designation (ODD) to SG for the treatment of metastatic TNBC. In addition, Everest announced in January 2021 that it submitted a New Drug Application (NDA) to the Health Sciences Authority (HSA) of Singapore for SG for the treatment of patients with metastatic TNBC who have received two prior therapies, at least one for metastatic disease. That application is currently under review.
In the United States, Trodelvy is indicated for the treatment of:
For more information, please visit its website at www.everestmedicines.com.
U.S. Prescribing Information for Trodelvy including BOXED WARNING, is available at www.gilead.com.
Trodelvy, Gilead and the Gilead logo are trademarks of Gilead Sciences, Inc., or its related companies.
For more information about Gilead, please visit the company’s website at www.gilead.com, f
View source version on businesswire.com: https://www.businesswire.com/news/home/20211110006163/en/
Source: Gilead Sciences, Inc.
Nov. 11, 2021 9:29 AM ET Gilead Sciences, Inc. (GILD) By: Dulan Lokuwithana, SA News Editor3 Comments
112021/11
05:57
12 November 2021
Download
Download pdf
Bagsværd, Denmark, 12 November 2021 – Novo Nordisk today announced that the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) has adopted a positive opinion recommending a label extension for the existing marketing authorisation for Ozempic®, a once-weekly glucagon-like peptide-1 (GLP-1) analogue, to introduce a new dose of 2.0 mg. Ozempic® is currently approved in the EU in 0.5 mg and 1.0 mg doses for the treatment of type 2 diabetes in adults.
The positive opinion is based on the results from the SUSTAIN FORTE trial. In the trial, people treated with semaglutide 2.0 mg achieved a statistically significant and superior reduction in HbA1c at week 40 compared to semaglutide 1.0 mg. In the trial, both doses of semaglutide appeared to have a safe and well-tolerated profile. The most common adverse events were gastrointestinal.
“The recommendation for the EU label extension of Ozempic® with the 2.0 mg dose is an important step towards offering additional options to people with type 2 diabetes who need treatment intensification to achieve their individualised glycaemic targets,” said Martin Lange, executive vice president, Development at Novo Nordisk.
Novo Nordisk expects a final approval by the European Commission within approximately two months, and Ozempic® 2.0 mg will be launched in the EU in the first half of 2022. The label expansion for semaglutide 2.0 mg is under regulatory review in the US. About the SUSTAIN clinical programme
The SUSTAIN clinical development programme for once-weekly subcutaneous semaglutide injection currently comprises 11 phase 3 global clinical trials, including a cardiovascular outcomes trial, involving more than 11,000 adults with type 2 diabetes. For more information about the SUSTAIN Forte trial, please read the headline results here.
Novo Nordisk is a leading global healthcare company, founded in 1923 and headquartered in Denmark. Our purpose is to drive change to defeat diabetes and other serious chronic diseases such as obesity and rare blood and endocrine disorders. We do so by pioneering scientific breakthroughs, expanding access to our medicines , and working to prevent and ultimately cure disease. Novo Nordisk employs about 47 , 0 00 people in 80 countries and markets its products in around 170 countries. Novo Nordisk's B shares are listed on Nasdaq Copenhagen (Novo-B). Its ADRs are listed on the New York Stock Exchange (NVO). For more information, visit novonordisk.com , Facebook, Twitter , LinkedIn and YouTube.
Nov. 12, 2021 6:20 AM ET
By: Aakash Babu, SA News Editor
Bagsværd , De n mark, 11 November 2021 – Novo Nordisk today announced that the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has adopted a positive opinion, recommending marketing authorisation for Wegovy™ for chronic weight management in adults with obesity.
Wegovy™ is a once-weekly semaglutide 2.4 mg injection for chronic weight management in adults living with obesity (initial BMI≥30 kg/m2) or overweight (initial BMI≥27 kg/m2) with at least one weight-related comorbidity such as high blood pressure, heart disease or type 2 diabetes.
The positive CHMP opinion is based on results from the STEP phase 3a clinical trial programme. Across the trials in people with obesity or overweight without type 2 diabetes, an average weight loss of 17-18%1 sustained over 68 weeks was reported for those treated with Wegovy™. Wegovy™ demonstrated a safe and well-tolerated profile across the programme, with the most common adverse events being gastrointestinal.
“With the unprecedented and sustained weight loss for an anti-obesity medication, Wegovy™ has the potential to transform obesity management and help millions of people living with obesity,” said Martin Holst Lange, executive vice president, Development at Novo Nordisk. “We are looking forward to driving change for people with obesity by introducing Wegovy™ in Europe next year.”
Novo Nordisk expects to receive final marketing authorisation from the European Commission in approximately two months.
Wegovy™ was launched in the US in June 2021 following approval by the U.S. Food and Drug Administration (FDA). In September 2021, Wegovy™ was approved by the UK Medicines and Health products Regulations Agency (MHRA). Novo Nordisk expects to launch Wegovy™ in Europe in the second half of 2022.
About Wegovy™ (s e maglutide 2.4 mg ) and STEP
Semaglutide 2.4 mg is an analogue of the human glucagon-like peptide-1 (GLP-1) hormone. It induces weight loss by reducing hunger, increasing feeling of fullness and thereby helping people eat less and reduce their calorie intake. Once-weekly semaglutide 2.4 mg injection is approved for the treatment of adults with obesity or overweight in the US and UK, as an adjunct to diet and exercise.
The approval is based on the results from the STEP (Semaglutide Treatment Effect in People with obesity) phase 3 clinical development programme. The global clinical phase 3a programme consists of four trials and enrolled approximately 4,500 adults with overweight or obesity.
For more information, visit novonordisk.com
Nov. 11, 2021 11:31 AM ET
By: Dulan Lokuwithana, SA News Editor2 Comments
November 8, 2021 at 7:00 AM EST Back
TARRYTOWN, N.Y., Nov. 8, 2021 /PRNewswire/ --
Single dose of REGEN-COV (1,200 mg subcutaneous) reduced the risk of COVID-19 by 81.6% during the pre-specified follow-up period (months 2-8), maintaining the 81.4% risk reduction previously reported during month 1
During the 8-month assessment period there were 0 hospitalizations for COVID-19 in the REGEN-COV group and 6 in the placebo group
The fully human antibodies in REGEN-COV were developed to provide long-lasting protective effects without any artificial mutations or sequences
Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) today announced additional positive results from a Phase 3 trial jointly run with the National Institute of Allergy and Infectious Diseases (NIAID), which assessed use of a single dose of investigational REGEN-COV® (1,200 mg administered via 4 subcutaneous injections) to prevent COVID-19 in uninfected individuals. The new analyses show REGEN-COV reduced the risk of contracting COVID-19 (i.e., laboratory-confirmed symptomatic SARS-CoV-2 infections) by 81.6% during the pre-specified follow-up period (months 2-8), maintaining the 81.4% risk reduction during the first month after administration, which was previously reported in The New England Journal of Medicine.
"Today's new data demonstrate how a single dose of REGEN-COV can help protect people from COVID-19 for many months after administration," said Myron S. Cohen, M.D., who leads the monoclonal antibody efforts for the NIH-sponsored COVID Prevention Network (CoVPN) and is Director of the Institute for Global Health & Infectious Diseases at the University of North Carolina at Chapel Hill. "These results demonstrate that REGEN-COV has the potential to provide long-lasting immunity from SARS-CoV-2 infection, a result particularly important to those who do not respond to COVID-19 vaccines including people who are immunocompromised."
In results previously published, the trial met its primary endpoint, reducing the risk of COVID-19 (i.e., laboratory-confirmed symptomatic SARS-CoV-2 infections) by 81.4% within 1 month of receiving REGEN-COV (p<0.0001). The new results released today describe a pre-specified analysis for the following 7 months, throughout which an additional 45 symptomatic infections occurred. During this time period, REGEN-COV continued to prevent infection, without requiring additional doses. Compared to placebo (n=842), people who received a single dose of REGEN-COV (n=841) experienced:
REGEN-COV is currently authorized in the U.S. to treat people who are at high risk of serious consequences from COVID-19 infection who are either already infected (non-hospitalized) or in certain post-exposure prophylaxis settings. In the U.S., REGEN-COV is not authorized as a substitute for vaccination against COVID-19, or for pre-exposure prophylaxis for prevention of COVID-19, or for use in patients who are hospitalized due to COVID-19 or require oxygen therapy, or for people currently using chronic oxygen therapy because of an underlying comorbidity who require an increase in baseline oxygen flow rate due to COVID-19. REGEN-COV has not been approved by the Food and Drug Administration (FDA), but is currently authorized for the duration of the declaration that circumstances exist justifying the authorization of the emergency uses under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner.
The development and manufacturing of REGEN-COV have been funded in part with federal funds from the Biomedical Advanced Research and Development Authority (BARDA), part of the U.S. Department of Health and Human Services' Office of the Assistant Secretary for Preparedness and Response, under OT number: HHSO100201700020C.
Regeneron is collaborating with Roche to increase global supply of the antibody cocktail, with Roche primarily responsible for development and distribution outside the U.S. Regeneron and Roche share a commitment to making the antibody cocktail available to COVID-19 patients around the globe and will support access in low- and lower-middle-income countries through drug donations to be made in partnership with public health organizations.
About the Trial
The Phase 3 double-blind, placebo-controlled trial enrolled people who lived in the same household as an individual who was diagnosed with SARS-CoV-2 within the prior 4 days. All participants were tested for SARS-CoV-2 at baseline using a RT-qPCR test from nasopharyngeal swabs and for the presence of antibodies using serum antibody testing. Participants were randomized (1:1) to receive either 1 dose of REGEN-COV (1,200 mg) or placebo, administered via 4 subcutaneous injections.
During the trial, participants were tested weekly for SARS-CoV-2 during the initial month (4 weeks), as part of the primary analyses. Following this, from months 2-8 (week 5 to week 32), participants were to be tested if they developed any COVID-19 symptoms.
The new analyses include results from 1,683 people who were not infected with SARS-CoV-2 and did not have antibodies for SARS-CoV-2 (seronegative) at baseline. Across both groups approximately 95% completed the trial. In total, 42% identified as Hispanic/Latino and 9% identified as Black/African American. In addition, 34% were obese and 37% were aged ≥50 years (median age: 43 years; range: 12-92 years).
About the REGEN-COV Antibody Cocktail
REGEN-COV (casirivimab and imdevimab) is a cocktail of two monoclonal antibodies that was designed specifically to block infectivity of SARS-CoV-2, the virus that causes COVID-19, using Regeneron's proprietary VelocImmune® and VelociSuite® technologies. The two potent, virus-neutralizing antibodies that form the cocktail bind non-competitively to the critical receptor binding domain of the virus's spike protein, which diminishes the ability of mutant viruses to escape treatment and protects against spike variants that have arisen in the human population, as detailed in Cell and Science.
REGEN-COV has not been approved by the FDA, but is currently authorized in the U.S. for the treatment and post-exposure prophylaxis in certain high risk individuals. This authorization is for the duration of the declaration that circumstances exist justifying the authorization of the emergency uses under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner. Additional information about REGEN-COV in the U.S. is below (authorized uses and important safety information).
In October, the U.S. FDA accepted for priority review the first of two Biologics License Applications (BLAs) for REGEN-COV to treat COVID-19 in non-hospitalized patients and as prophylaxis in certain individuals. The second BLA submission will focus on those hospitalized because of COVID-19, and is expected to be completed later this year.
Emergency or temporary pandemic use authorizations are currently in place in more than 40 countries, including the U.S., several European Union countries, India, Switzerland and Canada, and the antibody cocktail is fully approved in Japan and conditionally approved in the UK.
Treatment:
REGEN-COV is authorized for the treatment of mild to moderate coronavirus disease 2019 (COVID-19) in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death
Limitations of Authorized Use (Treatment)
For additional information about the company, please visit www.regeneron.com
View original content:https://www.prnewswire.com/news-releases/new-phase-3-analyses-show-that-a-single-dose-of-regen-cov-casirivimab-and-imdevimab-provides-long-term-protection-against-covid-19-301418179.html
SOURCE Regeneron Pharmaceuticals, Inc.
Nov. 08, 2021 7:25 AM ET Regeneron Pharmaceuticals, Inc. (REGN), RHHBY
By: Dulan Lokuwithana, SA News Editor
November 11, 2021 at 11:03 AM EST
TARRYTOWN, N.Y., Nov. 11, 2021 /PRNewswire/ -- Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) today announced that the European Medicines Agency's (EMA) Committee for Medicinal Products for Human Use (CHMP) has adopted a positive opinion for the casirivimab and imdevimab antibody cocktail, known as REGEN-COV® in the U.S. and Ronapreve™ in the European Union (EU) and other countries. The positive opinion is for people aged 12 years and older for the treatment of non-hospitalized patients (outpatients) with confirmed COVID-19 who do not require oxygen supplementation and who are at increased risk of progressing to severe COVID-19, and to prevent COVID-19. A final decision from the European Commission regarding the approval of the antibody cocktail is expected in the near future.
Tuesday, November 09, 2021 - 09:09am
NEW HAVEN, Conn., and NEW YORK, November 9, 2021 /PRNewswire/ -- Biohaven Pharmaceutical Holding Company Ltd. (NYSE: BHVN) and Pfizer Inc. (NYSE: PFE), today announced a strategic commercialization arrangement for rimegepant in markets outside of the United States upon approval. Rimegepant is commercialized as Nurtec® ODT in the U.S., and is indicated for the acute treatment of migraine attacks with or without aura and the preventive treatment of episodic migraine in adults. An application for the approval of rimegepant is currently under review by the European Medicines Agency and several additional regulatory authorities outside of the U.S.
“We believe this collaboration, which brings together the winning combination of Biohaven’s Neuroscience R&D with Pfizer’s industry-leading expertise and large global footprint will help accelerate access to rimegepant for patients around the world,” said Vlad Coric MD, Chief Executive Officer of Biohaven. “With this alliance, Biohaven Pharmaceutical and Pfizer believe there is an opportunity to change the paradigm in migraine treatment and potentially establish a new standard of care.”
Under the terms of the arrangement, Biohaven would remain primarily responsible for further clinical development of rimegepant and the parties will cooperate in regulatory activities to secure approval for the product. Biohaven will continue to solely commercialize Nurtec ODT in the U.S and Pfizer would commercialize rimegepant, upon approval, in all regions outside the U.S. Additionally, per the arrangement, Pfizer gains rights outside of the U.S. to zavegepant, a third generation, high affinity, selective and structurally unique, small molecule CGRP receptor antagonist, currently being studied in an intranasal delivery and a soft-gel formulation in Phase 3 clinical trials for migraine indications.
"We are excited to join forces with Biohaven in the fight against migraine and help those patients impacted by this debilitating neurological disease,” said Nick Lagunowich, Global President, Pfizer Internal Medicine. “We believe our legacy in pain and Women’s Health combined with our experienced customer-facing colleagues, will enable us to maximize this opportunity with Biohaven, potentially bringing a valuable new treatment option to patients living with migraine pain.”
Biohaven and Pfizer are entering into a collaboration and license agreement and related sublicense agreement pursuant to which Pfizer will acquire rights to commercialize rimegepant and zavegepant outside of the U.S. Biohaven will continue to lead research and development globally and Pfizer would execute commercialization globally, outside of the U.S. Under the financial terms of all transaction agreements, Pfizer will make an upfront payment of $500 million, consisting of $150 million cash and $350 million in the purchase of Biohaven equity at a 25 percent market premium. Biohaven is also eligible to receive up to $740 million in milestones. In addition to the tiered double-digit royalties owed to Biohaven on net sales outside of the U.S., Pfizer will compensate Biohaven for the related royalties on net sales outside of the U.S. owed under the Company’s license and funding agreements with Bristol-Myers Squibb Company and Royalty Pharma.
As noted above, in connection with the transaction, Pfizer will make a $350 million investment in the common shares of Biohaven.
Closing of the license agreements and equity purchase are contingent on completion of review under applicable antitrust laws, including the Hart-Scott-Rodino (HSR) Antitrust Improvements Act of 1976 in the U.S. and equivalents outside the U.S., and other customary closing conditions.
Biohaven and Pfizer global collaboration will be discussed on Biohaven 3Q Earnings Investor Call 8:00AM ET on November 9, 2021. To access the call, please dial 877-407-9120 (domestic) or 412-902-1009 (international). The conference call webcast and accompanying slide presentation can be accessed through the “Investors” section of Biohaven’s website at www.biohavenpharma.com.
Rimegepant targets a root cause of migraine by reversibly blocking CGRP receptors, thereby inhibiting the biologic cascade that results in a migraine attack. Rimegepant was approved by the U.S. Food and Drug Administration (FDA) under the trade name Nurtec® ODT for the acute treatment of migraine in February 2020 and for the preventive treatment of episodic migraine in May 2021. Nurtec ODT is the #1 prescribed migraine treatment in its class with a cumulative launch to date of U.S. net revenue of approximately $336 million and with more than one million prescriptions. A single dose of 75 mg Nurtec ODT provides fast pain relief, significant pain reduction and return to normal function, and has a lasting effect of up to 48 hours in some patients. Nurtec ODT is taken orally as needed, up to 18 doses/month to stop migraine attacks or taken every other day to help prevent migraine attacks and reduce the number of monthly migraine days. Nurtec ODT does not have addiction potential and is not associated with medication overuse headache or rebound headache.
Please click here for full Prescribing Information and Patient Information
More information about Biohaven is available at www.biohavenpharma.com
NURTEC and NURTEC ODT are registered trademarks of Biohaven Pharmaceutical Ireland DAC. Neuroinnovation is a trademark of Biohaven Pharmaceutical Holding Company Ltd.
Nov. 09, 2021 8:13 AM ET
Biohaven Pharmaceutical Holding Company Ltd. (BHVN), PFE
By: Jonathan M Block, SA News Editor
11/09/2021- Rimegepant, commercialized as Nurtec® ODT in the U.S., is the first and only oral CGRP (calcitonin gene-related peptide) receptor antagonist for the acute and preventive treatment of migraine- Biohaven to receive tiered double-digit royalties on ex-U.S. net sales as well as upfront and milestone payments of up to $1.24 billion- Biohaven and Pfizer global collaboration to be discussed on Biohaven 3Q Earnings Investor Call 8:00AM ET today
NEW HAVEN, Conn. and NEW YORK, Nov. 9, 2021 /PRNewswire/ -- Biohaven Pharmaceutical Holding Company Ltd. (NYSE: BHVN) and Pfizer Inc. (NYSE: PFE), today announced a strategic commercialization arrangement for rimegepant in markets outside of the United States upon approval. Rimegepant is commercialized as Nurtec® ODT in the U.S., and is indicated for the acute treatment of migraine attacks with or without aura and the preventive treatment of episodic migraine in adults. An application for the approval of rimegepant is currently under review by the European Medicines Agency and several additional regulatory authorities outside of the U.S.
https://www.biohavenpharma.com/investors/news-events/press-releases/11-09-2021-0
November 9, 2021
- Post-hoc analyses of the Phase 3 SELECT-PsA 1 and SELECT-PsA 2 trials show people with active psoriatic arthritis (PsA) demonstrated greater clinical responses related to axial involvement when treated with upadacitinib (RINVOQ®) compared to placebo at week 24[1]
- Results will be presented at American College of Rheumatology (ACR) Convergence 2021
NORTH CHICAGO, Ill., Nov. 9, 2021 /PRNewswire/ -- AbbVie (NYSE: ABBV) today announced results from new post-hoc analyses from the Phase 3 SELECT-PsA 1 and SELECT-PsA 2 trials assessing the efficacy of upadacitinib (RINVOQ®) on axial symptoms in adult patients with active psoriatic arthritis (PsA) and axial involvement. The analysis showed that patients with active PsA demonstrated numerically greater clinical responses related to their axial involvement with upadacitinib (15 mg, once daily) compared to placebo at week 24 across both studies and consistently numerically higher responses compared to HUMIRA® (adalimumab) at week 24 in SELECT-PsA 1.1
Axial involvement was defined by investigator assessment and patient-reported-outcome-based criteria (Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 and BASDAI Question 2 ≥4 at baseline).1 These results will be featured at the American College of Rheumatology (ACR) Convergence 2021, in an oral presentation on Tuesday, Nov. 9, from 3:30-3:45 p.m. CT (Abstract #1945).
"These data further add to the body of evidence that support the potential of upadacitinib to be an important treatment option that helps reduce the impact of the many disease manifestations of psoriatic arthritis," said Thomas Hudson, M.D., senior vice president, research and development, chief scientific officer, AbbVie. "We remain committed to advancing research across our portfolio of therapies to help improve care for more people living with rheumatic diseases, including psoriatic arthritis."
About SELECT-PsA 111,12
SELECT-PsA 1 is a Phase 3, multicenter, randomized, double-blind, active comparator- and placebo-controlled study designed to evaluate the safety and efficacy of RINVOQ compared to placebo and adalimumab in adult patients with moderately to severely active psoriatic arthritis who have a history of intolerance or inadequate response to at least one non-biologic DMARD. Patients were randomized to RINVOQ 15 mg, RINVOQ 30 mg, adalimumab 40 mg every other week or placebo followed by either RINVOQ 15 mg or RINVOQ 30 mg at week 24.
The primary endpoint was the percentage of subjects receiving RINVOQ 15 mg or 30 mg who achieved an ACR20 response after 12 weeks of treatment versus placebo. The long-term extension of the trial is ongoing. More information on this trial can be found at www.clinicaltrials.gov (NCT03104400).
About SELECT-PsA 211,13
SELECT-PsA 2 is a Phase 3, multicenter, randomized, double-blind, placebo-controlled study designed to evaluate the safety and efficacy of RINVOQ in adult patients with moderately to severely active psoriatic arthritis who have a history of intolerance or inadequate response to at least one biologic DMARD. Patients were randomized to RINVOQ 15 mg, RINVOQ 30 mg or placebo followed by either RINVOQ 15 mg or RINVOQ 30 mg at week 24.
The primary endpoint was the percentage of subjects achieving an ACR20 response after 12 weeks of treatment versus placebo. The long-term extension of the trial is ongoing. More information on this trial can be found at www.clinicaltrials.gov (NCT03104374).
About RINVOQ® (upadacitinib)
Discovered and developed by AbbVie scientists, RINVOQ is a selective JAK inhibitor that is being studied in several immune-mediated inflammatory diseases. Based on enzymatic and cellular assays, RINVOQ demonstrated greater inhibitory potency for JAK-1 vs JAK-2, JAK-3, and TYK-2.11 The relevance of inhibition of specific JAK enzymes to therapeutic effectiveness is not currently known. RINVOQ 15 mg is approved by the U.S. Food and Drug Administration (FDA) for adults with moderately to severely active rheumatoid arthritis. RINVOQ 15 mg is also approved by the European Commission for adults with moderate to severe active rheumatoid arthritis, adults with active psoriatic arthritis and adults with active ankylosing spondylitis. RINVOQ is approved by the European Commission for adults (15 mg and 30 mg) and adolescents (15 mg) with moderate to severe atopic dermatitis. Phase 3 trials of RINVOQ in rheumatoid arthritis, atopic dermatitis, psoriatic arthritis, axial spondyloarthritis, Crohn's disease, ulcerative colitis, giant cell arteritis and Takayasu arteritis are ongoing.14-21
Please click here for the Full Prescribing Information and Medication Guide.
For more information about AbbVie, please visit us at www.abbvie.com
Nov. 09, 2021 9:39 AM ET AbbVie Inc. (ABBV)
By: Jonathan M Block, SA News Editor2 Comments
November 4, 2021 6:40 am ET
U.K.’s Medicines and Healthcare Products Regulatory Agency Authorizes Molnupiravir for the Treatment of Mild-to-Moderate COVID-19 in Adults With a Positive SARS-CoV-2 Diagnostic Test and Who Have at Least One Risk Factor for Developing Severe Illness
Applications Remain Under Review by Other Regulatory Authorities, Including U.S. Food and Drug Administration and the European Medicines Agency
KENILWORTH, N.J. & MIAMI--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, and Ridgeback Biotherapeutics today announced that the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA) has granted authorization in the United Kingdom (U.K.) for molnupiravir (MK-4482, EIDD-2801), the first oral antiviral medicine authorized for the treatment of mild-to-moderate COVID-19 in adults with a positive SARS-CoV-2 diagnostic test and who have at least one risk factor for developing severe illness. In the U.K., LAGEVRIO® (lah-GEV-ree-oh) is the planned trademark for molnupiravir; the trademark for molnupiravir in other countries has not been approved. Merck announced its application with the U.S. Food and Drug Administration (FDA) for Emergency Use Authorization (EUA) of molnupiravir is under review and recently announced the European Medicines Agency has initiated a rolling review of the company’s Marketing Authorization Application. Merck is actively working to submit applications to other regulatory agencies around the world.
“The first global authorization of molnupiravir is a major achievement in Merck’s singular legacy of bringing forward breakthrough medicines and vaccines to address the world’s greatest health challenges. In pursuit of Merck’s unwavering mission to save and improve lives, we will continue to move with both rigor and urgency to bring molnupiravir to patients around the world as quickly as possible,” said Robert M. Davis, chief executive officer and president, Merck.
“As an oral therapeutic, molnupiravir offers an important addition to the vaccines and medicines deployed so far to counter the COVID-19 pandemic,” said Dr. Dean Y. Li, executive vice president and president, Merck Research Laboratories. “We are very grateful to the investigators, patients and their families for their critical contributions to the MOVe-OUT study that made this authorization possible.”
The authorization is based on positive results from a planned interim analysis from the Phase 3 MOVe-OUT clinical trial, which evaluated molnupiravir 800 mg twice-daily in non-hospitalized, unvaccinated adult patients with laboratory-confirmed mild-to-moderate COVID-19, symptom onset within five days of study randomization and at least one risk factor associated with poor disease outcomes (e.g., heart disease, diabetes).
“When we embarked on the journey to take molnupiravir from a hope to a reality, we believed we had a responsibility to move as quickly and safely as possible. We believed each day saved could save lives and limit severe disease and the global hardships of this pandemic,” said Wendy Holman, chief executive officer, Ridgeback Biotherapeutics. “It is gratifying to reach this milestone and show that the extraordinary effort of our collaborators, patients, physicians and team and the personal sacrifices made have now achieved that important goal. It is also gratifying to see the first global authorization occur in the U.K., the very place where we administered molnupiravir to the first brave human volunteer.”
About Molnupiravir
Molnupiravir (MK-4482, EIDD-2801) is an investigational, orally administered form of a potent ribonucleoside analog that inhibits the replication of SARS-CoV-2, the causative agent of COVID-19.
Molnupiravir was invented at Drug Innovations at Emory (DRIVE), LLC, a not-for-profit biotechnology company wholly owned by Emory University; Emory/DRIVE received some research funding from the U.S. Department of Defense and the National Institutes of Health. Molnupiravir is being developed by Merck & Co., Inc. in collaboration with Ridgeback Biotherapeutics. Ridgeback received an upfront payment from Merck and also is eligible to receive contingent payments dependent upon the achievement of certain developmental and regulatory approval milestones. Any profits from the collaboration will be split between the partners equally. Since licensed by Ridgeback, all funds used for the development of molnupiravir have been provided by Merck and by Wayne and Wendy Holman of Ridgeback.
Molnupiravir is also being evaluated for post-exposure prophylaxis in MOVe-AHEAD, a global, multicenter, randomized, double-blind, placebo-controlled Phase 3 study, which is evaluating the efficacy and safety of molnupiravir in preventing the spread of COVID-19 within households. For more information, please visit http://merckcovidresearch.com. Please visit the Merck media library for molnupiravir images and b-roll.
About the MOVe-OUT Study
The MOVe-OUT trial (MK-4482-002) (NCT04575597) is a global Phase 3, randomized, placebo-controlled, double-blind, multi-site study of non-hospitalized adult patients with laboratory-confirmed mild-to-moderate COVID-19. Patients enrolled in the study were unvaccinated against SARS-CoV-2, had at least one risk factor associated with poor disease outcomes, and symptom onset within five days prior to randomization. The primary efficacy objective of MOVe-OUT is to evaluate the efficacy of molnupiravir compared to placebo as assessed by the percentage of participants who are hospitalized and/or die from the time of randomization through Day 29.
The Phase 3 portion of the MOVe-OUT trial was conducted globally in countries including Brazil, Canada, Chile, Colombia, France, Germany, Guatemala, Mexico, Philippines, Russia, South Africa, Spain, Taiwan, Ukraine, the United Kingdom and the United States. For further information about the MOVe-OUT trial, please visit clinicaltrials.gov.
The most common risk factors for poor disease outcome included obesity, older age (>60 years), diabetes mellitus and heart disease. Delta, Gamma and Mu variants accounted for nearly 80% of the baseline viral variants that had been sequenced at the time of the interim analysis. Recruitment in Latin America, Europe and Africa accounted for 56%, 23% and 15% of the study population, respectively.
For more information, visit www.merck.com
Nov. 04, 2021 6:48 AM ET Merck & Co., Inc. (MRK) By: Dulan Lokuwithana, SA News Editor7 Comments
11/08/2021CATEGORY:
CheckMate -816 is the first Phase 3 trial with an immunotherapy-based combination to demonstrate improved event-free survival and pathologic complete response in the neoadjuvant setting of non-small cell lung cancer
Positive results reinforce the improved efficacy seen with Opdivo-based treatments in four Phase 3 clinical trials in earlier-stage cancers, including lung cancer, bladder cancer, esophageal/gastroesophageal junction cancer and melanoma
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) today announced the Phase 3 CheckMate -816 trial met the primary endpoint of improved event-free survival (EFS) in patients with resectable stage IB to IIIA non-small cell lung cancer (NSCLC). In a prespecified interim analysis, Opdivo (nivolumab) plus chemotherapy showed a statistically significant and clinically meaningful improvement in EFS compared to chemotherapy alone when given before surgery. This combination previously showed a significant improvement of pathologic complete response (pCR), the trial’s other primary endpoint. The safety profile of Opdivo plus chemotherapy was consistent with previously reported studies in NSCLC.
“While the intent of surgery is curative in resectable non-small cell lung cancer, between 30% to 55% of patients experience recurrence after surgery and ultimately succumb to the disease, presenting a strong need for additional options that can disrupt this cycle,” said Nicolas Girard, M.D., Ph.D., professor of respiratory medicine at Paris Saclay University and head of the Thorax Institute Curie Montsouris in Paris. “The positive event-free survival data seen with neoadjuvant nivolumab plus chemotherapy is groundbreaking and can have important implications for how we treat resectable non-small cell lung cancer.”
“CheckMate -816 is the first Phase 3 trial with an immunotherapy-based combination to demonstrate a statistically significant and clinically meaningful benefit as a neoadjuvant treatment for patients with non-metastatic non-small cell lung cancer. The combination of Opdivo plus chemotherapy first showed a statistically significant improvement in pathologic complete response rate without impacting surgical outcomes and has now extended the time patients live free of disease progression, recurrence or death,” said Abderrahim Oukessou, M.D., vice president, thoracic cancers development lead, Bristol Myers Squibb. “The event-free survival data from CheckMate -816 strengthen the evidence for the potential of Opdivo-based therapies to improve long-term clinical outcomes when used in the earlier stages of non-metastatic cancers.”
About CheckMate -816
CheckMate -816 is a Phase 3 randomized, open label, multi-center trial evaluating Opdivo plus chemotherapy compared to chemotherapy alone as neoadjuvant treatment in patients with resectable non-small cell lung cancer, regardless of PD-L1 expression. For the primary analysis, 358 patients were randomized to receive either Opdivo 360 mg plus histology-based platinum doublet chemotherapy every three weeks for three doses, or platinum doublet chemotherapy every three weeks for three doses, followed by surgery. The primary endpoints of the trial are pathologic complete response (pCR) and event-free survival (EFS). Secondary endpoints include overall survival (OS), major pathologic response (MPR), and time to death or distant metastases.
About Opdivo
Opdivo is a programmed death-1 (PD-1) immune checkpoint inhibitor that is designed to uniquely harness the body’s own immune system to help restore anti-tumor immune response. By harnessing the body’s own immune system to fight cancer, Opdivo has become an important treatment option across multiple cancers.
Opdivo’s leading global development program is based on Bristol Myers Squibb’s scientific expertise in the field of Immuno-Oncology, and includes a broad range of clinical trials across all phases, including Phase 3, in a variety of tumor types. To date, the Opdivo clinical development program has treated more than 35,000 patients. The Opdivo trials have contributed to gaining a deeper understanding of the potential role of biomarkers in patient care, particularly regarding how patients may benefit from Opdivo across the continuum of PD-L1 expression.
In July 2014, Opdivo was the first PD-1 immune checkpoint inhibitor to receive regulatory approval anywhere in the world. Opdivo is currently approved in more than 65 countries, including the United States, the European Union, Japan and China. In October 2015, the Company’s Opdivo and Yervoy combination regimen was the first Immuno-Oncology to receive regulatory approval for the treatment of metastatic melanoma and is currently approved in more than 50 countries, including the United States and the European Union.
INDICATIONS
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of patients with unresectable or metastatic melanoma.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of patients with unresectable or metastatic melanoma.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) whose tumors express PD-L1 (≥1%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab) and 2 cycles of platinum-doublet chemotherapy, is indicated for the first-line treatment of adult patients with metastatic or recurrent non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab) is indicated for the treatment of patients with metastatic non-small cell lung cancer (NSCLC) with progression on or after platinum-based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving OPDIVO.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with unresectable malignant pleural mesothelioma (MPM).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of patients with intermediate or poor risk advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab), in combination with cabozantinib, is indicated for the first-line treatment of patients with advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab) is indicated for the treatment of patients with advanced renal cell carcinoma (RCC) who have received prior anti-angiogenic therapy.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with classical Hodgkin lymphoma (cHL) that has relapsed or progressed after autologous hematopoietic stem cell transplantation (HSCT) and brentuximab vedotin or after 3 or more lines of systemic therapy that includes autologous HSCT. This indication is approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab) is indicated for the treatment of patients with recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN) with disease progression on or after platinum-based therapy.
OPDIVO® (nivolumab) is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
OPDIVO® (nivolumab), as a single agent, is indicated for the adjuvant treatment of patients with urothelial carcinoma (UC) who are at high risk of recurrence after undergoing radical resection of UC.
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of adult and pediatric (12 years and older) patients with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adults and pediatric patients 12 years and older with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of patients with melanoma with involvement of lymph nodes or metastatic disease who have undergone complete resection.
OPDIVO® (nivolumab) is indicated for the treatment of patients with unresectable advanced, recurrent or metastatic esophageal squamous cell carcinoma (ESCC) after prior fluoropyrimidine- and platinum-based chemotherapy.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of completely resected esophageal or gastroesophageal junction cancer with residual pathologic disease in patients who have received neoadjuvant chemoradiotherapy (CRT).
OPDIVO® (nivolumab), in combination with fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the treatment of patients with advanced or metastatic gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma.
Please see US Full Prescribing Information for OPDIVO and YERVOY.
For more information about Bristol Myers Squibb, visit us at BMS.com
Nov. 08, 2021 8:05 AM ET Bristol-Myers Squibb Company (BMY) By: Aakash Babu, SA News Editor2 Comments
https://www.nasdaq.com/market-activity/stocks/bmy/dividend-history
October 29, 2021
- Presbyopia, or age-related blurry near vision, is a common, progressive condition that reduces the eye's ability to focus on near objects, affecting most adults over the age 40
- Phase 3 clinical studies demonstrated VUITY works in as early as 15 minutes and lasts for up to 6 hours, as measured on day 30, to improve near and intermediate vision without impacting distance vision
- Allergan, an AbbVie company, continues legacy of eye care innovation with VUITY, a first-of-its-kind FDA-approved treatment for presbyopia
NORTH CHICAGO, Ill., Oct. 29, 2021 /PRNewswire/ -- Allergan, an AbbVie (NYSE: ABBV) company, today announced the U.S. Food and Drug Administration (FDA) approval of VUITY™ (pilocarpine HCl ophthalmic solution) 1.25% for the treatment of presbyopia, commonly known as age-related blurry near vision, in adults. VUITY is the first and only FDA-approved eye drop to treat this common and progressive eye condition that affects 128 million Americans, nearly half of the U.S. adult population.
"Most adults cope with presbyopia, or difficulty with near vision, as we age. Beginning around the age of 40, many find themselves using reading glasses, holding text further away, or even increasing the font size and lighting on screens to try to see more clearly," said Michael Severino, M.D., vice chairman and president, AbbVie. "We are proud to offer VUITY as a first-of-its-kind once-daily eye drop that we believe will change the way people and their eye doctors approach presbyopia. The FDA approval of VUITY exemplifies our continued pursuit of innovative new treatments that push the boundaries of what's possible in eye care."
VUITY is a daily, prescription eye drop that works in as early as 15 minutes and lasts up to 6 hours, as measured on day 30, to improve near and intermediate vision without impacting distance vision. Specifically designed for presbyopia, VUITY is an optimized formulation of pilocarpine, an established eye care therapeutic, delivered with pHast™ technology. The proprietary pHast™ technology allows VUITY to rapidly adjust to the physiologic pH of the tear film. VUITY uses the eye's own ability to reduce pupil size, improving near vision without affecting distance vision.
"As we age, the lenses of our eyes become less flexible, making it more difficult to focus on things up close. VUITY offers a novel, safe, well-tolerated and effective alternative to current options for managing age-related blurry near vision," said George O. Waring IV, M.D., FACS, medical director, Waring Vision Institute, South Carolina, and GEMINI 1 and GEMINI 2 principal study investigator. "I am particularly encouraged by the rapid onset of action and duration of efficacy for VUITY to improve near and intermediate vision without impacting distance vision with one drop daily, particularly for those with mild to moderate presbyopia."
The FDA approval of VUITY is based on data from two pivotal phase 3 clinical studies, GEMINI 1 and GEMINI 2, which evaluated the efficacy, safety and tolerability of VUITY for the treatment of presbyopia. In both studies, VUITY met the primary endpoint, reaching statistical significance in improvement in near vision in low light (mesopic) conditions without a loss of distance vision versus the vehicle (placebo) on day 30 at hour 3. Additionally, improvement was seen as early as 15 minutes and lasted through 6 hours. There were no serious adverse events observed in participants receiving VUITY in either the GEMINI 1 or GEMINI 2 study. The most common adverse events occurring at a frequency of >5% were headache and eye redness.
Highlights from the Phase 3 GEMINI 1 & GEMINI 2 Clinical Studies
Approved Use and Important Safety Information
VUITY™ (pilocarpine HCL ophthalmic solution) 1.25% is a prescription eyedrop to treat age-related blurry near vision (presbyopia) in adults.
What is the most important information I should know about VUITY™?
Please see full Prescribing Information.
For more information about AbbVie, please visit us at www.abbvie.com.
Oct. 29, 2021 1:53 PM ET AbbVie Inc. (ABBV)
By: Jonathan M Block, SA News Editor14 Comments
Friday, November 05, 2021 - 06:45am
NEW YORK--(BUSINESS WIRE)-- Pfizer Inc. (NYSE: PFE) today announced its investigational novel COVID-19 oral antiviral candidate,PAXLOVID™, significantly reduced hospitalization and death, based on an interim analysis of the Phase 2/3 EPIC-HR (Evaluation of Protease Inhibition for COVID-19 in High-Risk Patients) randomized, double-blind study of non-hospitalized adult patients with COVID-19, who are at high risk of progressing to severe illness. The scheduled interim analysis showed an 89% reduction in risk of COVID-19-related hospitalization or death from any cause compared to placebo in patients treated within three days of symptom onset (primary endpoint); 0.8% of patients who received PAXLOVID™ were hospitalized through Day 28 following randomization (3/389 hospitalized with no deaths), compared to 7.0% of patients who received placebo and were hospitalized or died (27/385 hospitalized with 7 subsequent deaths). The statistical significance of these results was high (p<0.0001). Similar reductions in COVID-19-related hospitalization or death were observed in patients treated within five days of symptom onset; 1.0% of patients who received PAXLOVID™ were hospitalized through Day 28 following randomization (6/607 hospitalized, with no deaths), compared to 6.7% of patients who received a placebo (41/612 hospitalized with 10 subsequent deaths), with high statistical significance (p<0.0001). In the overall study population through Day 28, no deaths were reported in patients who received PAXLOVID™ as compared to 10 (1.6%) deaths in patients who received placebo.
At the recommendation of an independent Data Monitoring Committee and in consultation with the U.S. Food and Drug Administration (FDA), Pfizer will cease further enrollment into the study due to the overwhelming efficacy demonstrated in these results and plans to submit the data as part of its ongoing rolling submission to the U.S. FDA for Emergency Use Authorization (EUA) as soon as possible.
“Today’s news is a real game-changer in the global efforts to halt the devastation of this pandemic. These data suggest that our oral antiviral candidate, if approved or authorized by regulatory authorities, has the potential to save patients’ lives, reduce the severity of COVID-19 infections, and eliminate up to nine out of ten hospitalizations,” said Albert Bourla, Chairman and Chief Executive Officer, Pfizer. “Given the continued global impact of COVID-19, we have remained laser-focused on the science and fulfilling our responsibility to help healthcare systems and institutions around the world while ensuring equitable and broad access to people everywhere.”
If approved or authorized, PAXLOVID™, which originated in Pfizer’s laboratories, would be the first oral antiviral of its kind, a specifically designed SARS-CoV-2-3CL protease inhibitor. Upon successful completion of the remainder of the EPIC clinical development program and subject to approval or authorization, it could be prescribed more broadly as an at-home treatment to help reduce illness severity, hospitalizations, and deaths, as well as reduce the probability of infection following exposure, among adults. It has demonstrated potent antiviral in vitro activity against circulating variants of concern, as well as other known coronaviruses, suggesting its potential as a therapeutic for multiple types of coronavirus infections.
About the Phase 2/3 EPIC-HR Study Interim Analysis
The primary analysis of the interim data set evaluated data from 1219 adults who were enrolled by September 29, 2021. At the time of the decision to stop recruiting patients, enrollment was at 70% of the 3,000 planned patients from clinical trial sites across North and South America, Europe, Africa, and Asia, with 45% of patients located in the United States. Enrolled individuals had a laboratory-confirmed diagnosis of SARS-CoV-2 infection within a five-day period with mild to moderate symptoms and were required to have at least one characteristic or underlying medical condition associated with an increased risk of developing severe illness from COVID-19. Each patient was randomized (1:1) to receive PAXLOVID™ or placebo orally every 12 hours for five days.
About the Phase 2/3 EPIC-HR Study Safety Data
The review of safety data included a larger cohort of 1881 patients in EPIC-HR, whose data were available at the time of the analysis. Treatment-emergent adverse events were comparable between PAXLOVID™ (19%) and placebo (21%), most of which were mild in intensity. Among the patients evaluable for treatment-emergent adverse events, fewer serious adverse events (1.7% vs. 6.6%) and discontinuation of study drug due to adverse events (2.1% vs. 4.1%) were observed in patients dosed with PAXLOVID™ compared to placebo, respectively.
About PAXLOVID™ (PF-07321332; ritonavir) and the EPIC Development Program
PAXLOVID™ isan investigational SARS-CoV-2 protease inhibitor antiviral therapy, specifically designed to be administered orally so that it can be prescribed at the first sign of infection or at first awareness of an exposure, potentially helping patients avoid severe illness which can lead to hospitalization and death. PF-07321332 is designed to block the activity of the SARS-CoV-2-3CL protease, an enzyme that the coronavirus needs to replicate. Co-administration with a low dose of ritonavir helps slow the metabolism, or breakdown, of PF-07321332 in order for it to remain active in the body for longer periods of time at higher concentrations to help combat the virus.
PF-07321332 inhibits viral replication at a stage known as proteolysis, which occurs before viral RNA replication. In preclinical studies, PF-07321332 did not demonstrate evidence of mutagenic DNA interactions.
Pfizer initiated the EPIC-HR study in July 2021 following positive Phase 1 clinical trial results and continues to evaluate the investigational antiviral in additional EPIC studies. In August 2021, Pfizer initiated the Phase 2/3 EPIC-SR (Evaluation of Protease Inhibition for COVID-19 in Standard-Risk Patients), to evaluate efficacy and safety in patients with a confirmed diagnosis of SARS-CoV-2 infection who are at standard risk (i.e., low risk of hospitalization or death). EPIC-SR includes a cohort of vaccinated patients who have an acute breakthrough symptomatic COVID-19 infection and who have risk factors for severe illness. In September, Pfizer initiated the Phase 2/3 EPIC-PEP (Evaluation of Protease Inhibition for COVID-19 in Post-Exposure Prophylaxis) to evaluate efficacy and safety in adults exposed to SARS-CoV-2 by a household member.
For more information on the EPIC Phase 2/3 clinical trials for PAXLOVID™, visit clinicaltrials.gov.
to learn more, please visit us on www.Pfizer.com
View source version on businesswire.com: https://www.businesswire.com/news/home/20211105005260/en/
Source: Pfizer Inc.
Nov. 05, 2021 7:03 AM ET Pfizer Inc. (PFE) By: Aakash Babu, SA News Editor4 Comments
Friday, October 29, 2021 - 05:45pm
NEW YORK & MAINZ, Germany--(BUSINESS WIRE)-- Pfizer Inc. (NYSE: PFE) and BioNTech SE (Nasdaq: BNTX) today announced that the U.S. Food and Drug Administration (FDA) has authorized for emergency use the Pfizer-BioNTech COVID-19 Vaccine for children 5 through 11 years of age (also referred to as 5 to <12 years). For this age group, the vaccine is to be administered in a two-dose regimen of 10-µg doses given 21 days apart. The 10-µg dose level was carefully selected based on safety, tolerability and immunogenicity data. This is the first COVID-19 vaccine authorized in the U.S. for individuals 5 through 11 years of age.
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20211029005549/en/
“This is a day so many parents, eager to protect their young children from this virus, have been waiting for,” said Albert Bourla, Chairman and Chief Executive Officer, Pfizer. “Over 6 million children in the U.S. have been diagnosed with COVID-19 since the start of this pandemic, and a high number of young people continue to be infected every week. With this FDA authorization, we have achieved another key marker in our ongoing effort to help protect families and communities, and to get this disease under control.”
“Today’s emergency use authorization is supported by clinical data showing a favorable safety profile and high vaccine efficacy in children, underlining its potential to address a current public health need,” said Ugur Sahin, M.D., CEO and Co-founder of BioNTech. “As children 5 through 11 get reacclimated to the new school year, both in and out of the classroom, our goal is to help keep them safe and protected and get them back to normalcy.”
The FDA based its decision on data from a Phase 2/3 randomized, controlled trial that included ~4,500 children 5 through 11 years of age (2,268 from the original group and 2,379 from the supplemental safety group). Results from this trial were reviewed by the FDA Vaccines and Related Biological Products Advisory Committee (VRBPAC). In the trial, the vaccine demonstrated a favorable safety profile, robust immune responses and a vaccine efficacy rate of 90.7% in participants without prior SARS-CoV-2 infection, measured from 7 days after the second dose. The Data Monitoring Committee for the study has reviewed the data and has not identified any serious safety concerns related to the vaccine.
The companies will begin shipping 10-µg pediatric doses immediately, as directed by the U.S. government (ages referred to as 5y to <12y on the vial and 5 to <12 years on the carton). Eligible U.S. residents will continue to receive the vaccine for free, consistent with the U.S. government’s commitment to free access to COVID-19 vaccines.
As a next step, the U.S. Centers for Disease Control and Prevention’s (CDC) Advisory Committee on Immunization Practices (ACIP) will meet next week to discuss a potential recommendation for the use and rollout of the vaccine to children 5 through 11 years of age. Pediatric vaccinations are anticipated to start, subject to, and after, CDC endorses the ACIP recommendation.
Pfizer and BioNTech have submitted requests for authorization of their COVID-19 vaccine in this age group to other regulators around the world, including the European Medicines Agency. Initial data from the other two age cohorts in the ongoing Pfizer-BioNTech clinical trial in children – those 2 to <5 years of age and those 6 months to <2 years of age – are expected as soon as fourth quarter 2021 or early first quarter 2022.
The Pfizer-BioNTech COVID-19 Vaccine, which is based on BioNTech’s proprietary mRNA technology, was developed by both BioNTech and Pfizer. BioNTech is the Marketing Authorization Holder in the United States, the European Union, the United Kingdom, Canada and the holder of emergency use authorizations or equivalents in the United States (jointly with Pfizer) and other countries. Submissions to pursue regulatory approvals in those countries where emergency use authorizations or equivalent were initially granted are planned or ongoing.
U.S. INDICATION & AUTHORIZED USE
HOW IS THE VACCINE GIVEN?
The vaccine will be given as an injection into the muscle.
Primary Series:
In individuals 5 years of age and older, the vaccine is administered as a 2-dose series, 3 weeks apart. In individuals 12 years of age and older, a third primary series dose may be administered at least 4 weeks after the second dose to individuals who are determined to have certain kinds of immunocompromise.
Booster Dose:
WHAT IS THE INDICATION AND AUTHORIZED USE?
The Pfizer-BioNTech COVID-19 Vaccine has received EUA from FDA to provide:
COMIRNATY® (COVID-19 Vaccine, mRNA) is an FDA-approved COVID-19 vaccine made by Pfizer for BioNTech.
Fact Sheets and Prescribing Information for individuals 12 years of age and older
Full Prescribing Information (16 years of age and older)
EUA Fact Sheet for Vaccination Providers (12 years of age and older), Purple Cap
EUA Fact Sheet for Vaccination Providers (12 years of age and older), Gray Cap
Recipients and Caregivers Fact Sheet (12 years of age and older)
Fact Sheets for individuals 5 through 11 years of age
EUA Fact Sheet for Vaccination Providers (5 through 11 years of age), Orange Cap
Recipients and Caregivers Fact Sheet (5 through 11 years of age)
Please see EUA Fact Sheets at www.cvdvaccine-us.com
to learn more, please visit us on www.Pfizer.com
For more information, please visit www.BioNTech.de
View source version on businesswire.com: https://www.businesswire.com/news/home/20211029005549/en/
Source: Pfizer Inc.
Nov. 01, 2021 12:28 PM ET
By: Jonathan M Block, SA News Editor16 Comments
Oct 29, 2021
Basel, October 29, 2021 — Novartis announced today that the US Food and Drug Administration (FDA) approved Scemblix® (asciminib) for the treatment of chronic myeloid leukemia (CML) in two distinct indications. The FDA granted Scemblix accelerated approval for adult patients with Philadelphia chromosome-positive CML in chronic phase (Ph+ CML-CP) previously treated with two or more tyrosine kinase inhibitors (TKIs), based on major molecular response (MMR) rate at 24 weeks; and full approval for adult patients with Ph+ CML-CP with the T315I mutation1. In accordance with the Accelerated Approval Program, continued approval for the first indication may be contingent upon verification and description of clinical benefit from confirmatory evidence1. Scemblix is the first FDA-approved CML treatment that works by binding to the ABL myristoyl pocket, and represents an important development for patients who experience resistance and/or intolerance to currently available TKI therapies1-3. Also known as a STAMP inhibitor in scientific literature, Scemblix is being studied across multiple treatment lines for CML-CP, including the ASC4FIRST Phase III study evaluating Scemblix as a first-line treatment2-18.
“The introduction of TKIs twenty years ago revolutionized treatment for CML; however, there remain many patients who do not respond adequately to at least two available treatments and often experience challenging side effects that add a burden to their daily lives,” said Lee Greenberger, Chief Scientific Officer at The Leukemia & Lymphoma Society. “The approval of Scemblix may offer hope to patients by addressing gaps in CML care.”
For many patients, current treatment for CML may be limited by intolerance or resistance, and sequential use of available TKIs is associated with increased failure rates19-26. In an analysis of patients with CML treated with two prior TKIs, approximately 55% reported intolerance to previous treatment27. Additionally, a pooled analysis in the second-line setting showed that up to 70% of patients are unable to achieve major molecular response (MMR) within two years of follow-up28-30. Moreover, patients who develop the T315I mutation are resistant to most available TKIs, leaving them at an increased risk of disease progression4.
“CML can be difficult to treat when currently available treatments fail patients, when treatment side effects cannot be tolerated, or sometimes both,” expressed Dr. Michael J. Mauro**, Hematologist and Myeloproliferative Neoplasms Program Leader at Memorial Sloan Kettering Cancer Center (MSK). “The addition of Scemblix into the CML treatment landscape gives us a novel approach to combat this blood cancer, helping address clinical challenges in patients struggling after switching to a second treatment, as well as in patients who develop the T315I mutation and face significantly worse outcomes.”
The FDA approval of Scemblix is based on results from the Phase III ASCEMBL trial and a Phase I (NCT02081378) study that included patients with Ph+ CML-CP with the T315I mutation.
In patients with Ph+ CML-CP who had experienced resistance or intolerance to at least two TKIs, the ASCEMBL trial showed that1-3:
“After more than two decades of reimagining CML care, we continue to boldly push the boundaries of innovation to transform the standard-of-care and help even more patients living with this disease,” said Susanne Schaffert, PhD, President, Novartis Oncology. “We would like to thank all those who have been involved in helping to advance this new and important breakthrough.”
Scemblix is currently available for physicians to prescribe to appropriate patients in the US.
Additional efficacy and safety details for Scemblix, including data on patients with the T315I mutation, and full Prescribing Information can be found at https://www.novartis.us/sites/www.novartis.us/files/scemblix.pdf.
About Scemblix® (asciminib)
Scemblix (asciminib) is indicated for the treatment of adult patients with Ph+ CML-CP pre-treated with two or more TKIs, as well as adult patients with Ph+ CML-CP with the T315I mutation1. The first indication is approved under the US FDA Accelerated Approval Program based on MMR rate at 24 weeks; continued approval for the first indication may be contingent upon verification and description of clinical benefit from confirmatory evidence.
Scemblix is the first FDA-approved CML treatment that binds to the ABL myristoyl pocket1. This novel mechanism of action, also known in scientific literature as a STAMP inhibitor, may help address resistance in patients with CML previously treated with two or more TKIs and overcome mutations at the defective BCR-ABL1 gene, which is associated with the over-production of leukemic cells2-11. Scemblix has also been shown to limit off-target activity in pre-clinical studies31.
Novartis has initiated regulatory filings for Scemblix in multiple countries and regions across the globe.
Scemblix represents an important development for patients who experience resistance and/or intolerance to currently available TKI therapies, and it is being studied across multiple treatment lines for CML-CP 2-18. Specifically, the ASC4FIRST Phase III study (NCT04971226) evaluates Scemblix as a first-line treatment and is in the recruitment phase13.
Indication
SCEMBLIX® (asciminib) tablets is a prescription medicine used to treat adults with Philadelphia chromosome-positive chronic myeloid leukemia (Ph+ CML) in chronic phase (CP), previously treated with 2 or more tyrosine kinase inhibitor (TKI) medicines. The effectiveness of SCEMBLIX in these patients is based on a study that measured major molecular response (MMR) rates. No clinical information is available to show if these patients treated with SCEMBLIX live longer or if their symptoms improve. Ongoing studies exist to find out how SCEMBLIX works over a longer period of time.
SCEMBLIX is also approved for use in adults with Ph+ CML in CP with the T315I mutation.
It is not known if SCEMBLIX is safe and effective in children.
Please see full Prescribing Information for SCEMBLIX, available at https://www.novartis.us/sites/www.novartis.us/files/scemblix.pdf.
Find out more at https://www.novartis.com.
Oct. 29, 2021 2:10 PM ET Novartis AG (NVS)
By: Jonathan M Block, SA News Editor
October 28, 2021
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced it has entered into a clinical trial collaboration and supply agreement with Merck (known as MSD outside of the United States and Canada) to evaluate the efficacy of Gilead’s Trop-2 targeting antibody-drug conjugate Trodelvy® (sacituzumab govitecan-hziy) in combination with Merck’s anti-PD-1 therapy, KEYTRUDA® (pembrolizumab), as a first-line treatment for patients with locally advanced or metastatic triple-negative breast cancer (TNBC).
Under the terms of the agreement, Gilead will sponsor a global Phase 3 clinical trial to evaluate Trodelvy in combination with KEYTRUDA compared to standard of care KEYTRUDA in combination with chemotherapy in first-line patients with locally advanced or metastatic TNBC.
“Trodelvy has already been established as a preferred treatment option in second-line metastatic TNBC,” said Merdad Parsey, MD, PhD, Chief Medical Officer, Gilead Sciences. “Looking ahead, we are excited about the opportunity to advance Trodelvy as a potential treatment for first-line metastatic TNBC. This helps further our ambition of displacing chemotherapy with Trodelvy to improve outcomes for people living with cancer.”
Metastatic TNBC has the worst survival rate among breast cancer subtypes, and there is an urgent need for new therapies that improve patient outcomes. Trodelvy is an antibody-drug conjugate that specifically targets Trop-2 expressing cells to enable local delivery of a cytotoxic payload that selectively kills the targeted cells. The combination of Trodelvy with an immune-stimulating agent such as KEYTRUDA could provide a novel regimen in first-line metastatic TNBC.
The Trodelvy U.S. Prescribing Information has a BOXED WARNING for severe or life-threatening neutropenia and severe diarrhea; see below for Important Safety Information.
The combination of Trodelvy and KEYTRUDA has not been approved by any regulatory agency in any treatment setting. The safety and efficacy of this combination is under investigation and has not been established.
KEYTRUDA® is a registered trademark of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
About Trodelvy
Trodelvy (sacituzumab govitecan-hziy) is a first-in-class antibody and topoisomerase inhibitor conjugate directed to the Trop-2 receptor, a protein overexpressed in multiple types of epithelial tumors, including metastatic TNBC and metastatic urothelial cancer (UC), where high expression is associated with poor survival and relapse. Beyond the approvals of Trodelvy in the United States, it is also approved in Australia, Canada, Great Britain and Switzerland for adults with metastatic TNBC. Trodelvy is also under multiple regulatory reviews worldwide, including the EU, as well as in Singapore and China through our partner Everest Medicines. Trodelvy continues to be developed for potential use in other TNBC and metastatic UC populations and is also being developed as an investigational treatment for hormone receptor-positive/human epidermal growth factor receptor 2-negative (HR+/HER2-) metastatic breast cancer and metastatic non-small cell lung cancer. Additional evaluation across multiple solid tumors is also underway.
In the United States, Trodelvy is indicated for the treatment of:
U.S. Prescribing Information for Trodelvy including BOXED WARNING, is available at www.gilead.com.
Trodelvy, Gilead and the Gilead logo are trademarks of Gilead Sciences, Inc., or its related companies.
For more information about Gilead, please visit the company’s website at www.gilead.com, follow Gilead on Twitter (@GileadSciences) or call Gilead Public Affairs at 1-800-GILEAD-5 or 1-650-574-3000.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211028006064/en/
Source: Gilead Sciences, Inc.
Oct. 28, 2021 4:19 PM ET Gilead Sciences, Inc. (GILD) MRKBy: Aakash Babu, SA News Editor
October 29, 2021
– Clinical Data Across a Diverse Range of People Living With HIV on Biktarvy Treatment in the International BICSTaR Study Showed High Effectiveness and High Levels of Adherence –
– Long-Term Switch Data Presented at EACS 2021 Further Establish the Robust and Durable Efficacy Profile of Biktarvy –
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced interim results from the ongoing, multinational, observational single-arm, non-comparative real-world cohort BICSTaR study, which is designed to evaluate the antiviral effectiveness and safety profile of Biktarvy® (bictegravir 50 mg/emtricitabine 200 mg/tenofovir alafenamide 25 mg tablets, B/F/TAF) in 1,135 people living with HIV. The BICSTaR study also collected patient-reported outcomes in routine clinical practice to better understand the impact of treatment on health-related quality of life in people living with HIV. These data were presented at the 18th European AIDS Conference (EACS 2021).
Gilead presented an analysis of patient-reported outcomes after 12 months of treatment with Biktarvy from the BICSTaR study. During the study, conducted at sites in Europe, Canada and Israel, people living with HIV who initiated treatment with Biktarvy between June 2018 and September 2020 completed questionnaires at baseline and 12 months. The questionnaires assessed patient-reported outcomes covering a range of measures. The results underline the importance of collecting patient-reported outcomes in order to understand the impact on mental health status, health-related quality of life and treatment satisfaction of people living with HIV.
“Despite advances in antiretroviral therapy, people living with HIV experience burdensome multidimensional symptoms and concerns requiring person-centered care,” said Fernando Bognar, MD, Vice President, Medical Affairs, HIV at Gilead Sciences. “The patient-reported outcomes observed in the BICSTaR study provide a first-hand assessment of the impact of HIV treatment and care on the quality of life that people living with HIV experience. As physicians and people living with HIV look to understand what long-term treatment means to them individually, these data presented at EACS also reinforce that Biktarvy can meet the specific treatment needs of diverse groups of people, including men and women aging with HIV and those with existing comorbid conditions.”
In a second analysis of the BICSTaR study, 97% (n=149/154) of treatment-naïve adults and 96% (n=771/800) of treatment-experienced adults achieved and maintained virologic suppression (HIV-1 RNA <50 copies/mL) after one year of treatment. Participants included adults 50 years of age or older, cisgender women, and late presenters (CD4 <200 cells/μl and/or ≥1 AIDS-defining event at baseline). Both treatment-naïve and treatment-experienced participants had high persistence with Biktarvy (91%, n=1032/1135) across both groups, Biktarvy was generally well-tolerated and no resistance to the components of Biktarvy emerged. 148 (13%) participants had any adverse event and 2 (<1%) had a serious adverse event. The most common drug-related adverse events observed to date in the BICSTaR study were weight increase (3%), nausea (1%), depression (1%), headache (1%), fatigue (1%), diarrhoea (1%) and sleep disorder (1%). These large cohort findings continue to reinforce the real-world effectiveness of Biktarvy across populations and are consistent with evidence from randomized clinical trials.
Additional Biktarvy data presented at EACS 2021 include a Phase 3 trial (Study 1878) that demonstrated the durable efficacy of Biktarvy. In the study, 99% of people living with HIV who switched to Biktarvy from a boosted protease inhibitor-based regimen maintained and achieved long-term viral suppression through a median of 101 weeks (n=525/532), including 98% of participants with pre-existing resistance (n=212/217; median of 108 weeks) and 98% of participants with viral blips (n=39/40; median of 109 weeks), with no treatment-emergent resistance to Biktarvy. A pooled analysis of five Phase 3 studies (1844, 1878, 4030, 4449, 4580) also found that regardless of pre-existing TAMS (thymidine analog-associated mutations M41L, D67N, K70R, L210W, T215Y/F, and K219Q/E), a high proportion of those on Biktarvy were able to maintain virologic suppression and had an absence of treatment-emergent resistance. These data support the continued evaluation of Biktarvy as a potential option for virologically suppressed people living with HIV with known resistance. The use of Biktarvy in individuals with a history of treatment failure or known resistance to the components of Biktarvy is investigational, and the safety and efficacy of Biktarvy for this use have not been established.
Please see below for the U.S. Indication and Important Safety Information, including Boxed Warning, for Biktarvy.
There is currently no cure for HIV or AIDS.
About BICSTaR
The Bictegravir Single Tablet Regimen (BICSTaR) Study is an ongoing, multinational, observational single-arm, non-comparative real-world cohort study, which aims to evaluate the effectiveness, safety, tolerability, and patient-reported outcomes of treatment with Biktarvy in treatment‐naïve and treatment‐experienced people living with HIV. Among the people living with HIV enrolled in the BICSTaR study, there is a high baseline prevalence of comorbidities.
About Biktarvy
Biktarvy is a complete HIV-1 treatment that combines three powerful medicines to form the smallest integrase strand transfer inhibitor (INSTI)-based single-tablet regimen (STR) available, offering simple once-daily dosing with or without food, with a limited drug interaction potential and a high barrier to resistance. Biktarvy combines the novel, unboosted INSTI bictegravir, with the Descovy® (emtricitabine 200 mg/tenofovir alafenamide 25 mg tablets, F/TAF) backbone. Biktarvy is a complete single-tablet regimen and should not be taken with other HIV-1 medicines.
In February 2018, the U.S. Food and Drug Administration (FDA) approved Biktarvy (bictegravir 50 mg/emtricitabine 200 mg/tenofovir alafenamide 25 mg tablets, B/F/TAF) as a once-daily single-tablet regimen for the treatment of HIV-1 infection in adults. In June 2019, the FDA approved labeling revisions to Biktarvy, expanding the patient population to include pediatric patients weighing at least 25 kg. In October 2021, the FDA approved a new low-dose tablet formulation of Biktarvy (bictegravir 30 mg/emtricitabine 120 mg/tenofovir alafenamide 15 mg tablets) for pediatric patients weighing at least 14 kg to less than 25 kg. For all patient populations, Biktarvy is only indicated for the treatment of HIV-1 infection in people who have no antiretroviral treatment history or to replace the current antiretroviral regimen in those who are virologically suppressed (HIV-1 RNA less than 50 copies per mL) on a stable antiretroviral regimen with no history of treatment failure and no known substitutions associated with resistance to the individual components of Biktarvy.
U.S. Indication for Biktarvy
Biktarvy is indicated as a complete regimen for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults and pediatric patients weighing at least 14 kg who have no antiretroviral treatment history or to replace the current antiretroviral regimen in those who are virologically-suppressed (HIV-1 RNA less than 50 copies per mL) on a stable antiretroviral regimen with no history of treatment failure and no known substitutions associated with resistance to the individual components of Biktarvy.
U.S. full Prescribing Information for Biktarvy and Descovy, including BOXED WARNINGS, are available at www.gilead.com.
Biktarvy, Descovy, Gilead and the Gilead logo are registered trademarks of Gilead Sciences, Inc., or its related companies. All other trademarks are the property of their respective owner(s).
For more information about Gilead, please visit the company’s website at www.gilead.com, follow Gilead on Twitter (@Gilead Sciences) or call Gilead Public Affairs at 1-800-GILEAD-5 or 1-650-574-3000.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211029005237/en/
Source: Gilead Sciences, Inc.
Oct. 29, 2021 8:43 AM ET Gilead Sciences, Inc. (GILD)
By: Ravikash, SA News Editor3 Comments
October 29, 2021
- In a Phase 3 clinical trial, Study 3111-301-001, cariprazine (VRAYLAR®) met its primary endpoint demonstrating statistically significant change from baseline to week six in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score in patients with major depressive disorder
- In a second Phase 3 clinical trial, Study 3111-302-001, cariprazine demonstrated numerical improvement in depressive symptoms from baseline to week six in MADRS total score compared with placebo but did not achieve statistical significance
- Safety data were consistent with the established safety profile of cariprazine across indications with no new safety signals identified
NORTH CHICAGO, Ill., Oct. 29, 2021 /PRNewswire/ -- AbbVie (NYSE: ABBV) today announced top-line results from two Phase 3 clinical trials, Study 3111-301-001 and Study 3111-302-001, evaluating the efficacy and safety of cariprazine (VRAYLAR®) as an adjunctive treatment for patients with major depressive disorder (MDD). In Study 3111-301-001, cariprazine showed a statistically significant change from baseline to week six in the Montgomery-Åsberg Depression Rating Scale (MADRS) total score compared with placebo. Patients treated with cariprazine at 1.5 mg/day achieved improved MADRS total score at week six compared to placebo (p-value=0.0050). Patients treated with cariprazine at 3.0 mg/day demonstrated improvement in MADRS total score at week six over placebo but did not meet statistical significance (p-value=0.0727). In Study 3111-302-001, cariprazine demonstrated numerical improvement in depressive symptoms from baseline to week six in MADRS total score compared with placebo but did not meet its primary endpoint for either the 1.5 mg/day or 3.0 mg/day dose.
In a previously published Phase 2/3 registration-enabling study, RGH-MD-75, patients treated with cariprazine flexible doses of 2.0–4.5 mg/day in addition to ongoing antidepressant therapy (ADT) met the primary endpoint and achieved improved MADRS total scores at week eight compared to placebo (p-value=0.0114).
Based on the positive results of studies 3111-301-001 and RGH-MD-75, and the totality of data reported, AbbVie intends to submit a supplemental New Drug Application (sNDA) with the U.S. Food and Drug Administration for the expanded use of cariprazine for the adjunctive treatment of MDD.
"When added to ongoing antidepressant treatment that has produced inadequate response in patients with major depressive disorder, cariprazine has now demonstrated that it can further improve depressive symptoms by providing statistically significant and clinically meaningful improvements compared to placebo in two large, well-controlled registrational clinical trials," said Michael Severino, M.D., vice chairman and president, AbbVie. "Major depressive disorder is one of the most common and serious mental illnesses, and more than half of these patients never experience satisfactory results from this debilitating condition. Based on the results, we believe cariprazine has the potential to benefit these patients as an adjunctive treatment."
The safety results of cariprazine in all three studies were consistent with its established safety profile across indications with no new safety signals identified. The most common adverse events occurring at >5% in the cariprazine groups during the six-week study period were akathisia, nausea, insomnia, headache and somnolence.
Full results from studies 3111-301-001 and 3111-302-001 will be presented at a future medical meeting.
MDD is a common condition with 19 million people of all ages affected in the United States.1 The World Health Organization lists depression as the third-leading cause of disability worldwide and as a major contributor to the overall global burden of disease. Symptoms can include depressed mood, loss of pleasure or interest in activities, changes in appetite or weight, changes in sleep, psychomotor agitation, loss of energy, feelings of worthlessness, indecisiveness, and current thoughts of death.2 In the United States, the mean age of onset for the first episode is 26 years old,3 and MDD represents an estimated $211 billion economic burden.4
Cariprazine is marketed as VRAYLAR in the United States and is FDA-approved to treat depressive, acute manic and mixed episodes associated with bipolar I disorder, as well as schizophrenia in adults. Cariprazine is being co-developed by AbbVie and Gedeon Richter Plc. More than 8,000 patients worldwide have been treated with cariprazine across more than 20 clinical trials evaluating the efficacy and safety of cariprazine for a broad range of psychiatric disorders.
About Studies 3111-301-001 and 3111-302-001
Study 3111-301-001 is a randomized, double-blind, placebo-controlled, multicenter trial with 759 participants conducted in United States, Bulgaria, Estonia, Germany, Hungary, Ukraine, and the United Kingdom. Study 311-302-001 is a randomized, double-blind, placebo-controlled, multicenter trial with 752 participants conducted in United States, Canada, Czech Republic, Finland, Poland, Serbia, and Slovakia. For both studies, following a screening period of up to 14 days, patients with an inadequate clinical response to their antidepressant monotherapy (ADT) were randomized into three treatment groups (1:1:1). The first group received cariprazine 1.5 mg/day + ADT, the second group received cariprazine 3.0 mg/day + ADT, and the third group received placebo + ADT. For six weeks, the medication was given once daily in addition to the ongoing ADT treatment, to which the patient had experienced inadequate clinical response.
About Study RGH-MD-75
Study RGH-MD-75 is a randomized, double-blind, placebo-controlled, flexible-dose, outpatient, multicenter trial with 808 participants, conducted in United States, Estonia, Finland, Slovakia, Ukraine and Sweden. After 7-14 days of screening and washout of prohibited medications, eligible patients entered an 8-week, double-blind treatment period in which they continued antidepressant treatment and were randomized (1:1:1) to adjunctive cariprazine 1-2 mg/day, cariprazine 2-4.5 mg/day, or placebo. After double-blind treatment, patients entered a 1-week safety follow-up period. Data from Study RGH-MD-75 were published in the Journal of Clinical Psychiatry.5
More information about studies 3111-301-001, 3111-302-001 and RGH-MD-75 is available at www.clinicaltrials.gov.
About VRAYLAR® (cariprazine)
VRAYLAR is an oral, once-daily atypical antipsychotic approved for the acute treatment of adults with manic or mixed episodes associated with bipolar I disorder (3 to 6 mg/day) and for the treatment of depressive episodes associated with bipolar I disorder (bipolar depression) in adults (1.5 or 3 mg/day). VRAYLAR is also approved for the treatment of schizophrenia in adults (1.5 to 6 mg/day).
While the mechanism of action of VRAYLAR is unknown, the efficacy of VRAYLAR could be mediated through a combination of partial agonist activity at central dopamine D₂ and serotonin 5-HT1A receptors and antagonist activity at serotonin 5-HT2A receptors. Pharmacodynamic studies with cariprazine have shown that it acts as a partial agonist with high binding affinity at dopamine D3, dopamine D2, and serotonin 5-HT1A receptors. Cariprazine demonstrated up to ~8-fold greater in vitro affinity for dopamine D3 vs D2 receptors. Cariprazine also acts as an antagonist at serotonin 5-HT2B and 5-HT2A receptors with high and moderate binding affinity, respectively as well as it binds to the histamine H1 receptors. VRAYLAR shows lower binding affinity to the serotonin 5-HT2C and α1A- adrenergic receptors and has no appreciable affinity for cholinergic muscarinic receptors. The clinical significance of these in vitro data is unknown.
VRAYLAR is being developed jointly by AbbVie and Gedeon Richter Plc, with AbbVie responsible for commercialization in the U.S., Canada, Japan, Taiwan and certain Latin American countries (including Argentina, Bolivia, Brazil, Chile, Colombia, Ecuador, Mexico, Peru and Venezuela).
Visit www.vraylar.com for more information.
Please see the full Prescribing Information, including Boxed Warnings, and Medication Guide.
For more information about AbbVie, please visit us at www.abbvie.com.
Oct. 29, 2021 8:03 AM ETAbbVie Inc. (ABBV)
By: Aakash Babu, SA News Editor3 Comments
Oct. 29, 2021 7:00 AM ETSanofi (SNY)
Q3: 2021-10-28 Earnings Summary
TranscriptEPS of - misses by $1.17 | Revenue of $12.19B (10.12% Y/Y) beats by $370.16M
MISSISSAUGA, ON, Oct. 29, 2021 /CNW/ - Libtayo® (cemiplimab) is now approved in Canada for the treatment of adults with locally advanced basal cell carcinoma (BCC) previously treated with a hedgehog pathway inhibitor (HHI).2 The approval of Libtayo follows the European Commission (EC) approval announced in June 2021 and the US in February 2021.3
"This is a game changer for patients with advanced basal cell carcinoma who no longer have treatment options," says Dr. Marcus Butler, University Health Network. "Again, we are finding that immunotherapy can benefit patients who previously had no options. Immunotherapy continues to be a pillar of cancer therapy."
With today's announcement, Libtayo is now offered as an immunotherapy option for three advanced cancers. In April 2019, Libtayo became the first immunotherapy option in Canada for adults with metastatic or locally advanced cutaneous squamous cell carcinoma (CSCC) who are not candidates for curative surgery or curative radiation. In October 2021, the availability of Libtayo was expanded to include adults with advanced non-small cell lung cancer expressing PD-L1 in ≥ 50% of tumour cells (Tumour Proportion Score [TPS] ≥ 50%), as determined by a validated test, with no EGFR, ALK or ROS1 aberrations who have locally advanced NSCLC who are not candidates for surgical resection or definitive chemoradiation, or metastatic NSCLC.4
"Canadians living with advanced basal cell carcinoma are in urgent need of access to new therapeutic options to treat this elusive yet serious skin cancer," says Kathy Barnard, skin cancer survivor and founder of Save Your Skin Foundation. "We applaud Health Canada's approval of Libtayo, which provides new hope for patients and their loved ones."
"Patients with locally advanced basal cell carcinoma face significant life-threatening challenges, including the physical and emotion devastation of the disease," says Annette Cyr, founder and chair of board of directors, Melanoma Network of Canada. "Beyond surgery and very limited therapeutic treatments, there are few options currently available. Today's approval of Libtayo by Health Canada provides a treatment option that gives our patient community and their families reason to hope."
About Study 1620
This Health Canada approval was based on data from Phase 2, Study 1620 that enrolled 132 patients with advanced BCC in an open-label, single arm trial.5 The major efficacy endpoints were confirmed objective response rate (ORR) and duration of response (DOR) as assessed by independent central review (ICR).6 For patients with externally visible target lesions, ORR was determined by a composite endpoint that integrated ICR assessments of radiologic data (RECIST 1.1) and digital medical photography (WHO criteria).7
"BCC affects a significant number of people across Canada," says Carrie McElroy, Interim General Manager, Sanofi Genzyme, and Interim Sanofi Canada Country Lead. "The approval of Libtayo as another treatment option for those with advanced BCC is an important milestone and we are proud to be able to make this available to patients."
About Libtayo
Libtayo is a fully human monoclonal antibody targeting the immune checkpoint receptor PD-1 on T-cells. By binding to PD-1, Libtayo has been shown to block cancer cells from using the PD-1 pathway to suppress T-cell activation.
The recommended dose of Libtayo is 350 mg administered as an intravenous infusion over 30 minutes every three weeks, until disease progression or unacceptable toxicity. Libtayo is available as a single-dose 350 mg vial.8
The extensive clinical program for Libtayo is focused on difficult-to-treat cancers. Current clinical development programs include Libtayo in combination with chemotherapy for advanced NSCLC irrespective of PD-L1 expression and Libtayo monotherapy for advanced cervical cancer. Libtayo is also being investigated in combination with either conventional or novel therapeutic approaches for other solid tumors and blood cancers. These potential uses are investigational, and their safety and efficacy have not been evaluated by any regulatory authority.
Libtayo is being jointly developed by Sanofi and Regeneron under a global collaboration agreement.
SOURCE Sanofi Canada
https://seekingalpha.com/symbol/REGN
https://seekingalpha.com/symbol/SNY
October 28, 2021
-- The pivotal Phase 3, randomized, double-blind, double-dummy, active-controlled study of continuous, subcutaneous infusion of ABBV-951 (foslevodopa/foscarbidopa) in patients with advanced Parkinson's disease met its primary endpoint in a 12-week study
-- Patients who received 24 hours/daily ABBV-951 showed statistically significant increases in hours of "On" time without troublesome dyskinesia, compared to oral levodopa/carbidopa. A significant reduction in hours of "Off" time was also observed
-- Systemic adverse events were generally consistent with the well-established safety profile of levodopa/carbidopa medications and infusion site adverse events were mostly non-serious and mild or moderate in severity
-- Data from this head-to-head superiority study will be a key component of global regulatory submissions
NORTH CHICAGO, Ill., Oct. 28, 2021 /PRNewswire/ -- AbbVie (NYSE: ABBV) today announced that continuous 24 hours/day subcutaneous infusion of ABBV-951 (foslevodopa/foscarbidopa) was statistically superior to oral levodopa/carbidopa in reducing motor fluctuations in patients with advanced Parkinson's disease (PD) in a Phase 3, randomized, double-blind, double-dummy, active-controlled study. The study met its primary endpoint of increase from baseline in "On" time (hours) without troublesome dyskinesia (involuntary movements) after 12 weeks based on the Parkinson's Disease Diary (PD Diary).1 These results will be a key component of global regulatory submissions.
The increase in "On" time at week 12 was 2.72 hours for ABBV-951 versus 0.97 hours for oral levodopa/carbidopa (LD/CD) (p= 0.0083).1 Improvements in "On" time were observed as early as the first week and persisted throughout the 12 weeks.1 It was also observed that an improvement from baseline in hours of average daily normalized "Off" time followed a similar pattern in reductions versus oral LD/CD after the first week and persisting through week 12.1 Decreases in "Off" time after 12 weeks were 2.75 hours for ABBV-951 versus 0.96 hours for oral LD/CD (p=0.0054).1
"Parkinson's disease is a progressive, irreversible neurological disease with debilitating symptoms that can make daily life challenging," said Michael Severino, M.D., vice chairman and president, AbbVie. "We're committed to addressing the continued needs of patients and are encouraged by these results that highlight a potential alternative treatment option for those affected by advanced Parkinson's disease."
"Patients need more therapeutic options to control their symptoms and troublesome dyskinesia for this debilitating disease," said Jason Aldred, M.D. FAAN of Selkirk Neurology, clinical associate professor at the University of Washington, clinical assistant professor at Washington State University Elson S. Floyd College of Medicine, and a principal investigator of the study. "These data are promising and demonstrate positive results on a key endpoint used to assess efficacy of treatments for patients with advanced Parkinson's."
The majority of the adverse events (AEs) reported were non-serious and mild to moderate in severity in the ABBV-951 group.1 Incidence of serious AEs were 8% and 6% in the ABBV-951 group and oral LD/CD group, respectively.1 There was one patient with a treatment-emergent AE leading to death in the oral LD/CD group and none in the ABBV-951 group.1 The most common AEs reported in ≥5% in the ABBV-951 group were infusion site AEs (erythema, pain, cellulitis, edema, bruising, hemorrhage, nodule, induration, infection, and pruritus), dyskinesia, "On" and "Off" phenomenon, fall, hallucinations (including visual hallucination), balance disorder, constipation and peripheral swelling.1 The incidence of infusion site AEs was higher in the ABBV-951 group than in the oral LD/CD group and most of them were non-serious, mild to moderate in severity, resolved with or without treatment and none led to systemic complications.1 Incidence of hallucination and psychosis AEs was higher in the ABBV-951 group than in the oral LD/CD group.1 These AEs were non-serious, mild to moderate in severity. Incidence of falls and associated injuries was lower in the ABBV-951 group compared to the oral LD/CD group.1 Adverse events led to study treatment discontinuation in 21.6% of patients in the ABBV-951 group and 1.5% in the oral LD/CD group.1
Full results from the Phase 3 study will be presented at a future medical meeting or submitted for publication in a peer-reviewed journal. ABBV-951 is an investigational therapy and it is not approved for use. The safety and efficacy of ABBV-951 have not been evaluated by regulatory authorities.
About the Phase 3 M15-736 Study2
The Phase 3 randomized, double-blind, double-dummy, active-controlled study compared the efficacy, safety and tolerability of ABBV-951 (foslevodopa/foscarbidopa) to oral LD/CD in advanced PD patients. Parkinson's disease patients were provided with a home diary (the PD Diary) to assess their motor status. The primary endpoint of "good" time (defined as "On" time without dyskinesia plus "On" time with non-troublesome dyskinesia), in contrast to "bad" time ("Off" time plus "On" time with troublesome dyskinesia) is collected and averaged over three consecutive days and normalized to a typical 16-hour waking period. Baseline values are defined as the average of normalized "good" time collected over the three PD Diary days before randomization. Approximately 130 adult participants with advanced PD were enrolled in the study across 80 sites worldwide. The study was comprised of two arms. In one arm, participants received the ABBV-951 solution as a continuous infusion under the skin plus oral placebo capsules for levodopa/carbidopa. In the second arm, participants received placebo solution for ABBV-951 as a continuous subcutaneous infusion plus oral capsules containing levodopa/carbidopa encapsulated tablets. The treatment duration was 12 weeks. More information on the study can be found on www.clinicaltrials.gov (NCT04380142).
About ABBV-951
ABBV-951 (foslevodopa/foscarbidopa) is a solution of levodopa and carbidopa prodrugs for continuous subcutaneous infusion that is being investigated for the treatment of advanced Parkinson's disease in patients whose motor symptoms are not controlled by oral medications.
For more information about AbbVie, please visit us at www.abbvie.com
Oct. 28, 2021 8:59 AM ET AbbVie Inc. (ABBV)
By: Aakash Babu, SA News Editor11 Comments
https://www.nasdaq.com/market-activity/stocks/abbv/dividend-history
October 27, 2021 6:45 am ET
Islatravir Combined with Doravirine Continued to Maintain Viral Suppression for People Who Had Not Previously Received Treatment
KENILWORTH, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, today announced Week 144 data from the Phase 2b dose-ranging study evaluating the antiretroviral activity, tolerability, and safety of islatravir in combination with doravirine compared to doravirine/lamivudine/tenofovir disoproxil fumarate (DOR/3TC/TDF) in antiretroviral treatment-naïve adults with HIV-1. Phase 2b data through 144 weeks demonstrated that islatravir combined with doravirine continued to maintain viral suppression, as measured by the number of study participants achieving HIV-1 RNA levels <50 copies/mL, similar to DOR/3TC/TDF. The data further characterized the tolerability and safety profile of islatravir in combination with doravirine. The Week 144 results are a follow-up to the Week 96 results and safety data presented at the virtual 2020 International Congress on Drug Therapy in HIV Infection (HIV Glasgow 2020) and the virtual International AIDS Society Conference on HIV Science (IAS 2021), respectively. The Week 144 data were consistent with the Week 96 results in the 0.75 mg dose group (the selected Phase 3 dose), and were shared as an oral presentation during the 18thEuropean AIDS Conference (EACS 2021) in London, U.K.
“We are encouraged by the Week 144 findings presented at EACS, which further support the potential of a two-drug doravirine/islatravir regimen for the treatment of HIV-1,” said Dr. Joan Butterton, vice president, global clinical development, infectious diseases, Merck Research Laboratories. “As we advance the ILLUMINATE clinical development program, we look forward to continuing to study this investigational treatment across diverse patient populations.”
Initial data from two pivotal ILLUMINATE Phase 3 clinical trials evaluating a combination product of doravirine/islatravir in virologically suppressed adults with HIV-1 switching from a stable antiretroviral regimen were also recently shared.
Islatravir is currently being evaluated across a variety of dosing regimens, for both the treatment of HIV-1 in combination with other antiretroviral agents and for the prevention of HIV-1 as a monotherapy. An overview of the islatravir treatment and prevention development program is available here.
Week 144 Efficacy and Safety Results from Phase 2b Study of Investigational Islatravir with Doravirine
In this international, multicenter clinical trial (NCT03272347), treatment-naïve adult participants with HIV-1 were randomly assigned (1:1:1:1) to one of four once-daily oral treatment groups: islatravir 0.25 mg (n=29), 0.75 mg (n=30), or 2.25 mg (n=31) in combination with doravirine (100 mg) and 3TC (300 mg) compared to DOR/3TC/TDF (n=31; Part 1). After a minimum of 24 weeks of treatment, participants in the islatravir treatment groups with HIV-1 RNA <50 copies/mL were transitioned to a two-drug regimen consisting of doravirine and islatravir, without 3TC (Part 2). Participants in the islatravir treatment groups then transitioned to 0.75 mg islatravir (the selected Phase 3 dose) plus doravirine between Weeks 60 to 84, and they continued the combination therapy through Week 144 (Part 3). At Week 144, participants switched to the fixed-dose combination of the selected dose of islatravir and doravirine as open-label treatment until the end of the trial at Week 192 (Part 4).
At Week 144, at all dose levels, islatravir combined with doravirine maintained virologic suppression as measured by the proportion of study participants achieving HIV-1 RNA levels <50 copies/mL: 72.4% (n=21/29), 83.3% (n=25/30), and 61.3% (n=19/31) of participants maintained virologic suppression in the 0.25 mg, 0.75 mg, and 2.25 mg islatravir combined with doravirine groups, respectively. Overall, 72.2% (n=65/90) of the combined islatravir with doravirine groups had HIV-RNA levels <50 copies/mL, which was similar to 77.4% (n=24/31) of the DOR/3TC/TDF group. Through Week 144, seven participants met the criteria for protocol-defined virologic failure (PDVF) (confirmed HIV-1 RNA ≥50 copies/mL) and discontinued treatment, all of whom had HIV-1 RNA levels <80 copies/mL. No participants met the criteria for clinically significant confirmed viremia (HIV-1 RNA ≥200 copies/mL) or viral drug resistance analysis.
The proportion of participants experiencing at least one adverse event (AE) at Week 144 was similar between the islatravir combined with doravirine and DOR/3TC/TDF groups. At Week 144, 89.7% (n=26/29), 90.0% (n=27/30), and 77.4% (n=24/31) of participants experienced AEs in the 0.25 mg, 0.75 mg, and 2.25 mg islatravir combined with doravirine groups, respectively. Additionally, 85.6% (n=77/90) of the combined islatravir combined with doravirine groups experienced AEs, as compared to 87.1% (n=27/31) in the DOR/3TC/TDF group. The most common drug-related AEs for the combined islatravir combined with doravirine groups versus DOR/3TC/TDF were diarrhea (1.1% [n=1/90] vs. 12.9% [n=4/31), nausea (3.3% [n=3/90] vs. 9.7% [n=3/31]), headache (2.2% [n=2/90] vs. 3.2% [n=1/31]), and abnormal dreams [2.2% [n=2/90] vs. 0% [n=0/31]). No additional islatravir combined with doravirine participants reported drug-related AEs after Week 48. The rate of discontinuations due to drug-related AEs was 2.2% (n=2/90) for the combined islatravir with doravirine groups and 3.2% (n=1/31) for DOR/3TC/TDF, all occurring before Week 48. There were no deaths or serious-drug-related AEs in the islatravir combined with doravirine groups.
About PIFELTRO™ and DELSTRIGO™
PIFELTRO™ (doravirine, 100 mg) is indicated in combination with other antiretroviral (ARV) agents for the treatment of HIV-1 infection in adult patients with no prior ARV treatment history or to replace the current ARV regimen in those who are virologically suppressed (HIV-1 RNA less than 50 copies per mL) on a stable ARV regimen with no history of treatment failure and no known substitutions associated with resistance to doravirine.
DELSTRIGO™ (doravirine, 100 mg/lamivudine 300 mg/tenofovir disoproxil fumarate, 300 mg) is indicated as a complete regimen for the treatment of HIV-1 infection in adult patients with no prior ARV treatment history or to replace the current ARV regimen in those who are virologically suppressed (HIV-1 RNA less than 50 copies per mL) on a stable ARV regimen with no history of treatment failure and no known substitutions associated with resistance to the individual components of DELSTRIGO. DELSTRIGO contains a boxed warning regarding post-treatment acute exacerbations of hepatitis B (HBV) infection. See Selected Safety Information below.
About Islatravir (MK-8591)
Islatravir (MK-8591) is Merck’s investigational nucleoside reverse transcriptase translocation inhibitor under evaluation in more than 10 clinical trials. For treatment, islatravir is being evaluated in combination with other antiretrovirals, including the ILLUMINATE clinical trials program for a once-daily regimen. In the IMPOWER clinical trials, islatravir is also being studied for pre-exposure prophylaxis (PrEP) of HIV-1 infection as a single agent across a variety of formulations, including an oral once-monthly regimen.
For more information, visit www.merck.com
Please see Prescribing Information for PIFELTRO (doravirine) at: https://www.merck.com/product/usa/pi_circulars/p/pifeltro/pifeltro_pi.pdf; and Patient Information for PIFELTRO (doravirine) at: https://www.merck.com/product/usa/pi_circulars/p/pifeltro/pifeltro_ppi.pdf
Please see Prescribing Information for DELSTRIGO (doravirine/3TC/TDF) at: https://www.merck.com/product/usa/pi_circulars/d/delstrigo/delstrigo_pi.pdf; and Patient Information for DELSTRIGO (doravirine/3TC/TDF) at: https://www.merck.com/product/usa/pi_circulars/d/delstrigo/delstrigo_ppi.pdf
Oct. 27, 2021 7:08 AM ET
By: Ravikash, SA News Editor
- Second Fast Track Designation for Nemvaleukin -
DUBLIN, Oct. 25, 2021 /PRNewswire/ -- Alkermes plc (Nasdaq: ALKS) today announced that the U.S. Food and Drug Administration (FDA) has granted Fast Track designation to nemvaleukin alfa (nemvaleukin), the company's novel, investigational, engineered interleukin-2 (IL-2) variant immunotherapy, in combination with pembrolizumab, an anti-PD-1 antibody, for the treatment of platinum-resistant ovarian cancer. The FDA previously granted Fast Track designation and Orphan Drug designation to nemvaleukin for the treatment of mucosal melanoma.
"This Fast Track designation in platinum-resistant ovarian cancer highlights the potential clinical utility of nemvaleukin in combination with pembrolizumab in this difficult-to-treat disease for which there is no approved immunotherapy and there remains significant need for new treatment options," said Craig Hopkinson, M.D., Chief Medical Officer and Executive Vice President of Research & Development at Alkermes. "We are excited to initiate our planned ARTISTRY-7 phase 3 trial in platinum-resistant ovarian cancer, as we advance nemvaleukin toward potential registration and seek to help patients living with this disease."
Fast Track is an FDA process designed to facilitate the development, and expedite the review, of potential therapies that seek to treat serious conditions and fill an unmet medical need. A drug candidate that receives Fast Track designation is eligible for more frequent communication with the FDA throughout the drug development process and a rolling and/or priority review of its marketing application if relevant criteria are met. For more information on Fast Track designation, please visit the FDA's website, available at https://www.fda.gov/patients/fast-track-breakthrough-therapy-accelerated-approval-priority-review/fast-track.
About Nemvaleukin Alfa (nemvaleukin)
Nemvaleukin is an investigational, novel, engineered fusion protein comprised of modified interleukin-2 (IL-2) and the high affinity IL-2 alpha receptor chain, designed to preferentially expand tumor-killing immune cells while avoiding the activation of immunosuppressive cells by selectively binding to the intermediate-affinity IL-2 receptor complex. The selectivity of nemvaleukin is designed to leverage the proven anti-tumor effects of existing IL-2 therapy while mitigating certain limitations.
About the ARTISTRY Clinical Development Program
ARTISTRY is an Alkermes-sponsored clinical development program evaluating nemvaleukin as a potential immunotherapy for cancer. The ARTISTRY program is comprised of multiple clinical trials evaluating intravenous and subcutaneous dosing of nemvaleukin, both as a monotherapy and in combination with the anti-PD-1 therapy KEYTRUDA® (pembrolizumab) in patients with advanced solid tumors. Ongoing trials in the ARTISTRY program include: ARTISTRY-1, ARTISTRY-2, ARTISTRY-3 and ARTISTRY-6.
View original content to download multimedia:https://www.prnewswire.com/news-releases/alkermes-receives-fda-fast-track-designation-for-nemvaleukin-alfa-in-combination-with-pembrolizumab-for-the-treatment-of-platinum-resistant-ovarian-cancer-301407169.html
SOURCE Alkermes plc
Oct. 25, 2021 7:10 AM ET Alkermes plc (ALKS) By: Aakash Babu, SA News Editor
New Analyses Suggest Favorable Results for STELARA® (ustekinumab) When Used as a First-Line Therapy for Bio-Naïve Patients with Moderately to Severely Active Crohn’s Disease and Ulcerative Colitis
STELARA when used as a first-line therapy was associated with longer time in clinical remission or clinical response, including the postponing of surgery, among adult patients with moderately to severely active ulcerative colitis compared with usage as a second- or third-line therapy in a modelled analysis
Bio-naïve patients with moderately to severely active Crohn’s disease started on STELARA showed higher rates of persistence at one year compared to adalimumab in a retrospective real-world evidence study
SPRING HOUSE, PENNSYLVANIA, October 25, 2021 – The Janssen Pharmaceutical Companies of Johnson & Johnson today announced data from two new analyses of STELARA® (ustekinumab) for the treatment of adults with moderately to severely active Crohn’s disease (CD) and ulcerative colitis (UC).1,2 In a modelled analysisa focused on treatment sequencing using data from randomized controlled trials, network meta-analysis and literature, results showed patient time spent in clinical remission or response was highest when STELARA was used as a first-line advanced therapy for bio-naïve patients with moderately to severely active UC relative to outcomes associated with second- or third-line use (P0540).1 Additionally, in a separate real-world claims analysis, a greater proportion of bio-naïve patients who started biologic therapy with STELARA (n=948) for moderately to severely active CD showed persistence at 12 months versus adalimumab (n=4,143) (P0525).2 These data are among 16 abstracts, including one oral presentation, presented at the 2021 American College of Gastroenterology Annual Scientific Meeting, which is taking place October 22-27 in Las Vegas, Nevada.1,2
“Data emerging from these analyses inform physicians with additional evidence to support STELARA as a first-line option for patients with moderately to severely active Crohn’s disease and ulcerative colitis,” said Christopher Gasink, M.D., Head of Immunology Medical Affairs, Gastroenterology, Janssen Scientific Affairs, LLC. “Many patients living with inflammatory bowel disease can cycle through different therapies as a result of loss or lack of treatment response. Studies like these are important in helping guide physicians to select an appropriate therapeutic option for their patients in a first-line setting.”
About (P0540): Identifying the Optimal Treatment Sequence for STELARA in Treatment Algorithms for Advanced Therapies in UC1
In the hybrid model described in the editor’s note, the treatment basket for first- and second-line UC was represented by infliximab (33 percent), adalimumab (33 percent), and vedolizumab (33 percent), and third-line treatment was comprised of vedolizumab (50 percent) and tofacitinib (50 percent). Patients moved to next line of treatment upon loss of response, and those failing the first three lines of advanced therapy moved to conventional treatment (e.g., aminosalicylates and/or immunosuppressants, corticosteroids). The model estimated time spent in remission, response, active UC (Mayo score 6-12), surgery, as well as occurrences of death over one, three and five years. Transition probabilities for remission, response and surgery were derived from randomized controlled trials, network meta-analysis, and the literature.These are modelled results based on input assumptions largely from clinical trials and a network meta-analysis.
About (P0525): Treatment Persistence Among Bio-Naïve Patients with CD Initiated on STELARA or adalimumab2
Bio-naïve adults with CD initiated on STELARA or adalimumab between September 23, 2016 and August 1, 2019 were selected from a de-identified health insurance claims data from the IQVIA PharMetrics® Plus. Bio-naïve patients were defined as patients with no medical or pharmacy claim for biologics indicated for CD during the baseline period (12 months before the initiation of the index agent). Baseline characteristics were balanced in weighted STELARA (n=948) and adalimumab (n=4,143) cohorts using inverse probability of treatment weights. Persistence on index agent was defined as absence of gaps >120 days (STELARA) or >60 days (adalimumab) between days of therapy supply. Composite endpoints of being persistent on index biologic and corticosteroid-free (<14 days of supply after day 90 post-index) and persistent and on monotherapy (no immunomodulators or non-index biologics) were assessed. All endpoints were estimated at 12 months post-index using weighted Kaplan-Meier and Cox's proportional hazards model analyses. Analyses of claims database depend on correct diagnosis, procedure, and drug codes, and misclassification may have occurred. All patients are assumed to have moderate to severe disease since they started biologic treatment.
About STELARA® (ustekinumab)10
STELARA® (ustekinumab) is a fully human monoclonal antibody and is the first biologic treatment to selectively inhibit the interleukin (IL)-12 and IL-23 pathways. STELARA is approved in the United States for the treatment of: 1) adults and children six years and older with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy; 2) adult patients (18 years or older) with active psoriatic arthritis, used alone or in combination with methotrexate (MTX); 3) adult patients (18 years and older) with moderately to severely active CD; 4) adult patients (18 years and older) with moderately to severely active UC.
The Janssen Pharmaceutical Companies of Johnson & Johnson maintain exclusive worldwide marketing rights to STELARA®.
Please read the full Prescribing Information and Medication Guide for STELARA® and discuss any questions you have with your doctor.
Oct. 25, 2021 8:48 AM ET Johnson & Johnson (JNJ)
By: Jonathan M Block, SA News Editor
Johnson & Johnson (NYSE:JNJ) says two analyses of Stelara (ustekinumab) indicate the biologic was very effective in patients who had not received prior treatment.
October 28, 2021 DownloadPDF Format (opens in new window)
- The MAA seeks approval of ruxolitinib cream for the treatment of adolescent and adult patients with non-segmental vitiligo with facial involvement
WILMINGTON, Del.--(BUSINESS WIRE)-- Incyte (Nasdaq:INCY) today announced the validation of the European Marketing Authorization Application (MAA) for ruxolitinib cream, a topical JAK inhibitor, as a potential treatment for adolescents and adults (age >12 years) with non-segmental vitiligo with facial involvement. The validation of the MAA by the European Medicines Agency (EMA) confirms that the submission is ready to enter the formal review process.
“The EMA’s validation of the MAA for ruxolitinib cream marks an important milestone for people with vitiligo, for whom there is often a significant impact on everyday life and who currently have limited treatment options,” said Jonathan Dickinson, General Manager Europe, Executive Vice President at Incyte. “We are committed to listening to the patient community to understand how we can help fulfill unmet needs and support healthcare providers to better manage this challenging disease. We look forward to working with the regulatory authorities, with the aim to bring this new potential therapy to eligible patients as soon as possible.”
The MAA is supported by data from the Phase 3 TRuE-V clinical trial program evaluating the safety and efficacy of ruxolitinib cream in more than 600 people (age >12 years) with vitiligo. Results from the Phase 3 program, recently presented at the 30th European Academy of Dermatology and Venereology (EADV) congress during a late-breaking research session, showed significant improvements in facial and total body repigmentation at 24 weeks of treatment with ruxolitinib cream in people with vitiligo. In the TRuE-V studies, patients using ruxolitinib cream did not report clinically significant application site reactions, and the overall safety profile was consistent with previous study data.
Vitiligo is a chronic autoimmune disease characterized by depigmentation of skin that results from the loss of pigment-producing cells known as melanocytes. Over-activity of the JAK signaling pathway has been shown to drive inflammation involved in the pathogenesis and progression of vitiligo. It affects approximately 0.5% to 2.0% of the population globally1 and there are no U.S. Food and Drug Administration (FDA) or EMA-approved drug therapies for regimentation in vitiligo. The disease can occur at any age, although many patients with vitiligo will experience initial symptoms before the age of 20.2
About TRuE-V
The TRuE-V clinical trial program includes two Phase 3 studies, TRuE-V1 (NCT04052425) and TRuE-V2 (NCT04057573), evaluating the safety and efficacy of ruxolitinib cream in patients with vitiligo.
The studies each enrolled approximately 300 patients (age ≥12 years) who have been diagnosed with non-segmental vitiligo and have depigmented areas including at least 0.5% of the body surface area (BSA) on the face, ≥0.5 facial vitiligo area severity index [F-VASI] score, at least 3% BSA on nonfacial areas, ≥3 total body Vitiligo Area Scoring Index [T-VASI] score and total BSA involvement (facial and nonfacial) of up to 10%. Participants were randomized into two arms: 1.5% ruxolitinib cream twice daily (BID) and vehicle control for the 24-week double-blind period. Patients who successfully completed baseline and Week 24 assessments, including those that received vehicle control during the double-blind phase, were offered treatment extension with 1.5% ruxolitinib cream BID for an additional 28 weeks.
The primary endpoint of both studies in the TRuE-V program is the proportion of patients achieving F-VASI75, defined as at least a 75% improvement from baseline in the F-VASI score at Week 24. Key secondary endpoints include: the percentage change from baseline in facial BSA (F-BSA) at Week 24, the proportion of patients achieving F-VASI50 (at least 50% improvement from baseline in the F-VASI), F-VASI90 (at least 90% improvement from baseline in the F-VASI) and T-VASI50 (at least 50% improvement from baseline in the T-VASI) at Week 24, the proportion of patients achieving F-VASI75, F-VASI90, T-VASI50 and T-VASI75 (at least 75% improvement from baseline in the T-VASI) at Week 52 and the proportion of patients achieving a Vitiligo Noticeability Scale (VNS) score of 4 (a lot less noticeable) or 5 (no longer noticeable) at Week 24. The studies also track the frequency, duration and severity of adverse events associated with the use of ruxolitinib cream.
For more information on the TRuE-V studies, please visit https://clinicaltrials.gov/ct2/show/NCT04052425 and https://clinicaltrials.gov/ct2/show/NCT04057573.
About Ruxolitinib Cream
Ruxolitinib cream is an investigational novel cream formulation of Incyte’s selective JAK1/JAK2 inhibitor ruxolitinib.
In September 2021, ruxolitinib cream (Opzelura™) was approved for use by the U.S. Food and Drug Administration (FDA) for the topical short-term and non-continuous chronic treatment of mild to moderate atopic dermatitis (AD) in non-immunocompromised patients 12 years of age and older, whose disease is not adequately controlled with topical prescription therapies, or when those therapies are not advisable.
Additionally, ruxolitinib cream is being investigated for the treatment of adolescents and adults with vitiligo in the Phase 3 TRuE-V clinical trial program. Results from this program were recently announced.
Incyte has worldwide rights for the development and commercialization of ruxolitinib cream, marketed in the United States as Opzelura.
Opzelura is a trademark of Incyte.
For additional information on Incyte, please visit Incyte.com
View source version on businesswire.com: https://www.businesswire.com/news/home/20211028005620/en/
Source: Incyte
Oct. 28, 2021 7:57 AM ET Incyte Corporation (INCY)
By: Aakash Babu, SA News Editor
Oct 27, 2021
Basel, October 27, 2021 — Novartis today announced that the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have accepted the company’s Supplemental Biologics License Application (sBLA) and Type II Variation, respectively, for Kymriah® (tisagenlecleucel) in adult patients with relapsed or refractory (r/r) follicular lymphoma (FL) after two prior lines of treatment. The FDA has also granted priority review to the company’s sBLA for Kymriah in adult patients with r/r FL. Kymriah was previously granted orphan medicinal product designation by the European Commission (EC) for FL. If approved in this potential third indication, Kymriah would have the opportunity to present an important treatment option for those patients with r/r FL in need of potentially definitive outcomes.
The regulatory submissions are based on positive data from the pivotal Phase II ELARA trial, which investigated the efficacy and safety of Kymriah in adult patients with r/r FL. The trial met the primary endpoint with robust responses observed in heavily pretreated patients. The safety profile was remarkable, with no patients experiencing grade 3 or higher cytokine release syndrome (CRS) related to Kymriah within the first 8 weeks following infusion1. Data from the trial was presented earlier this year as an oral presentation during the 2021 Annual American Society of Clinical Oncology (ASCO) Virtual Scientific Meeting.
“This is an important milestone in our mission to bring Kymriah to adult patients with relapsed or refractory follicular lymphoma. Receiving orphan drug designation from the EC as well as priority review from the FDA underscores the unmet need and urgency for these patients. With Kymriah demonstrating impressive results in the ELARA trial, we are hopeful that we can offer a unique and potentially definitive treatment that minimizes the burden,” said Jeff Legos, Executive Vice President, Global Head of Oncology & Hematology Development, Novartis.
Orphan drug designation is reserved for medicines that treat, prevent or diagnose a life-threatening or chronically debilitating rare disease with a prevalence in the EU of below 5 in 10,000 and with either no currently approved method of diagnosis, prevention or treatment or with significant benefit to those affected by the disease3. The decision follows a positive opinion from the Committee for Orphan Medicinal Products (COMP) of the EMA. Kymriah also has Orphan Drug designation from the FDA and the Japan Ministry of Health, Labour and Welfare (MHLW) for this disease.
Priority Review is granted to therapies that have the potential to provide significant improvements in the treatment, diagnosis or prevention of serious conditions, as determined by the FDA4.
Kymriah is currently approved by the FDA, EMA and other regulatory authorities for the treatment of r/r pediatric and young adult (up to and including 25 years of age) acute lymphoblastic leukemia (ALL), and r/r adult diffuse large B-cell lymphoma (DLBCL).
About the ELARA trial
ELARA is a Phase II, single-arm, multicenter, open-label trial investigating the efficacy and safety of Kymriah in adult patients with r/r FL after at least two prior therapies. This international trial has enrolled patients from over 30 sites in 12 countries worldwide. The primary endpoint is complete response rate (CRR) based on best response by central review (Lugano 2014 criteria). Patients evaluable for efficacy had measurable disease at infusion and more than six months of follow-up from infusion or discontinued early. After infusion, disease assessments were performed every three months. Secondary endpoints include overall response rate, duration of response, progression-free survival, overall survival and safety. Primary analysis data announced at ASCO 2021 showed Kymriah led to responses for the majority of patients treated, with 66% achieving a complete response (95% CI, 56-75). The overall response rate was 86% (95% CI, 78-92)1. Importantly, no patients in ELARA trial experienced grade 3 or higher cytokine release syndrome related to Kymriah within the first 8 weeks following infusion, the most common side effect associated with CAR-T therapy1.
Kymriah (tisagenlecleucel) is an autologous, immunocellular cancer therapy which involves reprogramming a patient's own T-cells with a transgene encoding a chimeric antigen receptor (CAR) to identify and eliminate CD19-expressing cells. It is administered as intravenous infusion.
Kymriah is indicated for the treatment of pediatric and young adult patients up to and including 25 years of age with B-cell acute lymphoblastic leukemia (ALL) that is refractory, in relapse post-transplant or in second or later relapse as well as for adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) after two or more lines of systemic therapy.
Kymriah must not be administered in case of hypersensitivity to the active substance or to any of the excipients of the product. In addition, contraindications of the lymphodepleting chemotherapy that is usually preceding the Kymriah infusion to prepare the patient's body, must be considered.
Find out more at https://www.novartis.com.
Oct. 27, 2021 8:17 AM ET
By: Dulan Lokuwithana, SA News Editor
October 25, 2021 at 12:59 AM EDT Back
TARRYTOWN, N.Y. and PARIS, Oct. 25, 2021 /PRNewswire/ --
Dupixent 300 mg weekly significantly improved the ability to swallow and reduced eosinophils in the esophagus compared to placebo, reinforcing positive results from first Phase 3 trial
Eosinophilic esophagitis is a progressive disease that damages the esophagus and impairs the ability to swallow; 90% of trial participants had at least one coexisting type 2 inflammatory condition such as asthma or atopic dermatitis
Dupixent is the only biologic medicine to show positive, clinically meaningful Phase 3 results in these patients; regulatory filings planned for 2022
Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) and Sanofi today announced results from a second Phase 3 trial assessing the investigational use of Dupixent® (dupilumab) in patients 12 years and older with eosinophilic esophagitis (EoE). The trial met its co-primary endpoints in patients taking Dupixent 300 mg weekly, showing significant improvements in clinical (Dysphagia Symptom Questionnaire) and histologic disease measures compared to placebo. In September 2020, the U.S. Food and Drug Administration (FDA) granted Breakthrough Therapy designation to Dupixent for the treatment of patients 12 years and older with EoE.
"This trial gives insight into how terrible this disease can be, with more than a third of patients having previously required invasive endoscopic dilations that can temporarily reduce symptoms but carry the risk of rupturing the esophagus," said George D. Yancopoulos, M.D., Ph.D., President and Chief Scientific Officer at Regeneron and a principal inventor of Dupixent. "Dupixent, which blocks the IL-4 and IL-13 pathways, has now shown compelling results across a spectrum of diseases where there has been great unmet need. In fact, our positive Phase 3 data in six different diseases help confirm our early hypothesis that IL-4 and IL-13 are the main drivers of allergic or type 2 inflammation and disease, whether manifested in the gastrointestinal tract as eosinophilic esophagitis, the respiratory tract as asthma or nasal polyps, or the skin as atopic dermatitis, chronic spontaneous urticaria, or prurigo nodularis."
EoE is a chronic and progressive type 2 inflammatory disease that damages the esophagus and prevents it from working properly. At times, swallowing the smallest quantity of food or taking a sip of water can be a painful and worrisome choking experience. Those with EoE live with anxiety and frustration from having a constantly evolving list of trigger foods to avoid. Dilation (physical expansion) of the esophagus, which is used to address narrowing, is often painful. In severe cases, a feeding tube is the only option to ensure proper caloric intake and weight gain. People with EoE may have poor quality of life and are more likely to experience depression, especially as they age, than people without EoE. In the U.S. there are approximately 160,000 patients with EoE who are currently treated, of whom approximately 48,000 have failed multiple treatments.
"The current standard of care for people with eosinophilic esophagitis may only provide limited relief of their symptoms. Efforts to develop a treatment that targets an underlying cause of the disease has eluded the field for some time, resulting in an incredible unmet need," said Naimish Patel, M.D. Head of Global Development, Immunology and Inflammation at Sanofi. "We are encouraged that Dupixent, which targets IL-4 and IL-13, was able to reduce inflammation in the esophagus and provided significant relief when swallowing for patients taking the weekly dose. We look forward to continuing to study Dupixent's potential role in addressing the underlying type 2 inflammation that can lead to eosinophilic esophagitis."
In this trial, 80 patients were enrolled into a Dupixent 300 mg weekly treatment group and 79 patients were enrolled into the placebo group. The co-primary endpoints at 24 weeks assessed patient-reported measures of difficulty swallowing (change from baseline in the Dysphagia Symptom Questionnaire, or DSQ), and esophageal inflammation (proportion of patients achieving peak esophageal intraepithelial eosinophil count of ≤6 eos/high power field [hpf]).
Patients treated with Dupixent 300 mg weekly experienced the following changes by week 24 compared to placebo:
About the Dupixent Eosinophilic Esophagitis Trial
The Phase 3, randomized, double-blind, placebo-controlled trial evaluated the efficacy and safety of Dupixent in adolescents and adults with EoE. The second trial (Part B) enrolled 240 patients aged 12 years and older with EoE, as determined by histological and patient-reported measures. Following the first Phase 3 trial (Part A), in which Dupixent 300 mg weekly was evaluated compared to placebo, the second confirmatory trial evaluated Dupixent 300 mg weekly or every two weeks compared to placebo for a 24-week treatment period.
The clinical trial program is ongoing, with patients from the first and second trials continuing into a 28-week long-term extension trial (Part C). Full results from this trial will be available in 2022.
About Dupixent
Dupixent, which was invented using Regeneron's proprietary VelocImmune® technology, is a fully human monoclonal antibody that inhibits the signaling of the interleukin-4 (IL-4) and interleukin-13 (IL-13) pathways and is not an immunosuppressant. IL-4 and IL-13 are key and central drivers of the type 2 inflammation that plays a major role in atopic dermatitis, asthma and chronic rhinosinusitis with nasal polyposis (CRSwNP).
Dupixent is currently approved in the U.S., Europe, Japan and other countries around the world for use in specific patients with moderate-to-severe atopic dermatitis, as well as certain patients with asthma or CRSwNP in different age populations. Dupixent is also approved in one or more of these indications in more than 60 countries around the world and more than 300,000 patients have been treated globally.
U.S. Indications
DUPIXENT is a prescription medicine used:
Please see accompanying full Prescribing Information including Patient Information.
For additional information about the company, please visit www.regeneron.com
SOURCE Regeneron Pharmaceuticals, Inc.
Oct. 25, 2021 6:45 AM ET Regeneron Pharmaceuticals, Inc. (REGN), SNY
By: Aakash Babu, SA News Editor
PUBLISHED25 October 2021
Positive high-level results from the TOPAZ-1 Phase III trial showed Imfinzi (durvalumab), in combination with standard-of-care chemotherapy, demonstrated a statistically significant and clinically meaningful overall survival (OS) benefit versus chemotherapy alone as a 1st-line treatment for patients with advanced biliary tract cancer (BTC).
At a predefined interim analysis, the Independent Data Monitoring Committee concluded that the trial met the primary endpoint by demonstrating an improvement in OS in patients treated with Imfinzi plus chemotherapy versus chemotherapy alone. The combination also demonstrated an improvement in progression-free survival (PFS) and overall response rate, key secondary endpoints.
Imfinzi plus chemotherapy was well tolerated, had a similar safety profile versus the comparator arm and did not increase the discontinuation rate due to adverse events compared to chemotherapy alone.
BTC is a group of rare and aggressive cancers that occur in the bile ducts and gallbladder.1,2 Incidence of BTC often depends on the prevalence of common risk factors for each type within a geographical region.
Approximately 50,000 people in the US, Europe and Japan and about 210,000 people worldwide are diagnosed with BTC each year.3-5 These patients have a poor prognosis, with approximately only 5% to 15% of all patients with BTC surviving five years.4 In December 2020, Imfinzi was granted Orphan Drug Designation in the US for the treatment of BTC.
Do-Youn Oh, MD, PhD, Professor, Division of Medical Oncology, Department of Internal Medicine at Seoul National University Hospital and Seoul National University College of Medicine, and principal investigator in the TOPAZ-1 Phase III trial, said: “Patients with advanced biliary tract cancer are in dire need of new treatments as progress in the 1st-line setting has remained largely stagnant for more than 10 years. TOPAZ-1 is the first Phase III trial to show that adding an immunotherapy to standard chemotherapy delivers a meaningful overall survival benefit for patients in this setting. Today’s exciting results are a major step forward in treating this disease and represent new hope for our patients.”
Susan Galbraith, Executive Vice President, Oncology R&D, AstraZeneca, said: “We are delighted TOPAZ-1 has been unblinded early due to clear evidence of efficacy for Imfinzi plus chemotherapy, which has also demonstrated a strong safety profile. We have now delivered two positive gastrointestinal cancer trials in a row for Imfinzi, following the HIMALAYA trial in liver cancer. We believe the significant survival benefit demonstrated marks a new era of immunotherapy treatment in this devastating disease, and it advances our commitment to improving long-term survival for patients across these cancers where treatment options are limited.”
The data will be presented at a forthcoming medical meeting and shared with health authorities.
TOPAZ-1
TOPAZ-1 is a randomised, double-blind, placebo controlled, multicentre, global Phase III trial of Imfinzi in combination with chemotherapy (gemcitabine plus cisplatin) versus placebo in combination with chemotherapy as a 1st-line treatment in 685 patients with unresectable advanced or metastatic BTC including intrahepatic and extrahepatic cholangiocarcinoma, and gallbladder cancer (ampullary carcinoma was excluded).
The trial is being conducted in more than 145 centres across 17 countries including in the US, Europe, South America and several countries in Asia including South Korea, Thailand, Japan, Taiwan and China. The primary endpoint is OS and key secondary endpoints include progression-free survival, objective response rate and safety.
Imfinzi
Imfinzi (durvalumab) is a human monoclonal antibody that binds to the PD-L1 protein and blocks the interaction of PD-L1 with the PD-1 and CD80 proteins, countering the tumour’s immune-evading tactics and releasing the inhibition of immune responses.
Imfinzi is the only approved immunotherapy in the curative-intent setting of unresectable, Stage III non-small cell lung cancer (NSCLC) in patients whose disease has not progressed after chemoradiation therapy and is the global standard of care in this setting based on the PACIFIC Phase III trial.
Imfinzi is also approved in the US, EU, Japan, China and many other countries around the world for the treatment of extensive-stage small cell lung cancer (ES-SCLC) based on the CASPIAN Phase III trial.
Imfinzi is also approved for previously treated patients with advanced bladder cancer in several countries. Since the first approval in May 2017, more than 100,000 patients have been treated with Imfinzi.
As part of a broad development programme, Imfinzi is being tested as a single treatment and in combinations with other anti-cancer treatments for patients with SCLC, NSCLC, bladder cancer, liver cancer, BTC, oesophageal cancer, gastric and gastroesophageal cancer, cervical cancer, ovarian cancer, endometrial cancer, and other solid tumours.
Please visit astrazeneca.com
Oct. 25, 2021 8:50 AM ET AstraZeneca PLC (AZN) By: Dulan Lokuwithana, SA News Editor2 Comments
AstraZeneca (NASDAQ:AZN) announced positive interim results from a Phase III trial for Imfinzi (durvalumab) plus standard-of-care chemotherapy as a first-line treatment for patients with advanced biliary tract cancer (BTC).
October 25, 2021 6:00 am ET
If Granted Marketing Authorization by the European Commission, Molnupiravir Could Be the First Oral Antiviral Medicine for the Treatment of COVID-19 in the European Union
KENILWORTH, N.J. & MIAMI--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, and Ridgeback Biotherapeutics today announced that the European Medicines Agency (EMA) has initiated a rolling review for molnupiravir, an investigational oral antiviral medicine, for the treatment of COVID-19 in adults. Merck plans to work with the EMA’s Committee for Medicinal Products for Human Use (CHMP) to complete the rolling review process to facilitate initiating the formal review of the Marketing Authorization Application. As previously announced, Merck has submitted an application for Emergency Use Authorization (EUA) to the U.S. Food and Drug Administration (FDA), and is actively working to submit applications to other regulatory agencies worldwide.
“This application to the EMA is another step in our efforts to bring molnupiravir forward to patients globally,” said Dr. Dean Y. Li, executive vice president and president, Merck Research Laboratories. “We believe that molnupiravir will be an important addition to the range of public health tools to fight COVID-19 – including the vaccines developed by the research-based pharmaceutical industry, which remain essential and are the first-line of defense against this pandemic.”
The submission is based on positive results from a planned interim analysis from the Phase 3 MOVe-OUT clinical trial, which evaluated molnupiravir in non-hospitalized adult patients with mild-to-moderate COVID-19 who were at increased risk for progressing to severe COVID-19 and/or hospitalization. At the interim analysis, molnupiravir 800 mg twice-daily reduced the risk of hospitalization or death by approximately 50%; 7.3% of patients who received molnupiravir were hospitalized through Day 29 following randomization (28/385), compared with 14.1% of placebo-treated patients (53/377) that were either hospitalized or died; p=0.0012. Through Day 29, no deaths were reported in patients who received molnupiravir, as compared to 8 deaths in patients who received placebo. The incidence of any adverse event was comparable in the molnupiravir and placebo groups (35% and 40%, respectively). The incidence of drug-related adverse events was also comparable (12% and 11%, respectively), and fewer patients in the molnupiravir group discontinued therapy due to an adverse event compared to the placebo group (1.3% and 3.4%, respectively).
“In the nearly two years since COVID-19 emerged, the global scientific community has made extraordinary strides in developing several critical vaccines and treatments, but we still have a need for an oral antiviral medicine that can be taken at home,” said Wendy Holman, chief executive officer, Ridgeback Biotherapeutics. “We believe that molnupiravir, with the exciting finding of reduction in hospitalization and death in the MOVe-OUT study, may help fill that need and look forward to working with the EMA on its review.”
About Merck’s Efforts to Enable Global Access to Molnupiravir, if it is Granted Authorization or Approval
In anticipation of the results from MOVe-OUT and the potential for regulatory authorization or approval, Merck has been producing molnupiravir at risk and expects to produce 10 million courses of treatment by the end of 2021, with even more courses expected to be produced in 2022.
On October 11, Merck and Ridgeback announced that Merck had submitted an application for EUA to the U.S. FDA for molnupiravir for the treatment of at-risk adults with mild-to-moderate COVID-19. Additional submissions to global regulatory agencies are underway.
Earlier this year, Merck entered into a procurement agreement with the U.S. Government under which the company will supply approximately 1.7 million courses of molnupiravir to the U.S. Government following EUA or approval from the U.S. FDA. Additionally, Merck has entered into supply and advance purchase agreements for molnupiravir with other governments worldwide, pending regulatory authorization, and is currently in discussions with additional governments.
Merck is committed to providing timely access to molnupiravir globally, if it is authorized or approved, and plans to implement a tiered pricing approach based on World Bank country income criteria that reflect countries’ relative ability to finance their health response to the pandemic.
As part of its commitment to widespread global access, Merck previously announced that the company has entered into non-exclusive voluntary licensing agreements for molnupiravir with established Indian generic manufacturers to accelerate availability of molnupiravir in more than 100 low- and middle-income countries (LMICs) following approvals or emergency authorization by local regulatory agencies. Merck continues to discuss additional measures and collaborations to accelerate broad, global access to molnupiravir.
About Molnupiravir
Molnupiravir (MK-4482 and EIDD-2801) is an investigational, orally administered form of a potent ribonucleoside analog that inhibits the replication of SARS-CoV-2, the causative agent of COVID-19. Molnupiravir has been shown to be active in several preclinical models of SARS-CoV-2, including for prophylaxis, treatment, and prevention of transmission. Additionally, pre-clinical and clinical data have shown molnupiravir to be active against the most common SARS-CoV-2 variants.
Molnupiravir was invented at Drug Innovations at Emory (DRIVE), LLC, a not-for-profit biotechnology company wholly owned by Emory University; Emory/DRIVE received some research funding from the U.S. Department of Defense and the U.S. National Institutes of Health. Molnupiravir is being developed by Merck & Co., Inc. in collaboration with Ridgeback Biotherapeutics. Ridgeback received an upfront payment from Merck and also is eligible to receive contingent payments dependent upon the achievement of certain developmental and regulatory approval milestones. Any profits from the collaboration will be split between the partners equally. Since licensed by Ridgeback, all funds used for the development of molnupiravir have been provided by Merck and by Wayne and Wendy Holman of Ridgeback.
Molnupiravir is also being evaluated for post-exposure prophylaxis in MOVe-AHEAD, a global, multicenter, randomized, double-blind, placebo-controlled Phase 3 study, which is evaluating the efficacy and safety of molnupiravir in preventing the spread of COVID-19 within households. For more information, please visit http://merckcovidresearch.com.
About the MOVe-OUT Study
The MOVe-OUT trial (MK-4482-002) (NCT04575597) is a global Phase 3, randomized, placebo-controlled, double-blind, multi-site study of non-hospitalized adult patients with laboratory-confirmed mild-to-moderate COVID-19. Patients enrolled in the study were unvaccinated against SARS-CoV-2, had at least one risk factor associated with poor disease outcomes, and symptom onset within five days prior to randomization. The primary efficacy objective of MOVe-OUT is to evaluate the efficacy of molnupiravir 800 mg twice daily for five days compared to placebo as assessed by the percentage of participants who are hospitalized and/or die from the time of randomization through Day 29.
The Phase 3 portion of the MOVe-OUT trial was conducted globally, including in more than 170 planned sites in countries including Argentina, Brazil, Canada, Chile, Colombia, Egypt, France, Germany, Guatemala, Israel, Italy, Japan, Mexico, Philippines, Poland, Russia, South Africa, Spain, Sweden, Taiwan, Ukraine, the United Kingdom and the United States. For further information about the MOVe-OUT trial, please visit clinicaltrials.gov.
The most common risk factors for poor disease outcome included obesity, older age (>60 years), diabetes mellitus and heart disease. Delta, Gamma and Mu variants accounted for nearly 80% of the baseline viral variants that had been sequenced at the time of the interim analysis. Recruitment in Latin America, Europe, and Africa accounted for 56%, 23% and 15% of the study population, respectively.
For more information, visit www.merck.com
Oct. 25, 2021 6:21 AM ET Merck & Co., Inc. (MRK) By: Aakash Babu, SA News Editor
PUBLISHED15 October 2021
Positive high-level results from the HIMALAYA Phase III trial showed a single, high priming dose of tremelimumab added to Imfinzi (durvalumab) demonstrated a statistically significant and clinically meaningful overall survival (OS) benefit versus sorafenib as a 1st-line treatment for patients with unresectable hepatocellular carcinoma (HCC) who had not received prior systemic therapy and were not eligible for localised treatment. This novel dose and schedule of tremelimumab, an anti-CTLA4 antibody, and Imfinzi is called the STRIDE regimen (Single Tremelimumab Regular Interval Durvalumab). The combination demonstrated a favourable safety profile, and the addition of tremelimumab to Imfinzi did not increase severe hepatic toxicity.
Imfinzi alone demonstrated non-inferior OS to sorafenib with a numerical trend in favour of Imfinzi and an improved tolerability profile compared to sorafenib.
Liver cancer, of which HCC is the most common type, is the third leading cause of cancer death and the sixth most commonly diagnosed cancer worldwide with approximately 900,000 people diagnosed each year.1-2 Only 7% of patients with advanced disease survive five years.3
Ghassan Abou-Alfa, MD, MBA, Attending Physician at Memorial Sloan Kettering Cancer Center and principal investigator in the HIMALAYA Phase III trial, said: “HIMALAYA is the first Phase III trial to add a novel single priming dose of an anti-CTLA4 antibody to another checkpoint inhibitor, durvalumab. This serves to boost the patient’s own immune system against their liver cancer, aiming to maximise long-term survival with minimal side effects. This is very exciting news for our patients.”
Susan Galbraith, Executive Vice President, Oncology R&D, AstraZeneca, said: “Inhibition of CTLA-4 has shown the ability to drive benefit particularly in the tail of the survival curve in several settings. This is the first time a dual immunotherapy regimen has improved overall survival as a 1st-line treatment for patients with unresectable liver cancer for whom treatment options are limited and long-term outcomes are poor.”
The data from the HIMALAYA Phase III trial will be presented at a forthcoming medical meeting.
Imfinzi and tremelimumab were granted Orphan Drug Designations in the US for the treatment of HCC in 2020. Tremelimumab was also granted orphan designation in the EU in HCC in 2020.
HIMALAYA
HIMALAYA was a randomised, open-label, multicentre, global Phase III trial of Imfinzi monotherapy and the STRIDE regimen, comprising a single priming dose of tremelimumab 300mg added to Imfinzi 1500mg followed by Imfinzi every four weeks versus sorafenib, a standard-of-care multi-kinase inhibitor, in a total of 1,324 patients with unresectable, advanced HCC who had not been treated with prior systemic therapy and were not eligible for locoregional therapy (treatment localised to the liver and surrounding tissue).
The trial was conducted in 190 centres across 16 countries, including in the US, Canada, Europe, South America and Asia. The primary endpoint was OS for STRIDE versus sorafenib and key secondary endpoints included OS for Imfinzi versus sorafenib, objective response rate and progression-free survival (PFS) for STRIDE and for Imfinzi alone.
Imfinzi
Imfinzi (durvalumab) is a human monoclonal antibody that binds to the PD-L1 protein and blocks the interaction of PD-L1 with the PD-1 and CD80 proteins, countering the tumour’s immune-evading tactics and releasing the inhibition of immune responses.
Imfinzi is the only approved immunotherapy in the curative-intent setting of unresectable, Stage III non-small cell lung cancer (NSCLC) in patients whose disease has not progressed after chemoradiation therapy and is the global standard of care in this setting based on the PACIFIC Phase III trial.
Imfinzi is also approved in the US, EU, Japan, China and many other countries around the world for the treatment of extensive-stage small cell lung cancer (ES-SCLC) based on the CASPIAN Phase III trial.
Imfinzi is also approved for previously treated patients with advanced bladder cancer in several countries. Since the first approval in May 2017, more than 100,000 patients have been treated with Imfinzi.
In 1st-line Stage IV NSCLC, positive results from the POSEIDON Phase III trial showed Imfinzi plus chemotherapy with a short course of tremelimumab 75mg demonstrated a statistically significant and clinically meaningful improvement in OS and PFS compared to chemotherapy.
As part of a broad development programme, Imfinzi is being tested as a single treatment and in combinations with other anti-cancer treatments for patients with NSCLC, SCLC, bladder cancer, HCC, biliary tract cancer (BTC), oesophageal cancer, gastric and gastroesophageal cancer, cervical cancer, ovarian cancer, endometrial cancer, and other solid tumours.
Tremelimumab
Tremelimumab is a human monoclonal antibody and potential new medicine that targets the activity of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). Tremelimumab blocks the activity of CTLA-4, contributing to T-cell activation, priming the immune response to cancer and fostering cancer cell death.
Tremelimumab is being tested in a clinical trial programme in combination with Imfinzi in NSCLC, SCLC, bladder cancer and liver cancer.
Please visit astrazeneca.com
Oct. 15, 2021 6:36 AM ET AstraZeneca PLC (AZN) By: Mamta Mayani, SA News Editor1 Comment
Friday, Oct 22, 2021
South San Francisco, CA -- October 22, 2021 --
Genentech, a member of the Roche Group (SIX: RO, ROG; OTCQX: RHHBY), today announced that the U.S. Food and Drug Administration (FDA) has approved Susvimo TM (ranibizumab injection) 100 mg/mL for intravitreal use via ocular implant for the treatment of people with wet, or neovascular, age-related macular degeneration (AMD) who have previously responded to at least two anti-vascular endothelial growth factor (VEGF) injections. Wet AMD is a potentially blinding condition that requires treatment with eye injections as often as once a month. Susvimo, previously called Port Delivery System with ranibizumab, is the first and only FDA-approved treatment for wet AMD that offers as few as two treatments per year.
“Susvimo represents a major advancement in the treatment of retinal disease, and is an important new option for patients with wet AMD,” said Carl Regillo, M.D., Chief of Retina Service at Wills Eye Hospital in Philadelphia and an Archway study investigator. “With Susvimo, my patients now have an option that can help them maintain their vision as well as anti-VEGF injections, but on a more manageable twice-yearly treatment schedule.”
Susvimo delivers ranibizumab continuously, offering people living with wet AMD an alternative to anti-VEGF eye injections needed as often as once a month. The implant is surgically inserted into the eye during a one-time, outpatient procedure and refilled every six months. If necessary, supplemental ranibizumab treatment can be given to the affected eye while the Susvimo implant is in place.
“We believe that Susvimo can help people with wet AMD preserve their vision while potentially alleviating the treatment burden associated with current standards of care,” said Levi Garraway, M.D., Ph.D., chief medical officer and head of Global Product Development. “Susvimo’s approval builds on Genentech's long-standing commitment to people living with vision-threatening conditions.”
The approval is based on positive results from the Phase III Archway study primary analysis, which showed wet AMD patients treated with Susvimo achieved and maintained vision gains equivalent to monthly ranibizumab injections – +0.2 and +0.5 eye chart letters from baseline, respectively – at weeks 36 and 40 of treatment. In addition, only 1.6% of Susvimo patients received supplemental ranibizumab treatment before their first refill, and more than 98% could go six months before their first refill.
In the Archway study, Susvimo was generally well-tolerated, with a favorable benefit-risk profile. However, the Susvimo implant has been associated with a three-fold higher rate of endophthalmitis than monthly intravitreal injections of ranibizumab. Many of these events were associated with conjunctival retractions or erosions. Appropriate conjunctiva management and early detection with surgical repair of conjunctival retractions or erosions may reduce the risk of endophthalmitis. In clinical trials, 2.0% of patients receiving a ranibizumab implant experienced at least one episode of endophthalmitis. The most common adverse events (AEs) were conjunctival hemorrhage, conjunctival hyperemia, iritis and eye pain. The safety profile of Susvimo in the clinical trial setting is well understood and will continue to be monitored closely.
Genentech has a robust Phase III clinical development program for Susvimo, including the Portal, Pagoda, Pavilion and Velodrome studies. Portal is an extension study evaluating the long-term safety and efficacy of Susvimo in wet AMD. Pagoda is evaluating Susvimo for the treatment of people with diabetic macular edema (DME), while Pavilion is a study of Susvimo in diabetic retinopathy without DME. Velodrome is evaluating Susvimo refilled every nine months in wet AMD. Susvimo is also currently under review for the treatment of wet AMD by the European Medicines Agency (EMA).
Susvimo will be available in the United States in the coming months. Genentech is committed to helping people access the medicines they are prescribed and will be offering comprehensive services for people prescribed Susvimo to help minimize barriers to access and reimbursement. Patients can call 833-EYE-GENE for more information. For people who qualify, Genentech plans to offer patient assistance programs through Genentech Access Solutions. More information is also available at (866) 4ACCESS/(866) 422-2377 or http://www.Genentech-Access.com.
About the Archway Study
Archway (NCT03677934) was a randomized, multicenter, open-label Phase III study evaluating the efficacy and safety of Susvimo (ranibizumab injection) 100 mg/mL for intravitreal use via ocular implant administered via the Susvimo eye implant, refilled every six months at fixed intervals, compared to monthly intravitreal injections of ranibizumab 0.5 mg in 415 people living with wet age-related macular degeneration (AMD). Patients enrolled in Archway were responders to prior treatment with anti-vascular endothelial growth factor (VEGF) therapy. In both study arms, patients were treated with at least three anti-VEGF injections within the six months prior to their Archway screening visit. The primary endpoint of the study was the change in best-corrected visual acuity (BCVA) score (the best distance vision a person can achieve – including with correction such as glasses – when reading letters on an eye chart) from baseline at the average of Week 36 and Week 40. Secondary endpoints include safety, overall change in BCVA from baseline and change from baseline in center point thickness over time.
According to pre-specified study criteria, Susvimo was shown to be non-inferior and equivalent to monthly ranibizumab injections. On average, patients had received five prior ranibizumab injections before their first study treatment visit. In the Susvimo arm of the study, patients gained an average of 0.2 eye chart letters in visual acuity from baseline compared with 0.5 eye chart letters for the monthly ranibizumab arm. During the first treatment interval, before the first scheduled refill, 1.6% of Susvimo patients assessed (n=4/246) received supplemental ranibizumab treatment, and 98.4% of patients (n=242/246) did not receive supplemental treatment.
In the Archway study, Susvimo was generally well-tolerated, with a favorable benefit-risk profile. However, the Susvimo implant has been associated with a three-fold higher rate of endophthalmitis than monthly intravitreal injections of ranibizumab. Many of these events were associated with conjunctival retractions or erosions. Appropriate conjunctiva management and early detection with surgical repair of conjunctival retractions or erosions may reduce the risk of endophthalmitis. In clinical trials, 2.0% of patients receiving a ranibizumab implant experienced at least one episode of endophthalmitis. The most common adverse events (AEs) were conjunctival hemorrhage, conjunctival hyperemia, iritis and eye pain. The safety profile of Susvimo in the clinical trial setting is well understood and will continue to be monitored closely.
About Susvimo™ (ranibizumab injection) 100 mg/mL for intravitreal use via ocular implant
Susvimo™ (ranibizumab injection) 100 mg/mL for intravitreal use via ocular implant is a refillable implant surgically inserted into the eye during a one-time, outpatient procedure. Susvimo continuously delivers a customized formulation of ranibizumab over time. Susvimo is indicated for intravitreal use via the Susvimo eye implant only. Ranibizumab is a vascular endothelial growth factor (VEGF) inhibitor designed to bind to and inhibit VEGF-A, a protein that has been shown to play a critical role in the formation of new blood vessels and the leakiness of the vessels.
Susvimo is different from the ranibizumab intravitreal injection, a medicine marketed as Lucentis® (ranibizumab injection), which is FDA-approved to treat wet age-related macular degeneration (AMD) and other retinal diseases.
Susvimo Indication
Susvimo (ranibizumab injection) 100 mg/mL for intravitreal use via ocular implant is indicated for the treatment of patients with neovascular (wet) age-related macular degeneration (AMD) who have previously responded to at least two intravitreal injections of a vascular endothelial growth factor inhibitor medication.
For additional information about the company, please visit http://www.gene.com.
Oct. 22, 2021 2:46 PM ET Roche Holding AG (RHHBY), RHHBF
By: Dulan Lokuwithana, SA News Editor
Genentech, a member of Roche Group (OTCQX:RHHBY) (OTCQX:RHHBF), has secured the FDA approval for Susvimo (ranibizumab injection), that the company calls the first wet AMD therapy in 15 years to give an alternative to frequent standard-of-care eye injections.
Friday, Oct 15, 2021
South San Francisco, CA -- October 15, 2021 --
Genentech, a member of the Roche Group (SIX: RO, ROG; OTCQX: RHHBY), today announced that the U.S. Food and Drug Administration (FDA) has approved Tecentriq® (atezolizumab) as adjuvant treatment following surgery and platinum-based chemotherapy for adults with Stage II-IIIA non-small cell lung cancer (NSCLC) whose tumors express PD-L1≥1%, as determined by an FDA-approved test.
“Tecentriq is now the first and only cancer immunotherapy available for adjuvant treatment of NSCLC, introducing a new era where people diagnosed with early lung cancer may have the opportunity to receive immunotherapy to increase their chances for cure,” said Levi Garraway, M.D., Ph.D., chief medical officer and head of Global Product Development. “Today’s landmark approval gives physicians and patients a new way to treat early lung cancer that has the potential to significantly reduce risk of cancer recurrence, after more than a decade with limited treatment advances in this setting.”
“Too many patients with early-stage lung cancer experience disease recurrence following surgery. Now, the availability of immunotherapy following surgery and chemotherapy offers many patients new hope and a powerful new tool to reduce their risk of cancer relapse,” said Bonnie Addario, Co-founder and Chair, GO2 Foundation for Lung Cancer. “With this approval, it is more important than ever to screen for lung cancer early and test for PD-L1 at diagnosis to help bring this advance to the people who can benefit.”
The approval is based on results from an interim analysis of the Phase III IMpower010 study that showed treatment with Tecentriq following surgery and platinum-based chemotherapy reduced the risk of disease recurrence or death by 34% (hazard ratio [HR]=0.66, 95% CI: 0.50-0.88) in people with Stage II-IIIA (UICC/AJCC 7th edition) NSCLC whose tumors express PD-L1≥1%, compared with best supportive care (BSC). Safety data for Tecentriq were consistent with its known safety profile and no new safety signals were identified. Fatal and serious adverse reactions occurred in 1.8% and 18%, respectively, of patients receiving Tecentriq. The most frequent serious adverse reactions (>1%) were pneumonia (1.8%), pneumonitis (1.6%), and pyrexia (1.2%).
The review of this application was conducted under the FDA’s Project Orbis initiative, which provides a framework for concurrent submission and review of oncology medicines among international partners. According to the FDA, collaboration among international regulators may allow patients with cancer to receive earlier access to products in other countries where there may be significant delays in regulatory submissions. Simultaneous applications were submitted to regulators in the United States, Switzerland, the United Kingdom, Canada, Brazil and Australia under Project Orbis. Additionally, the FDA reviewed and approved the supplemental application under its Real-Time Oncology Review pilot program, which aims to explore a more efficient review process to ensure safe and effective treatments are available to patients as early as possible.
Tecentriq has previously shown clinically meaningful benefit in various types of lung cancer, with six currently approved indications in the U.S. In addition to becoming the first approved cancer immunotherapy for adjuvant NSCLC, Tecentriq was also the first approved cancer immunotherapy for front-line treatment of adults with extensive-stage small cell lung cancer (SCLC) in combination with carboplatin and etoposide (chemotherapy). Tecentriq also has four approved indications in advanced NSCLC as either a single agent or in combination with targeted therapies and/or chemotherapies. Tecentriq is available in three dosing options, providing the flexibility to choose administration every two, three or four weeks.
Genentech has an extensive development program for Tecentriq, including multiple ongoing and planned Phase III studies across different lung, genitourinary, skin, breast, gastrointestinal, gynecological, and head and neck cancers. This includes studies evaluating Tecentriq both alone and in combination with other medicines, as well as studies in metastatic, adjuvant and neoadjuvant settings across various tumor types.
About the IMpower010 study
IMpower010 is a Phase III, global, multicenter, open-label, randomized study evaluating the efficacy and safety of Tecentriq compared with BSC, in participants with Stage IB-IIIA NSCLC (UICC/AJCC 7th edition), following surgical resection and up to 4 cycles of adjuvant cisplatin-based chemotherapy. The study randomized 1,005 people with a ratio of 1:1 to receive either Tecentriq for 1 year (16 cycles), unless disease recurrence or unacceptable toxicity occurred, or BSC. The primary endpoint is investigator-determined DFS in the PD-L1-positive Stage II-IIIA, all randomized Stage II-IIIA and intent-to-treat (ITT) Stage IB-IIIA populations. Key secondary endpoints include overall survival (OS) in the overall study population, ITT Stage IB-IIIA NSCLC.
About Tecentriq ® (atezolizumab)
Tecentriq is a monoclonal antibody designed to bind with a protein called PD-L1. Tecentriq is designed to bind to PD-L1 expressed on tumor cells and tumor-infiltrating immune cells, blocking its interactions with both PD-1 and B7.1 receptors. By inhibiting PD-L1, Tecentriq may enable the re-activation of T cells. Tecentriq may also affect normal cells.
Tecentriq U.S. Indications
Tecentriq is a prescription medicine used to treat adults with:
A type of lung cancer called non-small cell lung cancer (NSCLC).
A type of lung cancer called small cell lung cancer (SCLC).
It is not known if Tecentriq is safe and effective in children.
Please see full Prescribing Information and Medication Guide for additional Important Safety Information.
Please see http://www.Tecentriq.com for full Prescribing Information and additional Important Safety Information.
For more information visit http://www.gene.com/cancer-immunotherapy.
For additional information about the company, please visit http://www.gene.com.
Oct. 15, 2021 12:42 PM ET Roche Holding AG (RHHBY) By: Aakash Babu, SA News Editor
Summit, NJ, USA 14 Oct 2021
Seqirus, a global leader in influenza prevention, and a business of CSL Limited (ASX:CSL), today announced that The New England Journal of Medicine has published absolute efficacy data on the company’s cell-based quadrivalent influenza vaccine (QIVc) from a randomized controlled trial (RCT) which met its primary endpoint.1 The study indicates that the seasonal influenza vaccine was effective and produced a sufficient immune response against influenza in children and adolescents ≥2 to <18 years of age over three influenza seasons in the Southern (2017) and Northern (2017/18 and 2018/19) Hemispheres, compared to a non-influenza comparator.1 This represents the first absolute efficacy study of a cell-based influenza vaccine in children as young as two years of age.
“In this study, QIVc demonstrated absolute efficacy in children and adolescents, showing consistent benefit across three seasons and eight countries.1 This is particularly impactful given the disease burden in children as young as two years of age,” said Jonathan Edelman, MD, Vice President, Clinical Development at Seqirus and study author. “These data add to a growing body of evidence supporting the fact that our differentiated, cell-based seasonal influenza vaccine can help provide effective protection against flu.”
QIVc utilizes a cell-based influenza vaccine manufacturing process, an alternative to traditional egg-based manufacturing.2 Traditional egg-based vaccine production can cause the strain to mutate at several steps throughout the manufacturing process, which may lead to an antigenic mismatch between the circulating strains and the inactivated influenza strains contained within the seasonal influenza vaccine.2
Cell-based influenza vaccines are designed to produce an exact match to WHO-selected influenza virus strains by avoiding egg-adapted changes, and therefore have the potential for greater vaccine effectiveness.2,3 Cell-based influenza vaccine technology may offer additional advantages over the standard influenza manufacturing process, including increased scalability and production speed in the event of an influenza pandemic.2
“Young children are at a higher risk than adults for serious influenza-related complications.4 We’re particularly pleased with the results of this study because it supports the use of our differentiated, cell-based influenza vaccine technology as an effective means of influenza protection in children as young as two years old,” said Gregg Sylvester, MD, Chief Medical Officer at Seqirus.
The results of this study supported the recent U.S. Food and Drug Administration (FDA) approval for an expanded age indication for use of QIVc in children ≥2 years of age.5 QIVc is marketed in the United States as FLUCELVAX® QUADRIVALENT (Influenza Vaccine) and is currently under review by the U.S. Food and Drug Administration (FDA) for an expanded age indication for children as young as six months of age.5
The Centers for Disease Control and Prevention (CDC), recommends annual seasonal influenza vaccination for everyone six months of age and older without contraindications as the best way to prevent seasonal influenza.6 It is estimated that more than 52,000 hospitalizations occurred in children younger than 18 years of age during the 2019/20 U.S. influenza season, and there were approximately over 434 influenza-related deaths in this age group.7
In the U.S., Seqirus operates a state-of-the-art cell-based manufacturing facility in Holly Springs, North Carolina, purpose-built in partnership with the Biomedical Advanced Research and Development Authority (BARDA) to increase cell-based vaccine manufacturing capacity and combat pandemic influenza threats.8 Last year, Seqirus announced plans to build a new, world-class cell-based manufacturing facility in Australia, which will be the only cell-based influenza vaccine manufacturing facility in the Southern Hemisphere.9
About the Study
This phase III/IV multi-center, randomized, observer-blind study was conducted across eight countries (Australia, Philippines, Thailand, Estonia, Finland, Lithuania, Poland, and Spain) over three influenza seasons – Southern Hemisphere (2017) and Northern Hemisphere (2017/18 and 2018/19).1
The study was designed to demonstrate the efficacy and safety of QIVc in children ≥2 to <18 years of age, compared to a non-influenza comparator.1 A total of 4514 subjects (n= 2258 QIVc, n=2256 comparator (Menveo®, meningococcal [Serogroup ACYW-135] conjugate vaccine)) were enrolled.1 The primary endpoint was the first occurrence of laboratory-confirmed influenzas illness (by RT-PCR or viral culture) occurring between >14 days after last vaccination and the end of the influenza season.1 The influenza attack rate in the QIVc group was 175/2257 (7.8%; 6.5% to 10.2% over three seasons), compared with 364/2252 (16.2%; 15.2% to 17.4% over three seasons) cases in the control group.1
The efficacy of QIVc in children and adolescents against laboratory-confirmed influenza illness was 54.6% (95% CI 45.7 to 62.1), meeting the pre-specified endpoint for success and showing benefit across three seasons and eight countries.1
The safety profile of QIVc was comparable to the non-influenza comparator.1
For more information visit www.seqirus.com and www.csl.com.
What is FLUCELVAX® QUADRIVALENT (Influenza Vaccine)?
FLUCELVAX QUADRIVALENT is a vaccine that helps protect people aged 2 and older from the flu. Vaccination with FLUCELVAX QUADRIVALENT may not protect all people who receive the vaccine.
Before receiving this vaccine, please see the full US Prescribing Information for FLUCELVAX QUADRIVALENT. The information provided here does not include all that is known about FLUCELVAX QUADRIVALENT. To learn more, talk with your healthcare provider or pharmacist.
FLUCELVAX® QUADRIVALENT is a registered trademark of Seqirus UK Limited or its affiliates.
Oct. 15, 2021 11:55 AM ETCSL Limited (CMXHF)
By: Ravikash, SA News Editor
October 20, 2021 at 5:29 PM EDT Back
TARRYTOWN, N.Y. and PARIS, Oct. 20, 2021 /PRNewswire/ --
Dupixent is the only biologic medicine to improve lung function in children aged 6 to 11 years in a randomized Phase 3 trial, supporting potential as a best-in-class option
Only biologic medicine approved for children with oral corticosteroid-dependent asthma
Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) and Sanofi today announced that the U.S. Food and Drug Administration (FDA) has approved Dupixent® (dupilumab) as an add-on maintenance treatment of patients aged 6 to 11 years with moderate-to-severe asthma characterized by an eosinophilic phenotype or with oral corticosteroid-dependent asthma.
"Despite available treatments, moderate-to-severe asthma can severely impact children's developing airways, causing sleepless nights, persistent coughing and wheezing, and potentially life-threatening exacerbations that require the use of systemic steroids that can negatively affect growth," said George D. Yancopoulos, M.D., Ph.D., President and Chief Scientific Officer at Regeneron. "This approval means that Dupixent, a first-of-its-kind treatment with a well-established efficacy and safety profile, can now be used by younger children with certain types of moderate-to-severe asthma in the U.S. In our pivotal trial, Dupixent helped children aged 6 to 11 years breathe better, suffer fewer asthma attacks and improve health-related quality of life. We also continue to study Dupixent in patients with other dermatologic, respiratory and gastrointestinal conditions where type 2 inflammation may play a role."
Asthma is one of the most common chronic diseases in children. Approximately 75,000 children aged 6 to 11 years live with the uncontrolled moderate-to-severe form of the disease in the U.S., with many more worldwide. Despite treatment with current standard-of-care inhaled corticosteroids and bronchodilators, these children may continue to experience serious symptoms such as coughing, wheezing and difficulty breathing. They also may require the use of multiple courses of systemic corticosteroids that carry significant risks.
"This FDA approval brings new hope for children who may be suffering from life-threatening asthma attacks and poor lung function, affecting their ability to breathe, potentially into adulthood," said Naimish Patel, M.D., Head of Global Development in Immunology and Inflammation at Sanofi. "Dupixent has helped to make a difference to the lives of many patients and families across three diseases with underlying type 2 inflammation, with more than 300,000 patients treated globally. We now have the opportunity to offer a safe and effective treatment option to children as young as 6 years of age living with certain types of moderate-to-severe asthma."
The FDA approval is based on data from a Phase 3 randomized, double-blind, placebo-controlled trial that evaluated the efficacy and safety of Dupixent combined with standard-of-care asthma therapy in children with uncontrolled moderate-to-severe asthma. More than 90% of children in the trial had at least one concurrent type 2 inflammatory condition.
Among patients who entered the trial with high levels of a certain type of white blood cell (eosinophils [EOS] ≥300 cells/μl; n=259), those who added Dupixent (100 mg or 200 mg every two weeks, based on weight) to standard-of-care experienced:
Children with elevated fractional exhaled nitric oxide (FeNO ≥20 ppb), an airway biomarker of inflammation that plays a major role in asthma, were also evaluated. In this subgroup, children who added Dupixent to standard-of-care experienced a reduction in the rate of severe asthma attacks.
The safety results from the trial were generally consistent with the known safety profile of Dupixent in patients aged 12 years and older with uncontrolled moderate-to-severe asthma, with the addition of helminth infections which were reported in 2.2% of Dupixent patients and 0.7% of placebo patients. The overall rates of adverse events were 83% for Dupixent and 80% for placebo. The most common adverse events that were more commonly observed with Dupixent compared to placebo were injection site reactions (18% Dupixent, 13% placebo), viral upper respiratory tract infections (12% Dupixent, 10% placebo) and eosinophilia (6% Dupixent, 1% placebo).
Dupixent is currently under regulatory review for children aged 6 to 11 years with moderate-to-severe asthma in the European Union and other health authorities worldwide.
About the LIBERTY ASTHMA VOYAGE Trial
The Phase 3 randomized, double-blind, placebo-controlled trial evaluated the efficacy and safety of Dupixent combined with standard-of-care asthma therapy in 408 children aged 6 to 11 years with uncontrolled moderate-to-severe asthma. In this trial, 86% of children had markers of type 2 inflammation underlying their asthma.
The primary endpoint was the annualized rate of severe asthma exacerbations over one year, and the key secondary endpoint was the change from baseline in FEV1pp at week 12. The FEV1pp seeks to evaluate a patient's change in lung function compared to their predicted lung function based on age, height, sex and ethnicity to account for children's growing lung capacity at different stages of development. Additional secondary endpoints included mean change from baseline in responder rates as measured by a ≥0.5 improvement on the Asthma Control Questionnaire (ACQ) 7-Interviewer Administered.
About Dupixent
Dupixent is an injection under the skin (subcutaneous injection) at different injection sites. For pediatric patients aged 6 to 11 years, Dupixent dosing is based on weight (100 mg every two weeks or 300 mg every four weeks for children ≥15 to <30 kg and 200 mg every two weeks for children ≥30 kg) and is supplied as a pre-filled syringe. It is also available as a pre-filled pen for adolescents (12 to 17 years) and adults at 200 mg and 300 mg doses. Dupixent is intended for use under the guidance of a healthcare professional and can be given in a clinic or at home by self-administration after training by a healthcare professional. In children younger than 12 years of age, Dupixent should be administered by a caregiver if given at home.
In the U.S., Dupixent is approved as an add-on maintenance treatment of patients aged 6 years and older with moderate-to-severe asthma characterized by an eosinophilic phenotype or with oral corticosteroid-dependent asthma; in patients aged 6 years and older with uncontrolled moderate-to-severe atopic dermatitis; and for use with other medicines for the maintenance treatment of chronic rhinosinusitis with nasal polyposis (CRSwNP) in adults whose disease is not controlled.
Regeneron and Sanofi are committed to helping patients in the U.S. who are prescribed Dupixent gain access to the medicine and receive the support they may need with the DUPIXENT MyWay® program. For more information, please call 1-844-DUPIXENT (1-844-387-4936) or visit www.DUPIXENT.com.
Dupixent is also approved in Europe, Japan and other countries around the world for use in certain patients with asthma or CRSwNP in different age populations, as well as specific patients with moderate-to-severe atopic dermatitis. Dupixent is approved in one or more of these indications in more than 60 countries around the world, and more than 300,000 patients have been treated globally.
Dupixent, which was invented using Regeneron's proprietary VelocImmune® technology, is a fully human monoclonal antibody that inhibits the signaling of the interleukin-4 (IL-4) and interleukin-13 (IL-13) pathways and is not an immunosuppressant. IL-4 and IL-13 are key and central drivers of the type 2 inflammation that plays a major role in atopic dermatitis, asthma and CRSwNP.
U.S. Indications
DUPIXENT is a prescription medicine used:
Please see accompanying full Prescribing Information including Patient Information.
For additional information about the company, please visit www.regeneron.com
View original content:https://www.prnewswire.com/news-releases/fda-expands-approval-of-dupixent-dupilumab-to-include-children-aged-6-to-11-years-with-moderate-to-severe-asthma-301405141.html
SOURCE Regeneron Pharmaceuticals
Oct. 20, 2021 5:50 PM ETSanofi (SNY), REGNBy: Jonathan M Block, SA News Editor2 Comments
October 22, 2021 at 12:59 AM EDT
TARRYTOWN, N.Y. and PARIS, Oct. 22, 2021 /PRNewswire/ --
Pivotal trial met primary and all key secondary endpoints
Dupixent significantly reduced itch at 12 weeks, and nearly three times as many Dupixent patients experienced reductions in both itch and skin lesions at 24 weeks
Prurigo nodularis, the sixth disease where Dupixent has demonstrated positive Phase 3 results, is a chronic skin condition that causes extreme itch and inflammatory skin lesions (nodules)
Data reinforce impact of targeting IL-4 and IL-13, key and central drivers of type 2 inflammation, to address itch and skin lesions
Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) and Sanofi today announced positive pivotal Phase 3 results from a trial evaluating Dupixent® (dupilumab) in adults with uncontrolled prurigo nodularis, a chronic type 2 inflammatory skin disease that causes extreme itch and skin lesions. The trial met its primary and all key secondary endpoints, showing that Dupixent significantly reduced itch and skin lesions compared to placebo in this investigational setting. The impact of uncontrolled prurigo nodularis on quality of life is one of the highest among inflammatory skin diseases with intense, chronic itch.
SOURCE Regeneron Pharmaceuticals, Inc.
October 15, 2021
-- Recommendation is Based on the Phase 3 ASCENT Study Showing that Sacituzumab Govitecan Significantly Improved Overall Survival in 2L vs. Physician’s Choice of Chemotherapy in Metastatic Triple-Negative Breast Cancer --
-- Sacituzumab Govitecan Could Offer a New Treatment Option for an Aggressive Type of Metastatic Breast Cancer if Approved --
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced that the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion for sacituzumab govitecan as monotherapy indicated for adult patients with unresectable or metastatic triple-negative breast cancer (TNBC) who have received two or more prior systemic therapies, at least one of them for advanced disease. The final European Commission decision on the Marketing Authorization Application for sacituzumab govitecan is anticipated later in 2021.
TNBC is the most aggressive type of breast cancer and accounts for approximately 15% of all breast cancers. It is more frequently diagnosed in younger and premenopausal women and is more prevalent in Black and Hispanic women. The five-year survival rate for this sub-type is 12%, compared with 28% for other breast cancer types, and these poor outcomes are often coupled with a significant decrease in quality of life, especially in relapsed/refractory disease. Sacituzumab govitecan is a first-in-class Trop-2 directed antibody-drug conjugate. Trop-2 is a protein located on the surface of cells and is overexpressed in TNBC and many other tumors.
“Effective treatment options are extremely limited for patients with metastatic TNBC, especially once they progress. We are encouraged by this CHMP positive opinion for sacituzumab govitecan, as we are now one step closer to bringing this much needed treatment option to patients across Europe,” said Merdad Parsey, MD, PhD, Chief Medical Officer, Gilead Sciences. “We look forward to the final decision by the EMA and the potential for sacituzumab govitecan to become a new standard of care for use as a second-line option.”
The positive opinion is supported by results from the Phase 3 ASCENT study, where sacituzumab govitecan showed a statistically significant and clinically meaningful 57% reduction in the risk of disease worsening or death and improved median progression-free survival (PFS) to 4.8 months from 1.7 months seen with physician’s choice of chemotherapy alone among all randomized patients, which included those with and without brain metastases (HR: 0.43; 95% CI: 0.35-0.54; p<0.0001). Sacituzumab govitecan also reduced the risk of death by 49% and improved median overall survival to 11.8 months vs. 6.9 months with physician’s choice of chemotherapy (HR: 0.51; 95% CI: 0.41-0.62; p<0.0001). The most common Grade 3 or higher adverse reactions were neutropenia (49.5%), leukopenia (12.0%), diarrhea (10.7%), anemia (10.1%), febrile neutropenia (6.6%), fatigue (5.2%), hypophosphatemia (5.2%), nausea (4.1%) and vomiting (3.0%). The sacituzumab govitecan U.S. Prescribing Information has a BOXED WARNING for severe or life-threatening neutropenia and severe diarrhea; see below for Important Safety Information.
Sacituzumab govitecan (under the trade name Trodelvy®) is approved in Australia, Canada, Great Britain, Switzerland, and the United States in metastatic TNBC, and review is also underway in Singapore and China through Everest Medicines.
About the ASCENT Study
The ASCENT study is a global, open-label, randomized Phase 3 study that enrolled more than 500 patients across 230 study locations. The study evaluated the efficacy and safety of sacituzumab govitecan compared with a single-agent chemotherapy of the physician’s choice in patients with unresectable, locally advanced or metastatic TNBC who had received at least two prior systemic treatments. Patients were randomly allocated to receive either sacituzumab govitecan or a chemotherapy chosen by the patient’s treating physician. The primary endpoint was progression-free survival (PFS, as determined by blinded independent central review) in patients without brain metastases. Secondary endpoints included: PFS for full study population or intention-to-treat (ITT) population, overall survival in both the ITT population and in the subgroup without brain metastasis, independently determined objective response rate, duration of response, time to onset of response according to Response Evaluation Criteria in Solid Tumors (RECIST 1.1), quality of life and safety. More information about ASCENT is available at http://clinicaltrials.gov/show/NCT02574455.
About Sacituzumab Govitecan
Sacituzumab govitecan is a first-in-class antibody and topoisomerase inhibitor conjugate directed to the Trop-2 receptor, a protein overexpressed in multiple types of epithelial tumors, including metastatic TNBC and metastatic urothelial cancer (UC), where high expression is associated with poor survival and relapse. Beyond the approvals of sacituzumab govitecan in the United States, it is also approved for metastatic TNBC in Australia, Canada, Great Britain and Switzerland for adults with metastatic TNBC. Sacituzumab govitecan is also under multiple regulatory reviews worldwide, including the EU, as well as in Singapore and China through our partner Everest Medicines. Sacituzumab govitecan continues to be developed for potential use in other TNBC and metastatic UC populations and is also being developed as an investigational treatment for hormone receptor-positive/human epidermal growth factor receptor 2-negative (HR+/HER2-) metastatic breast cancer and metastatic non-small cell lung cancer. Additional evaluation across multiple solid tumors is also underway.
Trodelvy, Gilead and the Gilead logo are trademarks of Gilead Sciences, Inc., or its related companies.
For more information about Gilead, please visit the company’s website at www.gilead.com, follow Gilead on Twitter (@GileadSciences) or call Gilead Public Affairs at 1-800-GILEAD-5 or 1-650-574-3000.
View source version on businesswire.com: https://www.businesswire.com/news/home/20211015005276/en/
Source: Gilead Sciences, Inc.
Oct. 15, 2021 8:31 AM ET Gilead Sciences, Inc. (GILD) By: Jonathan M Block, SA News Editor1 Comment
October 22, 2021 6:45 am ET
KEYTRUDA Is Now Approved in Combination With Chemotherapy as First-Line Treatment for Patients With Locally Recurrent Unresectable or Metastatic TNBC Whose Tumors Express PD-L1 (CPS ≥10) and Who Have Not Received Prior Chemotherapy for Metastatic Disease
KEYTRUDA Is the First Anti-PD-1 Therapy in Combination With Chemotherapy to Demonstrate Statistically Significant Overall Survival in These Patients; Based on Results From the Phase 3 KEYNOTE-355 Study
KENILWORTH, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, today announced that the European Commission (EC) has approved KEYTRUDA, Merck’s anti-PD-1 therapy, in combination with chemotherapy for the first-line treatment of locally recurrent unresectable or metastatic triple-negative breast cancer (TNBC) in adults whose tumors express PD-L1 (Combined Positive Score [CPS] ≥10) and who have not received prior chemotherapy for metastatic disease. Triple-negative breast cancer is an aggressive type of breast cancer. This represents KEYTRUDA’s first approval in Europe in a breast cancer setting.
The approval is based on final analysis from the Phase 3 KEYNOTE-355 trial, in which KEYTRUDA in combination with chemotherapy (nab-paclitaxel, paclitaxel or gemcitabine/carboplatin) significantly improved overall survival (OS), reducing the risk of death by 27% (HR=0.73 [95% CI, 0.55-0.95]; p=0.0093), and progression-free survival (PFS), reducing the risk of disease progression or death by 34% (HR=0.66 [95% CI, 0.50-0.88]; p=0.0018) compared to chemotherapy alone in these patients. In this trial, 38% of enrolled patients had tumors expressing PD-L1 with CPS ≥10.
“This approval is an important milestone for appropriate patients with metastatic TNBC who are in need of new treatment options,” said Dr. Javier Cortés, head of the International Breast Cancer Center (IBCC), Quironsalud Group. “With this approval, patients in Europe with metastatic TNBC whose tumors express PD-L1 (CPS ≥10) have a new immunotherapy treatment option that can be used in combination with different chemotherapy agents.”
“At Merck, we are committed to improving outcomes for people with difficult-to-treat cancers, such as TNBC, around the world and are proud of this first European approval for KEYTRUDA in a breast cancer setting,” said Dr. Vicki Goodman, vice president, clinical research, Merck Research Laboratories. “Now patients with metastatic TNBC who have tumors that express PD-L1 (CPS ≥10) in Europe have the new option of KEYTRUDA in combination with chemotherapy, a regimen that has shown significant improvement in overall survival. Today marks an important step forward in the treatment of this aggressive disease.”
This approval allows marketing of the combination with KEYTRUDA in all 27 European Union member states plus Iceland, Lichtenstein, Norway and Northern Ireland.
Merck is committed to delivering meaningful advances in breast cancer and women’s cancers. The company is rapidly advancing a broad portfolio in gynecologic and breast cancers through an extensive clinical development program for KEYTRUDA and several other investigational and approved medicines across these areas.
Data Supporting the European Approval
The approval was based on data from KEYNOTE-355 (NCT02819518), a multicenter, randomized, placebo-controlled, Phase 3 trial that enrolled 847 patients with locally recurrent unresectable or metastatic TNBC who had not been previously treated with chemotherapy in the advanced setting. Patients were randomized 2:1 to receive KEYTRUDA (200 mg every three weeks) plus chemotherapy (investigator’s choice of paclitaxel, nab-paclitaxel or gemcitabine/carboplatin) or placebo plus chemotherapy. Treatment with KEYTRUDA or placebo, both in combination with chemotherapy, continued until disease progression, unacceptable toxicity or a maximum of 24 months. Patients could continue to be treated with chemotherapy, per standard of care. Patients could continue to be treated with KEYTRUDA beyond RECIST-defined disease progression if the patient was clinically stable and deriving clinical benefit as determined by the investigator. The dual primary efficacy outcome measures were OS and PFS. Secondary efficacy outcome measures included objective response rate and duration of response.
In the final analysis of the study, median OS was 23.0 months (95% CI, 19.0-26.3) with KEYTRUDA plus chemotherapy versus 16.1 months (95% CI, 12.6-18.8) with chemotherapy alone. Median PFS was 9.7 months (95% CI, 7.6-11.3) with KEYTRUDA plus chemotherapy versus 5.6 months (95% CI, 5.3-7.5) with chemotherapy alone.
About KEYTRUDA® (pembrolizumab) Injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of patients with melanoma with involvement of lymph node(s) following complete resection.
Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [tumor proportion score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [combined positive score (CPS) ≥1] as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy. KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Carcinoma
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
Non-muscle invasive Bladder Cancer
KEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with MSI-H central nervous system cancers have not been established.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC).
Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or GEJ (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
KEYTRUDA, in combination with chemotherapy, with or without bevacizumab, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma.
Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
For more information about our oncology clinical trials, visit www.merck.com/clinicaltrials.
For more information, visit www.merck.com
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf.
Oct. 22, 2021 7:00 AM ET Merck & Co., Inc. (MRK)
By: Aakash Babu, SA News Editor1 Comment
October 15, 2021 7:20 am ET
Positive Opinion Granted for Advanced Renal Cell Carcinoma Based on Significant Progression-Free Survival (PFS), Overall Survival (OS) and Objective Response Rate (ORR) Benefit Compared to Sunitinib in CLEAR/KEYNOTE-581 Trial
Positive Opinion Granted for Advanced Endometrial Carcinoma Based on Significant OS and PFS Benefit Compared to Chemotherapy in KEYNOTE-775/Study 309 Trial
KENILWORTH, N.J. & TOKYO--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, and Eisai today announced that the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency has adopted positive opinions recommending approval of the combination of KEYTRUDA, Merck’s anti-PD-1 therapy, plus LENVIMA (marketed as KISPLYX® in the European Union [EU] for the treatment of advanced renal cell carcinoma [RCC]), the orally available multiple receptor tyrosine kinase inhibitor discovered by Eisai, for two different indications. One positive opinion is for the first-line treatment of adult patients with advanced RCC, and the other is for the treatment of adult patients with advanced or recurrent endometrial carcinoma (EC) who have disease progression on or following prior treatment with a platinum-containing therapy in any setting and are not candidates for curative surgery or radiation. Decisions on the CHMP’s recommendations will be given by the European Commission for marketing authorization in the EU, and are expected in the fourth quarter of 2021. If approved, this would be the first combination of an anti-PD-1 therapy with a tyrosine kinase inhibitor approved for the treatment of two different types of cancer in the EU.
The positive CHMP opinions are based on data from two pivotal Phase 3 trials: CLEAR (Study 307)/KEYNOTE-581 evaluating the combination in adult patients with advanced RCC and KEYNOTE-775/Study 309 evaluating the combination in certain patients with advanced EC.
In CLEAR/KEYNOTE-581, KEYTRUDA plus LENVIMA demonstrated statistically significant improvements versus sunitinib in the efficacy outcome measures of overall survival (OS), reducing the risk of death by 34% (HR=0.66 [95% CI, 0.49-0.88]; p=0.0049) versus sunitinib, and progression-free survival (PFS), reducing the risk of disease progression or death by 61% (HR=0.39 [95% CI, 0.32-0.49]; p<0.0001) with a median PFS of 23.9 months versus 9.2 months for sunitinib. Additionally, the confirmed objective response rate was 71% (95% CI: 66-76) (n=252) for patients who received KEYTRUDA plus LENVIMA versus 36% with sunitinib (95% CI: 31-41) (n=129).
In KEYNOTE-775/Study 309, KEYTRUDA plus LENVIMA demonstrated statistically significant improvements in the study’s dual efficacy outcome measures of OS, reducing the risk of death by 38% (HR=0.62 [95% CI, 0.51-0.75]; p<0.0001) with a median OS of 18.3 months versus 11.4 months for chemotherapy (investigator’s choice of doxorubicin or paclitaxel), and PFS, reducing the risk of disease progression or death by 44% (HR=0.56 [95% CI, 0.47-0.66]; p<0.0001), with a median PFS of 7.2 months versus 3.8 months for chemotherapy (investigator’s choice of doxorubicin or paclitaxel).
“KEYTRUDA plus LENVIMA demonstrated a survival benefit for advanced renal cell carcinoma in the first-line setting and represents an important potential new treatment option for these patients. Additionally, KEYTRUDA plus LENVIMA is the first anti-PD-1 and tyrosine kinase inhibitor combination to demonstrate a survival benefit in advanced endometrial carcinoma patients, and the benefit was shown regardless of mismatch repair status,” said Dr. Gregory Lubiniecki, Vice President, Clinical Research, Merck Research Laboratories. “We are pleased that the CHMP has recognized the important role of the combination therapy in these difficult-to-treat cancers.”
“We appreciate the positive opinions rendered by the EU CHMP recommending approval of KEYTRUDA plus LENVIMA in advanced renal cell carcinoma and advanced endometrial carcinoma, underscoring the potential significance of the outcomes observed in the CLEAR/KEYNOTE-581 and KEYNOTE-775/Study 309 trials,” said Dr. Takashi Owa, President, Oncology Business Group at Eisai. “We are grateful to the patients who participated in these studies, their families and clinicians. Their commitment made these meaningful milestones possible.”
The safety of KEYTRUDA in combination with axitinib or LENVIMA in advanced RCC, and in combination with LENVIMA in advanced EC has been evaluated in a total of 1,456 patients with advanced RCC or advanced EC. In these patient populations, the most frequent adverse reactions were diarrhea (58%), hypertension (54%), hypothyroidism (46%), fatigue (41%), decreased appetite and nausea (40% each), arthralgia (30%), vomiting, weight decreased, dysphonia and abdominal pain (28% each), proteinuria (27%), palmar plantar erythrodysesthesia syndrome and rash (26% each), stomatitis and constipation (25% each), musculoskeletal pain and headache (23% each) and cough (21%).
About KEYTRUDA® (pembrolizumab) Injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of patients with melanoma with involvement of lymph node(s) following complete resection.
Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [tumor proportion score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [combined positive score (CPS ≥1)] as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy. KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Carcinoma
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
Non-muscle Invasive Bladder Cancer
KEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin (BCG)-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ (CIS) with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with MSI-H central nervous system cancers have not been established.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC).
Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction (GEJ) adenocarcinoma whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test, with disease progression on or after 2 or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, HER2/neu-targeted therapy.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or GEJ (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
KEYTRUDA, in combination with chemotherapy, with or without bevacizumab, is indicated for the treatment of patients with persistent, recurrent, or metastatic cervical cancer whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test.
Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
KEYTRUDA, in combination with LENVIMA, is indicated for the first-line treatment of adult patients with advanced RCC.
Endometrial Carcinoma
KEYTRUDA, in combination with LENVIMA, is indicated for the treatment of patients with advanced endometrial carcinoma that is not MSI-H or dMMR, who have disease progression following prior systemic therapy in any settings and are not candidates for curative surgery or radiation.
Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf.
About LENVIMA® (lenvatinib); available as 10 mg and 4 mg capsules
LENVIMA, discovered and developed by Eisai, is a multiple receptor tyrosine kinase inhibitor that inhibits the kinase activities of vascular endothelial growth factor (VEGF) receptors VEGFR1 (FLT1), VEGFR2 (KDR), and VEGFR3 (FLT4). LENVIMA inhibits other kinases that have been implicated in pathogenic angiogenesis, tumor growth, and cancer progression in addition to their normal cellular functions, including fibroblast growth factor (FGF) receptors FGFR1-4, the platelet derived growth factor receptor alpha (PDGFRα), KIT, and RET. The combination of LENVIMA and everolimus showed increased anti-angiogenic and anti-tumor activity as demonstrated by decreased human endothelial cell proliferation, tube formation, and VEGF signaling in vitro and tumor volume in mouse xenograft models of human renal cell cancer greater than each drug alone. In syngeneic mouse tumor models, the combination of lenvatinib with an anti-PD-1 monoclonal antibody decreased tumor-associated macrophages, increased activated cytotoxic T cells, and demonstrated greater antitumor activity compared to either treatment alone.
LENVIMA® (lenvatinib) Indications in the U.S.
Please see Prescribing Information for LENVIMA (lenvatinib) at http://www.lenvima.com/pdfs/prescribing-information.pdf .
About the Merck and Eisai Strategic Collaboration
In March 2018, Eisai and Merck, known as MSD outside the United States and Canada, through an affiliate, entered into a strategic collaboration for the worldwide co-development and co-commercialization of LENVIMA. Under the agreement, the companies will jointly develop, manufacture and commercialize LENVIMA, both as monotherapy and in combination with Merck’s anti-PD-1 therapy KEYTRUDA.
In addition to ongoing clinical studies evaluating the KEYTRUDA plus LENVIMA combination across several different tumor types, the companies have jointly initiated new clinical studies through the LEAP (LEnvatinib And Pembrolizumab) clinical program and are evaluating the combination in more than 10 different tumor types across more than 20 clinical trials.
For more information about Eisai, please visit www.eisai.com (for global headquarters: Eisai Co., Ltd.), us.eisai.com (for U.S. headquarters: Eisai, Inc.) or www.eisai.eu (for Europe, Middle East, Africa, Russia, Australia, and New Zealand headquarters: Eisai Europe Ltd.), and connect with us on Twitter (U.S. and global) and LinkedIn (for U.S. and EMEA).
For more information, visit www.merck.com
Oct. 15, 2021 7:44 AM ET Merck & Co., Inc. (MRK), ESALFESALY By: Mamta Mayani, SA News Editor
October 20, 2021 7:07 pm ET
Series Would be Routinely Recommended Both for Adults 65 Years and Older and for Adults Ages 19 to 64 at Increased Risk for Disease, Such as Those with Certain Underlying Medical Conditions
KENILWORTH, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, today announced the U.S. Centers for Disease Control and Prevention’s (CDC’s) Advisory Committee on Immunization Practices (ACIP) unanimously voted in favor of updates to pneumococcal vaccination recommendations for adults 65 years and older, and for adults ages 19 to 64 with certain underlying medical conditions (e.g., chronic conditions such as diabetes, chronic heart disease, chronic lung disease, or chronic liver disease, as well as HIV, an immunocompromising condition) or other disease risk factors (e.g., smoking, alcoholism). In both groups, the ACIP voted to provisionally recommend vaccination either with a sequential regimen of VAXNEUVANCEfollowed by PNEUMOVAX23, or with a single dose of 20-valent pneumococcal conjugate vaccine. These updates would apply to adults who have not previously received a pneumococcal conjugate vaccine or whose previous pneumococcal vaccination history is unknown.
Further details will be available from the CDC. These provisional recommendations will be reviewed by the director of the CDC and the Department of Health and Human Services, and final recommendations will become official when published in the CDC’s Morbidity and Mortality Weekly Report (MMWR).
VAXNEUVANCE is indicated for active immunization for the prevention of invasive disease caused by Streptococcus pneumoniae serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, 22F, 23F and 33F in adults 18 years of age and older. VAXNEUVANCE is contraindicated for individuals with a history of severe allergic reaction (e.g., anaphylaxis) to any component of VAXNEUVANCE or to diphtheria toxoid.
PNEUMOVAX 23 is indicated for active immunization for the prevention of pneumococcal disease caused by the 23 serotypes contained in the vaccine (1, 2, 3, 4, 5, 6B, 7F, 8, 9N, 9V, 10A, 11A, 12F, 14, 15B, 17F, 18C, 19F, 19A, 20, 22F, 23F and 33F), in persons 50 years of age or older and persons aged ≥2 years who are at increased risk for pneumococcal disease; PNEUMOVAX 23 will not prevent disease caused by capsular types of pneumococcus other than those contained in the vaccine. PNEUMOVAX 23 is contraindicated in individuals with a history of a hypersensitivity reaction to any component of PNEUMOVAX 23.
See additional Select Safety Information for each of these vaccines below.
“Today’s vote reinforces the potential for VAXNEUVANCE in series with PNEUMOVAX 23 to help address a significant unmet need in the U.S. for adult populations at increased risk of invasive pneumococcal disease (IPD). VAXNEUVANCE in series with PNEUMOVAX 23 elicits a strong immune response to the serotypes shared by both vaccines, and together this regimen can help protect against pneumococcal serotypes responsible for about two-thirds of IPD cases in adults at increased risk,” said Dr. Roy Baynes, senior vice president and head of global clinical development, chief medical officer, Merck Research Laboratories. “We look forward to the CDC’s final, published recommendations and applaud the ACIP and the CDC for their continued efforts to address the significant burden of IPD by continuously evaluating vaccination recommendations utilizing a comprehensive body of scientific evidence.”
For more information, visit www.merck.com
Please see Prescribing Information for VAXNEUVANCE at https://www.merck.com/product/usa/pi_circulars/v/vaxneuvance/vaxneuvance_pi.pdf.
and Patient Information at https://www.merck.com/product/usa/pi_circulars/v/vaxneuvance/vaxneuvance_ppi.pdf.
Please see Prescribing Information for PNEUMOVAX 23 at http://www.merck.com/product/usa/pi_circulars/p/pneumovax_23/pneumovax_pi.pdf and Patient Information for PNEUMOVAX 23 at http://www.merck.com/product/usa/pi_circulars/p/pneumovax_23/pneumovax_ppi.pdf.
Oct. 21, 2021 8:20 AM ET
By: Dulan Lokuwithana, SA News Editor1 Comment
Merck (NYSE:MRK) announced that an advisory committee of the U.S. Centers for Disease Control and Prevention (CDC) has unanimously voted to recommend its 15-valent pneumococcal vaccine named VAXNEUVANCE.
10/15/2021CATEGORY:
CHMP recommendation based on positive results from the Phase 3 True North study, in which Zeposia demonstrated clinically meaningful improvements in key clinical, endoscopic and mucosal healing endpoints
If approved by the European Commission, Zeposia will become the first and only oral sphingosine 1-phosphate (S1P) receptor modulator approved for ulcerative colitis in the European Union
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE:BMY) today announced the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) has recommended approval of Zeposia (ozanimod) for the treatment of adults with moderately to severely active ulcerative colitis (UC) who have had an inadequate response, lost response, or were intolerant to either conventional therapy or a biologic agent. Zeposia, an oral medication taken once daily,is a sphingosine 1-phosphate (S1P) receptor modulator that binds with high affinity selectively to S1P subtypes 1 (S1P1) and 5 (S1P5). The CHMP recommendation will now be reviewed by the European Commission (EC), which has the authority to approve medicines for the European Union (EU).
“Despite available therapies, many people living with ulcerative colitis struggle to effectively manage this often relentless and unpredictable disease, making the availability of different treatment options extremely important,” said Jonathan Sadeh, M.D., MSc., senior vice president of Immunology and Fibrosis Development, Bristol Myers Squibb. “With this positive recommendation from the CHMP, we are one step closer to bringing Zeposia, which has the potential to be a truly transformative medicine, andthe first and only oral S1P receptor modulator for ulcerative colitis, to appropriate patients in the EU living with the disease.”
The CHMP adopted this positive opinion based on data from True North, a pivotal Phase 3 trial evaluating Zeposia as an induction and maintenance therapy versus placebo in adult patients with moderately to severely active UC. The trial demonstrated significant improvements across all primary and secondary efficacy endpoints – including clinical remission, clinical response, endoscopic improvement and endoscopic histologic mucosal improvement – versus placebo at Week 10 and Week 52. The overall safety observed in True North was consistent with the known safety profile for Zeposia in approved labeling. The clinical findings from True North, entitled “Ozanimod as Induction and Maintenance Therapy for Ulcerative Colitis,” were published in the September 30th issue of The New England Journal of Medicine.
The U.S. Food and Drug Administration approved Zeposia for the treatment of adults with moderately to severely active UC on May 27, 2021.
Bristol Myers Squibb thanks the patients and investigators involved in the True North clinical trial.
About True North
True North is a Phase 3, multicenter, randomized, double-blind, placebo-controlled clinical trial assessing the efficacy and safety of Zeposia 0.92 mg in patients with moderately to severely active UC who had an inadequate response or were intolerant to any of the following: oral aminosalicylates, corticosteroids, immunomodulators or a biologic. Patients were to be receiving treatment with oral aminosalicylates and/or corticosteroids prior to and during the induction period. A total of 30% of patients had previously failed or were intolerant to TNF blockers. Of these patients, 63% received at least two biologics including TNF blockers. At study entry, mean age was 42 years, 60% were male and mean disease duration was 7 years; patient characteristics were well-balanced across treatment groups. In the 10-week induction study (UC Study 1), a total of 645 patients were randomized 2:1 to receive Zeposia (n=429) or placebo (n=216), of whom 94% and 89%, respectively, completed the induction study. No new safety signals were observed in the induction phase.
In maintenance, UC Study 2, a total of 457 patients who received Zeposia in either UC Study 1 or in an open-label arm and achieved clinical response at Week 10 were re-randomized 1:1 and were treated with either Zeposia 0.92 mg (n=230) or placebo (n=227) for 42 weeks (UC Study 2), for a total of 52 weeks of treatment. Concomitant aminosalicylates were required to remain stable through week 52. Patients on concomitant corticosteroids were to taper their dose upon entering the maintenance study. Of these, 80% and 54.6% of patients who received Zeposia and placebo, respectively, completed the study. In the maintenance phase, the overall safety profile was consistent with the known safety profile for Zeposia and patients with moderate-to-severe UC.
All eligible patients were rolled into an open-label extension trial, which is ongoing and designed to assess the longer-term profile of Zeposia for the treatment of moderately to severely active UC. More information can be found on www.clinicaltrials.gov, NCT02435992.
About Zeposia (ozanimod)
Zeposia (ozanimod) is an oral, sphingosine 1-phosphate (S1P) receptor modulator that binds with high affinity to S1P receptors 1 and 5. Zeposia reduces the capacity of lymphocytes to migrate from lymphoid tissue, reducing the number of circulating lymphocytes in peripheral blood. The mechanism by which Zeposia exerts therapeutic effects in ulcerative colitis is unknown but may involve the reduction of lymphocyte migration into the intestines.
Bristol Myers Squibb is continuing to evaluate Zeposia in an open-label extension trial, which is ongoing and designed to assess the longer-term profile of Zeposia for the treatment of moderately to severely active ulcerative colitis. The company is also investigating Zeposia for the treatment of moderately to severely active Crohn’s disease in the ongoing Phase 3 YELLOWSTONE clinical trial program.
The U.S. Food and Drug Administration (FDA) approved Zeposia for the treatment of adults with moderately to severely active UC on May 27, 2021. Zeposia was approved by the FDA for the treatment of adults with relapsing forms of multiple sclerosis (RMS) in March 2020. The European Commission approved Zeposia for the treatment of adult patients with relapsing remitting multiple sclerosis (RRMS) with active disease as defined by clinical or imaging features in May 2020.
FDA APPROVED INDICATIONS
ZEPOSIA (ozanimod) is indicated for the treatment of:
1. Relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults.
2. Moderately to severely active ulcerative colitis (UC) in adults.
For additional safety information, please see the fullPrescribing InformationandMedication Guide.
For more information about Bristol Myers Squibb, visit us at BMS.com
Oct. 15, 2021 7:09 AM ET Bristol-Myers Squibb Company (BMY)
By: Mamta Mayani, SA News Editor10 Comments
Friday, October 15, 2021 - 07:50am
NEW YORK--(BUSINESS WIRE)-- Pfizer Inc. (NYSE: PFE) today announced that the European Medicines Agency's (EMA) Committee for Medicinal Products for Human Use (CHMP) has adopted a positive opinion recommending the 100 mg and 200 mg doses of abrocitinib, an oral, once-daily, Janus kinase 1 (JAK1) inhibitor, for marketing authorization to treat moderate to severe atopic dermatitis (AD) in adults who are candidates for systemic therapy. The CHMP also adopted a positive opinion recommending an extension to the existing indications for XELJANZ® (tofacitinib) for the treatment of adults with active ankylosing spondylitis (AS) who have responded inadequately to conventional therapy.
“The CHMP’s positive recommendation brings us closer to our goal of helping people living with moderate to severe atopic dermatitis in Europe find relief,” said Michael Corbo, PhD, Chief Development Officer, Inflammation & Immunology, Pfizer Global Product Development. “We look forward to working with the European Commission and hope to soon provide abrocitinib to people in Europe and eventually to more people worldwide who are living with this debilitating disease, many of whom have limited treatment options today.”
“Atopic dermatitis can be a debilitating condition that impacts the daily lives of millions of people,” said Dr. Diamant Thaci, Comprehensive Center for Inflammation Medicine, University of Luebeck, Germany. “Abrocitinib has shown significant efficacy, including relief from the hallmark chronic itch, rapid improvements in skin clearance, extent and severity of disease versus placebo, and a favorable risk-benefit profile. If approved, abrocitinib may become an important new treatment option for patients living with moderate to severe atopic dermatitis.”
Based on these CHMP recommendations, a decision by the European Commission, which authorizes marketing approval in the European Union, is expected on the abrocitinib and XELJANZ applications later this year. If granted by the European Commission, the centralized marketing authorizations would be valid in all EU Member States as well as in Iceland, Liechtenstein, and Norway.
The recommendation for abrocitinib is based on the results of five Phase 3 studies and a long-term extension study from a robust clinical trial program including more than 3,100 patients.
The recommendation for XELJANZ is based on data from a Phase 3, multicenter, randomized, double-blind, placebo-controlled study that evaluated the efficacy and safety of XELJANZ twice daily versus placebo in 269 adult patients living with active AS.
About Abrocitinib
Abrocitinib is an oral small molecule that selectively inhibits Janus kinase (JAK) 1. Inhibition of JAK1 is thought to modulate multiple cytokines involved in pathophysiology of atopic dermatitis, including interleukin IL-4, IL-13, IL-31, IL-22, and thymic stromal lymphopoietin (TSLP).
CIBINQO® (abrocitinib) received marketing authorization from the UK Medicines and Healthcare products Regulatory Agency (MHRA) and the Japanese Ministry of Health, Labour and Welfare (MHLW) in September 2021.
About XELJANZ
XELJANZ is approved in the European Union in four indications: adults with moderately to severely active rheumatoid arthritis (RA) after disease modifying antirheumatic drug (DMARD) failure or intolerance, adults with active psoriatic arthritis (PsA) after DMARD failure or intolerance, adults with moderately to severely active ulcerative colitis (UC) who have had an inadequate response, lost response, or were intolerant to either conventional therapy or a biologic agent, and active polyarticular juvenile idiopathic arthritis (JIA) and juvenile PsA in patients two years of age and older who have responded inadequately to previous therapy with DMARDs. Limitations of Use below.
XELJANZ has been studied in more than 50 clinical trials worldwide and prescribed to more than 300,000 adult patients (the majority of whom were RA patients) worldwide since 2012.i,ii.
In June 2021, the Committee for Medicine Products for Human Use of the European Medicines Agency adopted a recommendation from the Pharmacovigilance Risk Assessment Committee (PRAC) following its review of XELJANZ in the European Union, which states that in patients over 65 years of age, patients who are current or past smokers, patients with other cardiovascular (CV) risk factors, and patients with other malignancy risk factors, XELJANZ should only be used if no suitable treatment alternatives are available. The CHMP-endorsed PRAC recommendation is applicable to all EU member states and has been implemented in the XELJANZ summary of product characteristics.
Pfizer is also continuing to work with the U.S. Food and Drug Administration (FDA) and other regulatory agencies to review the full results and analysis of the ORAL Surveillance data. Most recently in September 2021, the FDA issued a Drug Safety Communication (DSC) related to XELJANZ®/XELJANZ XR® and two other arthritis medicines in the same drug class, based on its completed review of the ORAL Surveillance trial.
FDA-APPROVED INDICATIONS
Rheumatoid Arthritis
Psoriatic Arthritis
Ulcerative Colitis
Polyarticular Course Juvenile Idiopathic Arthritis
In addition, to learn more, please visit us on www.Pfizer.com
View source version on businesswire.com: https://www.businesswire.com/news/home/20211015005319/en/
Source: Pfizer Inc.
Oct. 15, 2021 7:59 AM ET Pfizer Inc. (PFE)
By: Aakash Babu, SA News Editor1 Comment
October 14, 2021 at 7:21 AM EDT Back
European Medicines Agency also announced earlier this week it would review Marketing Authorization Application for the antibody cocktail
TARRYTOWN, N.Y., Oct. 14, 2021 /PRNewswire/ -- Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) today announced that the U.S. Food and Drug Administration (FDA) has accepted for priority review a Biologics License Application (BLA) for REGEN-COV® (casirivimab and imdevimab) to treat COVID-19 in non-hospitalized patients and as prophylaxis in certain individuals. The FDA has assigned a target action date of April 13, 2022 and informed us that they currently are planning to hold an advisory committee meeting to discuss this application in advance of that date.
The BLA is supported by two positive Phase 3 trials involving more than 6,000 patients that evaluated the efficacy and safety of REGEN-COV to treat non-hospitalized patients already infected with SARS-CoV-2, and to prevent symptomatic infection in asymptomatic household contacts of SARS-CoV-2 infected individuals (both uninfected and infected contacts). A second BLA submission focusing on the treatment of patients hospitalized due to COVID-19 is expected to be submitted later this year.
Regulatory submissions are also progressing in the European Union (EU). Earlier this week, the European Medicines Agency (EMA) accepted for review the Marketing Authorization Application for the same antibody cocktail, known as Ronapreve™ in the EU and other countries outside of the U.S., for use in certain people either as a treatment in infected non-hospitalized patients, or as prophylaxis. The submission approach for the EMA is similar to that used for the FDA, with a Type II Variation planned in those already hospitalized because of COVID-19.
In the U.S., REGEN-COV has not been approved by the FDA, but is currently authorized under an Emergency Use Authorization (EUA) to treat people with mild to moderate COVID-19 who are at high risk of serious consequences from COVID-19 infection who are either already infected (non-hospitalized) or in certain post-exposure prophylaxis settings. REGEN-COV is available for free to eligible people as part of a U.S. government funded program, and in September Regeneron announced a new agreement with the U.S. government to supply an additional 1.4 million 1,200 mg doses of REGEN-COV by January 2022. Information on how to access REGEN-COV throughout the U.S. is available from the Department of Health and Human Services and the National Infusion Center Association.
The development and manufacturing of REGEN-COV have been funded in part with federal funds from the Biomedical Advanced Research and Development Authority, part of the U.S. Department of Health and Human Services' Office of the Assistant Secretary for Preparedness and Response, under OT number: HHSO100201700020C.
About the REGEN-COV Antibody Cocktail
REGEN-COV (casirivimab and imdevimab) is a cocktail of two monoclonal antibodies that was designed specifically to block infectivity of SARS-CoV-2, the virus that causes COVID-19, using Regeneron's proprietary VelocImmune® and VelociSuite® technologies. The two potent, virus-neutralizing antibodies that form the cocktail bind non-competitively to the critical receptor binding domain of the virus's spike protein, which diminishes the ability of mutant viruses to escape treatment and protects against spike variants that have arisen in the human population, as detailed in Cell and Science.
Multiple analyses have shown that the antibody cocktail retains potency against the main variants of concern, including Delta (first identified in India), Gamma (first identified in Brazil), Beta (first identified in South Africa) and Mu (first identified in Colombia), with information available in the Fact Sheet for Healthcare Providers. Regeneron will continue actively monitoring the potency of REGEN-COV against emerging variants.
REGEN-COV has not been approved by the FDA, but is currently authorized for emergency use for the treatment and post-exposure prophylaxis in certain high risk individuals. Post-exposure prophylaxis with REGEN-COV is not a substitute for vaccination against COVID-19. REGEN-COV is not authorized for pre-exposure prophylaxis for prevention of COVID-19 or for use in patients who are hospitalized due to COVID-19 or require oxygen therapy, or for people currently using chronic oxygen therapy because of an underlying comorbidity who require an increase in baseline oxygen flow rate due to COVID-19. This authorization is for the duration of the declaration that circumstances exist justifying the authorization of the emergency uses under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner. Additional information about REGEN-COV in the U.S. is below (authorized uses and important safety information).
REGEN-COV is an investigational medicine that has emergency or temporary pandemic authorizations currently in place in more than 40 countries, including the U.S., several countries in the EU, India, Switzerland and Canada, and the antibody cocktail is fully approved in Japan and conditionally approved in the UK.
Regeneron invented REGEN-COV and is collaborating with Roche to increase global supply, with Roche primarily responsible for development and distribution outside the U.S. Regeneron and Roche share a commitment to making the antibody cocktail available around the globe and will support access in low- and lower-middle-income countries through drug donations to be made in partnership with public health organizations.
Treatment:
REGEN-COV is authorized for the treatment of mild to moderate coronavirus disease 2019 (COVID-19) in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death
Limitations of Authorized Use (Treatment)
For additional information about the company, please visit www.regeneron.com
View original content:https://www.prnewswire.com/news-releases/fda-accepts-regen-cov-casirivimab-and-imdevimab-for-priority-review-for-treatment-and-prophylaxis-of-covid-19-301400260.html
SOURCE Regeneron Pharmaceuticals, Inc.
Oct. 14, 2021 7:37 AM ET Regeneron Pharmaceuticals, Inc. (REGN) By: Mamta Mayani, SA News Editor2 Comments
Tuesday, Oct 12, 2021
South San Francisco, CA -- October 12, 2021 --
Genentech, a member of the Roche Group (SIX: RO, ROG; OTCQX: RHHBY), today announced new long-term data that reinforce the benefit of early initiation and ongoing treatment of Ocrevus® (ocrelizumab) on disability progression in relapsing multiple sclerosis (RMS) and primary progressive MS (PPMS), as well as safety outcomes for an analysis of a shorter two-hour infusion in minority populations. Ocrevus data from all clinical trials consistently show a favorable benefit-risk profile over eight years. Genentech and research partners will also present four late-breaking abstracts to share the latest data regarding COVID-19 and vaccine response in patients treated with Ocrevus. These data are being presented virtually at the 37th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS).
“Many neurologists have had first-hand experience with Ocrevus over eight years in clinical trials and witnessed the consistently favorable efficacy and safety outcomes in RMS and PPMS, especially the reductions in progression to disability when given early in the disease,” said Levi Garraway, M.D., Ph.D. chief medical officer and head of Global Product Development. “Additionally, the new safety analysis of the shorter two-hour Ocrevus infusion is encouraging particularly for groups that are often underrepresented in clinical trials. We continue our commitment to diversity and health equity in clinical trial participation and access to treatment.”
Phase III OPERA I and OPERA II open-label extension (OLE): Sustained reduction in disability progression and low relapse rates in RMS
Long-term Ocrevus treatment continues to demonstrate sustained reduction in disability progression and suppression of disease activity in people with RMS. Earlier intervention with Ocrevus resulted in a 35% reduction in the risk of patients with RMS needing a walking aid over 7.5 years compared with patients who switched from interferon beta-1a to Ocrevus after the 96-week double-blind period (5.2% vs. 7.0%, respectively; 95% CI: 0.65 [0.44–0.97]; p=0.034). The risk was measured by the length of time until a person reached a score on the Expanded Disability Status Scale of 6 or greater (EDSS≥6) that was sustained for at least 48 weeks in a post-hoc analysis. Data also showed that switching from interferon beta-1a to Ocrevus at the start of the OLE period was associated with a rapid and robust reduction in annualized relapse rate (ARR) that was maintained through the 5.5-year OLE period. ARR was 0.2 pre-switch, 0.1 after one year of Ocrevus treatment and 0.03 after 5.5 years of Ocrevus treatment in the OLE. Ocrevus continuers maintained a low ARR of 0.03 after 7.5 years of Ocrevus treatment.
Phase III ORATORIO OLE: Sustained reduction in overall and upper limb disability progression in PPMS
After eight years, outcomes continue to favor early and ongoing treatment with Ocrevus to slow disability progression in people with PPMS. Earlier intervention with Ocrevus resulted in a 29% reduction in 48-week confirmed disability progression (CDP) in patients with PPMS over eight years compared with patients who switched to Ocrevus from placebo after the double-blind period of at least 120 weeks (95% CI: 0.71 [0.57–0.87]; p=0.001). A 24% (95% CI: 0.76 [0.62–0.92]; p=0.005) reduced risk of recurrent 48-week CDP (re-baselining EDSS after onset of CDP event) was seen in patients who were continuously treated with Ocrevus compared with those who switched from placebo. Many people with PPMS eventually transition into a wheelchair; therefore, maintaining the ability to use their hands and arms is important for these patients. Upper limb disability progression, measured by the nine-hole peg test (9-HPT), was also reduced in patients who were continuously treated with Ocrevus compared with those who switched from placebo (95% CI: 0.66 [0.50–0.86] respectively; p=0.002).
About Ocrevus® (ocrelizumab)
Ocrevus is the first and only therapy approved for both RMS (including RRMS and active, or relapsing, secondary progressive MS [SPMS], in addition to clinically isolated syndrome [CIS] in the U.S.) and PPMS. Ocrevus is a humanized monoclonal antibody designed to target CD20-positive B cells, a specific type of immune cell thought to be a key contributor to myelin (nerve cell insulation and support) and axonal (nerve cell) damage. This nerve cell damage can lead to disability in people with MS. Based on preclinical studies, Ocrevus binds to CD20 cell surface proteins expressed on certain B cells, but not on stem cells or plasma cells, suggesting that important functions of the immune system may be preserved. Ocrevus is administered by intravenous infusion every six months. The initial dose is given as two 300 mg infusions given two weeks apart. Subsequent doses are given as single 600 mg infusions.
Indications and Important Safety Information
What is Ocrevus?
Ocrevus is a prescription medicine used to treat:
It is not known if Ocrevus is safe or effective in children.
For more information, go to http://www.Ocrevus.com or call 1-844-627-3887.
For additional safety information, please see the full Prescribing Information and Medication Guide.
For additional information about the company, please visit http://www.gene.com.
Oct. 13, 2021 5:49 AM ET Roche Holding AG (RHHBY) By: Mamta Mayani, SA News Editor1 Comment
October 15, 2021
- CHMP positive opinion is based on data from two Phase 3 studies, KEEPsAKE-1 and KEEPsAKE-2[1],[2]
- If approved, this will mark the second indication for risankizumab in the EU[3]
- Psoriatic arthritis is a systemic inflammatory disease that impacts the skin and joints, affecting approximately 30 percent of patients with psoriasis[4],[5],[6],[7]
NORTH CHICAGO, Ill., Oct. 15, 2021 /PRNewswire/ -- AbbVie (NYSE: ABBV) today announced the European Medicines Agency's (EMA) Committee for Medicinal Products for Human Use (CHMP) recommended the approval of risankizumab (SKYRIZI®, 150 mg, subcutaneous injection at week 0, week 4 and every 12 weeks thereafter) alone or in combination with methotrexate (MTX), for the treatment of active psoriatic arthritis in adults who have had an inadequate response or who have been intolerant to one or more disease-modifying antirheumatic drugs (DMARDs). The CHMP positive opinion is a scientific recommendation for marketing authorization to the European Commission, which authorizes marketing approval in the European Union.
This CHMP positive opinion was supported by data from two pivotal Phase 3 studies, KEEPsAKE-1 and KEEPsAKE-2, which evaluated risankizumab in adults with active psoriatic arthritis including those who had responded inadequately or were intolerant to biologic therapy and/or non-biologic disease-modifying antirheumatic drugs (DMARDs).1,2 Additionally, the efficacy and safety profile of risankizumab with up to 52 weeks of exposure was consistent with the profile observed up to 24 weeks.8
"Many patients with psoriatic arthritis experience uncontrolled skin and joint symptoms despite the availability of existing therapies. For this reason, it is important to have multiple treatment options available for physicians to effectively manage their patients' condition," said Thomas Hudson, senior vice president, research and development, AbbVie. "The CHMP's recommendation to approve risankizumab in psoriatic arthritis is an important step in bringing treatment to more patients in need."
Across the Phase 3 KEEPsAKE-1 and KEEPsAKE-2 clinical studies, risankizumab met the primary endpoint of ACR20 response at week 24 versus placebo.1,2 In both studies, risankizumab also met ranked secondary endpoints including, but not limited to improvements in several clinical manifestations of psoriatic arthritis such as skin clearance (as measured by at least a 90 percent improvement in Psoriasis Area Severity Index [PASI 90]), physical function (as measured by the Health Assessment Questionnaire Disability Index [HAQ-DI]) and minimal disease activity (MDA) at week 24. In both KEEPsAKE-1 and KEEPsAKE-2, the most common adverse reactions associated with risankizumab were upper respiratory infections, headache, fatigue, injection site reactions and tinea infections.1,2
If the CHMP recommendation is accepted by the European Commission, this will mark the second indication for risankizumab in the European Union, which was approved in 2019 for the treatment of adult plaque psoriasis. The Marketing Authorization will be valid in all member states of the European Union, as well as Iceland, Liechtenstein, Norway and Northern Ireland.
Risankizumab (SKYRIZI®) is part of a collaboration between Boehringer Ingelheim and AbbVie, with AbbVie leading development and commercialization globally.
Use of risankizumab in psoriatic arthritis is not approved and its safety and efficacy are under evaluation by regulatory authorities.
About KEEPsAKE-1 and KEEPsAKE-21,2,3,8
KEEPsAKE-1 and KEEPsAKE-2 are both Phase 3, multicenter, randomized, double-blind, placebo-controlled studies designed to evaluate the safety and efficacy of risankizumab in adult patients with active psoriatic arthritis. KEEPsAKE-1 evaluated risankizumab in patients who had an inadequate response or intolerance to at least one DMARD. KEEPsAKE-2 evaluated risankizumab in patients who had an inadequate response or intolerance to biologic therapy and/or DMARDs. Patients were randomized to risankizumab 150 mg or placebo followed by risankizumab 150 mg at week 24. Patients randomized to risankizumab received four maintenance doses a year, following two initiation doses.
The primary endpoint for both studies was the achievement of ACR20 response at week 24. Ranked secondary endpoints included, but were not limited to, change from baseline in HAQ-DI, as well as the achievement of PASI 90 and minimal disease activity (MDA) at week 24. The studies are ongoing, and the long-term extension remains blinded to evaluate the long-term safety, tolerability and efficacy of risankizumab in patients who have completed the placebo-controlled period.
More information on these trials can be found at www.clinicaltrials.gov (KEEPsAKE-1: NCT03675308; KEEPsAKE-2: NCT03671148).
About SKYRIZI® (risankizumab)3
SKYRIZI is an interleukin-23 (IL-23) inhibitor that selectively blocks IL-23 by binding to its p19 subunit.3,9 IL-23, a cytokine involved in inflammatory processes, is thought to be linked to a number of chronic immune-mediated diseases, including psoriasis.9 The approved dose for SKYRIZI is 150 mg (two 75 mg injections), administered by subcutaneous injection at week 0 and 4, and every 12 weeks thereafter. Phase 3 trials of SKYRIZI in psoriasis, Crohn's disease, ulcerative colitis and psoriatic arthritis are ongoing.10,11,12,13
For more information about AbbVie, please visit us at www.abbvie.com.
Oct. 15, 2021 8:46 AM ET
By: Aakash Babu, SA News Editor2 Comments
October 13, 2021Download PDFAdding Verzenio to endocrine therapy demonstrated a significant and clinically meaningful reduction in the risk of recurrence in patients with HR+ HER2-, node-positive, high risk early breast cancer and a Ki-67 score of ≥20%
Verzenio is the first addition to adjuvant endocrine therapy approved by the FDA in nearly two decades for the treatment of HR+ HER2- early breast cancer
Updated data supporting this new indication will be featured in the October 14 European Society for Medical Oncology (ESMO) Virtual Plenary
INDIANAPOLIS, Oct. 13, 2021 /PRNewswire/ -- The U.S. Food and Drug Administration (FDA) has approved Eli Lilly and Company's (NYSE: LLY) Verzenio® (abemaciclib), in combination with endocrine therapy (tamoxifen or an aromatase inhibitor), for the adjuvant treatment of adult patients with hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2-), node-positive, early breast cancer (EBC) at high risk of recurrence and a Ki-67 score of ≥20% as determined by an FDA-approved test. Ki-67 is a marker of cellular proliferation. Verzenio is the first and only CDK4/6 inhibitor approved for this patient population.
"Over time, the collective results of the Verzenio clinical development program have demonstrated a differentiated CDK4/6 inhibitor profile, and the landmark data from the monarchE trial that supported this new indication in HR+ HER2- early breast cancer represent another important step forward for people who are in need of new treatment options," said Jacob Van Naarden, senior vice president, CEO of Loxo Oncology at Lilly and president, Lilly Oncology. "We are pleased with this initial approval in the adjuvant setting and as these data continue to mature, we look forward to further opportunities to work with health authorities to expand the use of Verzenio in this setting."
The Verzenio Phase 3 monarchE trial is a randomized (1:1), open-label, two cohort, multicenter study in adult women and men with HR+ HER2-, node-positive, resected EBC with clinical and pathological features consistent with a high risk of disease recurrence. In the trial, patients were randomized to receive two years of Verzenio 150 mg twice daily plus physician's choice of standard endocrine therapy, or standard endocrine therapy alone. Patients in both treatment arms were instructed to continue to receive adjuvant endocrine therapy for up to 5-10 years as recommended by their clinician. The primary endpoint of the study is invasive disease-free survival (IDFS) and was met at a pre-specified interim analysis in the intent-to-treat (ITT) population, with a statistically significant improvement in IDFS for patients treated with Verzenio plus ET compared to those treated with ET alone. Consistent with expert guidelines, IDFS was defined as the length of time before breast cancer comes back, any new cancer develops, or death.
Having achieved the study's primary endpoint in the entire enrolled population, a pre-specified analysis of IDFS was also conducted in patients with high-risk clinical and pathological factors and a Ki-67 score ≥20%. This subgroup analysis (N=2,003) included patients with ≥4 positive axillary lymph nodes (ALN), or 1-3 positive ALN with either Grade 3 disease and/or tumor size ≥5 cm, and whose tumors had a Ki-67 score of ≥20%. There was also a statistically significant improvement in IDFS for this pre-specified subgroup of patients receiving Verzenio plus ET compared to those who received ET alone (HR=0.643, 95% CI: 0.475, 0.872, p=0.0042).1,3
This approval is based on efficacy results from an analysis of this subgroup with additional follow-up, conducted post-hoc. In this analysis, Verzenio given in combination with ET continued to demonstrate a clinically meaningful benefit, with a 37 percent decrease in the risk of breast cancer recurrence or death compared to standard adjuvant ET alone for patients with high risk clinical and pathological features and a Ki-67 score ≥20% (HR: 0.626 [95% CI: 0.49-0.80]), and an absolute benefit in IDFS event rate of 7.1 percent at three years. The number of IDFS events at the time of this analysis was 104 with Verzenio plus ET compared to 158 with ET alone. Overall survival data were not mature and additional follow up is ongoing.
Adverse reactions from monarchE were consistent with the known safety profile for Verzenio.2 Safety and tolerability were evaluated in 5,591 patients. The most common adverse reactions reported (>10%) in the Verzenio plus ET (tamoxifen or an aromatase inhibitor) arm, and >2% higher than the ET arm alone, were diarrhea, infections, fatigue, nausea, headache, vomiting, stomatitis, decreased appetite, dizziness, rash, and alopecia.3 The most common laboratory abnormalities (all grades ≥10%) were creatinine increased, white blood cell count decreased, neutrophil count decreased, anemia, lymphocyte count decreased, platelet count decreased, ALT increased, AST increased, and hypokalemia.
This FDA approval builds on the established body of evidence for Verzenio, which is already approved for the treatment of certain types of HR+ HER2- advanced or metastatic breast cancer. Concurrent with this approval, the FDA has expanded the use of Verzenio in all indications, when given in combination with endocrine therapy, to include men. Verzenio is available in tablet strengths of 200 mg, 150 mg, 100 mg, and 50 mg.
The labelling for Verzenio contains warnings and precautions for diarrhea, neutropenia, interstitial lung disease (ILD/pneumonitis), hepatotoxicity, venous thromboembolism, and embryo-fetal toxicity. Instruct patients at the first sign of loose stools to initiate antidiarrheal therapy, increase oral fluids, and notify their healthcare provider. Perform complete blood counts and liver function tests prior to the start of Verzenio treatment, every two weeks for the first two months, monthly for the next two months and as clinically indicated. Based on results, Verzenio may require dose modification. Monitor patients for signs and symptoms of thrombosis and pulmonary embolism and treat as medically appropriate. Advise patients of potential risk to a fetus and to use effective contraception.
See Important Safety Information below and full Prescribing Information for additional information.
Click here to view the early breast cancer infographic.
Click here to view the monarchE clinical trial infographic.
Click to view the Verzenio product photos: 50 mg, 100 mg, 150 mg, 200 mg.
About Verzenio® (abemaciclib)
Verzenio® abemaciclib is a targeted treatment known as a CDK4/6 inhibitor. Verzenio is a non-chemotherapy oral tablet.
Verzenio works inside the cell to block CDK4/6 activity and help stop the growth of cancer cells, so they may eventually die (based on preclinical studies).* Cyclin-dependent kinases (CDK)4/6 are activated by binding to D-cyclins. In estrogen receptor-positive (ER+) breast cancer cell lines, cyclin D1 and CDK4/6 promote phosphorylation of the retinoblastoma protein (Rb), cell cycle progression, and cell proliferation.
In vitro, continuous exposure to Verzenio inhibited Rb phosphorylation and blocked progression from G1 to S phase of the cell cycle, resulting in senescence and apoptosis (cell death). Preclinically, Verzenio dosed daily without interruption resulted in reduction of tumor size. Inhibiting CDK4/6 in healthy cells can result in side effects, some of which may be serious. Clinical evidence also suggests that Verzenio crosses the blood-brain barrier. In patients with advanced cancer, including breast cancer, concentrations of Verzenio and its active metabolites (M2 and M20) in cerebrospinal fluid are comparable to unbound plasma concentrations.
Verzenio is Lilly's first solid oral dosage form to be made using a faster, more efficient process known as continuous manufacturing. Continuous manufacturing is a new and advanced type of manufacturing within the pharmaceutical industry, and Lilly is one of the first companies to use this technology.
INDICATIONS FOR VERZENIO
Verzenio® (abemaciclib) in combination with endocrine therapy (ET) is indicated for the adjuvant treatment of adult patients with hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2-), node-positive, early breast cancer (EBC) at high risk of recurrence and a Ki-67 score of ≥20% as determined by an FDA-approved test.
Verzenio is indicated for the treatment of HR+ HER2- advanced or metastatic breast cancer:
Please see full Prescribing Information for Verzenio.
Verzenio® is a trademark owned by or licensed to Eli Lilly and Company, its subsidiaries, or affiliates.
View original content to download multimedia:https://www.prnewswire.com/news-releases/fda-approves-verzenio-abemaciclib-as-the-first-and-only-cdk46-inhibitor-for-certain-people-with-hr-her2--high-risk-early-breast-cancer-301398910.html
SOURCE Eli Lilly and Company
Oct. 13, 2021 7:09 AM ET Eli Lilly and Company (LLY) By: Mamta Mayani, SA News Editor
October 14, 2021Download PDFAdjuvant Verzenio in combination with endocrine therapy (ET) continued to demonstrate a significant improvement in reducing the risk of recurrence in patients with HR+ HER2-, node-positive, high risk early breast cancer
With additional follow up, the treatment benefit of Verzenio plus ET was maintained over time and extended beyond the two-year treatment period; mature safety analysis was also consistent with previous results
Consistent Verzenio treatment benefit in reducing risk of recurrence observed in patients regardless of Ki-67 score
INDIANAPOLIS, Oct. 14, 2021 /PRNewswire/ -- Eli Lilly and Company (NYSE: LLY) today announced updated data from the positive Phase 3 monarchE trial evaluating the investigational use of Verzenio® (abemaciclib) in combination with standard adjuvant endocrine therapy (ET) for the treatment of hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2-), node-positive, high risk early breast cancer (EBC). These data were presented at today's ESMO Virtual Plenary and simultaneously published in the Annals of Oncology.
As previously published in the Journal of Clinical Oncology,1 monarchE met its primary endpoint of a statistically significant improvement in invasive disease-free survival (IDFS) in the intent-to-treat (ITT) population for patients treated with adjuvant Verzenio plus ET compared to those treated with ET alone. Consistent with expert guidelines, IDFS was defined as the length of time before breast cancer comes back, any new cancer develops, or death.
The trial included women and men with HR+ HER2-, node-positive EBC who had a high risk of disease recurrence based on clinical and pathological features (N=5,637). Patients were assigned to one of two cohorts. Cohort 1 enrolled patients with ≥4 positive axillary lymph nodes (ALN), or 1-3 positive ALN and either Grade 3 disease or tumor size ≥5 cm. Cohort 2 enrolled patients with 1-3 positive ALN and centrally determined Ki-67 score of ≥20% (defined in the study as "Ki-67 high"). Ki-67 is a marker of cellular proliferation. Ki-67 score was also determined centrally in Cohort 1 patients with a suitable sample, but Ki-67 determination was not required for enrollment in this cohort. The ITT population included both Cohort 1 and Cohort 2.
About the monarchE Study
monarchE is a global, randomized, open-label, two cohort, multicenter Phase 3 study in adult women and men with HR+ HER2-, node-positive resected EBC with clinical and pathological features consistent with a high risk of disease recurrence. A total of 5,637 patients were randomized (1:1) to receive two years of Verzenio 150 mg twice daily plus physician's choice of standard endocrine therapy, or standard endocrine therapy alone. Patients in both treatment arms were instructed to continue to receive adjuvant endocrine therapy for up to 5-10 years as recommended by their clinician. Cohort 1 enrolled patients with ≥4 positive axillary lymph nodes (ALN), or 1-3 positive ALN and either Grade 3 disease or tumor size ≥5 cm. Cohort 2 enrolled patients with 1-3 positive ALN and centrally determined Ki-67 score of ≥20%. The primary endpoint was IDFS in the ITT population (Cohorts 1 & 2). Secondary endpoints were IDFS in patients with high Ki-67 score (in the ITT population and in the Cohort 1 population), DRFS, overall survival, and safety.1,2
Wednesday, Oct 13, 2021
South San Francisco, CA -- October 13, 2021 --
Genentech, a member of the Roche Group (SIX: RO, ROG; OTCQX: RHHBY), today announced new longer-term efficacy and safety data for Enspryng™ (satralizumab-mwge). The data show Enspryng has a favorable benefit-risk profile and is effective in reducing relapses over four years of treatment in people with anti-aquaporin-4 antibody (AQP4-IgG) seropositive NMOSD, a rare debilitating disease that affects the central nervous system. Efficacy and safety results from the open-label extension (OLE) periods of the SAkuraStar and SAkuraSky studies, in addition to the design of SAkuraBONSAI, a new study in people with AQP4-IgG seropositive NMOSD who are treatment-naïve, or where prior rituximab (or biosimilar) treatment has failed, will be presented at the 37th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS).
“The positive longer-term efficacy and safety results for Enspryng are important for physicians as they consider Enspryng as a treatment option for their patients,” said Dr. Ingo Kleiter, Ruhr University Bochum, Germany. “Just one NMOSD relapse can lead to lifelong disability. An early accurate diagnosis followed by an effective treatment is vital to conserving the quality of life of people with this chronic disease.”
The pivotal SAkuraStar and SAkuraSky four year OLE data found that 73% and 71% of people with AQP4-IgG seropositive NMOSD treated with Enspryng remained relapse-free after 192 weeks (3.7 years), respectively, and 90% and 91% remained free from severe relapse.1 These results demonstrate that the robust efficacy observed in the studies’ double-blind periods is sustained longer-term for Enspryng as both a monotherapy and in combination with immunosuppressive therapy.
The data also demonstrate a favorable safety and tolerability profile for Enspryng in the overall Enspryng treatment period of up to seven years, comparable to the double-blind treatment periods in both SAkuraStar and SAkuraSky studies. Rates of adverse events and serious adverse events during the overall treatment periods were consistent with Enspryng and placebo in the double-blind periods. The most common adverse reactions observed were: headache, arthralgia, white blood cell count decrease, hyperlipidemia, and injection-related reactions. No new safety signals were observed.
About SAkuraStar and SAkuraSky in NMOSD
Enspryng has been investigated in two pivotal Phase III studies in neuromyelitis optica spectrum disorder (NMOSD), with the primary endpoint of both studies being time to first protocol-defined relapse (PDR) adjudicated by an independent review committee in the double-blind period. In the open-label extension (OLE) periods of the SAkura studies, PDRs were determined by the investigator.
The Phase III SAkuraStar study evaluated the efficacy and safety of Enspryng monotherapy administered to adults with NMOSD. In the anti-aquaporin-4 antibody (AQP4-IgG) seropositive subgroup, 83% treated with Enspryng remained relapse-free at 48 weeks, compared with 55% of those treated with placebo. At 96 weeks, 77% of those treated with Enspryng remained relapse-free, compared with 41% with placebo.
The Phase III SAkuraSky study evaluated the efficacy and safety of Enspryng in combination with baseline immunosuppressive therapy in adults and adolescents with NMOSD. Overall, 92% of AQP4-IgG seropositive participants receiving Enspryng in combination with immunosuppressive therapy remained relapse-free at 48 and 96 weeks, compared with 60% and 53% with placebo, respectively.
Enspryng showed a favorable safety and tolerability profile in the Phase III studies. The most common adverse reactions observed were: headache, arthralgia, white blood cell count decrease, hyperlipidemia and injection-related reactions.
About EnspryngTM (satralizumab-mwge)
Enspryng, which was designed by Chugai, a member of the Roche Group, is a humanized monoclonal antibody that targets interleukin-6 (IL-6) receptor activity. The cytokine IL-6 is believed to be a key driver in NMOSD disease processes, triggering the inflammation cascade and leading to damage and disability. Enspryng was designed using novel recycling antibody technology. When compared to conventional antibodies, Enspryng’s recycling antibody technology enables the medicine to remain in the bloodstream for a longer period of time and bind repeatedly to its target (the IL-6 receptor) - maximally sustaining IL-6 suppression in a chronic disease like NMOSD and enabling subcutaneous dosing every four weeks.
Positive Phase III results for Enspryng, as both monotherapy and in combination with baseline immunosuppressive therapy, suggest that IL-6 inhibition is an effective therapeutic approach for NMOSD. The Phase III clinical development program for Enspryng includes two studies: SAkuraStar and SAkuraSky.
Enspryng is currently approved in 58 countries, including the United States, Canada, Japan, South Korea and the European Union.
Enspryng has been designated as an orphan drug in the United States, Europe, Japan and Russia. In addition, it was granted Breakthrough Therapy Designation for the treatment of NMOSD by the U.S. Food and Drug Administration (FDA) in December 2018, which is given to treatments that may demonstrate substantial improvement over other available options.
Indications and Important Safety Information
Patients should not take Enspryng if they:
Please see the full Prescribing Information for additional Important Safety Information.
For additional information about the company, please visit http://www.gene.com.
Oct. 14, 2021 1:46 AM ET Roche Holding AG (RHHBY)
By: Mamta Mayani, SA News Editor
Oct 13, 2021
Basel, October 13, 2021 — Novartis today announced that the US Food and Drug Administration (FDA) has accepted the company’s supplemental Biologics License Application (sBLA) and that the European Medicines Agency (EMA) has validated the type-II variation application for Beovu® (brolucizumab) 6 mg for the treatment of diabetic macular edema (DME). Additionally, the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) accepted an application for Beovu in the treatment of DME. Regulatory decisions for Beovu in DME are expected in mid-2022 for the US and Europe.
If approved, DME would be the second indication for Beovu following its approval for wet age-related macular degeneration in October 2019 (FDA) and February 2020 (European Commission)5,6. DME is the leading cause of blindness in adults in developed countries, affecting 12% of people with type 1 diabetes and 28% of those with type 2 diabetes1. Consistently high blood sugar levels associated with diabetes can damage small blood vessels in the eye, causing them to leak fluid1. Unmet needs in DME include improving fluid resolution and addressing the burden of frequent treatment schedules1-3.
“People living with diabetes often need to manage multiple comorbidities related to diabetes and there is a significant need to provide better disease management. If approved, Beovu has the potential to provide better fluid resolution and fewer injections during the loading phase and throughout maintenance treatment,” said Jill Hopkins, SVP and Global Development Unit Head, Ophthalmology, Novartis Pharmaceuticals. “We look forward to bringing this potential new treatment option that may help to address unmet needs in the DME patient population.”
The regulatory applications are based on year one data from the Phase III, randomized, double-masked KESTREL and KITE* studies, which met their primary endpoint of non-inferiority in change in best corrected visual acuity (BCVA) from baseline versus aflibercept at year one4. In KESTREL and KITE, following the loading phase, over half of patients in the Beovu 6 mg arm remained on a 12-week dosing interval through year one4. Fewer eyes treated with Beovu had intraretinal and/or subretinal fluid (IRF/SRF) at week 32 and week 52 versus eyes treated with aflibercept4. The KESTREL and KITE trials are the first pivotal trials to assess an anti-VEGF treatment on six-week dosing intervals in the loading phase, suggesting Beovu may offer fewer injections from the start of treatment4.
Overall, Beovu demonstrated a favorable benefit-risk profile in KESTREL and KITE4. The most common ocular and non-ocular adverse events (≥5%) in KESTREL and KITE were conjunctival hemorrhage, nasopharyngitis and hypertension4. IOI rates in KESTREL were 4.7% for brolucizumab 3 mg (including 1.6% retinal vasculitis), 3.7% for Beovu 6 mg (including 0.5% retinal vasculitis), and 0.5% for aflibercept 2 mg4. IOI rates in KITE were equivalent (1.7%) between the Beovu 6 mg and aflibercept 2 mg arms with no retinal vasculitis reported4. Retinal vascular occlusion was reported in KESTREL for brolucizumab 3 mg (1.1%) and 6 mg (0.5%), and in KITE for brolucizumab and aflibercept (0.6% each)4. The majority of these events were manageable and resolved with or without treatment4.
About the KESTREL and KITE clinical trials
KESTREL and KITE are global, randomized, double-masked, Phase III, two-year studies comparing the safety and efficacy of Beovu and aflibercept in the treatment of DME4,7,8.
KESTREL and KITE involved 926 patients in 36 countries7,8. In the loading phase of both trials, patients in the Beovu arms were treated every six weeks for a total of five doses; patients in the aflibercept arms were treated every four weeks for a total of five doses, in line with its label at the start of the studies7.8. Following the loading phase, patients in the Beovu arms were subsequently treated every 12 weeks, with those demonstrating disease activity moved to dosing every eight weeks for the remainder of the study7,8.
At week 72 of KITE, Beovu patients dosed every 12 weeks could be extended to dosing every 16 weeks, and patients dosed every eight weeks could be extended to every 12 weeks8. As in year one, those demonstrating disease activity were moved to dosing every eight weeks for the remainder of the study8. Through the entirety of both two-year trials, patients in the aflibercept arms were treated every eight weeks7,8.
About Beovu (brolucizumab) 6 mg
Beovu (brolucizumab, also known as RTH258) 6 mg is approved for the treatment of wet age-related macular degeneration (AMD) in more than 70 countries, including in the US, EU, UK, Japan, Canada and Australia5,6,11-13. Additional trials, which study the effects of brolucizumab in patients with wet AMD, diabetic macular edema (DME), and proliferative diabetic retinopathy (PDR), are currently ongoing.
Find out more at https://www.novartis.com.
Oct. 14, 2021 2:59 AM ETNovartis AG (NVS)By: Mamta Mayani, SA News Editor
October 9, 2021 at 11:23 AM EDT
ROCKVILLE, Md., Oct. 9, 2021 /PRNewswire/ --
REGENXBIO Inc. (Nasdaq: RGNX) today announced initial data from the ongoing Phase II ALTITUDE™ trial of RGX-314 for the treatment of diabetic retinopathy (DR) without center-involved diabetic macular edema (CI-DME) using in-office suprachoroidal delivery. The data is being presented at the American Society of Retina Specialists (ASRS) Annual Meeting by Dennis Marcus, M.D., F.A.S.R.S., President, Southeast Retina Center. RGX-314 is a potential one-time gene therapy in clinical development for the treatment of wet age-related macular degeneration and DR.
"We are pleased to share initial data from the Phase II ALTITUDE trial, and we are encouraged to see treatment effect particularly at this early time point of three months after the one-time, in-office administration of RGX-314," said Steve Pakola, M.D., Chief Medical Officer of REGENXBIO. "RGX-314 is the first gene therapy in clinical trials for DR using suprachoroidal delivery, and has the potential to provide sustainable, long-term anti-VEGF protein production in the eye for the treatment of DR, which affects approximately eight million people in the United States alone. We look forward to providing additional updates on this trial next year."
"DR can start in young adulthood and often progresses quickly, leading to vision-threatening complications, including diabetic macular edema (DME) and neovascularization that can lead to vision loss," said Dr. Marcus. "Current treatment options include 'watchful waiting,' treatment requiring repeated anti-VEGF injections, retinal laser or surgical treatment. This first look at the ALTITUDE trial data is promising, as it shows not only clinical improvement in disease severity as measured by the ETDRS-DRSS, but also that this treatment is well tolerated in patients."
Study Design and Safety Update from Phase II ALTITUDE Trial of RGX-314 for the Treatment of DR Using Suprachoroidal Delivery
ALTITUDE is a multi-center, open-label, randomized, controlled dose-escalation trial that will evaluate the efficacy, safety and tolerability of suprachoroidal delivery of RGX-314 using the SCS Microinjector® in patients with a DR diagnosis of moderately severe or severe nonproliferative diabetic retinopathy (NPDR) or mild proliferative diabetic retinopathy (PDR). Twenty patients in Cohort 1 were randomized to receive RGX-314 at a dose level of 2.5x1011 genomic copies per eye (GC/eye) versus observational control at a 3:1 ratio. Cohort 2 will include 20 patients randomized to receive RGX-314 at an increased dose level of 5x1011 GC/eye versus observational control at a 3:1 ratio. Cohort 3 is designed to evaluate RGX-314 at the same dose level as Cohort 2 in 20 patients who are neutralizing antibody (NAb) positive. Enrollment is ongoing in Cohorts 2 and 3. Patients in this trial do not receive prophylactic immune suppressive corticosteroid therapy before or after administration of RGX-314.
As of September 29, 2021, RGX-314 was reported to be well tolerated in the 15 patients dosed with RGX-314 in Cohort 1. One serious adverse event was reported in one patient dosed with RGX-314, which occurred in the patient's untreated fellow eye and is considered not related to RGX-314. Among patients in Cohort 1 dosed with RGX-314, no intraocular inflammation was observed on slit-lamp examination. One patient experienced a mild case of episcleritis that resolved with topical corticosteroids. Common ocular treatment emergent adverse events in the study eye were not considered drug-related and were predominantly mild. These included conjunctival hemorrhage and conjunctival hyperemia.
About RGX-314
RGX-314 is being investigated as a potential one-time treatment for wet AMD, diabetic retinopathy, and other chronic retinal conditions. RGX-314 consists of the NAV AAV8 vector, which encodes an antibody fragment designed to inhibit vascular endothelial growth factor (VEGF). RGX-314 is believed to inhibit the VEGF pathway by which new, leaky blood vessels grow and contribute to the accumulation of fluid in the retina.
REGENXBIO is advancing research in two separate routes of administration of RGX-314 to the eye, through a standardized subretinal delivery procedure as well as delivery to the suprachoroidal space. REGENXBIO has licensed certain exclusive rights to the SCS Microinjector® from Clearside Biomedical, Inc. to deliver gene therapy treatments to the suprachoroidal space of the eye.
View original content to download multimedia:https://www.prnewswire.com/news-releases/regenxbio-presents-positive-initial-data-from-phase-ii-altitude-trial-of-rgx-314-for-the-treatment-of-diabetic-retinopathy-using-suprachoroidal-delivery-at-american-society-of-retina-specialists-annual-meeting-301396478.html
SOURCE REGENXBIO Inc.
Oct. 11, 2021 5:09 AM ET
By: Mamta Mayani, SA News Editor1 Comment
10/13/2021CATEGORY:
Treatment with Zeposia demonstrated a low annualized relapse rate (ARR) of 0.103 and more than 70% of patients were relapse-free at four years
Safety was consistent with prior findings and the established safety profile of Zeposia in up to 5 years of follow-up
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) today announced interim results from the Phase 3 open-label extension trial DAYBREAK, demonstrating the long-term efficacy and safety profile of Zeposia (ozanimod) in patients with relapsing forms of multiple sclerosis (MS). These data (Presentation #P737) and five additional abstracts from company-sponsored studies will be presented at the 37th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS), taking place virtually October 13-15, 2021.
“Early and effective intervention can significantly impact physical and cognitive results over time, with low relapse rates an important indicator of patient outcomes,” said Bruce Cree, M.D., Ph.D., M.A.S., study investigator and Professor of Clinical Neurology, University of California San Francisco (UCSF) Weill Institute for Neurosciences and clinical research director, UCSF MS Center. “These data from the DAYBREAK trial provide a clear picture of the long-term safety and efficacy profile of Zeposia, and reinforce its potential when used early in the treatment process for people living with relapsing forms of MS.”
In the DAYBREAK extension study, safety was consistent with prior findings and no new safety signals emerged during the reporting period with long-term use of Zeposia. Treatment with Zeposia demonstrated a low annualized relapse rate (ARR) of 0.103. At months 36 and 48, 75% and 71% of participants were relapse-free and 3- and 6-month confirmed disability progression was observed in 13.9% and 11.4% of participants in the trial, respectively.
“Our presentations at ECTRIMS 2021 bolster the growing body of evidence underscoring the long-term efficacy and safety of Zeposia to treat relapsing forms of MS and demonstrate our focus on delivering meaningful innovations to help preserve the body and mind of people living with MS,” said Jonathan Sadeh, M.D., MSc., head of Immunology and Fibrosis Development, Bristol Myers Squibb. “We are committed to advancing our knowledge of this unpredictable, debilitating disease, with the goal of transforming medicine through science.”
In the DAYBREAK trial, of 2,494 participants exposed to Zeposia for an average of 46.8 months, 2,143 participants (85.9%) had any treatment-emergent adverse event (TEAE), 298 (11.9%) had a serious TEAE and 75 (3.0%) discontinued the study due to a TEAE. The most common TEAEs were nasopharyngitis (19.6%), headache (15.8%), upper respiratory tract infection (11.1%) and lymphopenia (10.3%). Data from this long-term observational study of patients treated for up to 62.7 months are consistent with the established safety profile of Zeposia and with sustained control of disease activity and disability progression.
At the ECTRIMS 2021 Congress, Bristol Myers Squibb and collaborators will present multiple abstracts that reinforce the company’s growing body of research in MS and commitment to people living with the disease. Accepted abstracts are available on the ECTRIMS 2021 Congress website.
Visit this page on BMS.com for more information on Bristol Myers Squibb’s scientific approach and resources on MS.
Summary of Presentations:
Bristol Myers Squibb studies featured at the ECTRIMS 2021 Congress include:
About DAYBREAK
DAYBREAK is a Phase 3, multi-center, long-term open-label extension (OLE), randomized, double-blind, double-dummy, active-controlled, parallel group study to evaluate the safety and efficacy of Zeposia (ozanimod) administered orally to patients with relapsing forms of multiple sclerosis (MS).
Eligible patients from the RADIANCE, SUNBEAM and RPC01-1001 trials diagnosed with relapsing forms of MS are enrolled to receive treatment until the end of the DAYBREAK trial or until the development program is discontinued. Patients in the trial are receiving Zeposia 0.92 mg (equivalent to ozanimod HCl 1 mg). In total, 2,639 participants completed the parent clinical trials, and this interim analysis (data cutoff February 2021), includes a total of 2,494 participants with mean (range) Zeposia exposure of 46.8 (0.03–62.7) months in the OLE.
The primary objective of the trial is to evaluate safety in the overall population. Bristol Myers Squibb thanks the patients and investigators who are participating in this clinical trial.
About Zeposia (ozanimod)
Zeposia (ozanimod) is an oral, sphingosine 1-phosphate (S1P) receptor modulator that binds with high affinity to S1P receptors 1 and 5. Zeposia blocks the capacity of lymphocytes to egress from lymph nodes, reducing the number of lymphocytes in peripheral blood. The mechanism by which Zeposia exerts therapeutic effects in multiple sclerosis is unknown but may involve the reduction of lymphocyte migration into the central nervous system.
Zeposia was approved by the U.S. Food and Drug Administration (FDA) for the treatment of adults with relapsing forms of multiple sclerosis in March 2020. The European Commission approved Zeposia for the treatment of adult patients with relapsing remitting multiple sclerosis with active disease as defined by clinical or imaging features in May 2020. The FDA approved Zeposia for the treatment of adults with moderately to severely active ulcerative colitis on May 27, 2021.
U.S. FDA-APPROVED INDICATIONS FOR ZEPOSIA
ZEPOSIA (ozanimod) is indicated for the treatment of:
1. Relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults.
2. Moderately to severely active ulcerative colitis (UC) in adults.
For additional safety information, please see the full
Prescribing InformationandMedication Guide.
For more information about Bristol Myers Squibb, visit us at BMS.com
Oct. 13, 2021 7:37 AM ET Bristol-Myers Squibb Company (BMY)
By: Mamta Mayani, SA News Editor2 Comments
Friday, Oct 8, 2021
South San Francisco, CA -- October 8, 2021 --
Genentech, a member of the Roche Group (SIX: RO, ROG; OTCQX: RHHBY), today announced that gantenerumab, an anti-amyloid beta antibody developed for subcutaneous administration, has been granted Breakthrough Therapy Designation by the U.S. Food and Drug Administration (FDA) for the treatment of people living with Alzheimer’s disease (AD). This designation is based on data showing that gantenerumab significantly reduced brain amyloid plaque, a pathological hallmark of AD, in the ongoing SCarlet RoAD and Marguerite RoAD open-label extension trials, as well as other studies. Learnings from these studies have been incorporated into the optimized design of two ongoing parallel, global, placebo-controlled and randomized Phase III trials, GRADUATE 1 and 2. The pivotal trials are evaluating gantenerumab in more than 2,000 participants for more than two years and are expected to be completed in the second half of 2022.
“For more than a decade, we’ve been committed to advancing the science of Alzheimer’s as well as our investigational medicine gantenerumab, and we look forward to delivering a comprehensive and robust data set that furthers our collective understanding of this devastating disease,” said Levi Garraway, M.D., Ph.D., chief medical officer and head of Global Product Development. “This Breakthrough Therapy Designation reinforces our confidence in gantenerumab, which would be the first subcutaneous medicine for the treatment of Alzheimer’s disease with the potential for at-home administration.”
Breakthrough Therapy Designation is designed to accelerate the development and review of medicines intended to treat serious or life-threatening conditions with preliminary evidence that indicates they may demonstrate a substantial improvement over available therapies that have received full FDA approval. This designation for gantenerumab marks the 39th Breakthrough Therapy Designation for Genentech’s portfolio of medicines.
AD is a progressive, fatal disease of the brain characterized by a decline in memory, language, and other thinking skills as well as changes in mood and behavior. Biological changes in the brain are believed to start decades before clinical symptoms of AD become evident. Alzheimer's is the most common form of dementia, which currently affects more than 55 million people worldwide, and is projected to reach 78 million by 2030. An enormous and growing public health challenge, it is predicted to cost the global economy a cumulative $20 trillion over the next decade, or the U.S. $2.8 trillion per year by 2030. Approximately 10 million people are diagnosed with AD each year. Given the medical and societal complexities of AD, several tools and treatment options will likely be required to meet the multiple and diverse needs of people living with the disease.
Genentech is continuing to explore multiple approaches and molecules that may address key pathways of AD, including amyloid beta and tau, as well as innovative tools designed to more effectively diagnose AD and support clinicians in monitoring disease progression.
About gantenerumab and its clinical program
Gantenerumab is an investigational IgG1 antibody designed to bind to aggregated forms of amyloid beta and remove brain amyloid plaques, a pathological hallmark of Alzheimer’s disease (AD). Gantenerumab significantly lowered brain amyloid plaques in patients with sporadic AD in the SCarlet RoAD and Marguerite RoAD open-label extension studies (OLEs) and in patients with dominantly inherited AD in the DIAN-TU-001 study. Learnings from these studies have been incorporated into the optimized design of two ongoing parallel, global, placebo-controlled and randomized Phase III trials, GRADUATE 1 and 2.
The pivotal GRADUATE trials are investigating the effect of gantenerumab on amyloid load and downstream biomarkers of disease progression, as well as the safety and efficacy of gantenerumab in people with early (prodromal-to-mild) AD. The studies include more than 2,000 participants treated for more than two years in up to 350 study centers in more than 30 countries worldwide. It is evaluating a monthly target dose of 1,020 mg with an optimized titration, aimed at maximizing exposure and minimizing dose interruption throughout the study period for better detection of a potential clinical benefit. Data from both trials are expected in the second half of 2022.
Other studies evaluating gantenerumab in AD include:
For additional information about the company, please visit http://www.gene.com.
Oct. 08, 2021 11:52 AM ET Roche Holding AG (RHHBY) By: Aakash Babu, SA News Editor5 Comments
Oct 10, 2021 5:00 PM
Marks BRUKINSA’s second approved indication in Australia, on the heels of its recent initial approval in Waldenström’s macroglobulinemia
To date, BRUKINSA is approved in mantle cell lymphoma in nine countries
SYDNEY & CAMBRIDGE, Mass. & BEIJING--(BUSINESS WIRE)-- BeiGene (NASDAQ: BGNE; HKEX: 06160), a global, science-driven biotechnology company focused on developing innovative and affordable medicines to improve treatment outcomes and access for patients worldwide, today announced that BRUKINSA® (zanubrutinib) has been approved in Australia for the treatment of adult patients with mantle cell lymphoma (MCL) who have received at least one prior therapy. On October 7, 2021, BRUKINSA received its initial approval in Australia for the treatment of adult patients with Waldenström’s macroglobulinemia (WM) who have received at least one prior therapy or in first line treatment for patients unsuitable for chemo-immunotherapy.1
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20211010005045/en/
(Photo: Business Wire)
Following registration of BRUKINSA with the Therapeutic Goods Administration (TGA) in both approved indications, these patients will have immediate access to BRUKINSA through the BeiGene sponsored post-approval, pre-reimbursement access program.
“Mantle cell lymphoma is an uncommon form of non-Hodgkin lymphoma that is generally considered incurable. While the majority of patients respond well to their initial treatment, virtually all will develop progressive lymphoma over time. Existing therapies for patients with recurrent or refractory MCL are often ineffective or have side effects that can lead to treatment discontinuation,” said Professor Stephen Opat, Director of Clinical Haematology at Monash Health and a principal investigator in the zanubrutinib clinical program. “I’m encouraged that zanubrutinib – a highly selective BTK inhibitor with promising clinical results from two trials in relapsed or refractory MCL – will provide a new treatment option for these patients living in Australia.”
“Australia has some of the highest rates of non-Hodgkin’s lymphoma in the world, and these patients need options for treatment beyond those that exist today,” said Sharon Winton, CEO, Lymphoma Australia. “MCL patients will certainly welcome the news that BeiGene is providing access to BRUKINSA by sponsoring a pre-reimbursement program, as new therapies are critical, especially to those diagnosed later in life when it may be challenging to tolerate more aggressive types of treatment.”
BeiGene submitted for reimbursement of BRUKINSA to the Pharmaceutical Benefits Advisory Committee (PBAC), with MCL recommended for listing in July 2021.
“BRUKINSA was designed to provide deep and durable responses while reducing off-target side effects compared to first-generation BTK inhibitors,” said Jane Huang, M.D., Chief Medical Officer, Hematology at BeiGene. “Our early BRUKINSA clinical trials started in Australia and coming off the heels of BRUKINSA’s TGA registration for the treatment of WM, we are delighted to be able to provide BRUKINSA to more Australians in need of new treatment options.”
More than 6,000 people are diagnosed with non-Hodgkin’s lymphoma (NHL) in Australia each year, making it the sixth most common cancer in adults.2 MCL is a B-cell NHL that develops in the outer edge of a lymph node called the mantle zone.3 MCL usually has a poor prognosis, with a median survival of three to six years, and is often diagnosed at a later stage of disease.3
The Australian registration for BRUKINSA in MCL is based on efficacy results from two single-arm clinical trials. Across both trials, as assessed by independent review committee (IRC) per 2014 Lugano Classification, BRUKINSA achieved an overall response rate (ORR) of 83.7%, defined as the combined rate of complete responses (CRs) and partial responses (PRs).
About BRUKINSA® (zanubrutinib)
BRUKINSA is a small molecule inhibitor of Bruton’s tyrosine kinase (BTK) discovered by BeiGene scientists that is currently being evaluated globally in a broad clinical program as a monotherapy and in combination with other therapies to treat various B-cell malignancies. Because new BTK is continuously synthesised, BRUKINSA was specifically designed to deliver complete and sustained inhibition of the BTK protein by optimising bioavailability, half-life, and selectivity. With differentiated pharmacokinetics compared to other approved BTK inhibitors, BRUKINSA has been demonstrated to inhibit the proliferation of malignant B cells within a number of disease relevant tissues.
BRUKINSA is approved in the United States, China, Australia, Canada, and other international markets in selected indications and under development for additional approvals globally.
To learn more about BeiGene, please visit www.beigene.com.au
View source version on businesswire.com: https://www.businesswire.com/news/home/20211010005045/en/
Source: BeiGene
Oct. 10, 2021 11:50 PM ET BeiGene, Ltd. (BGNE)
By: Mamta Mayani, SA News Editor
PUBLISHED11 October 2021
Positive high-level results from the TACKLE Phase III COVID-19 treatment trial showed AstraZeneca's AZD7442, a long acting antibody (LAAB) combination, achieved a statistically significant reduction in severe COVID-19 or death compared to placebo in non-hospitalised patients with mild-to-moderate symptomatic COVID-19.
A total of 90% of participants enrolled were from populations at high risk of progression to severe COVID-19, including those with co-morbidities.
The trial met the primary endpoint, with a dose of 600mg of AZD7442 given by intramuscular (IM) injection reducing the risk of developing severe COVID-19 or death (from any cause) by 50% compared to placebo in outpatients who had been symptomatic for seven days or less. The trial recorded 18 events in the AZD7442 arm (18/407) and 37 in the placebo arm (37/415). The LAAB was generally well tolerated in the trial.
In a prespecified analysis of participants who received treatment within five days of symptom onset, AZD7442 reduced the risk of developing severe COVID-19 or death (from any cause) by 67% compared to placebo, with nine events in the AZD7442 arm (9/253) and 27 in the placebo arm (27/251).
AZD7442 is the first LAAB with Phase III data to demonstrate benefit in both prophylaxis and treatment of COVID-19 and is easily administered by IM injection.
Hugh Montgomery, Professor of Intensive Care Medicine at University College London, and TACKLE principal investigator, said: “With continued cases of serious COVID-19 infections across the globe, there is a significant need for new therapies like AZD7442 that can be used to protect vulnerable populations from getting COVID-19 and can also help prevent progression to severe disease. These positive results show that a convenient intramuscular dose of AZD7442 could play an important role in helping combat this devastating pandemic.”
Mene Pangalos, Executive Vice President, BioPharmaceuticals R&D, AstraZeneca, said: “These important results for AZD7442, our long-acting antibody combination, add to the growing body of evidence for use of this therapy in both prevention and treatment of COVID-19. An early intervention with our antibody can give a significant reduction in progression to severe disease, with continued protection for more than six months.”
TACKLE included 903 participants in a 1:1 randomisation AZD7442 to placebo. The primary analysis was based on 822 participants.
AstraZeneca will be discussing the data with health authorities. On 5 October 2021, the Company announced that it had submitted a request to the US Food and Drug Administration for Emergency Use Authorisation for AZD7442 for prophylaxis of COVID-19.
Full results from TACKLE will be submitted for publication in a peer-reviewed medical journal and presented at a forthcoming medical meeting.
AZD7442
AZD7442 is a combination of two LAABs - tixagevimab (AZD8895) and cilgavimab (AZD1061) - derived from B-cells donated by convalescent patients after SARS-CoV-2 virus. Discovered by Vanderbilt University Medical Center and licensed to AstraZeneca in June 2020, the human monoclonal antibodies bind to distinct sites on the SARS-CoV-2 spike protein1 and were optimised by AstraZeneca with half-life extension and reduced Fc receptor and complement C1q binding. The half-life extension more than triples the durability of its action compared to conventional antibodies and could afford up to 12 months of protection from COVID-19 following a single administration2-4; data from the Phase I trial show high neutralising antibody titres for at least nine months.5 The reduced Fc receptor binding aims to minimise the risk of antibody-dependent enhancement of disease - a phenomenon in which virus-specific antibodies promote, rather than inhibit, infection and/or disease.6
In August 2021, AstraZeneca announced AZD7442 demonstrated a statistically significant reduction in the risk of developing symptomatic COVID-19 in the PROVENT Phase III pre-exposure prevention trial.
AZD7442 is also being studied as a potential treatment for hospitalised COVID-19 patients as part of the National Institute of Health’s ACTIV-3 trial and in an additional collaborator hospitalisation treatment trial.
AZD7442 is being developed with support from the US Government, including federal funds from the Department of Health and Human Services; Office of the Assistant Secretary for Preparedness and Response; Biomedical Advanced Research and Development Authority in partnership with the Department of Defense; Joint Program Executive Office for Chemical, Biological, Radiological and Nuclear Defense, under Contract No. W911QY-21-9-0001.
In preclinical experiments, data show the LAABs were able to block the binding of the SARS-CoV-2 virus to host cells and protect against infection in cell and animal models of disease.7 Additional in vitro findings demonstrate AZD7442 neutralises recent emergent SARS-CoV-2 viral variants, including the Delta and Mu variants.8
Under the terms of the licensing agreement with Vanderbilt, AstraZeneca will pay single-digit royalties on future net sales.
Please visit astrazeneca.com
Oct. 11, 2021 6:51 AM ET AstraZeneca PLC (AZN)
By: Mamta Mayani, SA News Editor
RETHYMIC is the first and only FDA-approved treatment indicated for immune reconstitution in pediatric patients with congenital athymia
Children with congenital athymia are born without a thymus causing severe immunodeficiency and immune dysregulation – with only supportive care they typically die by age two or three
RETHYMIC clinical trials included 105 patients, a data set encompassing 797 patient years of data and long-term survival to date up to 25.5 years
CAMBRIDGE, Mass. & BASEL, Switzerland, October 8, 2021 (GLOBE NEWSWIRE) – Enzyvant today announced the U.S. Food and Drug Administration (FDA) approval of RETHYMIC® (allogeneic processed thymus tissue-agdc), a one-time regenerative tissue-based therapy for immune reconstitution in pediatric patients with congenital athymia.
“For too long, families have faced a reality that the brutal journey for pediatric congenital athymia patients receiving supportive care only would end tragically. The FDA approval of RETHYMIC will help patients access this desperately needed therapy beyond clinical study,” said Rachelle Jacques, CEO of Enzyvant. “We are deeply grateful to the 105 patients who participated in clinical trials, their families, and all of the stakeholders who contributed to this pioneering regenerative medicine research program.”
Pediatric congenital athymia is ultra-rare with an estimated incidence of about 17 to 24 live births each year in the United States. Children who have this condition are born without a thymus and therefore have profound immunodeficiency, life-threatening immune dysregulation, and high susceptibility to potentially fatal infections. With only supportive care, children with congenital athymia typically die by age two or three.
“This therapy is the result of more than 25 years of research aimed at increasing survival for patients who previously had very little hope,” said Louise Markert, M.D, Ph.D., principal investigator for RETHYMIC clinical trials and Professor of Pediatrics and Immunology at the Duke University School of Medicine. “Our research program was inspired each and every day by the possibilities that exist for children who have congenital athymia with an FDA-approved treatment for this devastating condition.”
With this FDA approval, Enzyvant has obtained a Priority Review Voucher (PRV) under the Rare Pediatric Disease Program.
RETHYMIC Clinical Trial Data
Ten prospective single-arm, open-label studies with patient enrollment from 1993 to 2020 form the basis of the RETHYMIC data set. One hundred and five patients were surgically implanted with RETHYMIC under one of 10 Institutional Review Board (IRB)-approved protocols. Ninety-five patients were included in the Efficacy Analysis Set (EAS) and 105 patients were included in the Safety Analysis Set.
Survival rates were analyzed with the longest follow-up period of 25.5 years. In the EAS, Kaplan-Meier estimated survival rates (95% CI) were 77% (0.670–0.841) at one year and 76% (0.658–0.832) at two years. For patients who were alive at one year post implantation, the Kaplan-Meier estimated long-term survival rate was 94% at a median follow-up time of 10.7 years. For the patients in the clinical trials, naïve T-cell levels were measured using flow cytometry at six, 12, and 24 months after implantation with RETHYMIC. Patients in the clinical trials started out with very few naïve T cells but naïve CD4+ and CD8+ T cells began to reconstitute over the first year, with a durable increase through year two. Reductions in the number of infections over time during the first two years after treatment were statistically significant (p<0.001).
About RETHYMIC
RETHYMIC (allogeneic processed thymus tissue-agdc) is a novel one-time tissue-based regenerative therapy used for immune reconstitution in pediatric patients with congenital athymia. RETHYMIC is engineered human thymus tissue designed to regenerate the thymic function children with congenital athymia are missing and does not require donor-recipient matching. RETHYMIC has been studied across 10 clinical trials for more than 25 years and was granted multiple U.S. Food and Drug Administration (FDA) designations including Regenerative Medicine Advanced Therapy (RMAT), Breakthrough Therapy, Rare Pediatric Disease, and Orphan Drug. It also has been granted the Orphan Drug designation and the Advanced Therapy Medicinal Product (ATMP) designation by the European Medicines Agency (EMA). RETHYMIC is the first and only treatment approved by the FDA for immune reconstitution in pediatric patients with congenital athymia.
Please see full prescribing information.
Indication
RETHYMIC® (allogeneic processed thymus tissue–agdc) is indicated for immune reconstitution in pediatric patients with congenital athymia.
Limitations of Use:
RETHYMIC is not indicated for the treatment of patients with severe combined immunodeficiency (SCID).
For more information about Enzyvant, visit Enzyvant.com.
For more information, please visit https://www.sumitovant.com.
Oct. 11, 2021 4:50 AM ET Myovant Sciences Ltd. (MYOV)
By: Mamta Mayani, SA News Editor
Enzyvant announces the FDA approval of RETHYMIC (allogeneic processed thymus tissue-agdc), a one-time regenerative tissue-based therapy for immune reconstitution in pediatric patients with congenital athymia.
Broadest KRAS G12C Development Program Evaluating Multiple Combination Therapy Approaches
THOUSAND OAKS, Calif., Oct. 7, 2021 /PRNewswire/ -- Amgen (NASDAQ: AMGN) today announced new combination study results from the Phase 1b CodeBreaK 101 study, a comprehensive global master protocol trial evaluating the safety and efficacy of LUMAKRAS™ (sotorasib), the first and only approved KRASG12C inhibitor, in more than 10 different investigational combination regimens for the treatment of patients with KRAS G12C-mutated cancers. Results from two arms of the study — LUMAKRAS with afatinib, a pan-ErbB tyrosine kinase inhibitor, and LUMAKRAS with trametinib, a mitogen-activated protein kinase inhibitor (MEKi) — will be presented at the plenary session titled 'Drugging Difficult Targets' during the AACR-NCI-EORTC 2021 Virtual International Conference on Molecular Targets and Cancer Therapeutics on Saturday, Oct. 9, 2021.
"A critical component of cancer drug development is to interrogate multiple pathways to understand whether different combinations can meaningfully advance cancer care. For this reason, Amgen has undertaken the broadest and most comprehensive global clinical development program for patients with the KRAS G12C mutation, exploring multiple combinations that will allow us to understand where we can best serve patients," said David M. Reese, M.D., executive vice president of Research and Development at Amgen. "Consistent with our master protocol clinical trial design, which allows us to rapidly add, expand or remove cohorts to quickly understand what combinations work best for patients, Amgen will take these afatinib and trametinib results into account as we prioritize which combinations to move forward within our comprehensive LUMAKRAS development program. We look forward to presenting additional data, including PD-1 and SHP2 combination datasets, in the coming months."
LUMAKRAS in Combination with Afatinib (Abstract LBA6581)
The LUMAKRAS and afatinib combination arm enrolled 33 heavily pre-treated patients with KRAS G12C-mutated non-small cell lung cancer (NSCLC), including five patients previously treated with LUMAKRAS monotherapy. Ten patients received 20 mg of afatinib/960 mg of sotorasib (cohort 1; 4 patients with prior LUMAKRAS experience) and 23 patients received 30 mg of afatinib/960 mg of sotorasib (cohort 2; 1 patient with prior LUMAKRAS experience). The objective response rate (ORR) was 20% in cohort 1 and 35% in cohort 2, and the disease control rate was 70% and 74% in the two cohorts, respectively.
The most common treatment-related adverse events (TRAEs) for this study were diarrhea, nausea, and vomiting. TRAEs of grade 3 occurred in 30% of patients in both dose groups with diarrhea being the most common.
LUMAKRAS in Combination with Trametinib (Abstract LBA6580)
In CodeBreaK 101, the combination of LUMAKRAS and trametinib showed antitumor activity in heavily pre-treated patients with KRAS G12C-mutated solid tumors, including those with prior KRASG12C inhibitor treatment. A total of 41 patients were enrolled in the Phase 1b study with 18 patients with NSCLC, 18 patients with colorectal cancer (CRC) and five patients with other solid tumors. The maximum tolerated dose tested was 2 mg trametinib/960 mg sotorasib administered daily.
In patients with CRC who were KRASG12C inhibitor naïve, 9% achieved partial response (1 of 11), and 82% achieved disease control (9 of 11). In patients who were previously treated with a KRASG12C inhibitor, 14% achieved partial response (1 of 7), and 86% achieved disease control (6 of 7).
In patients with NSCLC who were KRASG12C inhibitor naïve, 20% achieved partial response (3 of 15) and 87% achieved disease control (13 of 15). In patients who were previously treated with a KRASG12C inhibitor, 67% achieved disease control (2 of 3).
The most common TRAEs for this study were diarrhea, rash, dermatitis acneiform, nausea and vomiting. No new safety concerns were identified.
About LUMAKRASTM (sotorasib)
Amgen took on one of the toughest challenges of the last 40 years in cancer research by developing LUMAKRAS, a KRASG12C inhibitor.1 LUMAKRAS has demonstrated a positive benefit-risk profile with rapid, deep and durable anticancer activity in patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) harboring the KRAS G12C mutation with a once daily oral formulation.2
In May 2021, LUMAKRAS was the first KRASG12C inhibitor to receive regulatory approval anywhere in the world with its approval in the U.S., under accelerated approval. LUMAKRAS is also approved in the United Arab Emirates, and in Canada and Great Britain under Project Orbis.
Amgen is progressing the largest and broadest global KRASG12C inhibitor development program with unparalleled speed and exploring more than 10 sotorasib combination regimens, including triplets, with clinical trial sites spanning five continents. To date, LUMAKRAS has treated over 3,000 patients around the world through the clinical development program and commercial use.
In the U.S., LUMAKRAS was reviewed by the FDA under its Real-Time Oncology Review (RTOR), a pilot program that aims to explore a more efficient review process that ensures safe and effective treatments are made available to patients as early as possible. Amgen is participating in the FDA's Project Orbis initiative and through the initiative, has Marketing Authorization Applications (MAAs) for sotorasib in review in Australia and Brazil. Additionally, Amgen has submitted an MAA in the European Union, Japan, Switzerland, South Korea, Singapore, Israel, Turkey, Taiwan, Colombia, Thailand, Mexico and Hong Kong.
LUMAKRAS is also being studied in multiple other solid tumors.1
For information, please visit www.hcp.codebreaktrials.com.
LUMAKRASTM (sotorasib) U.S. Indication
LUMAKRASTM is indicated for the treatment of adult patients with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.
This indication is approved under accelerated approval based on overall response rate (ORR) and duration of response (DOR). Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
Please see LUMAKRASTM full Prescribing Information.
For more information, visit www.amgen.com
View original content to download multimedia:https://www.prnewswire.com/news-releases/amgen-announces-new-lumakras-sotorasib-combination-data-from-phase-1b-codebreak-101-study-in-patients-with-kras-g12c-mutated-cancers-at-aacr-nci-eortc-2021-301394818.html
SOURCE Amgen
Oct. 07, 2021 10:13 AM ET Amgen Inc. (AMGN) By: Jonathan M Block, SA News Editor
Application utilizes extrapolation-based strategy across existing breadth of STELARA data in patients living with this chronic inflammatory disease
![]()
NEWS PROVIDED BY
Janssen Pharmaceutical Companies of Johnson & Johnson
Oct 08, 2021, 08:04 ET
HORSHAM, Pa., Oct. 8, 2021 /PRNewswire/ -- The Janssen Pharmaceutical Companies of Johnson & Johnson today announced the submission of a supplemental Biologics License Application (sBLA) to the U.S. Food and Drug Administration (FDA) seeking expanded approval of STELARA® (ustekinumab) to treat pediatric patients ages 5 years and older with juvenile psoriatic arthritis (jPsA).
The filing is supported by extrapolation of data from nine studies across both adult trials in active PsA and adult and pediatric studies in moderate to severe plaque psoriasis, totaling 3,997 patients evaluated across these closely associated diseases. Data extrapolation is the process of estimating response, trends or effects based on previous observations from patients with closely related conditions. With the limited availability of pediatric patients for clinical trial inclusion, researchers can extrapolate data from trials with adults to determine the potential efficacy and tolerability of a treatment for a pediatric population. A decision from the U.S. FDA is anticipated in late 2022.
"As children and their families manage the debilitating symptoms of juvenile psoriatic arthritis, it is critical that their physicians have a breadth of treatment options to consider," said Alyssa Johnsen, M.D., Ph.D., Vice President, Rheumatology Disease Area Leader, Janssen Research & Development, LLC. "With this latest submission, we're excited to work with the U.S. FDA to evaluate this potential therapeutic option that could help meet the needs of children living with psoriatic arthritis."
STELARA is the first and only biologic targeting both cytokines interleukin (IL)-12 and IL-23, both of which play an important role in inflammation associated with immune-mediated diseases like PsA. Since receiving approval in September 2009 for the treatment of adults living with moderate to severe plaque psoriasis, STELARA has received approval for four additional indications: children (ages 6 and older) with moderate to severe plaque psoriasis, adults with active PsA, adults with moderately to severely active Crohn's disease and adults with moderately to severely active ulcerative colitis.
About STELARA® (ustekinumab)
STELARA (ustekinumab), a human IL-12 and IL-23 antagonist, is approved in the United States for the treatment of: 1) adults and children 6 years and older with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy; 2) adult patients (18 years or older) with active PsA, used alone or in combination with methotrexate; 3) adult patients (18 years and older) with moderately to severely active Crohn's disease; 4) adult patients (18 years and older) with moderately to severely active ulcerative colitis.
Please read the full Prescribing Information and Medication Guide for STELARA® and discuss any questions you have with your doctor.
Learn more at www.janssen.com.
Oct. 08, 2021 8:28 AM ET
By: Jonathan M Block, SA News Editor2 Comments
− If approved by the FDA, maribavir will be the first and only treatment indicated for adults in this patient population
− CMV is one of the most common infections experienced by transplant recipients, with an estimated incidence rate of around 16–56% in solid organ transplant (SOT) recipients and 30–70% in HSCT recipients3,7
− Regulatory submission is based on the Phase 2 and Phase 3 TAK-620-303 (SOLSTICE) trial of maribavir
− Maribavir is one of four Wave 1 pipeline new molecular entities that Takeda has submitted for regulatory review to date
October 07, 2021 06:15 PM Eastern Daylight Time
OSAKA, Japan & CAMBRIDGE, Mass.--(BUSINESS WIRE)--Takeda Pharmaceutical Company Limited (TSE:4502/NYSE:TAK) (“Takeda”) today announced the U.S. Food and Drug Administration (FDA) Antimicrobial Drugs Advisory Committee (AMDAC) voted unanimously to recommend use of maribavir (TAK-620) for the treatment of refractory cytomegalovirus (CMV) infection and disease with genotypic resistance to ganciclovir, valganciclovir, foscarnet or cidofovir in transplant recipients. The committee also voted unanimously to recommend use of maribavir for the treatment of refractory CMV infection and disease without genotypic resistance to ganciclovir, valganciclovir, foscarnet or cidofovir in transplant recipients. Both recommendations were based on the results of the Phase 2 and Phase 3 TAK-620-303 (SOLSTICE) trials.
About maribavir
Maribavir, an orally bioavailable anti-CMV compound, is the only antiviral agent presently in Phase 3 development for the treatment of adult post-transplant patients with CMV in SOT or HSCT. Maribavir is an investigational treatment that has not been approved for use by the U.S. Food and Drug Administration (FDA), European Medicines Agency (EMA), or any other regulatory authorities. Maribavir is the only CMV antiviral drug that targets and inhibits the UL97 protein kinase and its natural substrates.14–17
Maribavir has been granted Orphan Drug Designation by the European Commission as a treatment of CMV disease in patients with impaired cell mediated immunity and by the FDA for treatment of clinically significant CMV viremia and disease in at-risk patients. Orphan status is granted to certain investigational medicines intended for the treatment or prevention of a rare, life-threatening disease. The FDA has also granted maribavir Breakthrough Therapy Designation as a treatment for CMV infection and disease in transplant patients resistant or refractory to prior therapy. Breakthrough Therapy Designation expedites the development and review of investigational treatments for serious conditions with preliminary clinical evidence indicating that the drug may demonstrate substantial improvement over available therapy. Most recently, the FDA has granted priority review of maribavir for the treatment of adult post-transplant recipients with CMV infection in those resistant and/or refractory to prior anti-CMV treatment. These designations and NDA acceptance do not guarantee that the EMA or FDA will approve maribavir for the treatment of CMV infections in adult transplant patients, and the timing of any such approval is uncertain.
For more information, visit https://www.takeda.com.
Takeda Pharmaceutical Company Limited (TSE: 4502/NYSE: TAK)
Oct. 08, 2021 5:23 AM ET
Takeda Pharmaceutical Company Limited (TAK)
By: Mamta Mayani, SA News Editor1 Comment
Oct 07, 2021 4:05 PM
Represents BRUKINSA’s second recent approval in the Asia-Pacific region, following October 1 approval in Singapore for treatment of patients with mantle cell lymphoma
The TGA approval is based on results from ASPEN, an Australia-inclusive head-to-head clinical trial evaluating BRUKINSA compared to ibrutinib in patients with Waldenström’s macroglobulinemia
SYDNEY & CAMBRIDGE, Mass. & BEIJING--(BUSINESS WIRE)-- BeiGene (NASDAQ: BGNE; HKEX: 06160), a global, science-driven biotechnology company focused on developing innovative and affordable medicines to improve treatment outcomes and access for patients worldwide, today announced that BRUKINSA® (zanubrutinib) has been approved in Australia for the treatment of adult patients with Waldenström’s macroglobulinemia (WM) who have received at least one prior therapy or in first line treatment for patients unsuitable for chemo-immunotherapy.1 Following registration of BRUKINSA with the Therapeutic Goods Administration (TGA), these patients will have immediate access to BRUKINSA through a BeiGene sponsored post-approval, pre-reimbursement access program.
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20211007005999/en/
(Photo: Business Wire)
In addition, BRUKINSA recently received approval from the Singapore Health Sciences Authority (HSA) for the treatment of adult patients with mantle cell lymphoma (MCL) who have received at least one prior therapy.
“BTK inhibition is an established mode of treatment for patients with WM, and the ASPEN trial showed that BRUKINSA is highly effective and has improved tolerability compared to the first-generation BTK inhibitor,” said Professor Con Tam, MBBS, M.D., Disease Group Lead for Low Grade Lymphoma and Chronic Lymphocytic Leukemia at the Peter MacCallum Cancer Centre and a principal investigator on the BRUKINSA clinical program. “BeiGene first began clinical trials of BRUKINSA in Australia in 2013, and since that time, many Australians have benefitted from treatment as part of ongoing clinical studies. We hope this therapy will offer new hope for people living with WM in Australia.”
In Australia, more than 6,000 people are diagnosed with non-Hodgkin’s lymphoma (NHL) each year, making it the sixth most common cancer in adults.2 WM is a rare, slow-growing lymphoma that occurs in less than two percent of patients with NHL.3 The disease usually affects older adults and is primarily found in the bone marrow, although it may also impact lymph nodes and the spleen.3
“While WM is a slow-growing lymphoma, not all patients fully respond to existing therapies and many discontinue treatment due to side effects,” commented David Young, the National Team Leader at the WMozzies. “We are pleased to hear that people living with WM in Australia will have immediate access to this next-generation BTK inhibitor that has demonstrated clinical benefit with potential to improve treatment outcomes.”
BeiGene has submitted for reimbursement of WM to the Pharmaceutical Benefits Advisory Committee (PBAC). In a first for the PBAC, BeiGene expects to enter a facilitated resolution pathway in order to seek a listing date for the WM indication.
“BRUKINSA has been shown to induce deep and durable responses with reduced off-target side effects, suggesting improved clinical benefit compared to standard BTK inhibitor therapy,” said Jane Huang, M.D., Chief Medical Officer, Hematology at BeiGene. “We are grateful to the Australian investigators, patients and families who participated in clinical trials contributing to TGA approval. Our ability to offer BRUKINSA to people in Australia impacted by WM is another step toward fulfilling our goal of increasing affordable access to oncology medicines around the world.”
“This approval in Australia, and our recent approval in Singapore, represent BRUKINSA’s continued expansion in the APAC region,” added Adam Roach, Vice President and Head of Commercial for APAC (ex-Greater China) at BeiGene. “We have been building commercial teams in these markets to support our goal of bringing this potential best-in-class BTK inhibitor to patients who need them globally.”
The Australian registration for BRUKINSA in WM is based on efficacy results from the ASPEN clinical trial, a Phase 3 randomised, open-label, multicentre trial (NCT03053440) that evaluated BRUKINSA compared to ibrutinib in patients with relapsed or refractory (R/R) or treatment-naïve (TN) WM who harbor a MYD88 mutation (MYD88MUT). In the ASPEN trial, BRUKINSA demonstrated a numerically higher very good partial response (VGPR) rate (28.4%, 95% CI: 20, 38) compared to ibrutinib (19.2%, 95% CI: 12, 28), although the primary endpoint of statistical superiority related to deep response (VGPR or better) was not met.
In the ASPEN trial, of the 101 patients with WM randomized and treated with BRUKINSA, 5% of patients discontinued due to adverse events, including cardiomegaly, neutropenia, plasma cell myeloma, and subdural haemorrhage. Adverse events leading to dose reduction occurred in 14.9% of patients, with the most common being neutropenia (3.0%) and diarrhea (2.0%).
The recommended dose of BRUKINSA is either 160 mg twice daily or 320 mg once daily, taken orally with or without food. The dose may be adjusted for adverse reactions and reduced for patients with severe hepatic impairment and certain drug interactions.
About BRUKINSA® (zanubrutinib)
BRUKINSA is a small molecule inhibitor of Bruton’s tyrosine kinase (BTK) discovered by BeiGene scientists that is currently being evaluated globally in a broad clinical program as a monotherapy and in combination with other therapies to treat various B-cell malignancies. Because new BTK is continuously synthesised, BRUKINSA was specifically designed to deliver complete and sustained inhibition of the BTK protein by optimising bioavailability, half-life, and selectivity. With differentiated pharmacokinetics compared to other approved BTK inhibitors, BRUKINSA has been demonstrated to inhibit the proliferation of malignant B cells within a number of disease relevant tissues.
BRUKINSA is approved in the United States, China, Australia, Canada, and other international markets in selected indications and under development for additional approvals globally.
To learn more about BeiGene, please visit www.beigene.com.au
View source version on businesswire.com: https://www.businesswire.com/news/home/20211007005999/en/
Source: BeiGene
Oct. 07, 2021 4:13 PM ET BGNE By: Manshi Mamtora, CFA
Oct 07, 2021PDF Version
NEW YORK, Oct. 07, 2021 (GLOBE NEWSWIRE) -- Y-mAbs Therapeutics, Inc. (“Y-mAbs” or the “Company”) (NASDAQ: YMAB), a commercial-stage biopharmaceutical company focused on the development and commercialization of novel, antibody-based therapeutic products for the treatment of cancer, today announced that the U.S. Food and Drug Administration (“FDA”) has granted Rare Pediatric Disease Designation (“RPDD”) for the Company’s lutetium labelled omburtamab antibody program for the treatment of medulloblastoma.
177Lu-omburtamab-DTPA, a monoclonal B7-H3 antibody that has been radiolabeled with lutetium-177, is currently in a multicenter Phase 1 clinical trial in pediatric patients with refractory medulloblastoma, and in a multicenter Phase 1 clinical trial targeting B7-H3 positive CNS/LM tumors in adults. We believe that both indications address clear unmet medical needs.
“The RPDD makes us eligible for a Priority Review Voucher (“PRV”) upon potential approval of the biologics license application for this rare pediatric cancer. Among our leading compounds under development, four now have RPDDs, and this designation for 177Lu-omburtamab-DTPA further increase our chances of ultimately receiving multiple PRVs,” said Thomas Gad, founder, Chairman and President.
Dr. Claus Moller, Chief Executive Officer, further notes, “We are dedicated to bring 177Lu-omburtamab-DTPA to patients who desperately need alternative methods of treatment. We are very pleased by this recognition by the FDA and look forward to expanding the ongoing Phase 1 studies with 177Lu-omburtamab-DTPA into separate Phase 2 arms.”
Researchers at Memorial Sloan Kettering Cancer Center (“MSK”) developed omburtamab, which is exclusively licensed by MSK to Y-mAbs. As a result of this licensing arrangement, MSK has institutional financial interests in the product.
“DANYELZA” and “Y-mAbs” are registered trademarks of Y-mAbs Therapeutics, Inc.
Oct. 07, 2021 9:47 AM ETY-mAbs Therapeutics, Inc. (YMAB)By: Ravikash, SA News Editor
PUBLISHED4 October 2021
The Food and Drug Administration (FDA) has granted Enhertu (trastuzumab deruxtecan), Breakthrough Therapy Designation (BTD) in the US for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received one or more prior anti-HER2-based regimens. Enhertu is a HER2-directed antibody drug conjugate (ADC) jointly developed by AstraZeneca and Daiichi Sankyo Company, Limited (hereafter, Daiichi Sankyo).
The FDA granted BTD based on data from the DESTINY-Breast03 Phase III trial presented during the European Society for Medical Oncology (ESMO) Congress 2021. This is the second BTD for Enhertu in breast cancer and now brings the total number of BTDs to four for this medicine.
The US FDA’s BTD is designed to accelerate the development and regulatory review of potential new medicines that are intended to treat a serious condition and address a significant unmet medical need. The new medicine needs to have shown encouraging preliminary clinical results that demonstrate substantial improvement on a clinically significant endpoint over available medicines.
Breast cancer remains the most common cancer worldwide, with more than two million cases diagnosed in 2020, resulting in nearly 685,000 deaths globally.1 Approximately one in five cases of breast cancer are considered HER2-positive.2
Despite initial treatment with trastuzumab and a taxane, patients with HER2-positive metastatic breast cancer will often experience disease progression.3 More effective options are needed to further delay progression and extend survival.3-5
Susan Galbraith, Executive Vice President, Oncology R&D, AstraZeneca, said: “This is an important step in bringing Enhertu as a potential new option in earlier lines of treatment for HER2-positive metastatic breast cancer, given the urgent need to improve outcomes. This recognition by the FDA underscores the transformative possibility of Enhertu seen with the remarkable DESTINY-Breast03 results presented at ESMO just two weeks ago.”
Ken Takeshita, Global Head, R&D, Daiichi Sankyo, said: “By granting a fourth Breakthrough Therapy Designation to Enhertu, the FDA continues to recognise the significant potential of this medicine across multiple HER2-targetable tumours. With the unprecedented data recently reported from the DESTINY-Breast03 trial, we look forward to working closely with the FDA to bring Enhertu to patients who have been previously treated for HER2-positive metastatic breast cancer as soon as possible.”
In DESTINY-Breast03, Enhertu demonstrated a 72% reduction in the risk of disease progression or death compared to T-DM1 (hazard ratio [HR] 0.28; 95% confidence interval [CI] 0.22-0.37; p=7.8x10-22) in patients with HER2-positive unresectable and/or metastatic breast cancer previously treated with trastuzumab and a taxane. Nearly all patients treated with Enhertu were alive at one year (94.1%) compared to 85.9% of patients treated with T-DM1. Confirmed objective response rate (ORR) more than doubled in the Enhertu arm versus the T-DM1 arm (79.7% vs. 34.2%). The safety profile of Enhertu was consistent with previous clinical trials, with no new safety concerns identified and no Grade 4 or 5 treatment-related interstitial lung disease events.
Previous BTDs for Enhertu were in late-line HER2-positive metastatic breast cancer in 2017 and HER2-mutant metastatic non-small cell lung cancer (NSCLC) and HER2-positive metastatic gastric cancer in 2020.
Enhertu is approved for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received two or more prior anti-HER2-based regimens in the metastatic setting in the US, Japan, the EU and several other countries based on the results from the DESTINY-Breast01 trial.
Enhertu is being further assessed in a comprehensive clinical development programme evaluating efficacy and safety across multiple HER2-targetable cancers, including breast, gastric, lung and colorectal cancers.
DESTINY-Breast03
DESTINY-Breast03 is a global head-to-head, randomised, open-label, registrational Phase III trial evaluating the safety and efficacy of Enhertu (5.4mg/kg) versus T-DM1 in patients with HER2-positive unresectable and/or metastatic breast cancer previously treated with trastuzumab and a taxane.
The primary efficacy endpoint of DESTINY-Breast03 is progression-free survival (PFS) based on blinded independent central review. Secondary efficacy endpoints include overall survival, objective response rate, duration of response, PFS based on investigator assessment and safety.
DESTINY-Breast03 enrolled approximately 500 patients at multiple sites in Asia, Europe, North America, Oceania and South America. For more information about the trial, visit ClinicalTrials.gov.
Enhertu
Enhertu is a HER2-directed ADC. Designed using Daiichi Sankyo’s proprietary DXd ADC technology, Enhertu is the lead ADC in the oncology portfolio of Daiichi Sankyo and the most advanced programme in AstraZeneca’s ADC scientific platform. Enhertu consists of a HER2 monoclonal antibody attached to a topoisomerase I inhibitor payload, an exatecan derivative, via a stable tetrapeptide-based cleavable linker.
Enhertu (5.4mg/kg) is approved in Canada, the EU, Israel, Japan, the UK and the US for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have received two or more prior anti-HER2-based regimens in the metastatic setting based on the results from the DESTINY-Breast01 trial.
Enhertu (6.4mg/kg) is also approved in Israel, Japan and the US for the treatment of adult patients with locally advanced or metastatic HER2-positive gastric or gastroesophageal junction adenocarcinoma who have received a prior trastuzumab-based regimen based on the results from the DESTINY-Gastric01 trial.
Daiichi Sankyo collaboration
Daiichi Sankyo and AstraZeneca entered into a global collaboration to jointly develop and commercialise Enhertu (a HER2-directed ADC) in March 2019, and datopotamab deruxtecan (DS-1062; a TROP2-directed ADC) in July 2020, except in Japan where Daiichi Sankyo maintains exclusive rights. Daiichi Sankyo is responsible for manufacturing and supply of Enhertu and datopotamab deruxtecan.
Please visit astrazeneca.com
Oct. 04, 2021 7:19 AM ET AstraZeneca PLC (AZN), DSKYFDSNKY By: Mamta Mayani, SA News Editor1 Comment
October 7, 2021
- In Study 1 of the Phase 3 SELECT-AXIS 2 clinical trial, RINVOQ® (upadacitinib) met the primary endpoint of ASAS40 at week 14 versus placebo (45 percent compared to 18 percent) in patients with ankylosing spondylitis and inadequate response to bDMARD therapy[1]
- All ranked secondary endpoints were met[1]
- Safety data were consistent with SELECT-AXIS 1, previous Phase 3 studies in other indications and the known safety profile of RINVOQ, with no new risks identified[1-6]
- RINVOQ, a selective and reversible JAK inhibitor discovered and developed by AbbVie, is an oral therapy approved in the EU for the treatment of active ankylosing spondylitis[6]
NORTH CHICAGO, Ill., Oct. 7, 2021 /PRNewswire/ -- AbbVie (NYSE: ABBV) today announced positive top-line results from the first of two studies of the Phase 3 SELECT-AXIS 2 clinical trial evaluating the efficacy and safety of RINVOQ® (upadacitinib; 15 mg, once daily) in patients with active ankylosing spondylitis (AS) who had an inadequate response to biologic DMARD therapy. In this study, RINVOQ met its primary endpoint of Assessment in SpondyloArthritis International Society (ASAS) 40 response and all ranked secondary endpoints at week 14.1 Significantly more RINVOQ-treated patients achieved ASAS40 response at week 14 compared to placebo (45 percent versus 18 percent; p<0.0001).1
The results of SELECT-AXIS 1, a Phase 2/3 study in adult patients with ankylosing spondylitis who were naïve to bDMARDs and had an inadequate response or intolerance to nonsteroidal anti-inflammatory drugs (NSAIDs), were used to support the European Commission approval of RINVOQ for the treatment of active ankylosing spondylitis in January 2021.6
AbbVie will also announce the positive results of the second study of SELECT-AXIS 2 in adults with non-radiographic axial spondyloarthritis (nr-axSpA) later today.1
"Ankylosing spondylitis is a debilitating disease that can cause severe pain, stiffness, restricted mobility and lasting structural damage impacting patients' everyday life," said Michael Severino, M.D., vice chairman and president, AbbVie. "AbbVie is committed to improving standards of care for patients with rheumatic diseases. We are encouraged by these results that show RINVOQ was able to provide significant improvements in signs and symptoms, as well as other measures of disease activity, for patients living with ankylosing spondylitis who have already failed treatment with a biologic."
Treatment with RINVOQ resulted in statistically significant reductions in signs and symptoms of AS, including back pain and inflammation, as well as improvements in physical function and disease activity at week 14.1 Significantly more patients treated with RINVOQ achieved Ankylosing Spondylitis Disease Activity Score (ASDAS) Low Disease Activity compared to those treated with placebo (44 percent versus 10 percent).1 A statistically significantly greater improvement in Magnetic Resonance Imaging (MRI) Spondyloarthritis Research Consortium of Canada (SPARCC) Score (Spine) as measured by mean change from baseline was reported in the RINVOQ group versus the placebo group (-3.95 versus -0.04).1 Patients on RINVOQ experienced a significantly greater mean decrease from baseline in Patient's Assessment of Total Back Pain at week 14 than those on placebo (-3.00 versus -1.47).1 Additionally, patients treated with RINVOQ experienced significantly greater improvement in physical function as assessed by mean change from baseline in Bath Ankylosing Spondylitis Functional Index (BASFI) compared to patients on placebo (-2.26 versus -1.09).1 All ranked secondary endpoints achieved p-values of <0.0001 versus placebo.1
About SELECT-AXIS 2 – Study 11,7
SELECT-AXIS 2 was conducted as a master study protocol that contains two standalone studies with randomization, data collection, analysis and reporting conducted independently. The Phase 3, randomized, placebo-controlled, double-blind studies are evaluating the efficacy and safety of RINVOQ compared with placebo on reduction of signs and symptoms in adult participants with active axSpA including bDMARD-IR AS (Study 1) and nr-axSpA (Study 2). Study 1 enrolled 420 who were randomized to receive RINVOQ for 104 weeks or placebo for 14 weeks followed by RINVOQ for 90 weeks. More information on this trial can be found at www.clinicaltrials.gov (NCT04169373).
About Axial Spondyloarthritis (axSpA)
Axial spondyloarthritis is a chronic inflammatory disease that affects the spine, causing back pain, limited mobility, and structural damage.8 It consists of two subsets that have been clinically defined as ankylosing spondylitis (AS) and non-radiographic axial spondyloarthritis (nr-axSpA).8 In ankylosing spondylitis, patients have definitive structural damage of the sacroiliac joints visible on x-rays.8 Non-radiographic axial spondyloarthritis is clinically defined by the absence of definitive x-ray evidence of structural damage to the sacroiliac (SI) joint by plain x-ray.8
About RINVOQ® (upadacitinib)
Discovered and developed by AbbVie scientists, RINVOQ is a selective and reversible JAK inhibitor that is being studied in several immune-mediated inflammatory diseases.1,6,7,9-15 In human cellular assays, RINVOQ preferentially inhibits signaling by JAK1 or JAK1/3 with functional selectivity over cytokine receptors that signal via pairs of JAK2.6 RINVOQ is approved by the European Commission for adults (15 mg and 30 mg) and adolescents (15 mg) with moderate to severe atopic dermatitis. RINVOQ 15 mg is approved by the European Commission for adults with moderate to severe active rheumatoid arthritis, adults with active psoriatic arthritis and adults with active ankylosing spondylitis. RINVOQ 15 mg is also approved by the U.S. Food and Drug Administration (FDA) for adults with moderately to severely active rheumatoid arthritis. Phase 3 trials of RINVOQ in rheumatoid arthritis, atopic dermatitis, psoriatic arthritis, axial spondyloarthritis, Crohn's disease, ulcerative colitis, giant cell arteritis and Takayasu arteritis are ongoing.9-15 Use of RINVOQ in non-radiographic axial spondyloarthritis is not approved and its safety and efficacy have not been established by regulatory authorities.
EU Indications and Important Safety Information About RINVOQ® (upadacitinib)6
Rheumatoid arthritis
RINVOQ is indicated for the treatment of moderate to severe active rheumatoid arthritis in adult patients who have responded inadequately to, or who are intolerant to one or more disease-modifying anti-rheumatic drugs (DMARDs). RINVOQ may be used as monotherapy or in combination with methotrexate.
Psoriatic arthritis
RINVOQ is indicated for the treatment of active psoriatic arthritis in adult patients who have responded inadequately to, or who are intolerant to one or more DMARDs. RINVOQ may be used as monotherapy or in combination with methotrexate.
Ankylosing spondylitis
RINVOQ is indicated for the treatment of active ankylosing spondylitis in adult patients who have responded inadequately to conventional therapy.
Atopic dermatitis
RINVOQ is indicated for the treatment of moderate to severe atopic dermatitis in adults and adolescents 12 years and older who are candidates for systemic therapy.
Contraindications
RINVOQ is contraindicated in patients hypersensitive to the active substance or to any of the excipients, in patients with active tuberculosis (TB) or active serious infections, in patients with severe hepatic impairment, and during pregnancy.
See RINVOQ full summary of product characteristics (SmPC) at www.ema.europa.eu. Globally, prescribing information varies; refer to the individual country product label for complete information
For more information about AbbVie, please visit us at www.abbvie.com.
https://seekingalpha.com/symbol/ABBV
https://www.nasdaq.com/market-activity/stocks/abbv/dividend-history
Issued: 6 October, London UK
GlaxoSmithKline (GSK) plc welcomes and applauds the WHO recommendation for the broader deployment of GSK’s RTS,S malaria vaccine to reduce childhood illness and deaths from malaria in children living in sub-Saharan Africa and other regions with moderate to high transmission as defined by WHO. RTS,S is the first and only malaria vaccine to have been shown in pivotal long-term clinical trials to significantly reduce malaria in children. The vaccine is the result of over 30 years of research led by GSK, with PATH and other partners.
Thomas Breuer, Chief Global Health Officer, GSK, said: “GSK is proud that RTS,S, our ground-breaking malaria vaccine, developed over decades by our teams and partners, can now be made available to children in sub-Saharan Africa and other regions with moderate to high malaria transmission. This long-awaited landmark decision can reinvigorate the fight against malaria in the region at a time when progress on malaria control has stalled. Both real world evidence and clinical trial data show that RTS,S, alongside other malaria prevention measures, has the potential to save hundreds of thousands of lives.”
In anticipation of the decision and wider roll-out beyond the pilot programmes in Malawi, Kenya and Ghana, GSK is working with partners to develop solutions to ensure equitable and long-term access to the RTS,S vaccine for the people who need it. GSK has committed to donate up to 10 million RTS,S doses for use in the pilots, and to supply up to 15 million doses annually, following a recommendation and funding for wider use. A Product Transfer, including technology transfer for long-term antigen production, is also underway with Bharat Biotech of India. GSK will now work closely with partners, funders and governments to support additional supply of the vaccine, and has committed to make the 15 million annual doses available at no more than 5% above cost of production.
This recommendation from WHO, informed by data generated from the pilot programme, is a second key milestone for the RTS,S malaria vaccine in recent weeks. In August, data from a study of 6,000 children by the London School Hygiene and Tropical Medicine, published in the New England Journal of Medicine, showed that after three years the combination of seasonal administration of antimalarials (known as Seasonal Malaria Chemoprevention/SMC) and RTS,S vaccination lowered clinical episodes of malaria, hospital admissions with WHO-defined severe malaria, and deaths from malaria by about 70% compared to SMC alone.[1]These data indicate that the impact of RTS,S vaccination can be increased to further reduce mortality, especially when combined with other recommended malaria control interventions in a seasonal setting.
Since the launch of the malaria vaccine pilots in 2019, 3 countries (Ghana, Kenya and Malawi) have led the introduction of the vaccine in selected areas of moderate to high malaria transmission, reaching more than 800,000 children with at least 1 dose of the vaccine. More than 2.3 million vaccine doses have been administered to date. Community demand for the vaccine is strong and evidence shows it can effectively be delivered through the routine child immunization platform.
For further information please visit www.gsk.com/about-us.
Oct. 06, 2021 12:11 PM ET GlaxoSmithKline plc (GSK)
By: Dulan Lokuwithana, SA News Editor1 Comment
Issued: 4 October, London UK
ViiV Healthcare, the global specialist HIV company majority-owned by GSK, with Pfizer Inc. and Shionogi Limited as shareholders, has announced it has made a regulatory submission to the U.S. Food and Drug Administration (FDA) for approval of a new dispersible tablet formulation of the fixed dose combination of abacavir, dolutegravir and lamivudine and, to extend its current approval for Triumeq (abacavir/ dolutegravir/ lamivudine) to lower the minimum weight at which a child can be prescribed this medicine, from 40kg and above to 14kg and above. If approved, this approval will result in further treatment options for younger children living with HIV.
Paediatric HIV remains a global issue, with children disproportionately affected by the HIV epidemic. Latest statistics show there are 1.7 million children living with HIV globally,[1] with most AIDS-related deaths among this group occurring during the first five years of life.[2]
Major obstacles persist for children, such as the availability of HIV testing, continued vertical transmission, slow initiation of treatment and, poor availability of optimised paediatric formulations of antiretrovirals (ARVs).[2],[3] The availability of age-appropriate treatment options is essential in ensuring children around the world can access optimal care.
About Triumeq
Two essential steps in the HIV life cycle are replication – when the virus turns its RNA copy into DNA – and integration – the moment when viral DNA becomes part of the host cell’s DNA. These processes require two enzymes called reverse transcriptase and integrase. NRTIs and integrase inhibitors interfere with the action of the two enzymes to prevent the virus from replicating and further infecting cells.
Trademarks are owned by or licensed to the ViiV Healthcare group of companies.
Important Safety Information (ISI) for TRIUMEQ (abacavir, dolutegravir, and lamivudine) tablets
INDICATION
TRIUMEQ is indicated for the treatment of human immunodeficiency virus type 1 (HIV-1) infection in adults and in pediatric patients weighing at least 40 kg.
Limitations of Use:
TRIUMEQ alone is not recommended in patients with resistance-associated integrase substitutions or clinically suspected INSTI resistance because the dose of dolutegravir in TRIUMEQ is insufficient in these subpopulations. See full prescribing information for TIVICAY (dolutegravir).
Please see full prescribing information available at: US Prescribing Information, including Boxed Warning.
About ViiV Healthcare
ViiV Healthcare is a global specialist HIV company established in November 2009 by GlaxoSmithKline (LSE: GSK) and Pfizer (NYSE: PFE) dedicated to delivering advances in treatment and care for people living with HIV and for people who are at risk of becoming infected with HIV. Shionogi joined as shareholders in October 2012. The company’s aims are to take a deeper and broader interest in HIV and AIDS than any company has done before and take a new approach to deliver effective and innovative medicines for HIV treatment and prevention, as well as support communities affected by HIV.
For more information on the company, its management, portfolio, pipeline, and commitment, please visit www.viivhealthcare.com.
About GSK
GSK is a science-led global healthcare company. For further information please visit https://www.gsk.com/en-gb/about-us/.
Oct. 04, 2021 3:37 PM ET
PFE, SGIOYBy: Jonathan M Block, SA News Editor
The Study Met its Primary Objective of Demonstrating a Statistically Greater Improvement From Baseline in Eczema Area and Severity Index Score at 16 Weeks Versus Placebo
Patients Receiving AMG 451/KHK4083 Showed Statistically Greater Improvements in Additional Secondary Efficacy Endpoints Versus Placebo
AMG 451/KHK4083 Also Showed Progressive Improvement in Efficacy Beyond 16 Weeks
THOUSAND OAKS, Calif. and TOKYO, Oct. 2, 2021 /PRNewswire/ -- Amgen (NASDAQ: AMGN) and Kyowa Kirin Co., Ltd. (TSE:4151) today announced that positive data from a Phase 2 study of AMG 451/KHK4083 were presented at the European Academy of Dermatology and Venereology 30th Virtual Congress on Oct. 2, 2021. AMG 451/KHK4083 is a potential first-in-class anti-OX40 fully human monoclonal antibody in development for the treatment of moderate-to-severe atopic dermatitis.
The Phase 2, multicenter, randomized, double-blind, placebo-controlled trial investigated the efficacy and safety of AMG 451/KHK4083 in adults with moderate-to-severe atopic dermatitis who were not adequately controlled with topical agents. The study met the primary objective, showing statistically greater improvements from baseline in Eczema Area and Severity Index (EASI) score at 16 weeks with all four subcutaneous doses of AMG 451/KHK4083 compared with placebo (600 mg every two weeks (Q2W) = -57.4%; 600 mg Q4W = -49.7%; 300 mg Q2W = -61.1%; 150 mg Q4W = -48.3% vs. placebo = -15%; P<0.001).
All treatment groups of patients receiving AMG 451/KHK4083 generally achieved improvements compared to placebo at week 16 for key secondary endpoints, such as achieving at least a 75% reduction from baseline in EASI score (EASI-75), an Investigator Global Assessment (IGA) score of 0 (clear) or 1 (almost clear) with at least 2-point reduction from baseline (IGA 0/1) and at least a 4-point reduction from baseline in pruritus Numerical Rating Scale (NRS) score (PNRS-4). Efficacy measures continued to improve after week 16 for all AMG 451/KHK4083 doses.
The most commonly reported adverse events that occurred in at least 5% of patients were pyrexia, nasopharyngitis, worsening of atopic dermatitis and chills. The events of pyrexia and chills were mild to moderate in intensity and did not lead to treatment discontinuations.
"The Phase 2 results are both positive and exciting. They show improvement across all 4 dose groups compared to placebo, and highlight the potential of OX40 antagonism to help patients," said the lead investigator of this study, Dr. Emma Guttman-Yassky, MD./PhD., system chair for the Department of Dermatology and Waldman Professor of Dermatology and Immunology, Icahn School of Medicine at Mount Sinai and Director of the Center for Excellence in Eczema, and the Laboratory of Inflammatory Skin Diseases at Mount Sinai. "I hope that future clinical development data will further elucidate the significance and potential of AMG 451/KHK4083 in the treatment of moderate-to-severe atopic dermatitis."
"We are very pleased to present data from our Phase 2 study assessing the efficacy and safety of AMG 451/KHK4083 in chronic, recurrent, moderate-to-severe atopic dermatitis at the EADV congress," said Yoshifumi Torii, Ph.D., executive officer, vice president, head of R&D Division of Kyowa Kirin. "The results show inhibition and deletion of the OX40-expressing cells may provide an important new approach to treating moderate-to-severe atopic dermatitis, with the potential to help patients maintain responses."
"Atopic dermatitis affects nearly 30 million people a year and is known to have an extremely negative impact on patients' lives," said David M. Reese, M.D., executive vice president of Research and Development at Amgen. "These data provide strong evidence of the potential of AMG 451/KHK4083 for patients, and we look forward to studying this treatment further in Phase 3 clinical trials, which we expect to begin in the first half of 2022."
About the AMG 451/KHK4083 Phase 2 Study
The Phase 2, multicenter, randomized, double-blind, placebo-controlled trial (NCT03703102) investigated the efficacy and safety of AMG 451/KHK4083 in adults with moderate-to-severe atopic dermatitis who were not adequately controlled with topical agents. The study randomized 274 patients in the U.S., Japan, Canada and Germany across four dose-ranging active treatment groups, which received subcutaneous AMG 451/KHK4083 (600mg Q2W, 600mg Q4W, 300mg Q2W, 150mg Q4W), and a comparator placebo arm.
The primary endpoint was percentage change from baseline in EASI score at week 16. Additional endpoints include achievement of ≥75% reduction (improvement) from baseline in EASI score, IGA score of 0 (clear) or 1 (almost clear) with ≥ 2 points reduction from baseline, and ≥ 4 points reduction from baseline in the pruritus numeric rating scale (NRS) score. Patients in the study were followed up to week 56.
The presentation slides are available on the EADV website: https://www.eadvcongress2021.org/.
Dr. Emma Guttman-Yassky is the lead investigator of the study and a paid consultant for the AMG 451/KHK4083 development by Kyowa Kirin.
About AMG 451/KHK4083
AMG 451/KHK4083 is an anti-OX40 fully human monoclonal antibody engineered with Kyowa Kirin's patented POTELLIGENT® defucosylation technology to enhance its antibody-dependent cellular cytotoxicity (ADCC) activity. The initial AMG 451/KHK4083 antibody was discovered in collaboration between Kyowa Kirin US Research and La Jolla Institute for Immunology.
AMG 451/KHK4083 targets and inhibits the activity of the OX40 receptor expressed on the surface of activated effector T-cells, and has been shown to enhance the depletion of activated OX40+ T-cells by ADCC. It has been reported that effector T-cells expressing OX40 are present in the lesions of patients with atopic dermatitis and are critical in the disease pathophysiology.
Amgen and Kyowa Kirin Collaboration
On June 1, 2021, Amgen and Kyowa Kirin entered into an agreement to jointly develop and commercialize AMG 451/KHK4083. Under the terms of the agreement, Amgen will lead the development, manufacturing, and commercialization for AMG 451/KHK4083 for all markets globally, except Japan, where Kyowa Kirin will retain all rights. If approved, the companies will co-promote the asset in the United States and Kyowa Kirin has opt-in rights to co-promote in certain other markets including Europe and Asia.
For more information, visit www.amgen.com
View original content to download multimedia:https://www.prnewswire.com/news-releases/amgen-and-kyowa-kirin-present-positive-late-breaking-data-from-phase-2-study-of-amg-451khk4083-in-adult-patients-with-moderate-to-severe-atopic-dermatitis-at-eadv-congress-301391185.html
SOURCE Amgen
Oct. 04, 2021 6:01 AM ET Amgen Inc. (AMGN), KYKOF By: Mamta Mayani, SA News Editor1 Comment
041021 Diversity Full Recruitment Press Release _FINAL
Mechelen, Belgium; 4 October 2021, 07.01 CET; Galapagos NV (Euronext & NASDAQ: GLPG) today announced randomization of the last patient into the multi-center, global DIVERSITY Phase 3 study. The study is designed to evaluate the efficacy and safety of filgotinib, a JAK1 preferential inhibitor, in the induction and maintenance of remission in patients with Crohn’s Disease (CD).
The DIVERSITY study enrolled 1,374 participants with moderately to severely active CD, including biologic-naïve and biologic-experienced patients. The study evaluates the safety and efficacy of 100mg and 200mg filgotinib versus placebo on clinical remission and endoscopic response, in a 10-week induction phase, followed by a 47-week maintenance phase. Topline results of the DIVERSITY study are anticipated in H1 2023.
Dr. Walid Abi-Saab, Chief Medical Officer, Galapagos NV said: “This is an important milestone in the DIVERSITY program, as it brings us closer to delivering robust evidence to assess the use of our JAK1 preferential inhibitor as a potentially new class of medicine in the treatment of patients with Crohn's Disease. I would like to thank the patients and the clinical trial centers for participating in this important program, especially during the recent COVID-19 pandemic, which has been a particularly challenging time for the health services and society as a whole.”
The DIVERSITY clinical program design was informed by results from the Phase 2 FITZROY study, with filgotinib, which provided positive results for the use of this JAK1 inhibitor in patients with active CD. Full results were reported in The Lancet.1
The use of filgotinib for CD is investigational and is not approved anywhere globally.
About the DIVERSITY Phase 3 Study
DIVERSITY consists of a combined, double-blind, placebo-controlled Phase 3 study, enrolling 1,374 patients from 369 centers worldwide. The study compares the efficacy of filgotinib 100mg or 200mg once-daily oral treatment versus placebo in the induction and maintenance of clinical remission measured by Crohn’s Disease Activity Index (CDAI) score and endoscopic response measured as simple endoscopic score for Crohn’s Disease (SES-CD) at week 10 and week 58, in biologically-naive and biologically-experienced patients with moderately to severely active CD. There are EU-specific co-primary objectives that evaluate clinical remission measured by Patient Reported Outcome (PR02) and endoscopic response (SES-CD) at Week-10 and Week-58. In addition to clinical endpoints the study will also evaluate the effects on Health-Related Quality of Life (HRQoL) scores and Health Care Resource Utilization (HCRU) at Week-10 and Week-58. Safety will be evaluated by assessment of clinical laboratory tests, physical examination, vital signs measurements at various timepoints during the study, and by the documentation of Adverse Events.
For DIVERSITY study information visit: ClinicalTrials.gov Identifier NCT02048618
About the filgotinib collaboration
Gilead and Galapagos NV are partners in a global collaboration to develop and commercialize filgotinib, which is approved and marketed as Jyseleca® in the European Union, Great Britain, and Japan for the treatment of adults with moderate to severe active rheumatoid arthritis (RA) who have responded inadequately or are intolerant to one or more disease modifying anti-rheumatic drugs (DMARDs). Galapagos will be responsible for the commercialization of filgotinib in Europe (transition from Gilead to Galapagos anticipated to be completed by end of 2021), while Gilead will remain responsible for filgotinib outside of Europe, including in Japan, where filgotinib is co-marketed with Eisai. Applications to extend the approved indication of filgotinib to include ulcerative colitis have been filed in the European Union, Great Britain, and Japan, and a global Phase 3 program is ongoing in Crohn’s Disease. More information about clinical trials can be accessed at https://www.clinicaltrials.gov.
The European Summary of Product Characteristics for filgotinib, which includes contraindications and special warnings and precautions, is available at www.ema.europa.eu. The interview form from the Japanese Ministry of Health, Labour and Welfare is available at www.info.pmda.go.jp. The individual Great Britain and Northern Ireland Summary of Product Characteristics can be found at www.medicines.org.uk/emc and www.emcmedicines.com/en-GB/northernireland respectively.
Jyseleca® is a trademark of Galapagos NV and Gilead Sciences, Inc. or its related companies.
More information at www.glpg.com.
Oct. 04, 2021 4:14 PM ET Galapagos NV (GLPG)
By: Aakash Babu, SA News Editor
October 5, 2021 at 7:30 AM EDTPDF Version
-- Demonstrated efficacy and safety consistent with global SOPHIA study
-- Company expects to file BLA in China in advanced HER2+ breast cancer by approximately year end 2021
SHANGHAI and SAN FRANCISCO, Oct. 05, 2021 (GLOBE NEWSWIRE) -- Zai Lab Limited (NASDAQ: ZLAB; HKEX: 9688), an innovative commercial-stage biopharmaceutical company, today announced that the bridging study of margetuximab plus chemotherapy in advanced, previously treated HER2+ breast cancer met its primary endpoint, with acceptable safety and tolerability. The study showed that efficacy of this combination in Chinese patients was consistent with that seen in the global population in the SOPHIA trial conducted by Zai Lab’s partner MacroGenics, Inc.
The study was a randomized, open-label, multi-center, Phase II clinical study to evaluate the efficacy and safety of margetuximab plus chemotherapy compared with trastuzumab plus chemotherapy in 123 Chinese patients in mainland China, Hong Kong, and Taiwan with advanced HER2+ breast cancer who had received at least two prior lines of anti-HER2-directed therapy in the metastatic setting, including trastuzumab. The primary endpoint of the study was median progression-free survival (mPFS) evaluated by blinded independent central review (BICR) as defined by the achievement of at least 50% of the efficacy of margetuximab plus chemotherapy in the SOPHIA study (hazard ratio (HR) < 0.88). The secondary endpoints included overall survival (OS), mPFS evaluated by investigator, and objective response rate (ORR).
In this study, the HR for PFS in the intent-to-treat population evaluated by BICR was 0.69 favoring the margetuximab combination, thus achieving the primary endpoint. The safety profile of margetuximab plus chemotherapy was acceptable and consistent with the safety profile of margetuximab plus chemotherapy seen in the SOPHIA trial. Zai Lab is planning to present the detailed study results at an upcoming medical conference. Based on these positive results, Zai Lab expects to file a BLA in China for this indication by approximately year end 2021.
“We are pleased to see that the results of our bridging study are consistent with those of the SOPHIA trial that were the basis for the approval of Margenza® in the United States,” said Alan Sandler, M.D., President and Head of Global Development, Oncology. “Both trials support the potential use of margetuximab as another treatment option for a very difficult-to-treat patient population. The successful completion of our bridging study further demonstrates Zai Lab’s capabilities to produce clinical data of global quality to support regulatory approval in China in collaboration with our partners.”
For additional information about the company, please visit www.zailaboratory.com
http://www.zailaboratory.com/pipeline/list.aspx
Oct. 05, 2021 8:08 AM ET
By: Jonathan M Block, SA News Editor
October 1, 2021 6:00 am ET
At the Interim Analysis, 7.3 Percent of Patients Who Received Molnupiravir Were Hospitalized Through Day 29, Compared With 14.1 Percent of Placebo-Treated Patients Who were Hospitalized or Died
Merck Plans to Seek Emergency Use Authorization in the U.S. as Soon as Possible and to Submit Applications to Regulatory Agencies Worldwide
If Authorized, Molnupiravir Could be the First Oral Antiviral Medicine for COVID-19
KENILWORTH, N.J. & MIAMI--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, and Ridgeback Biotherapeutics today announced that molnupiravir (MK-4482, EIDD-2801), an investigational oral antiviral medicine, significantly reduced the risk of hospitalization or death at a planned interim analysis of the Phase 3 MOVe-OUT trial in at risk, non-hospitalized adult patients with mild-to-moderate COVID-19. At the interim analysis, molnupiravir reduced the risk of hospitalization or death by approximately 50%; 7.3% of patients who received molnupiravir were either hospitalized or died through Day 29 following randomization (28/385), compared with 14.1% of placebo-treated patients (53/377); p=0.0012. Through Day 29, no deaths were reported in patients who received molnupiravir, as compared to 8 deaths in patients who received placebo. At the recommendation of an independent Data Monitoring Committee and in consultation with the U.S. Food and Drug Administration (FDA), recruitment into the study is being stopped early due to these positive results. Merck plans to submit an application for Emergency Use Authorization (EUA) to the U.S. FDA as soon as possible based on these findings and plans to submit marketing applications to other regulatory bodies worldwide.
“More tools and treatments are urgently needed to fight the COVID-19 pandemic, which has become a leading cause of death and continues to profoundly affect patients, families, and societies and strain health care systems all around the world. With these compelling results, we are optimistic that molnupiravir can become an important medicine as part of the global effort to fight the pandemic and will add to Merck’s unique legacy of bringing forward breakthroughs in infectious diseases when they are needed most. Consistent with Merck’s unwavering commitment to save and improve lives, we will continue to work with regulatory agencies on our applications and do everything we can to bring molnupiravir to patients as quickly as possible,” said Robert M. Davis, chief executive officer and president, Merck. “On behalf of all of us at Merck, I thank our network of clinical investigators and patients for their essential contributions to the development of molnupiravir.”
“With the virus continuing to circulate widely, and because therapeutic options currently available are infused and/or require access to a healthcare facility, antiviral treatments that can be taken at home to keep people with COVID-19 out of the hospital are critically needed,” said Wendy Holman, chief executive officer of Ridgeback Biotherapeutics. “We are very encouraged by the results from the interim analysis and hope molnupiravir, if authorized for use, can make a profound impact in controlling the pandemic. Our partnership with Merck is critical to ensuring rapid global access if this medicine is approved, and we appreciate the collaborative effort to reach this important stage of development.”
About Merck’s Efforts to Enable Access to Molnupiravir, if it is Granted EUA or Approval
In anticipation of the results from MOVe-OUT, Merck has been producing molnupiravir at risk. Merck expects to produce 10 million courses of treatment by the end of 2021, with more doses expected to be produced in 2022.
Earlier this year, Merck entered into a procurement agreement with the U.S. Government under which Merck will supply approximately 1.7 million courses of molnupiravir to the U.S. government, upon EUA or approval from the U.S. FDA. Additionally, Merck has entered into supply and purchase agreements for molnupiravir with other governments worldwide, pending regulatory authorization, and is currently in discussions with other governments.
Merck is committed to providing timely access to molnupiravir globally, if it is authorized or approved, and plans to implement a tiered pricing approach based on World Bank country income criteria to reflect countries’ relative ability to finance their health response to the pandemic.
As part of its commitment to widespread global access, Merck previously announced that the company has entered into non-exclusive voluntary licensing agreements for molnupiravir with established generic manufacturers to accelerate availability of molnupiravir in more than 100 low- and middle-income countries (LMICs) following approvals or emergency authorization by local regulatory agencies.
More About the MOVe-OUT Study
The MOVe-OUT trial (MK-4482-002) (NCT04575597) was a global Phase 3, randomized, placebo-controlled, double-blind, multi-site study of non-hospitalized adult patients with laboratory-confirmed mild to moderate COVID-19, at least one risk factor associated with poor disease outcomes, and symptom onset within five days prior to randomization. The primary efficacy objective of MOVe-OUT is to evaluate the efficacy of molnupiravir compared to placebo as assessed by the percentage of participants who are hospitalized and/or die from the time of randomization through Day 29.
The Phase 3 portion of the MOVe-OUT trial was conducted globally, including in more than 170 planned sites in countries including Argentina, Brazil, Canada, Chile, Colombia, Egypt, France, Germany, Guatemala, Israel, Italy, Japan, Mexico, Philippines, Poland, Russia, South Africa, Spain, Sweden, Taiwan, Ukraine, the United Kingdom and the United States. For further information about the MOVe-OUT trial, please visit clinicaltrials.gov.
The most common risk factors for poor disease outcome included obesity, older age (>60 years), diabetes mellitus, and heart disease. To date, the Delta, Gamma, and Mu variants have accounted for nearly 80% of the evaluable cases in the trial. Recruitment in Latin America, Europe, and Africa accounted for 55%, 23% and 15% of the study population, respectively.
About Molnupiravir
Molnupiravir (MK-4482/EIDD-2801) is an investigational, orally administered form of a potent ribonucleoside analog that inhibits the replication of SARS-CoV-2, the causative agent of COVID-19. Molnupiravir has been shown to be active in several preclinical models of SARS-CoV-2, including for prophylaxis, treatment, and prevention of transmission. Additionally, pre-clinical and clinical data have shown molnupiravir to be active against the most common SARS-CoV-2 variants. Molnupiravir was invented at Drug Innovations at Emory (DRIVE), LLC, a not-for-profit biotechnology company wholly owned by Emory University, and is being developed by Merck & Co., Inc. in collaboration with Ridgeback Biotherapeutics. Ridgeback received an upfront payment from Merck and also is eligible to receive contingent payments dependent upon the achievement of certain developmental and regulatory approval milestones. Any profits from the collaboration will be split between the partners equally. Since licensed by Ridgeback, all funds used for the development of molnupiravir have been provided by Merck and by Wayne and Wendy Holman of Ridgeback.
Molnupiravir is also being evaluated for post-exposure prophylaxis in MOVe-AHEAD, a global, multicenter, randomized, double-blind, placebo-controlled Phase 3 study, which is evaluating the efficacy and safety of molnupiravir in preventing the spread of COVID-19 within households. For more information, please visit http://merckcovidresearch.com.
For more information, visit www.merck.com
Oct. 01, 2021 6:23 AM ET Merck & Co., Inc. (MRK) By: Mamta Mayani, SA News Editor52 Comments
BY TYLER DURDENFRIDAY, OCT 01, 2021 - 07:45 AM
Looks like Merck just beat Pfizer to the punch.
Merck announced Friday that an experimental COVID pill it has developed reduced hospitalizations and deaths by 50% in people recently infected with COVID.
Oct 04, 2021United States
First examination of safety events in the 60 and over sub-population across STELARA indications, including inflammatory bowel disease where relatively little biologics safety data exists, are generally consistent with placebo
Janssen presents 13 abstracts, including four oral presentations and two late breakers
SPRING HOUSE, PENNSYLVANIA, October 4, 2021 – Today, the Janssen Pharmaceutical Companies of Johnson & Johnson announced a new analysis of STELARA® (ustekinumab) pooled safety data from 13 clinical studies across approved indications, showing rates of key safety events among adults 60 years and older treated with STELARA for up to five yearsa were similar to rates observed with placebo during the control phase of these trials.1,b Approved indications included adults with moderately to severely active Crohn’s disease (CD), moderately to severely active ulcerative colitis (UC), moderate to severe plaque psoriasis (PsO) and active psoriatic arthritis (PsA) (Oral Presentation OP198).1 These data represent an important patient population as patients 60 years old and older are at a higher risk of disease and therapy-associated morbidity, which can result in disease management challenges.1
“Little has been intentionally explored about the safety profile of biologics in patients aged 60 and older with inflammatory bowel disease, as this population is often limited in number in clinical trials,” said Professor Subrata Ghosh, Chair and Head of Department of Medicine, University College Cork, Ireland and lead study investigator of the pooled safety analysis.c “This analysis arms physicians with data to consider when treating older patients with STELARA given the safety profile observed across all approved indications.”
Pooled Safety (Oral Presentation OP198) data show:1
Data from 13 Phase 2 and 3 studies, including six studies in CD/UC and seven studies in PsO/PsA for STELARA, were pooled. Of patients 60 years old or older with moderate to severe CD or UC, 214 received STELARA, the equivalent of 311 patient years (PYs) of follow up, and 120 received placebo, the equivalent of 97 PYs. Across additional approved indications, 811 received STELARA (1,590 PYs) and 272 received placebo (143 PYs). Number of events across pooled indications per 100 PYs were as follows:
“Continuing to follow occurrences of safety events in older adults who are treated with our therapies is valuable information that can help physicians evaluate treatment options for their patients,” said Jan Wehkamp, M.D., Ph.D., Vice President, Gastroenterology Disease Area Leader, Janssen Research & Development, LLC. “These data build on the body of evidence for the safety profile of STELARA and underscore our commitment to developing meaningful therapies for people of all ages who are living with an untreated or undertreated immune-mediated disease.”
These data are among 13 total abstracts, including three other oral presentations (OP122, OP153, OP199), two posters of week 12 data for TREMFYA® (guselkumab) in the treatment of adults with moderately to severely active CD from the GALAXI study (P0402, P0403), and two late breakers Janssen is presenting at UEGW. TREMFYA is not currently approved for the treatment of CD in the United States.2 Late-breaking presentations are:
The Pharmacokinetics and Immunogenicity of Ustekinumab and Adalimumab in Patients with Moderate to Severe Crohn’s Disease: Results from the SEAVUE Study (LB15)
Clinical and Endoscopic Outcomes with Ustekinumab in Patients with Crohn’s Disease: Results from the Long-Term Extension Period of the STARDUST Trial (LB14)
About STELARA® (ustekinumab)10
STELARA® (ustekinumab) is a fully human monoclonal antibody and is the first biologic treatment to selectively inhibit the interleukin (IL)-12 and IL-23 pathways. STELARA is approved in the United States for the treatment of: 1) adults and children six years and older with moderate to severe plaque psoriasis who are candidates for phototherapy or systemic therapy; 2) adult patients (18 years or older) with active psoriatic arthritis, used alone or in combination with methotrexate (MTX); 3) adult patients (18 years and older) with moderately to severely active Crohn’s disease; 4) adult patients (18 years and older) with moderately to severely active ulcerative colitis.
The Janssen Pharmaceutical Companies of Johnson & Johnson maintain exclusive worldwide marketing rights to STELARA®.
IMPORTANT SAFETY INFORMATION
STELARA® is a prescription medicine that affects your immune system. STELARA® can increase your chance of having serious side effects including:
Please read the full Prescribing Information and Medication Guide for STELARA® and discuss any questions you have with your doctor.
Learn more at www.janssen.com.
Oct. 04, 2021 8:23 AM ETJohnson & Johnson (JNJ)By: Mamta Mayani, SA News Editor

10/01/2021CATEGORY:
If approved, mavacamten would be the first cardiac myosin inhibitor for the treatment of obstructive hypertrophic cardiomyopathy
Application based on positive results from Phase 3 EXPLORER-HCM trial
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) today announced that the European Medicines Agency (EMA) has validated its Marketing Authorization Application (MAA) for mavacamten, an investigational, first-in-class cardiac myosin inhibitor, for the treatment of patients with obstructive hypertrophic cardiomyopathy (obstructive HCM). Validation of the application confirms the submission is complete, and the EMA’s centralized procedure with Committee for Medicinal Products for Human Use (CHMP)’s assessment begins.
“Despite the global prevalence of obstructive HCM and its debilitating symptoms and cardiac dysfunction, there is yet to be an approved therapy that targets the underlying cause of this devastating disease. Currently prescribed medicines largely provide only symptom relief. Mavacamten could potentially provide a treatment option that addresses the unmet needs of people living with obstructive HCM around the world,” said Roland Chen, M.D., senior vice president, Cardiovascular Development, Bristol Myers Squibb. “Today’s acceptance of the dossier by the EMA is a step forward in bringing this important targeted therapeutic approach to patients and physicians in Europe, and we thank the patients and investigators who have been involved in the EXPLORER-HCM trial.”
The application is based on the results of the pivotal Phase 3 EXPLORER-HCM trial, which evaluated mavacamten in patients with symptomatic obstructive HCM versus placebo. Results from the trial showed mavacamten demonstrated a clear treatment effect, with clinically meaningful improvements in symptoms, functional status and quality of life, as well as the ability to relieve left ventricular outflow tract obstruction. In the EXPLORER-HCM study all primary and secondary endpoints were met with statistical significance.
About the Phase 3 EXPLORER-HCM Trial
The EXPLORER-HCM Phase 3 trial enrolled a total of 251 patients with symptomatic (NYHA Class II or III) obstructive hypertrophic cardiomyopathy. All participants had measurable left ventricular outflow tract (LVOT) gradient (resting and/or provoked) ≥50 mmHg at baseline.
The primary endpoint for EXPLORER-HCM was a composite functional analysis designed to capture mavacamten’s effect on both symptoms and function. Secondary endpoints were changes from baseline to week 30 in postexercise LVOT gradient, pVO2, proportion of patients with at least one NYHA class improvement, and measures of patient reported outcomes. Additional endpoints included changes from baseline to Week 30 in echocardiographic indices, circulating biomarkers, cardiac rhythm patterns and accelerometry.
About Mavacamten
Mavacamten is a first-in-class, oral, allosteric modulator of cardiac myosin being investigated for the treatment of conditions caused by excessive cardiac contractility and impaired diastolic filling of the heart, including hypertrophic cardiomyopathy (HCM) and heart failure with preserved ejection fraction (HFpEF).
In clinical studies, mavacamten has demonstrated significant efficacy in reducing cardiac muscle contractility by reducing excess actin-myosin cross-bridging, leading to less hypercontractility and improved relaxation.
In obstructive HCM specifically, it is the first and only selective cardiac myosin inhibitor that has the potential to treat the underlying pathophysiology of the disease.
Mavacamten is an investigational therapy and is not approved for use in any country.
For more information about Bristol Myers Squibb, visit us at BMS.com
Oct. 01, 2021 7:16 AM ETBristol-Myers Squibb Company (BMY)By: Mamta Mayani, SA News Editor1 Comment
10/01/2021CATEGORY:
The Phase 2/3 RELATIVITY-047 trial served as the basis for this application, in which a statistically significant and clinically meaningful benefit was observed with relatlimab and nivolumab over Opdivo monotherapy
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) today announced that the European Medicines Agency (EMA) has validated its Marketing Authorization Application (MAA) for the LAG-3-blocking antibody relatlimab and nivolumab fixed-dose combination for first-line treatment of adult and pediatric patients (12 years and older and weighing at least 40 kg) with advanced (unresectable or metastatic) melanoma. This validation confirms completion of the submission and begins the EMA’s centralized review process.
“Melanoma can be a devastating disease and cases have been on the rise for years. The results from the RELATIVITY-047 trial demonstrate the potential that the relatlimab and nivolumab fixed-dose combination holds for people with advanced forms of this cancer,” said Jonathan Cheng, senior vice president and head of oncology development, Bristol Myers Squibb. “If approved, this treatment would become the first of its kind available to patients in the European Union. We look forward to working with the EMA as they evaluate this combination – which includes our third distinct checkpoint inhibitor – and are proud of this first step toward making it available for patients with advanced melanoma.”
The filing was based on the efficacy and safety results from the Phase 2/3 RELATIVITY-047 trial, which is the first trial to demonstrate a statistically significant and clinically meaningful progression-free survival benefit of a combination therapy over standard of care anti-PD-1 monotherapy in metastatic melanoma.
Primary results from the RELATIVITY-047 trial were presented in an oral abstract session and selected for the official press program for the American Society of Clinical Oncology (ASCO) Annual Meeting in June 2021. Data were also presented in an oral presentation during the European Society for Medical Oncology (ESMO) Annual Meeting in September 2021.
The U.S. Food Drug Administration (FDA) has also accepted for priority review the Biologics License Application (BLA) for the relatlimab and nivolumab fixed-dose combination.
The fixed-dose combination of relatlimab and nivolumab is an investigational therapy and is not approved for use in any country.
Bristol Myers Squibb thanks the patients and investigators involved in the RELATIVITY-047 clinical trial.
About RELATIVITY-047 (CA224-047)
RELATIVITY-047 (CA224-047) is a global, randomized, double-blind Phase 2/3 study evaluating the fixed-dose combination of relatlimab and nivolumab in patients with previously untreated metastatic or unresectable melanoma versus Opdivo alone. The primary endpoint of the trial is progression-free survival (PFS) by Blinded Independent Central Review (BICR) and the secondary endpoints are overall survival (OS) and objective response rate (ORR). A total of 714 patients were randomized 1:1 to receive a fixed-dose combination of relatlimab 160 mg and nivolumab 480 mg or Opdivo 480 mg by intravenous infusion every four weeks until disease recurrence, unacceptable toxicity or withdrawal of consent. Follow-up for the secondary endpoints of OS and ORR is ongoing.
About LAG-3
Lymphocyte-activation gene 3 (LAG-3) is a cell-surface molecule expressed on effector T cells and regulatory T cells (Tregs) and functions to control T-cell response, activation and growth. Preclinical studies indicate that inhibition of LAG-3 may restore effector function of exhausted T cells and potentially promote an anti-tumor response. Early research demonstrates that targeting LAG-3 in combination with other potentially complementary immune checkpoints may be a key strategy to more effectively potentiate anti-tumor immune activity.
Bristol Myers Squibb is evaluating relatlimab, its LAG-3-blocking antibody, in clinical trials in combination with other agents in a variety of tumor types.
About Opdivo
Opdivo is a programmed death-1 (PD-1) immune checkpoint inhibitor that is designed to uniquely harness the body’s own immune system to help restore anti-tumor immune response. By harnessing the body’s own immune system to fight cancer, Opdivo has become an important treatment option across multiple cancers.
Opdivo’s leading global development program is based on Bristol Myers Squibb’s scientific expertise in the field of Immuno-Oncology and includes a broad range of clinical trials across all phases, including Phase 3, in a variety of tumor types. To date, the Opdivo clinical development program has treated more than 35,000 patients. The Opdivo trials have contributed to gaining a deeper understanding of the potential role of biomarkers in patient care, particularly regarding how patients may benefit from Opdivo across the continuum of PD-L1 expression.
In July 2014, Opdivo was the first PD-1 immune checkpoint inhibitor to receive regulatory approval anywhere in the world. Opdivo is currently approved in more than 65 countries, including the United States, the European Union, Japan, and China. In October 2015, the Company’s Opdivo and Yervoy combination regimen was the first Immuno-Oncology combination to receive regulatory approval for the treatment of metastatic melanoma and is currently approved in more than 50 countries, including the United States and the European Union.
INDICATIONS
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of patients with unresectable or metastatic melanoma.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of patients with unresectable or metastatic melanoma.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) whose tumors express PD-L1 (≥1%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab) and 2 cycles of platinum-doublet chemotherapy, is indicated for the first-line treatment of adult patients with metastatic or recurrent non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab) is indicated for the treatment of patients with metastatic non-small cell lung cancer (NSCLC) with progression on or after platinum-based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving OPDIVO.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with unresectable malignant pleural mesothelioma (MPM).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of patients with intermediate or poor risk advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab), in combination with cabozantinib, is indicated for the first-line treatment of patients with advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab) is indicated for the treatment of patients with advanced renal cell carcinoma (RCC) who have received prior anti-angiogenic therapy.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with classical Hodgkin lymphoma (cHL) that has relapsed or progressed after autologous hematopoietic stem cell transplantation (HSCT) and brentuximab vedotin or after 3 or more lines of systemic therapy that includes autologous HSCT. This indication is approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab) is indicated for the treatment of patients with recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN) with disease progression on or after platinum-based therapy.
OPDIVO® (nivolumab) is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
OPDIVO® (nivolumab), as a single agent, is indicated for the adjuvant treatment of patients with urothelial carcinoma (UC) who are at high risk of recurrence after undergoing radical resection of UC.
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of adult and pediatric (12 years and older) patients with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adults and pediatric patients 12 years and older with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of patients with melanoma with involvement of lymph nodes or metastatic disease who have undergone complete resection.
OPDIVO® (nivolumab) is indicated for the treatment of patients with unresectable advanced, recurrent or metastatic esophageal squamous cell carcinoma (ESCC) after prior fluoropyrimidine- and platinum-based chemotherapy.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of completely resected esophageal or gastroesophageal junction cancer with residual pathologic disease in patients who have received neoadjuvant chemoradiotherapy (CRT).
OPDIVO® (nivolumab), in combination with fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the treatment of patients with advanced or metastatic gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma.
For more information about Bristol Myers Squibb, visit us at BMS.com
Oct. 01, 2021 7:27 AM ET Bristol-Myers Squibb Company (BMY)
By: Mamta Mayani, SA News Editor2 Comments
September 28, 2021Download PDFERBITUX® is the first and only anti-EGFR antibody approved, in combination with encorafenib, specifically for adults with previously treated metastatic CRC with a BRAF V600E mutation
INDIANAPOLIS, Sept. 28, 2021 /PRNewswire/ -- Eli Lilly and Company (NYSE: LLY) today announced that the U.S. Food and Drug Administration (FDA) has granted approval of a new indication for ERBITUX® (cetuximab injection) in combination with BRAFTOVI® (encorafenib), marketed by Pfizer, Inc., for the treatment of adult patients with metastatic colorectal cancer (CRC) with a BRAF V600E mutation, as detected by an FDA-approved test, after prior therapy.1 ERBITUX is the first and only anti-EGFR antibody approved, in combination with encorafenib, for this indication and is based on results from Pfizer's BEACON CRC trial, the only Phase 3 trial to specifically study patients with previously treated metastatic CRC with a BRAF V600E mutation. With this approval, ERBITUX has now received seven FDA approvals to treat certain types of CRC and squamous cell carcinoma of the head and neck.
"The BEACON study showed that the combination of ERBITUX and encorafenib significantly improved overall survival in patients with metastatic colorectal cancer with a BRAF V600E mutation – a subtype that typically has worse outcomes compared to those without the mutation," said David Hyman, M.D., chief medical officer, oncology at Lilly. "We are grateful to Pfizer for their collaboration as we've worked to bring this treatment regimen to patients."
Based on results from the BEACON CRC trial, ERBITUX plus encorafenib showed a median overall survival (OS) of 8.4 months (95% CI: 7.5, 11.0), compared to 5.4 months (95% CI: 4.8, 6.6) for the control arm (irinotecan with ERBITUX or FOLFIRI with ERBITUX) ([HR 0.60, (95% CI: 0.45, 0.79), p=0.0003]). Additionally, ERBITUX plus encorafenib showed an objective response rate (ORR) of 20% (95% CI: 13%, 29%), compared to 2% (95% CI: 0%, 7%) for the control arm (p<0.0001), and a median progression-free survival (mPFS) of 4.2 months (95% CI: 3.7, 5.4), compared to 1.5 months for the control arm (95% CI: 1.4, 1.7) ([HR 0.40, (95% CI: 0.31, 0.52), p<0.0001]).
About the BEACON CRC Study
Encorafenib in combination with ERBITUX was evaluated in the randomized, active-controlled, open-label, multicenter, Phase 3 BEACON CRC trial. Eligible patients were required to have BRAF V600E mutant metastatic CRC, as detected by an FDA-approved test, with disease progression after one or two prior regimens. Patients were randomized 1:1:1 to one of the following treatment arms:
The major efficacy outcome measure was OS. Additional efficacy outcome measures included PFS, ORR, and duration of response (DoR) as assessed by blinded independent central review (BICR). OS and PFS were assessed in all randomized patients. ORR and DoR were assessed in the subset of the first 220 patients included in the randomized portion of the encorafenib/ERBITUX and control arm of the study. A total of 220 patients were randomized to the encorafenib/ERBITUX arm and 221 to the control arm. The trial was conducted at over 200 investigational sites in North America, South America, Europe and the Asia Pacific region.
Indications and Usage for ERBITUX® (cetuximab) injection
Head and Neck Cancer
ERBITUX (cetuximab) is approved:
Metastatic Colorectal Cancer
ERBITUX is indicated for the treatment of KRAS wild-type, epidermal growth factor receptor (EGFR)-expressing, metastatic colorectal cancer (mCRC) as determined by an FDA-approved test for this use:
Limitations of Use: ERBITUX is not indicated for treatment of RAS-mutant colorectal cancer or when the results of the RAS mutation tests are unknown
Please see full Prescribing Information for ERBITUX, including Boxed Warnings regarding infusion reactions and cardiopulmonary arrest.
To learn more about Lilly's commitment to people with cancer, please visit www.LillyOncology.com.
To learn more about Lilly, please visit us at lilly.com
PP-CE-US-1115 09/2021 © Lilly USA, LLC 2021. ALL RIGHTS RESERVED.
ERBITUX® is a registered trademark owned by or licensed to Eli Lilly and Company, its subsidiaries, or affiliates.
BRAFTOVI® is a registered trademark of Pfizer, Inc.
View original content to download multimedia:https://www.prnewswire.com/news-releases/fda-expands-lillys-erbitux-cetuximab-label-with-combination-of-braftovi-encorafenib-for-the-treatment-of-braf-v600e-mutation-positive-metastatic-colorectal-cancer-crc-after-prior-therapy-301387268.html
SOURCE Eli Lilly and Company
Sep. 29, 2021 2:10 AM ET Eli Lilly and Company (LLY)Pfizer Inc. (PFE) By: Mamta Mayani, SA News Editor
What is BRAFTOVI + MEKTOVI?
BRAFTOVI (encorafenib) and MEKTOVI (binimetinib) are prescription medicines used together to treat people with a type of skin cancer called melanoma:
BRAFTOVI should not be used to treat people with wildtype BRAF melanoma. Your healthcare provider will perform a test to make sure that BRAFTOVI + MEKTOVI is right for you.
It is not known if BRAFTOVI or MEKTOVI is safe and effective in children.
https://www.braftovimektovi.com/
-- sBLA Filing Based on Landmark ZUMA-7 Study, the First Randomized Clinical Trial to Evaluate CAR T Against Standard of Care in the Second-Line Setting --
-- If Approved, Yescarta Would Become First CAR T-Cell Therapy for Adults with Large B-Cell Lymphoma Who Relapse After or Are Refractory to First-Line Therapy –
September 30, 2021 04:50 PM Eastern Daylight Time
SANTA MONICA, Calif.--(BUSINESS WIRE)--Kite, a Gilead Company (Nasdaq: GILD), today announced that it has submitted a supplemental Biologics License Application (sBLA) to the U.S. Food and Drug Administration (FDA) for Yescarta® (axicabtagene ciloleucel) to expand its current indication to include the treatment of adults with relapsed or refractory large B-cell lymphoma (LBCL) in the second-line setting.
The sBLA filing is based on data from the ZUMA-7 study with the longest follow-up – over two years – of any Phase 3 CAR T-cell therapy trial. Top-line results from the primary analysis of ZUMA-7 were recently reported and showed superiority of Yescarta compared to standard of care (SOC) in second-line relapsed or refractory LBCL. With a median follow-up of two years, the study met the primary endpoint of event-free survival (EFS; hazard ratio 0.398, p <0.0001). This represents a clinically meaningful 60% reduction in risk of EFS events versus standard of care. The study also met the key secondary endpoint of objective response rate (ORR). The interim analysis of overall survival (OS) showed a trend favoring Yescarta; however, the data are immature at this time, and further analyses are planned for the future.
Detailed results from ZUMA-7 have been submitted for presentation at an upcoming medical congress. Kite is in discussions with global health authorities regarding submissions to expand the currently approved indications for Yescarta.
“Yescarta demonstrated an impressive clinical benefit over the current standard of care in the ZUMA-7 study, and these findings highlight the potential of this transformative therapy to help even more patients,” said Frank Neumann, MD, PhD, Kite’s Global Head of Clinical Development. “Approximately 40% of adult patients diagnosed with LBCL require second-line treatment, and we are committed to working with the FDA to provide a new treatment option for these patients.”
About ZUMA-7 Study Design
ZUMA-7 is a randomized, open-label, global, multicenter, Phase 3 study evaluating the safety and efficacy of Yescarta versus current standard of care for second-line therapy (platinum-based salvage combination chemotherapy regimen followed by high-dose therapy and autologous stem cell transplant in those who respond to salvage chemotherapy) in adult patients with relapsed or refractory LBCL within 12 months of first-line therapy. In the study, 359 patients in 77 centers around the world were randomized (1:1) to receive a single infusion of Yescarta or current standard-of-care second-line therapy. The primary endpoint is event free survival (EFS) as determined by blinded central review, and defined as the time from randomization to the earliest date of disease progression per Lugano Classification, commencement of new lymphoma therapy, or death from any cause. Key secondary endpoints include objective response rate (ORR) and overall survival (OS). Additional secondary endpoints include progression-free survival (PFS), patient reported outcomes (PROs) and safety.
ZUMA-7 was conducted under a Special Protocol Agreement (SPA) with the U.S. Food and Drug Administration whereby the trial design, clinical endpoints and statistical analysis were agreed in advance with the Agency.
About Yescarta
Yescarta is a CD19-directed genetically modified autologous T cell immunotherapy indicated for the treatment of:
Limitations of Use: Yescarta is not indicated for the treatment of patients with primary central nervous system lymphoma.
Further information is available at www.YescartaTecartusREMS.com or 1-844-454-KITE (5483).
Please see full Prescribing Information, including BOXED WARNING and Medication Guide.
For more information on Kite, please visit www.kitepharma.com.
U.S. Prescribing Information for Yescarta including BOXED WARNING, is available at www.kitepharma.com and www.gilead.com.
Kite, the Kite logo, Yescarta, Tecartus, XLP and GILEAD are trademarks of Gilead Sciences, Inc. or its related companies.
For more information on Kite, please visit the company’s website at www.kitepharma.com or call Gilead Public Affairs at 1-800-GILEAD-5 or 1-650-574-3000.
Sep. 30, 2021 5:08 PM ETGilead Sciences, Inc. (GILD)
By: Dulan Lokuwithana, SA News Editor
September 30, 2021 at 1:00 AM EDT Back
TARRYTOWN, N.Y., Sept. 30, 2021 /PRNewswire/ --
Trial met primary endpoint, showing REGEN-COV significantly reduced viral load within 7 days of treatment; trial conducted in patients hospitalized with COVID-19 who did not require high-flow oxygen or mechanical ventilation at baseline
Numeric improvements with REGEN-COV observed for all clinical endpoints, including a 36% reduced risk of death by day 29 in the overall population, increasing to 56% reduced risk in patients who were seronegative at baseline
Similar efficacy observed with both doses (2,400 mg and 8,000 mg); U.S. FDA is currently reviewing request to add treatment in hospital settings to REGEN-COV authorization
Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) today announced that a trial assessing investigational REGEN-COV™ (casirivimab and imdevimab) in patients hospitalized with COVID-19 met its primary endpoint. Trial results will be presented at IDWeek 2021 today, and show that REGEN-COV significantly reduced viral load in patients hospitalized with COVID-19 who entered the trial without having mounted their own antibody response (seronegative) and required low-flow or no supplemental oxygen (p=0.0172). The trial also had clinical results supportive of the much larger UK RECOVERY trial in hospitalized patients, with numeric improvements observed across all clinical endpoints assessed.
"COVID-19 continues to have a devastating impact on patients, our communities and healthcare systems, and has so far killed more than one in every 500 Americans," said Eleftherios Mylonakis, M.D., Ph.D., primary investigator of the trial and Professor of Medicine, Molecular Microbiology and Immunology, and Director of Infectious Disease at Brown University and the Lifespan hospitals. "We need a multi-faceted approach to best manage the virus' impact, including vaccination and effective treatment when patients become ill. These data show that REGEN-COV can benefit certain patients even after they are hospitalized, reducing the amount of virus and clinical consequences. Taken together with results announced earlier this year from the RECOVERY trial and other studies, these results have the potential to inform personalized care for hospitalized patients with this protean disease that presents with such high clinical variability."
REGEN-COV is an investigational medicine authorized by the U.S. Food and Drug Administration (FDA) under an emergency use authorization to treat people who are at high risk of serious consequences from COVID-19 infection who are either already infected (non-hospitalized) or in certain post-exposure prophylaxis settings. In the U.S., it is not currently authorized in patients who are hospitalized due to COVID-19 infection.
The trial, which was stopped due to slow enrollment after recruiting just over one third the patients originally planned, found that patients who received REGEN-COV (2,400 mg or 8,000 mg) in addition to standard-of-care (SOC) experienced numeric improvements across all clinical endpoints assessed, compared to SOC alone (placebo). Researchers did not observe any clinical difference between the two REGEN-COV doses (2,400 mg or 8,000 mg), or any serious or dose-dependent safety signals in REGEN-COV treated patients. In a safety analysis involving 2,007 patients (REGEN-COV=1,340, placebo=667) serious adverse events occurred in 21% REGEN-COV patients (n=285) and 26% placebo patients (n=174). Infusion-related reactions and hypersensitivity reactions that were grade ≥2 occurred more commonly among REGEN-COV patients (2% and 1% respectively) than placebo patients (1% and <0.5% respectively). The trial originally assessed a broader group of patients; however in late 2020 the trial was adjusted to exclude patients who were on mechanical ventilation or high-flow oxygen at baseline based on a potential safety signal identified by an Independent Data Monitoring Committee in 199 patients on mechanical ventilation or high flow-oxygen, a finding that was not replicated in the much larger RECOVERY trial that enrolled hospitalized patients with a broad range of severe COVID-19, including these patient groups.
"These new results, combined with the nearly 10,000-patient RECOVERY trial, further validate how REGEN-COV can change the course of illness for patients even after they are hospitalized with COVID-19," said George D. Yancopoulos, M.D., Ph.D., President and Chief Scientific Officer at Regeneron. "Patients who received REGEN-COV in this trial experienced a 36% reduced risk of dying within 29 days of receiving treatment, and in patients who were seronegative when they entered the trial the risk was reduced by 56%. It's important to remember that while results from this and the RECOVERY trial indicate that patients unable to develop their own antibodies against COVID-19 historically had the poorest prognosis – and hence the greatest benefit from REGEN-COV treatment – both were largely conducted before widespread vaccination or the emergence of variants such as Delta. Consequently, serostatus may be less informative for treatment decisions in the future because it might not be practical to assess whether patients' antibodies are for their current SARS-CoV-2 infection."
The robust REGEN-COV development program has reported positive Phase 3 trial results across the spectrum of COVID-19 infection, from prevention to hospitalization:
Multiple analyses have shown that the antibody cocktail retains potency against the main variants of concern circulating within the U.S., including Delta (first identified in India), Gamma (first identified in Brazil), Beta (first identified in South Africa) and Mu (first identified in Colombia), with information available in the Fact Sheet for Healthcare Providers. Consequently, REGEN-COV remains available for use across the U.S., and Regeneron will continue actively monitoring the potency of REGEN-COV against emerging variants.
In the U.S., REGEN-COV is available for free to eligible people, as part of a U.S. government funded program and earlier this month Regeneron announced a new agreement with the U.S. government to supply an additional 1.4 million 1,200 mg doses of REGEN-COV. Information on how to access REGEN-COV throughout the U.S. is available from the Department of Health and Human Services and the National Infusion Center Association.
The development and manufacturing of REGEN-COV have been funded in part with federal funds from the Biomedical Advanced Research and Development Authority, part of the U.S. Department of Health and Human Services' Office of the Assistant Secretary for Preparedness and Response, under OT number: HHSO100201700020C.
About the REGEN-COV Antibody Cocktail
REGEN-COV (casirivimab and imdevimab) is a cocktail of two monoclonal antibodies that was designed specifically to block infectivity of SARS-CoV-2, the virus that causes COVID-19, using Regeneron's proprietary VelocImmune® and VelociSuite® technologies. The two potent, virus-neutralizing antibodies that form the cocktail bind non-competitively to the critical receptor binding domain of the virus's spike protein, which diminishes the ability of mutant viruses to escape treatment and protects against spike variants that have arisen in the human population, as detailed in Cell and Science.
REGEN-COV has not been approved by the FDA, but is currently authorized in the U.S. for the treatment and post-exposure prophylaxis in certain high risk individuals. Post-exposure prophylaxis with REGEN-COV is not a substitute for vaccination against COVID-19. REGEN-COV is not authorized for pre-exposure prophylaxis for prevention of COVID-19 or for use in patients who are hospitalized due to COVID-19 or require oxygen therapy, or for people currently using chronic oxygen therapy because of an underlying comorbidity who require an increase in baseline oxygen flow rate due to COVID-19. This authorization is for the duration of the declaration that circumstances exist justifying the authorization of the emergency uses under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb-3(b)(1), unless the authorization is terminated or revoked sooner. Additional information about REGEN-COV in the U.S. is below (authorized uses and important safety information).
In August, Regeneron submitted the first of two Biologics License Applications (BLAs) for REGEN-COV. The initial submission included data on the efficacy and safety of REGEN-COV to treat and prevent SARS-CoV-2 infection in non-hospitalized people. The second BLA submission will focus on those hospitalized because of COVID-19, and is expected to be completed later this year.
Emergency or temporary pandemic use authorizations are currently in place in more than 40 countries, including the U.S., several European Union countries, India, Switzerland and Canada, and the antibody cocktail is fully approved in Japan and conditionally approved in the UK.
Regeneron invented REGEN-COV and is collaborating with Roche to increase global supply, with Roche primarily responsible for development and distribution outside the U.S. Regeneron and Roche share a commitment to making the antibody cocktail available to COVID-19 patients around the globe and will support access in low- and lower-middle-income countries through drug donations to be made in partnership with public health organizations.
AUTHORIZED USES AND IMPORTANT SAFETY INFORMATION
Treatment:
REGEN-COV is authorized for the treatment of mild to moderate coronavirus disease 2019 (COVID-19) in adults and pediatric patients (12 years of age and older weighing at least 40 kg) with positive results of direct SARS-CoV-2 viral testing, and who are at high risk for progression to severe COVID-19, including hospitalization or death
Limitations of Authorized Use (Treatment)
For additional information about the company, please visit www.regeneron.com
View original content:https://www.prnewswire.com/news-releases/new-regen-cov-casirivimab-and-imdevimab-data-show-supportive-results-in-patients-hospitalized-with-covid-19-301388370.html
SOURCE Regeneron Pharmaceuticals, Inc.
Sep. 30, 2021 1:52 AM ET Regeneron Pharmaceuticals, Inc. (REGN) By: Mamta Mayani, SA News Editor2 Comments
September 30, 2021
- Analyses include long-term efficacy and safety data from the open-label extension period of KEEPsAKE-1 and KEEPsAKE-2 evaluating risankizumab in psoriatic arthritis[1]
- The primary endpoint of ACR20 response at week 24 was maintained through one year (52 weeks)[1]
- Continuous treatment with risankizumab improved signs and symptoms of psoriatic arthritis, with efficacy maintained through one year (52 weeks) of treatment[1]
- Results were presented at the 30th European Academy of Dermatology and Venereology (EADV) Virtual Congress
NORTH CHICAGO, Ill., Sept. 30, 2021 /PRNewswire/ -- AbbVie (NYSE: ABBV) today presented results from new Phase 3 data analyses of KEEPsAKE-1 and KEEPsAKE-2, evaluating risankizumab (SKYRIZI®, 150 mg) in adults with active psoriatic arthritis for one year (52 weeks).1 These results were featured during the "Late Breaking News, Reviews and Updates" session at the 30th European Academy of Dermatology and Venereology (EADV) Virtual Congress.
KEEPsAKE-1 included adult patients with active psoriatic arthritis who responded inadequately to non-biologic disease-modifying anti-rheumatic drugs (DMARDs). KEEPsAKE-2 included adult patients with active psoriatic arthritis who had responded inadequately or were intolerant to biologic therapy and/or non-biologic disease-modifying anti-rheumatic drugs (DMARDs).2-5 In the first phase of the studies (Period 1), patients were randomized to risankizumab or placebo until week 24.1 At week 24, the open label extension (Period 2) began, and all patients were treated with risankizumab.1,4,5
The new long-term data from the open-label extension period showed that 70 and 58 percent of patients initially treated with risankizumab achieved American College of Rheumatology 20 (ACR20) response in KEEPsAKE-1 and KEEPsAKE-2 respectively at one year, where patients with missing data were categorized as non-responders.1 Among patients initially treated with risankizumab, 43 percent in KEEPsAKE-1 and 32 percent in KEEPsAKE-2 achieved ACR50 response, and 26 percent in KEEPsAKE-1 and 17 percent in KEEPsAKE-2 achieved ACR70 response at one year.1
Additionally, at one year, 68 and 64 percent of patients initially treated with risankizumab and with a body surface area ≥3 percent at baseline achieved a 90 percent reduction in the Psoriasis Area and Severity Index (PASI 90) in KEEPsAKE-1 and KEEPsAKE-2, respectively.1
"Millions of people still suffer daily with symptoms of psoriatic arthritis, driving us to advance treatment options for these patients," said Thomas Hudson, senior vice president of research and development, chief scientific officer, AbbVie. "These new analyses at one year support the potential of risankizumab to maintain improvements across multiple manifestations of psoriatic arthritis."
In terms of improvement in physical function (as measured by the Health Assessment Questionnaire Disability Index [HAQ-DI]), patients initially randomized to risankizumab reported mean reduction (i.e., improvement) of 0.41 and 0.26 from baseline in HAQ-DI score for KEEPsAKE-1 and KEEPsAKE-2, respectively, at week 52.1
In addition, pooled results from KEEPsAKE-1 and KEEPsAKE-2 showed that 76 and 55 percent of patients achieved the resolution of dactylitis and resolution of enthesitis, respectively, at week 52.1
"Dactylitis is a common symptom in psoriatic arthritis that can cause severe swelling in the fingers and toes that result in everyday tasks becoming difficult," said Lars Erik Kristensen, M.D., Ph.D., consultant and head of science at the Parker Institute Copenhagen Denmark, associate professor, Lund Sweden, SUS University Hospital. "These results provide important insights on how both biologic-naïve and experienced patients may benefit from treatment with risankizumab."
Both KEEPsAKE-1 and KEEPsAKE-2 demonstrated consistent long-term safety profiles with those shared at week 24, with no new safety findings observed from week 24 through week 52.1 Serious treatment-emergent adverse events (TEAE) occurred at 7.4 events/100 patient-years (E/100 PYs) and 9.4 E/100 PYs in KEEPsAKE-1 and KEEPsAKE-2, respectively.1 Rates of serious infections in KEEPsAKE-1 and KEEPsAKE-2 were 2.8 and 2.0 E/100 PYs, respectively.1 The rates of TEAEs leading to discontinuation of the study drug in KEEPsAKE-1 was 2.3 E/100 PYs and 1.6 E/100 PYs in KEEPsAKE-2.1 In KEEPsAKE-1, there were two deaths and both were not related to the study drug per investigator.1 There were no deaths reported in KEEPsAKE-2.1 In KEEPsAKE-2, three major adverse cardiac events (MACE) were reported, which were not related to the study drug per the investigator.1 No MACE were reported in KEEPsAKE-1.1
In January 2021, AbbVie announced positive top-line results from the Phase 3 KEEPsAKE-1 and KEEPsAKE-2 studies, showing that risankizumab (150 mg) achieved the primary endpoint of ACR20 response versus placebo during the 24-week double-blinded, placebo-controlled, parallel-group period (period 1).2,3
Risankizumab (SKYRIZI) is part of a collaboration between Boehringer Ingelheim and AbbVie, with AbbVie leading development and commercialization globally.
Use of risankizumab in psoriatic arthritis is not approved and its safety and efficacy have not been evaluated by regulatory authorities.
About KEEPsAKE-1 and KEEPsAKE-22-5
KEEPsAKE-1 and KEEPsAKE-2 are both Phase 3, multicenter, randomized, double-blind, placebo-controlled studies designed to evaluate the safety and efficacy of risankizumab in adult patients with active psoriatic arthritis. KEEPsAKE-1 evaluated risankizumab in patients who had an inadequate response or intolerance to at least one DMARD. KEEPsAKE-2 evaluated risankizumab in patients who had an inadequate response or intolerance to biologic therapy and/or DMARDs. Patients were randomized to risankizumab 150 mg or placebo followed by risankizumab 150 mg at week 24.
The primary endpoint for both studies was the achievement of ACR20 response at week 24 from the treatment with the study medication. Ranked secondary endpoints included change from baseline in HAQ-DI, as well as the achievement of PASI 90 and MDA at week 24. Other secondary endpoints included ACR50 and ACR70 (not controlled for multiplicity) at week 24. The studies are ongoing, and the long-term extension remains blinded to evaluate the long-term safety, tolerability and efficacy of risankizumab in patients who have completed the placebo-controlled period.
More information on these trials can be found at www.clinicaltrials.gov (KEEPsAKE-1: NCT03675308; KEEPsAKE-2: NCT03671148).
About risankizumab (SKYRIZI®)
SKYRIZI is an interleukin-23 (IL-23) inhibitor that selectively blocks IL-23 by binding to its p19 subunit.8,9 IL-23, a cytokine involved in inflammatory processes, is thought to be linked to a number of chronic immune-mediated diseases, including psoriasis.8 In April 2019 SKYRIZI was approved by the European Commission for the treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy. The approved dose for SKYRIZI is 150 mg, administered by subcutaneous injection prefilled pen or prefilled syringe at week 0 and 4, and every 12 weeks thereafter. Phase 3 trials of SKYRIZI in psoriasis, Crohn's disease, ulcerative colitis and psoriatic arthritis are ongoing.10-12
This is not a complete summary of all safety information. See SKYRIZI full summary of product characteristics (SmPC) at www.ema.europa.eu.
Sep. 30, 2021 12:48 PM ET
By: Jonathan M Block, SA News Editor2 Comments
Sep 30, 2021
ACTON, Mass.--(BUSINESS WIRE)--Sep. 30, 2021-- Insulet Corporation (NASDAQ: PODD) (Insulet or the Company), the global leader in tubeless insulin pump technology with its Omnipod® brand of products, has presented positive pivotal trial extension phase results for the Omnipod® 5 Automated Insulin Delivery System (Omnipod 5). Omnipod 5, the world’s first tubeless, wearable system that continuously adapts insulin delivery based on glucose levels and trends, significantly improved time in range and reduced HbA1c in children, adolescents, and adults, aged 6-70 years, with type 1 diabetes over a period of 12 months. The data was presented at EASD 2021, the annual meeting of the European Association for the Study of Diabetes.
”Reducing burden and making diabetes management easier through innovative technology is our number one goal at Insulet,” said Dr. Trang Ly MBBS, FRACP, PhD, Insulet Senior Vice President and Medical Director. “We are very pleased that the positive safety and efficacy outcomes resulting from our pivotal study continued for those patients who used Omnipod 5 for a total of 12 months. This product is having an enduring positive impact on trial participants’ health, and we couldn’t be more proud.”
Insulet also presented new clinical outcomes data for people with type 1 diabetes using the Omnipod DASH® System (Omnipod DASH) for 90 days, as well as overall positive self-management behaviors among individuals using Omnipod DASH, Continuous Glucose Monitoring (CGM) and a cloud-based diabetes data management system.
Omnipod 5 System Extension Data Overview
Following completion of the 3-month pivotal trial, the results of which were previously shared, 95% of study participants chose to continue use of Omnipod 5 in an ongoing extension study. Insulet presented its data in two groups of extension study participants with type 1 diabetes: 114 adults and adolescents between 14 and 70 years old, and 110 children aged 6 to 13.9 years. The participants used Omnipod 5 at home for a period of 12 months after a 14-day period using their standard therapy, which included both pump therapy and multiple daily injections. The results were presented in 3-month intervals: months 1-3 (the pivotal study), months 4-6, months 7-9, and months 10-12.
After 3 months of system use, adults and adolescents had a decrease in HbA1c from 7.2% to 6.8%. This decrease was maintained after a total of 12 months of use, with mean HbA1c remaining at 6.8%. Time in range (TIR) increased from 63.6% to 73.8% in the first 3 months of use, and persisted at 72.7% in months 10-12 of use, corresponding to an additional 2.3 hours in target range. Median time <70 mg/dL (<3.9 mmol/L) decreased from 2.1% to 1.1% after 3 months of use and remained at 1.1% in months 10-12 of use.
Children had a decrease in HbA1c from 7.7% to 7.0% after 3 months of system use. This decrease was maintained after a total of 12 months of system use, with mean HbA1c remaining at 7.0%. TIR increased from 52.4% to 67.9% in the first 3 months of use, and persisted at 66.8% in months 10-12 of use, corresponding to an additional 3.5 hours per day in target range. Median time <70 mg/dL (<3.9 mmol/L) remained low at 1.5% in the first 3 months of use and 1.4% in months 10-12 of use.
“The pivotal study results were certainly impressive, but what’s even more exciting is that these outcomes were maintained over an entire year of use,” said Dr. Anders Carlson, Medical Director of the International Diabetes Center. “Insulet has shown that these positive clinical outcomes can be maintained in real-world conditions, with a frequency of visits very similar to the usual standard of care.”
The Omnipod 5 System received breakthrough device designation from the U.S. Food and Drug Administration and is currently under premarket review. The Company expects to launch Omnipod 5 in limited release in the U.S. late in the fourth quarter 2021. The device is currently not CE marked or available in Europe.
Omnipod DASH Study Results
Insulet also presented two studies of people living with type 1 diabetes using Omnipod DASH. The first study evaluated glycemic improvement after 90 days of use in 4,738 individuals ranging in age from under 2 to over 65 years. The study was divided into two groups: children and adolescents under 18 years of age and adults 18 years or older.
Overall, there was a significant decrease in HbA1c of -0.9% and a reduction in self-reported hypoglycemic events (<70 mg/dL [<3.9 mmol/L]) from 2.9 episodes to 1.3 episodes per week in adults and from 2.8 to 1.5 in children and adolescents. These patient-reported outcomes provide positive evidence that the use of Omnipod DASH was associated with significant reductions in HbA1c and hypoglycemic events after the initial 90 days of use for children, adolescents, and adults living with type 1 diabetes.
The second presentation covered the first real-world study of 2,586 people living with type 1 diabetes on Omnipod DASH with CGM and cloud-based data management. Participants included Omnipod DASH users with at least 90 days of insulin pump data in a cloud-based data management system between July 2018 and August 2021, as well as a minimum of 14 days of CGM readings over a 3-month period. The study resulted in positive glycemic outcomes, with mean estimated HbA1c ranging from 7.2 to 7.7%, and a low percentage of time in hypoglycemia across all age groups. Overall, the mean TIR (70-180 mg/dl [3.9 – 10.0 mmol/L]) was 59.6%, with median 1.3% of time below 70mg/dL (<3.9 mmol/L) and 0.2% of time below 54 mg/dL (<3.0 mmol/L).
These real-world data demonstrate positive self-management behaviors among a large cohort of Omnipod DASH users, ranging from young children to older adults.
For more information, please visit: insulet.com and omnipod.com
©2021 Insulet Corporation. Omnipod, Omnipod 5, and DASH are registered trademarks of Insulet Corporation in the United States of America and other various jurisdictions. All rights reserved.
View source version on businesswire.com: https://www.businesswire.com/news/home/20210930005162/en/
Source: Insulet Corporation
Sep. 30, 2021 8:09 AM ET
Insulet Corporation (PODD) By: Mamta Mayani, SA News Editor
Insulet (NASDAQ:PODD) presents positive pivotal trial extension phase results for the Omnipod 5 Automated Insulin Delivery System.
September 30, 2021Download PDFParticipants taking tirzepatide 15 mg experienced 47.11% relative reduction in liver fat content compared to 11.17% for insulin degludec
Volume of abdominal adipose tissue decreased with tirzepatide, compared to an increase with insulin degludec
INDIANAPOLIS, Sept. 30, 2021 /PRNewswire/ -- Tirzepatide led to greater improvements in liver fat content and abdominal adipose tissue compared to titrated insulin degludec in adults with type 2 diabetes in an MRI sub-study of Eli Lilly and Company's (NYSE: LLY) phase 3 SURPASS-3 clinical trial.1 The results were presented today at the 57th European Association for the Study of Diabetes (EASD) Annual Meeting in an EASD-sponsored symposium.
The MRI (magnetic resonance imaging) sub-study achieved its primary and secondary endpoints. As evaluated by MRI scans, the sub-study showed all three doses of tirzepatide (5 mg, 10 mg, 15 mg) led to greater reductions in liver fat content compared to insulin degludec and reductions in volume of visceral adipose tissue and abdominal subcutaneous adipose tissue compared to increases in volume of both measurements with insulin degludec at 52 weeks.
"Increased ectopic fat – such as liver fat or visceral adipose tissue – is commonly seen in adults with type 2 diabetes and is associated with an inflammatory response and increased cardiometabolic risk," said Amalia Gastaldelli, Ph.D., Research Director of Cardiometabolic Risk Unit, Institute of Clinical Physiology, CNR, Pisa Italy. "We are encouraged by the robust reductions in liver fat content and abdominal adipose tissue observed with all three doses of tirzepatide in this population of adults with type 2 diabetes and elevated liver fat content."
Tirzepatide is a novel investigational once-weekly dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist that integrates the actions of both incretins into a single molecule, representing a new class of medicines being studied for the treatment of type 2 diabetes.
SURPASS-3 was a 52-week, multi-center, randomized, phase 3, open-label trial evaluating the efficacy and safety of tirzepatide compared to titrated insulin degludeci in adults with type 2 diabetes who have inadequate glycemic control on stable doses of metformin with or without an SGLT-2 inhibitor. Study participants were insulin-naïve and had a mean duration of diabetes of 8.4 years, a baseline A1C of 8.17 percent and a baseline weight of 94.3 kg.
The SURPASS-3 MRI sub-study compared the effect of tirzepatide to titrated insulin degludec on liver fat content (LFC), volume of visceral adipose tissue (VAT) and abdominal subcutaneous adipose tissue (ASAT) in 296 participants as evaluated by MRI scans at baseline and at 52 weeks. The subpopulation of adults with type 2 diabetes who participated in this sub-study had an overall baseline LFC of 15.7 percent.
About tirzepatide
Tirzepatide is a once-weekly dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist that integrates the actions of both incretins into a single novel molecule. GIP is a hormone that may complement the effects of GLP-1. In preclinical models, GIP has been shown to decrease food intake and increase energy expenditure therefore resulting in weight reductions, and when combined with a GLP-1 receptor agonist, may result in greater effects on glucose and body weight. Tirzepatide is in phase 3 development for blood glucose management in adults with type 2 diabetes and for chronic weight management. It is also being studied as a potential treatment for non-alcoholic steatohepatitis (NASH) and heart failure with preserved ejection fraction (HFpEF).
For the latest updates, visit http://www.lillydiabetes.com
o learn more about Lilly, please visit us at lilly.com
View original content to download multimedia:https://www.prnewswire.com/news-releases/lillys-tirzepatide-led-to-greater-improvements-in-liver-fat-content-compared-to-insulin-degludec-in-adults-with-type-2-diabetes-in-surpass-3-mri-sub-study-301388229.html
SOURCE Eli Lilly and Company
Sep. 30, 2021 6:29 AM ET
By: Mamta Mayani, SA News Editor
Wednesday, September 29, 2021 - 06:45am
NEW YORK--(BUSINESS WIRE)-- Pfizer Inc. (NYSE:PFE) today announced positive top-line results from a Phase 3 study (B7471004) evaluating the safety and immunogenicity of PREVNAR 20™ (Pneumococcal 20-valent Conjugate Vaccine) in adults 65 years of age or older when administered at the same time as the seasonal influenza vaccine (SIIV, Fluad Quadrivalent [adjuvanted], 2020/2021 strains). Responses elicited by PREVNAR 20 for all 20 serotypes and by seasonal influenza vaccine when given together were noninferior (the study’s primary immunogenicity objectives) to those elicited by the vaccines when administered one month apart. The safety profile of PREVNAR 20 was similar when the vaccines were coadministered as compared to when each vaccine was administered separately, one month apart.
“We are encouraged by these results showing that these two vaccines can be administered at the same time without affecting the immune protection provided by either vaccine or changing the safety profile,” said Kathrin U. Jansen, Ph.D., Senior Vice President and Head of Vaccine Research & Development, Pfizer. “This study adds to the body of evidence further supporting that pneumococcal conjugate vaccines may be coadministered with influenza vaccines, this time studied with the adjuvanted influenza vaccine. We are committed to vaccine development to help address needs across many respiratory diseases.”
Across 66 investigator sites in the United States, a total of 1,796 participants were enrolled and randomized, with 1,727 of participants completing the study.
“Both PREVNAR 20 and the influenza vaccine are important for helping protect adults against pneumococcal pneumonia and the flu respectively; however, vaccination rates decline when someone needs to make multiple appointments to receive these vaccines,” said Luis Jodar, Ph.D., Senior Vice President and Chief Medical Officer, Pfizer Vaccines. “The results of this trial supports current CDC clinical guidance allowing coadministration during a single doctor or pharmacy appointment, so that more adults are able to help protect themselves against both of these respiratory diseases.”
Pfizer will seek to present and publish detailed outcomes from this clinical trial at a future date. Additionally, in May 2021, Pfizer announced the initiation of a Phase 3 clinical trial exploring coadministration of PREVNAR 20 and a booster dose of COMIRNATY® (COVID-19 Vaccine, mRNA) in adults ages 65 or older.
About PREVNAR 20
PREVNAR 20 is Pfizer’s next-generation pneumococcal conjugate vaccine that includes capsular polysaccharide conjugates for the 13 serotypes (1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F and 23F) already included in Prevnar 13® (Pneumococcal 13-valent Conjugate Vaccine [Diphtheria CRM197 Protein]). The vaccine also contains capsular polysaccharide conjugates for seven additional serotypes (8, 10A, 11A, 12F, 15B, 22F and 33F) that cause invasive pneumococcal disease (IPD),1,2,3,4,5 and have been associated with high case-fatality rates,6,7,8,9 antibiotic resistance,10,11,12 and/or meningitis.13,14 PREVNAR 20 contains the broadest serotype coverage and helps protect against more strains of the bacteria that cause pneumococcal pneumonia than any other conjugate vaccine available.
On June 8, 2021, Pfizer announced the U.S. Food and Drug Administration (FDA) approved, based on accelerated approval and priority review, PREVNAR 20 for the prevention of invasive disease and pneumonia in adults age 18 years or older. On February 26, 2021, the European Medicines Agency (EMA) accepted for review Pfizer’s Marketing Authorization Application (MAA) for the 20-valent pneumococcal conjugate vaccine candidate, as submitted for the prevention of invasive disease and pneumonia caused by S. pneumoniae serotypes in the vaccine in adults ages 18 years and older. The formal review process by the EMA’s Committee for Medicinal Products for Human Use (CHMP) currently is ongoing.
PREVNAR 20™ U.S. Indications
Please see full prescribing information for PREVNAR 20™.
Please click here for full Prescribing Information (16+ years of age). Please click here for Fact Sheet for Vaccination Providers (12+ years of age). Please click here for the Recipients and Caregivers Fact Sheet.
to learn more, please visit us on www.Pfizer.com
View source version on businesswire.com: https://www.businesswire.com/news/home/20210929005276/en/
Source: Pfizer Inc.
Sep. 29, 2021 7:28 AM ET Pfizer Inc. (PFE)
By: Mamta Mayani, SA News Editor1 Comment
September 28, 2021
- QULIPTA demonstrated statistically significant, clinically meaningful rapid and continuous reductions in mean monthly migraine days among adults with episodic migraine compared to placebo across the 12-week treatment period with significant reductions seen in weeks 1-4¹
- The pivotal trial showed that when taking QULIPTA, the majority of patients experienced between a 50-100% reduction in monthly migraine days across 12 weeks¹
- AbbVie is the only pharmaceutical company to offer three treatments across the full spectrum of migraine to help patients living with this debilitating disease
NORTH CHICAGO, Ill., Sept. 28, 2021 /PRNewswire/ -- AbbVie (NYSE: ABBV) today announced that the U.S. Food and Drug Administration (FDA) approved QULIPTA™ (atogepant) for the preventive treatment of episodic migraine in adults.1 QULIPTA is the first and only oral calcitonin gene-related peptide (CGRP) receptor antagonist (gepant) specifically developed for the preventive treatment of migraine.2
Experience the interactive Multichannel News Release here: https://www.multivu.com/players/English/8940451-abbvie-qulipta-atogepant-fda-approval/
"Millions of people living with migraine often lose days of productivity each month because attacks can be debilitating. QULIPTA can help by reducing monthly migraine days with a once-daily, oral dose that works quickly and continuously," said Michael Severino, M.D., vice chairman and president, AbbVie. "We are proud that AbbVie is now the only pharmaceutical company to offer three products across the full spectrum of migraine treatment, which include preventive therapies for chronic and episodic migraine and an acute treatment for migraine attacks."
The approval is supported by data from a robust clinical program evaluating the efficacy, safety and tolerability of QULIPTA in nearly 2,000 patients who experienced 4 to 14 migraine days per month, including the pivotal Phase 3 ADVANCE study — which was published in The New England Journal of Medicine — the pivotal Phase 2b/3 study, and the Phase 3 long-term safety study.1,2
"When I have a migraine attack, my 5-year-old daughter doesn't understand why I can't take her to a birthday party or to the park. It's heartbreaking when I have to tell her I need to be away from her because my eyes feel like they're going to explode out of my head," said Kelsi Owens, an ADVANCE trial participant who has lived with migraine for nearly three decades. "During the trial while taking QULIPTA, I had many fewer migraine days. For the first time ever, I don't have difficulty doing my daily activities and I don't have to worry as much that a migraine attack will cause me to miss important events with family and friends."
Migraine is a complex disease with recurrent attacks that are often incapacitating and characterized by severe, throbbing headache pain as well as compounding associated symptoms like extreme sensitivity to light, sound or nausea.4 It is highly prevalent, affecting more than 1 billion people worldwide, including 39 million people in the U.S. alone,5 and is the highest cause of disability worldwide for people under 50 years of age.6,7
"This approval reflects a broader shift in the treatment and management paradigm for the migraine community. QULIPTA provides a simple oral treatment option specifically developed to prevent migraine attacks and target CGRP, which is believed to be crucially involved in migraine in many patients," said Peter J. Goadsby, M.D., Ph.D., D.Sc., neurologist and professor at University of California, Los Angeles, and King's College, London, who earned the prestigious Brain Prize in 2021 for his revolutionary research about CGRP's role in migraine attacks and co-authored the ADVANCE study.
"I'm particularly encouraged by the convenience of the oral daily use of QULIPTA, its rapid onset of significant efficacy, and its safety and tolerability as well as its high patient response rates. This is a milestone in preventive migraine treatment that I hope will help many patients for years to come," Goadsby said.
Highlights from the clinical program supporting the approval and additional data analysis include:
More information about the clinical program can be found on www.clinicaltrials.gov (NCT03777059, NCT02848326 and NCT03700320).
About QULIPTA™
QULIPTA™ (atogepant) is the first and only oral calcitonin gene-related peptide (CGRP) receptor antagonist (gepant) specifically developed for the preventive treatment of episodic migraine. CGRP and its receptors are expressed in regions of the nervous system associated with migraine pathophysiology, and studies have shown that CGRP levels are elevated during migraine attacks. QULIPTA blocks CGRP through a once-daily dose and is available in three strengths – 10 mg, 30 mg and 60 mg.
QULIPTA will be available in early October 2021. AbbVie believes people living with migraine should have access to all new medications for this debilitating disease. Visit www.QULIPTA.com for more information.
What is QULIPTA?
QULIPTA is a prescription medicine used for the preventive treatment of episodic migraine in adults.
IMPORTANT SAFETY INFORMATION
Before taking QULIPTA, tell your healthcare provider about all your medical conditions, including if you:
Please see full Prescribing Information.
For more information about AbbVie, please visit us at www.abbvie.com
Sep. 29, 2021 1:53 AM ET AbbVie Inc. (ABBV)
By: Mamta Mayani, SA News Editor14 Comments
September 27, 2021
– Antibody-Drug Conjugate Trodelvy is First Treatment to Show Survival Benefit versus Standard of Care in Metastatic Triple-Negative Breast Cancer –
– Project Orbis is a Collaborative Review Program Intended for High-Impact Oncology Products –
– Canada Joins Australia, Great Britain, Switzerland, and the United States in Approval of Trodelvy as a Second-Line Treatment Option for Adults with Metastatic TNBC –
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced that Health Canada has approved Trodelvy® (sacituzumab govitecan-hziy) for the treatment of adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (TNBC) who have received two or more prior therapies, at least one of them for metastatic disease. Canada joins Australia, Great Britain, Switzerland, and the United States among the countries that have approved Trodelvy for use under Project Orbis. Project Orbis is an initiative of the U.S. Food and Drug Administration (FDA) Oncology Center of Excellence (OCE) with international regulatory authorities as a global collaborative review program for high impact oncology marketing applications across participating countries.
Trodelvy is a first-in-class Trop-2 directed antibody-drug conjugate. Trop-2, a protein located on the surface of cells, is overexpressed in TNBC as well as other solid tumors. Beyond the Project Orbis regulatory approvals, the European Medicines Agency validated a Marketing Authorization Application for Trodelvy in March and regulatory review is also underway in Kazakhstan and Saudi Arabia, as well as Singapore via licensing partner, Everest Medicines.
“Because Trodelvy is the first and only targeted treatment to show benefit in overall survival in 2L metastatic TNBC versus chemotherapy, ensuring that it is accessible to eligible patients is imperative,” said Merdad Parsey, MD, PhD, Chief Medical Officer, Gilead Sciences. “We pursued innovative regulatory pathways, such as those made possible by Project Orbis, to help make Trodelvy available to patients as rapidly as possible.”
These approvals were supported by data from the Phase 3 ASCENT study, in which Trodelvy showed a statistically significant and clinically meaningful 57% reduction in the risk of disease worsening or death (progression-free survival (PFS)) and improved median PFS in patients regardless of brain metastasis to 4.8 months from 1.7 months with chemotherapy (HR: 0.43; 95% CI: 0.35-0.54; p<0.0001). Trodelvy also improved median overall survival to 11.8 months versus 6.9 months with chemotherapy (HR: 0.51; 95% CI: 0.41-0.62; p<0.0001), representing a 49% reduction in the risk of death. In the study of 2L+ TNBC patients, the most frequent Grade ≥3 treatment-related adverse events compared to single-agent chemotherapy were neutropenia (52% versus 34%), diarrhea (11% versus 1%), leukopenia (11% versus 6%) and anemia (9% versus 6%). The Trodelvy U.S. Prescribing Information has a BOXED WARNING for severe or life-threatening neutropenia and severe diarrhea; see below for Important Safety Information.
About the ASCENT Study
The ASCENT study is a global, open-label, randomized Phase 3 study that enrolled more than 500 patients across 230 study locations. The study evaluated the efficacy and safety of Trodelvy compared with a single-agent chemotherapy of the physician’s choice in patients with unresectable, locally advanced or metastatic TNBC who had received at least two prior systemic treatments. Patients were randomized to receive either Trodelvy or a chemotherapy chosen by the patients’ treating physicians. The primary endpoint was progression-free survival (PFS, as determined by blinded independent central review) in patients without brain metastases. Secondary endpoints included: PFS for full study population or intention-to-treat (ITT) population, overall survival in both the ITT population and in the subgroup without brain metastasis, independently determined objective response rate, duration of response, time to onset of response according to Response Evaluation Criteria in Solid Tumors (RECIST 1.1), quality of life and safety. More information about ASCENT is available at http://clinicaltrials.gov/show/NCT02574455.
Please see full Prescribing Information, including BOXED WARNING.
About Trodelvy
Trodelvy (sacituzumab govitecan-hziy) is a first-in-class antibody and topoisomerase inhibitor conjugate directed to the Trop-2 receptor, a protein overexpressed in multiple types of epithelial tumors, including metastatic TNBC and metastatic urothelial cancer (UC), where high expression is associated with poor survival and relapse. Beyond the approvals of Trodelvy in the United States, it is also approved for metastatic TNBC in Australia, Canada, Great Britain and Switzerland for adults with metastatic TNBC. Trodelvy is also under multiple regulatory reviews worldwide, including the EU, as well as in Singapore through our partner Everest Medicines. Trodelvy is also being developed as an investigational treatment for hormone receptor-positive/human epidermal growth factor receptor 2-negative (HR+/HER2-) metastatic breast cancer and metastatic non-small cell lung cancer. Additional evaluation across multiple solid tumors is also underway.
In the United States, Trodelvy is indicated for the treatment of:
U.S. Prescribing Information for Trodelvy including BOXED WARNING, is available at www.gilead.com.
Trodelvy, Gilead and the Gilead logo are trademarks of Gilead Sciences, Inc., or its related companies.
For more information about Gilead, please visit the company’s website at www.gilead.com, follow Gilead on Twitter (@GileadSciences) or call Gilead Public Affairs at 1-800-GILEAD-5 or 1-650-574-3000.
View source version on businesswire.com: https://www.businesswire.com/news/home/20210927005700/en/
Source: Gilead Sciences, Inc.
Sep. 27, 2021 4:43 PM ET Gilead Sciences, Inc. (GILD) By: Aakash Babu, SA News Editor
Foster City, Calif., September 27, 2021 – Gilead Sciences issued the following statement on the recent update to the NCCN Guidelines® for Breast Cancer. The updated NCCN Guidelines include sacituzumab govitecan-hziy (Trodelvy®) as a preferred regimen1 for the treatment of adult patients with metastatic triple-negative breast cancer (TNBC) who received at least two prior therapies, with at least one line for metastatic disease. The NCCN Guidelines now recommend sacituzumab govitecan-hziy in second-line and subsequent therap
TARRYTOWN, N.Y., Sept. 28, 2021 /PRNewswire/ --
European Union regulatory submission planned by end of 2021
Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) today announced that the U.S. Food and Drug Administration (FDA) has accepted for priority review the supplemental Biologics License Application (sBLA) for PD-1 inhibitor Libtayo® (cemiplimab-rwlc) to treat patients with recurrent or metastatic cervical cancer whose disease progressed on or after chemotherapy. The target action date for the FDA decision is January 30, 2022. The sBLA is also being reviewed under the FDA's Project Orbis initiative, which will allow for concurrent review by participating health authorities in Australia, Brazil, Canada and Switzerland. Additional global regulatory submissions are planned, including in the European Union (EU) by the end of 2021.
The sBLA is supported by results from a Phase 3 trial that enrolled patients irrespective of
PD-L1 expression status and is being conducted with The GOG Foundation, Inc. (GOG), the European Network for Gynaecological Oncological Trial groups (ENGOT) and NRG Oncology-Japan. Detailed results were first presented as part of a European Society for Medical Oncology (ESMO) Virtual Plenary in May 2021.
Cervical cancer is the fourth leading cause of cancer death in women worldwide and is most frequently diagnosed in those between the ages of 35 and 44. Almost all cases are caused by human papillomavirus (HPV) infection, with approximately 80% classified as squamous cell carcinoma (SCC; arising from cells lining the bottom of the cervix) and the remainder being largely adenocarcinomas (arising from glandular cells in the upper cervix). Cervical cancer is often curable when detected early and effectively managed, but treatment options are more limited in advanced stages.
It is estimated that approximately 570,000 people are diagnosed with cervical cancer worldwide each year, with deaths exceeding 250,000. In the U.S. there are 14,500 new cases diagnosed annually and approximately 4,000 die each year.
Libtayo, which was invented using Regeneron's proprietary VelocImmune® technology, is being jointly developed by Regeneron and Sanofi under a global collaboration agreement. The use of Libtayo in advanced cervical cancer is investigational, and its safety and efficacy have not been fully evaluated by any regulatory authority.
About the Phase 3 Trial
The Phase 3, open-label, multi-center trial, known as EMPOWER-Cervical 1, is the largest randomized clinical trial in advanced cervical cancer. The trial investigated Libtayo monotherapy versus an investigator's choice of chemotherapy in patients with recurrent or metastatic cervical cancer who had progressed on platinum-based chemotherapy. Patients were enrolled regardless of tumor PD-L1 expression status or histology in 14 countries, including the U.S., Japan, Taiwan, South Korea, Canada, Russia, Poland, Spain, Brazil, Australia, the UK, Italy, Greece and Belgium.
Patients (median age: 51 years) were randomized to receive Libtayo monotherapy (350 mg every three weeks) or commonly used chemotherapy regimens (pemetrexed, vinorelbine, topotecan, irinotecan or gemcitabine). The primary endpoint for the trial was overall survival, analyzed first among patients with SCC histology, then in the total population.
About Libtayo
Libtayo is a fully human monoclonal antibody targeting the PD-1 immune checkpoint receptor on T-cells. By binding to PD-1, Libtayo has been shown to block cancer cells from using the PD-1 pathway to suppress T-cell activation.
The extensive clinical program for Libtayo is focused on difficult-to-treat cancers. Libtayo is currently being investigated in trials as a monotherapy, as well as in combination with either conventional or novel therapeutic approaches for other solid tumors and blood cancers. These potential uses are investigational, and their safety and efficacy have not been evaluated by any regulatory authority.
What is Libtayo?
Libtayo is a prescription medicine used to treat people with a type of skin cancer called cutaneous squamous cell carcinoma (CSCC) that has spread or cannot be cured by surgery or radiation.
Libtayo is a prescription medicine used to treat people with a type of skin cancer called basal cell carcinoma that cannot be removed by surgery (locally advanced BCC) and have received treatment with a hedgehog inhibitor (HHI), or cannot receive treatment with an HHI.
Libtayo is a prescription medicine used to treat people with a type of skin cancer called basal cell carcinoma that has spread (metastatic BCC) and have received treatment with an HHI, or cannot receive treatment with an HHI. This use is approved based on how many patients responded to treatment and how long they responded. Studies are ongoing to provide additional information about clinical benefit.
Libtayo is a prescription medicine used to treat people with a type of lung cancer called non-small cell lung cancer (NSCLC). Libtayo may be used as your first treatment when your lung cancer has not spread outside your chest (locally advanced lung cancer) and you cannot have surgery or chemotherapy with radiation, or your lung cancer has spread to other areas of your body (metastatic lung cancer), and your tumor tests positive for high "PD-L1" and your tumor does not have an abnormal "EGFR"," ALK "or "ROS1" gene.
It is not known if Libtayo is safe and effective in children.
Please see full Prescribing Information, including Medication Guide.
For additional information about the company, please visit www.regeneron.com
View original content:https://www.prnewswire.com/news-releases/fda-accepts-libtayo-cemiplimab-rwlc-for-priority-review-for-advanced-cervical-cancer-301386422.html
SOURCE Regeneron Pharmaceuticals, Inc.
Sep. 28, 2021 7:41 AM ET
Regeneron Pharmaceuticals, Inc. (REGN)
By: Mamta Mayani, SA News Editor
PUBLISHED28 September 2021
AstraZeneca’s Saphnelo (anifrolumab) has been approved in Japan for the treatment of adult patients with systemic lupus erythematosus (SLE), a serious autoimmune disease, who show insufficient response to currently available treatment.
The approval by the Japanese Ministry of Health, Labour and Welfare (MHLW) was based on efficacy and safety data from the Saphnelo clinical development programme, including the TULIP Phase III trials and the MUSE Phase II trial. In these trials, more patients treated with Saphnelo experienced a reduction in overall disease activity across organ systems, including skin and joints, and achieved sustained reduction in oral corticosteroid (OCS) use compared to placebo, with both groups receiving standard therapy.1,2,3
This decision marks the first regulatory approval by the MHLW for a type I interferon (type I IFN) receptor antagonist in Japan. Type I IFN plays a central role in the pathophysiology in lupus and increased type I IFN signalling has been shown to be associated with increased disease activity and severity.4,5,6,7,8
Dr. Yoshiya Tanaka, Professor in the First Department of Internal Medicine at the University of Occupational and Environmental Health, Kitakyushu, Japan and an investigator in the TULIP-2 trial, said: “Compared with other rheumatic diseases, there are limited treatments available for systemic lupus erythematosus and outcomes remain poor for patients in Japan and around the world. Through our own local experience with anifrolumab in clinical trials, we have observed impressive efficacy and improved patient outcomes.”
Dr. Tsutomu Takeuchi, Emeritus Professor of Keio University, Tokyo, Japan and an investigator in the TULIP-2 trial, said: “Our treatment goals in systemic lupus erythematosus are to reduce disease activity, improve quality of life and prevent organ damage from either the disease or the medications used to treat it, especially corticosteroids. Innovative treatments are needed to address these goals. The anifrolumab clinical programme provided compelling evidence that blocking type I interferon is a promising new strategy in the treatment of systemic lupus erythematosus reducing both disease activity across organ systems and corticosteroid use.”
Mene Pangalos, Executive Vice President, BioPharmaceuticals R&D, said: “Saphnelo is the first and only type I interferon receptor antagonist to receive approval in Japan and represents a major advance in treating systemic lupus erythematosus, a complex heterogenous disease that is challenging to treat and often debilitating for patients. We will now work to bring this medicine to patients in Japan living with this disease as quickly as possible.”
The adverse reactions that occurred more frequently in patients who received Saphnelo in clinical trials included upper respiratory tract infection, bronchitis, infusion-related reactions, hypersensitivity reactions and herpes zoster.1,2,3
SLE can affect any organ, and people often experience inadequate disease control, long-term organ damage and poor health-related quality of life.9,10,11,12 SLE is designated as an Intractable Disease in Japan through a programme that incentivises research and development of drugs to treat rare diseases that lack effective treatments, and also helps reduce cost burden on patients.13 There are approximately 60,000 registered patients with SLE in Japan, and the majority are women diagnosed before age 45.14,15
Results from the TULIP-2 Phase III trial were published in The New England Journal of Medicine, results from the TULIP-1 Phase III trial were published in The Lancet Rheumatology and results from the MUSE Phase II trial were published in Arthritis & Rheumatology. The sub-analysis of patients in Japan enrolled in TULIP-2 was presented at the Japan College of Rheumatology annual meeting in April 2021. Efficacy and safety results from the sub-analysis were consistent with the overall trial populations.16
Saphnelo is approved in the US for the treatment of moderate to severe SLE and is under regulatory review for SLE in the EU. A Phase III trial in SLE using subcutaneous delivery has been initiated and additional Phase III trials are planned for lupus nephritis, cutaneous lupus erythematosus and myositis.
https://www.saphnelo.com/hcp.html
TULIP-1, TULIP-2 and MUSE
All three trials for Saphnelo (TULIP-1, TULIP-2 and MUSE) were randomised, double-blinded, placebo-controlled trials in patients with moderate to severe SLE who were receiving standard therapy. Standard therapy included at least one of the following: OCS, antimalarials and immunosuppressants (methotrexate, azathioprine or mycophenolate mofetil).1,2,3
The pivotal TULIP (Treatment of Uncontrolled Lupus via the Interferon Pathway) Phase III programme included two trials, TULIP-1 and TULIP-2, that evaluated the efficacy and safety of Saphnelo versus placebo.1,2 TULIP-2 demonstrated superiority across multiple efficacy endpoints versus placebo with both arms receiving standard therapy. In the trial, 362 eligible patients, including 43 patients in Japan, were randomised (1:1) and received a fixed-dose intravenous infusion of 300mg Saphnelo or placebo every four weeks. TULIP-2 assessed the effect of Saphnelo in reducing disease activity as measured by the BILAG-Based Composite Lupus Assessment (BICLA) scale.1 In TULIP-1, 457 eligible patients were randomised (1:2:2) and received a fixed-dose intravenous infusion of 150mg Saphnelo, 300mg Saphnelo or placebo every four weeks, in addition to standard therapy. The trial did not meet its primary endpoint based on the SLE Responder Index 4 (SRI4) composite measure.2
The MUSE Phase II trial evaluated the efficacy and safety of two doses of Saphnelo versus placebo. In MUSE, 305 adults were randomised and received a fixed-dose intravenous infusion of 300mg Saphnelo, 1,000mg Saphnelo or placebo every four weeks, in addition to standard therapy, for 48 weeks. The trial showed improvement versus placebo across multiple efficacy endpoints with both arms receiving standard therapy.3
In SLE, along with the pivotal TULIP Phase III programme, Saphnelo continues to be evaluated in a long-term extension Phase III trial and a Phase III trial assessing subcutaneous delivery.21,22 In addition, AstraZeneca is exploring the potential of Saphnelo in a variety of diseases where type I IFN plays a key role, including lupus nephritis, cutaneous lupus erythematosus and myositis.
Saphnelo
Saphnelo (anifrolumab) is a fully human monoclonal antibody that binds to subunit 1 of the type I IFN receptor, blocking the activity of type I IFN.3 Type I IFNs such as IFN-alpha, IFN-beta and IFN-kappa are cytokines involved in regulating the inflammatory pathways implicated in SLE.4,6,7,8,23,24 The majority of adults with SLE have increased type I IFN signalling, which is associated with increased disease activity and severity.4,5
Please visit astrazeneca.com
Sep. 28, 2021 6:11 AM ET AstraZeneca PLC (AZN)
By: Mamta Mayani, SA News Editor
September 27, 2021 6:45 am ET
In Phase 3 KEYNOTE-394 Study, KEYTRUDA Showed Statistically Significant Improvement in OS Versus Placebo
KENILWORTH, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, today announced that the Phase 3 KEYNOTE-394 trial investigating KEYTRUDA, Merck’s anti-PD-1 therapy, in Asian patients with advanced hepatocellular carcinoma (HCC) previously treated with sorafenib met its primary endpoint of overall survival (OS). The study found that treatment with KEYTRUDA plus best supportive care resulted in a statistically significant improvement in OS compared with placebo plus best supportive care. KEYNOTE-394 also met its key secondary endpoints of progression-free survival (PFS) and objective response rate (ORR), with statistically significant improvements for KEYTRUDA compared with placebo. No new safety signals were observed. These results will be presented at an upcoming medical meeting.
“Frequently diagnosed at an advanced stage, hepatocellular carcinoma has one of the highest mortality rates of solid cancers. Despite recent progress, there remains an unmet need for anti-PD-1 monotherapy after sorafenib, where KEYTRUDA is an established treatment option for patients,” said Dr. Scot Ebbinghaus, vice president, clinical research, Merck Research Laboratories. “It is very encouraging that KEYTRUDA significantly improved overall survival in this study, and we look forward to engaging with regulatory authorities as quickly as possible.”
KEYTRUDA was granted accelerated approval in November 2018 for the treatment of patients with HCC who have been previously treated with sorafenib, based on ORR and durability of response data from KEYNOTE-224. A subsequent study, KEYNOTE-240, did not meet its dual primary endpoints of OS and PFS. This accelerated approval for KEYTRUDA was discussed during the FDA’s Oncologic Drugs Advisory Committee (ODAC) meeting on April 29, 2021 that voted 8-0 in favor of maintaining accelerated approval of KEYTRUDA for this indication. KEYNOTE-394 was discussed at the ODAC meeting as a potential confirmatory trial that could verify the clinical benefit of KEYTRUDA for these patients.
Merck is dedicated to advancing research in HCC and has a global development program of seven clinical trials that have enrolled or are expected to enroll approximately 3,000 patients. In HCC, KEYTRUDA is being studied across multiple settings and lines of therapy as monotherapy and in combination with other treatments, including therapies through our collaborations.
About KEYNOTE-394
KEYNOTE-394 is a randomized, double-blind, Phase 3 trial (ClinicalTrials.gov, NCT03062358) evaluating KEYTRUDA plus best supportive care versus placebo plus best supportive care in Asian patients with advanced HCC previously treated with sorafenib or oxaliplatin chemotherapy. The primary endpoint is OS and secondary endpoints include PFS, ORR, duration of response and disease control rate. The study enrolled 453 patients who were randomized to receive either KEYTRUDA (intravenously every three weeks for up to 35 cycles of treatment [up to approximately two years]) plus best supportive care (including pain management and management of other potential complications including ascites per local standards of care) or placebo plus best supportive care.
About KEYTRUDA® (pembrolizumab) Injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of patients with melanoma with involvement of lymph node(s) following complete resection.
Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [tumor proportion score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [combined positive score (CPS ≥1)] as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy. KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Carcinoma
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
KEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with MSI-H central nervous system cancers have not been established.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC).
Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or GEJ (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma.
Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
For more information about our oncology clinical trials, visit www.merck.com/clinicaltrials.
For more information, visit www.merck.com
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf
Source: Merck & Co., Inc.
Sep. 27, 2021 7:48 AM ETMerck & Co., Inc. (MRK)By: Dulan Lokuwithana, SA News Editor3 Comments
09/27/2021
- Enfortumab vedotin is the first and only antibody-drug conjugate (ADC) approved in Japan for patients with advanced urothelial cancer -
TOKYO & BOTHELL, Wash.--(BUSINESS WIRE)-- Astellas Pharma Inc. (TSE:4503, President and CEO: Kenji Yasukawa, Ph.D., “Astellas”) and Seagen Inc. (Nasdaq:SGEN) today announced that Japan's Ministry of Health, Labour and Welfare (MHLW) has approved PADCEV®(enfortumab vedotin) for radically unresectable urothelial carcinoma that has progressed after anti-cancer chemotherapy. The New Drug Application received priority review.
Radically unresectable urothelial carcinoma is urothelial cancer that cannot be treated by surgical removal of the urinary bladder or the kidney and the ureter due to tumor growth.
“Unfortunately, advanced urothelial cancer has a relatively poor prognosis and can be challenging to treat with currently available therapies,” said Andrew Krivoshik, M.D., Ph.D., Senior Vice President and Head of Development Therapeutic Areas, Astellas. “The MHLW’s review of enfortumab vedotin in just six months, supported by overall survival data from a pivotal Phase 3 clinical trial, reflects the seriousness of this condition and the potential benefit of enfortumab vedotin for patients in Japan.”
The approval is primarily based on the global Phase 3 EV-301 clinical trial, which included sites in Japan. The trial evaluated enfortumab vedotin versus chemotherapy in adult patients with locally advanced or metastatic urothelial cancer who were previously treated with platinum-based chemotherapy and a PD-1/L1 inhibitor. At the time of pre-specified interim analysis, patients who received enfortumab vedotin (n=301) in the trial lived a median of 3.9 months longer than those who received chemotherapy (n=307). Median overall survival was 12.9 vs. 9.0 months, respectively [Hazard Ratio=0.70 (95% Confidence Interval [CI]: 0.56, 0.89), p=0.001]. The most common (≥20%) adverse reactions included alopecia, peripheral sensory neuropathy, pruritus, fatigue, decreased appetite, diarrhea, dysgeusia and nausea.
Each year in Japan, more than 24,300 people are diagnosed with bladder cancer and an estimated 9,500 die from the disease.1 Enfortumab vedotin is the subject of a robust clinical development program aimed at addressing unmet needs across the continuum of urothelial cancer and in other solid tumors.
About the EV-301 Trial
The EV-301 trial (NCT03474107) was a global, multicenter, open-label, randomized Phase 3 trial designed to evaluate enfortumab vedotin versus physician's choice of chemotherapy (docetaxel, paclitaxel or vinflunine) in 608 patients with locally advanced or metastatic urothelial cancer who were previously treated with a PD-1/L1 inhibitor and platinum-based therapies. The primary endpoint was overall survival, and secondary endpoints included progression-free survival, overall response rate, duration of response and disease control rate, as well as assessment of safety/tolerability and quality-of-life parameters. Results were published in the New England Journal of Medicine.
About Enfortumab Vedotin
Enfortumab vedotin is an antibody-drug conjugate (ADC) that is directed against Nectin-4, a protein located on the surface of cells and highly expressed in bladder cancer.3,4 Nonclinical data suggest the anticancer activity of enfortumab vedotin is due to its binding to Nectin-4 expressing cells followed by the internalization and release of the anti-tumor agent monomethyl auristatin E (MMAE) into the cell, which result in the cell not reproducing (cell cycle arrest) and in programmed cell death (apoptosis).4 PADCEV is co-developed by Astellas and Seagen.
For more information, please visit our website at https://www.astellas.com/en.
For more information on our marketed products and robust pipeline, visit www.seagen.com
About the Astellas and Seagen Collaboration
Astellas and Seagen are co-developing enfortumab vedotin under a 50:50 worldwide development and commercialization collaboration. In the United States, Astellas and Seagen co-promote enfortumab vedotin. In the Americas outside the US, Seagen holds responsibility for commercialization activities and regulatory filings. Outside of the Americas, including in Japan, Astellas holds responsibility for commercialization activities and regulatory filings.
View source version on businesswire.com: https://www.businesswire.com/news/home/20210927005268/en/
Source: Seagen Inc.
Sep. 27, 2021 3:36 AM ET Seagen Inc. (SGEN),
ALPMY Astellas Pharma Inc. (ALPMF)
By: Mamta Mayani, SA News Editor
PUBLISHED24 September 2021
Positive high-level results from the PROpel Phase III trial showed AstraZeneca and MSD’s Lynparza (olaparib) in combination with abiraterone demonstrated a statistically significant and clinically meaningful improvement in radiographic progression-free survival (rPFS) versus standard-of-care abiraterone as a 1st-line treatment for men with metastatic castration-resistant prostate cancer (mCRPC) with or without homologous recombination repair (HRR) gene mutations.
At a planned interim analysis, the Independent Data Monitoring Committee (IDMC) concluded that the trial met the primary endpoint of rPFS in men with mCRPC who had not received treatment in the 1st-line setting including with new hormonal agents (NHAs) or chemotherapy.
The trial also showed a trend at this interim analysis towards improved overall survival (OS). However, the data are still immature and the trial will continue to assess OS as a key secondary endpoint. The safety and tolerability were consistent with the known profiles of each medicine.
Prostate cancer is the second-most common cancer in men and despite an increase in the number of available treatments for men with mCRPC, five-year survival remains low.1
Susan Galbraith, Executive Vice President, Oncology R&D, said: “Today, men with metastatic castration-resistant prostate cancer have limited options in the 1st-line setting, and sadly often the disease progresses after initial treatment with current standards of care. These exciting results demonstrate the potential for Lynparza with abiraterone to become a new 1st-line option for patients regardless of their biomarker status and reach a broad population of patients living with this aggressive disease. We look forward to discussing the results with global health authorities as soon as possible.”
Roy Baynes, Senior Vice President and Head of Global Clinical Development, Chief Medical Officer, MSD Research Laboratories, said: “We are encouraged by the PROpel results and the clinical benefit Lynparza in combination with abiraterone demonstrated versus abiraterone alone as a 1st-line treatment option for men with metastatic castration-resistant prostate cancer. Today’s results build on MSD and AstraZeneca’s commitment to bring Lynparza earlier in lines of treatment and to more patients with advanced prostate cancer.”
The data will be presented at an upcoming medical meeting.
Metastatic castration-resistant prostate cancer
Metastatic prostate cancer is associated with a significant mortality rate.2 Development of prostate cancer is often driven by male sex hormones called androgens, including testosterone.3
In patients with mCRPC, their prostate cancer grows and spreads to other parts of the body despite the use of androgen-deprivation therapy to block the action of male sex hormones.3 Approximately 10-20% of men with advanced prostate cancer will develop CRPC within five years, and at least 84% of these men will have metastases at the time of CRPC diagnosis.4
Of men with no metastases at CRPC diagnosis, 33% are likely to develop metastases within two years.4 Despite advances in treatment for men with mCRPC, five-year survival is low and extending survival remains a key treatment goal.4
Lynparza
Lynparza (olaparib) is a first-in-class PARP inhibitor and the first targeted treatment to block DNA damage response (DDR) in cells/tumours harbouring a deficiency in homologous recombination repair (HRR), such as those with mutations in BRCA1 and/or BRCA2, or those where deficiency is induced by other agents (such as NHAs).
Inhibition of PARP with Lynparza leads to the trapping of PARP bound to DNA single-strand breaks, stalling of replication forks, their collapse and the generation of DNA double-strand breaks and cancer cell death. Lynparza is being tested in a range of PARP-dependent tumour types with defects and dependencies in the DDR pathway.
Lynparza is currently approved in a number of countries, including those in the EU, for the maintenance treatment of platinum-sensitive relapsed ovarian cancer. It is approved in the US, the EU, Japan, China, and several other countries as 1st-line maintenance treatment of BRCA-mutated advanced ovarian cancer following response to platinum-based chemotherapy.
It is also approved in the US, EU and Japan as a 1st-line maintenance treatment with bevacizumab for patients with HRD-positive advanced ovarian cancer (BRCAm and/or genomic instability).
Lynparza is approved in the US, Japan, and a number of other countries for germline BRCA-mutated, HER2-negative, metastatic breast cancer, previously treated with chemotherapy; in the EU, this includes locally advanced breast cancer.
It is also approved in the US, the EU, Japan and several other countries for the treatment of germline BRCAm metastatic pancreatic cancer. Lynparza is approved in the US for HRR gene-mutated metastatic castration-resistant prostate cancer (BRCAm and other HRR gene mutations) and in the EU and Japan for BRCAm metastatic castration-resistant prostate cancer.
Regulatory reviews are underway in several countries for ovarian, breast, pancreatic and prostate cancers.
Lynparza, which is being jointly developed and commercialised by AstraZeneca and MSD, has been used to treat over 40,000 patients worldwide. Lynparza has the broadest and most advanced clinical trial development programme of any PARP inhibitor, and AstraZeneca and MSD are working together to understand how it may affect multiple PARP-dependent tumours as a monotherapy and in combination across multiple cancer types.
Lynparza is the foundation of AstraZeneca's industry-leading portfolio of potential new medicines targeting DDR mechanisms in cancer cells.
The AstraZeneca and MSD strategic oncology collaboration
In July 2017, AstraZeneca and Merck & Co., Inc., Kenilworth, NJ, US, known as MSD outside the US and Canada, announced a global strategic oncology collaboration to co-develop and co-commercialise Lynparza, the world’s first PARP inhibitor, and Koselugo (selumetinib), a mitogen-activated protein kinase (MEK) inhibitor, for multiple cancer types.
Working together, the companies will develop Lynparza and Koselugo in combination with other potential new medicines and as monotherapies. Independently, the companies will develop Lynparza and Koselugo in combination with their respective PD-L1 and PD-1 medicines.
Please visit astrazeneca.com
Sep. 24, 2021 4:20 AM ET AstraZeneca PLC (AZN) Merck & Co., Inc. (MRK)By: Mamta Mayani, SA News Editor
09/27/2021CATEGORY:
Applications based on Phase 3 CheckMate -648 trial, in which both Opdivo-based combinations demonstrated a significant overall survival benefit over chemotherapy alone
The U.S. Food and Drug Administration assigned a target action date of May 28, 2022
CheckMate -648 is one of four positive Phase 3 trials in which Opdivo or Opdivo-based treatment combinations demonstrated a significant benefit for patients with upper gastrointestinal cancers
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) today announced that the U.S. Food and Drug Administration (FDA) has accepted the supplemental Biologics License Applications (sBLA) for both Opdivo® (nivolumab) in combination with Yervoy (ipilimumab) and Opdivo in combination with fluoropyrimidine- and platinum-containing chemotherapy as first-line treatments for adult patients with unresectable advanced, recurrent or metastatic esophageal squamous cell carcinoma (ESCC), based on results from the CheckMate -648 trial. The FDA assigned a Prescription Drug User Fee Act (PDUFA) goal date of May 28, 2022.
“Last year, over 19,000 people were diagnosed with esophageal cancer in the United States, and 15,000 people died as a result of this very aggressive disease,” said Ian M. Waxman, M.D., development lead, gastrointestinal cancers, Bristol Myers Squibb. “Additional treatment options are needed to improve upon outcomes achieved with the current standard of care. We are confident that our immunotherapy-based combinations can provide further clinical benefit and address this critical need.”
The filings were based on results from the pivotal Phase 3 CheckMate -648 trial, marking the fourth global trial in which Opdivo or Opdivo-based treatment combinations demonstrated a significant benefit for patients with upper gastrointestinal cancers. In CheckMate -648, both Opdivo-based treatment combinations — Opdivo plus Yervoy and Opdivo plus chemotherapy — demonstrated a statistically significant and clinically meaningful overall survival (OS) benefit compared to chemotherapy in patients with unresectable advanced or metastatic ESCC with tumor cell PD-L1 expression ≥1%, as well as in the all-randomized population. Opdivo plus Yervoy is the first dual immunotherapy combination to demonstrate a superior survival benefit versus chemotherapy in this setting.The safety profiles of Opdivo plus Yervoy and Opdivo plus chemotherapy were consistent with the known safety profiles of the individual components.
Results from CheckMate -648 were presented during the 2021 American Society of Clinical Oncology (ASCO) Annual Meeting and were selected for the official ASCO press program.
About CheckMate -648
CheckMate -648 is a randomized Phase 3 study evaluating Opdivo plus Yervoy or Opdivo plus fluorouracil and cisplatin against fluorouracil plus cisplatin alone in patients with unresectable advanced or metastatic esophageal squamous cell carcinoma.
The primary endpoints of the trial are overall survival (OS) and progression-free survival (PFS) by blinded independent central review (BICR) in patients whose tumors express PD-L1 ≥1% for both Opdivo-based combinations versus chemotherapy. Secondary endpoints of the trial include OS and PFS by BICR in the all-randomized population.
In the Opdivo plus Yervoy arm, patients received treatment with Opdivo 3 mg/kg every 2 weeks and Yervoy 1 mg/kg every 6 weeks up to 24 months or until disease progression or unacceptable toxicity.
In the Opdivo plus chemotherapy arm, patients received treatment with Opdivo 240 mg on Day 1 and Day 15, fluorouracil 800 mg/m²/day on Day 1 through Day 5 (for 5 days), and cisplatin 80 mg/m² on Day 1 of four-week cycle. Patients received Opdivo for up to 24 months or until disease progression or unacceptable toxicity, and chemotherapy until disease progression or unacceptable toxicity.
About Opdivo
Opdivo is a programmed death-1 (PD-1) immune checkpoint inhibitor that is designed to uniquely harness the body’s own immune system to help restore anti-tumor immune response. By harnessing the body’s own immune system to fight cancer, Opdivo has become an important treatment option across multiple cancers.
Opdivo’s leading global development program is based on Bristol Myers Squibb’s scientific expertise in the field of Immuno-Oncology and includes a broad range of clinical trials across all phases, including Phase 3, in a variety of tumor types. To date, the Opdivo clinical development program has treated more than 35,000 patients. The Opdivo trials have contributed to gaining a deeper understanding of the potential role of biomarkers in patient care, particularly regarding how patients may benefit from Opdivo across the continuum of PD-L1 expression.
In July 2014, Opdivo was the first PD-1 immune checkpoint inhibitor to receive regulatory approval anywhere in the world. Opdivo is currently approved in more than 65 countries, including the United States, the European Union, Japan, and China. In October 2015, the Company’s Opdivo and Yervoy combination regimen was the first Immuno-Oncology combination to receive regulatory approval for the treatment of metastatic melanoma and is currently approved in more than 50 countries, including the United States and the European Union.
About Yervoy
Yervoy is a recombinant, human monoclonal antibody that binds to the cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4). CTLA-4 is a negative regulator of T-cell activity. Yervoy binds to CTLA-4 and blocks the interaction of CTLA-4 with its ligands, CD80/CD86. Blockade of CTLA-4 has been shown to augment T-cell activation and proliferation, including the activation and proliferation of tumor infiltrating T-effector cells. Inhibition of CTLA-4 signaling can also reduce T-regulatory cell function, which may contribute to a general increase in T-cell responsiveness, including the anti-tumor immune response. On March 25, 2011, the U.S. Food and Drug Administration (FDA) approved Yervoy 3 mg/kg monotherapy for patients with unresectable or metastatic melanoma. Yervoy is approved for unresectable or metastatic melanoma in more than 50 countries. There is a broad, ongoing development program in place for Yervoy spanning multiple tumor types.
INDICATIONS
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of patients with unresectable or metastatic melanoma.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of patients with unresectable or metastatic melanoma.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) whose tumors express PD-L1 (≥1%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab) and 2 cycles of platinum-doublet chemotherapy, is indicated for the first-line treatment of adult patients with metastatic or recurrent non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab) is indicated for the treatment of patients with metastatic non-small cell lung cancer (NSCLC) with progression on or after platinum-based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving OPDIVO.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with unresectable malignant pleural mesothelioma (MPM).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of patients with intermediate or poor risk advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab), in combination with cabozantinib, is indicated for the first-line treatment of patients with advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab) is indicated for the treatment of patients with advanced renal cell carcinoma (RCC) who have received prior anti-angiogenic therapy.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with classical Hodgkin lymphoma (cHL) that has relapsed or progressed after autologous hematopoietic stem cell transplantation (HSCT) and brentuximab vedotin or after 3 or more lines of systemic therapy that includes autologous HSCT. This indication is approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab) is indicated for the treatment of patients with recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN) with disease progression on or after platinum-based therapy.
OPDIVO® (nivolumab) is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
OPDIVO® (nivolumab), as a single agent, is indicated for the adjuvant treatment of patients with urothelial carcinoma (UC) who are at high risk of recurrence after undergoing radical resection of UC.
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of adult and pediatric (12 years and older) patients with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adults and pediatric patients 12 years and older with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of patients with melanoma with involvement of lymph nodes or metastatic disease who have undergone complete resection.
OPDIVO® (nivolumab) is indicated for the treatment of patients with unresectable advanced, recurrent or metastatic esophageal squamous cell carcinoma (ESCC) after prior fluoropyrimidine- and platinum-based chemotherapy.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of completely resected esophageal or gastroesophageal junction cancer with residual pathologic disease in patients who have received neoadjuvant chemoradiotherapy (CRT).
OPDIVO® (nivolumab), in combination with fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the treatment of patients with advanced or metastatic gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma.
Please see US Full Prescribing Information for OPDIVO and YERVOY
For more information about Bristol Myers Squibb, visit us at BMS.com
Source: Bristol Myers Squibb
Sep. 27, 2021 7:54 AM ET Bristol-Myers Squibb Company (BMY) By: Aakash Babu, SA News Editor
September 22, 2021 DownloadPDF Format (opens in new window)
- Jakafi is approved for treatment of chronic GVHD after failure of one or two lines of systemic therapy in adult and pediatric patients 12 years and older
- Today’s approval marks the fourth FDA-approved indication for Jakafi, which received FDA-approval in 2019 for steroid-refractory acute GVHD in adult and pediatric patients 12 years and older
WILMINGTON, Del.--(BUSINESS WIRE)-- Incyte (Nasdaq:INCY) today announced that the U.S. Food and Drug Administration (FDA) has approved Jakafi® (ruxolitinib) for treatment of chronic graft-versus-host disease (GVHD) after failure of one or two lines of systemic therapy in adult and pediatric patients 12 years and older.
“GVHD is the leading cause of morbidity and mortality in patients following an allogeneic stem cell transplant, yet there historically have been limited treatment options available beyond first-line systemic therapies,” stated Steven Stein, M.D., Chief Medical Officer, Incyte. “Incyte is proud to have contributed to the overall scientific understanding of GVHD through our REACH program, which has led to important treatment advances on behalf of patients and the medical community, including today’s approval of Jakafi for certain people who develop chronic GVHD.”
The FDA approval was based on the REACH3 study, a Phase 3, randomized, open-label, multicenter study of Jakafi in comparison to best available therapy (BAT) for treatment of steroid-refractory chronic GVHD after allogeneic stem cell transplantation. The primary endpoint of overall response rate (ORR) at Week 24 (i.e., Cycle 7 Day 1) was 49.7% for Jakafi compared to 25.6% for BAT (P<0.0001)1. Furthermore, the ORR through Cycle 7 Day 1 was 70% for Jakafi compared to 57% for BAT2. The most common hematologic adverse reactions (incidence > 35%) were anemia and thrombocytopenia. The most common nonhematologic adverse reactions (incidence ≥ 20%) were infections (pathogen not specified) and viral infection. Full results from the REACH3 study were published in the New England Journal of Medicine (NEJM).
Incyte is committed to supporting patients and removing barriers to access medicines. Eligible patients in the U.S. who are prescribed Jakafi have access to IncyteCARES (Connecting to Access, Reimbursement, Education and Support), a comprehensive program offering patient support, including financial assistance and ongoing education and resources to eligible patients. More information about IncyteCARES is available by visiting www.incytecares.com or calling 1-855-452-5234.
About REACH3
REACH3 (NCT03112603), a randomized, open-label, multicenter Phase 3 study co-sponsored by Novartis and Incyte, evaluated the safety and efficacy of ruxolitinib compared with best available therapy (BAT) in patients with steroid-refractory chronic GVHD.
For more information about the study, please visit https://clinicaltrials.gov/ct2/show/NCT03112603.
About Jakafi® (ruxolitinib)
Jakafi is a JAK1/JAK2 inhibitor approved by the U.S. FDA for treatment of chronic GVHD after failure of one or two lines of systemic therapy in adult and pediatric patients 12 years and older.
Jakafi is also indicated for treatment of polycythemia vera (PV) in adults who have had an inadequate response to or are intolerant of hydroxyurea, intermediate or high-risk myelofibrosis (MF), including primary MF, post-polycythemia vera MF and post-essential thrombocythemia MF in adults, and for treatment of steroid-refractory acute GVHD in adult and pediatric patients 12 years and older2.
Jakafi is marketed by Incyte in the U.S. and by Novartis as Jakavi® (ruxolitinib) outside the U.S. Jakafi is a registered trademark of Incyte. Jakavi is a registered trademark of Novartis AG in countries outside the U.S.
For additional information on Incyte, please visit Incyte.com
View source version on businesswire.com: https://www.businesswire.com/news/home/20210922005863/en/
Source: Incyte
Sep. 22, 2021 12:48 PM ET
By: Aakash Babu, SA News Editor3 Comments
09/20/2021
- TIVDAK is a First-in-Class Antibody-Drug Conjugate Directed to Tissue Factor, a Protein Expressed on Cervical Cancer Cells -
- New Monotherapy Approved for Use in a Cancer with Limited Treatment Options -
BOTHELL, Wash. & COPENHAGEN, Denmark--(BUSINESS WIRE)-- Seagen Inc. (Nasdaq: SGEN) and Genmab A/S (Nasdaq: GMAB) today announced that the U.S. Food and Drug Administration (FDA) has granted accelerated approval to TIVDAK™ (tisotumab vedotin-tftv), the first and only approved antibody-drug conjugate (ADC) for the treatment of adult patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy. TIVDAK is approved under the FDA’s Accelerated Approval Program based on tumor response and the durability of the response. Continued approval may be contingent upon verification and description of clinical benefit in confirmatory trials.1
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20210920005921/en/
40 mg vial and carton (Photo: Business Wire)
“Once recurrent or metastatic cervical cancer progresses, there is a need for more options for these patients,” said Robert L. Coleman, M.D., Chief Scientific Officer, US Oncology Research and lead investigator of the innovaTV 204 clinical trial. “This is an important development for patients with recurrent or metastatic cervical cancer.”
In the innovaTV 204 clinical trial, TIVDAK was evaluated in 101 patients with recurrent or metastatic cervical cancer who had received no more than two prior systemic regimens in the recurrent or metastatic setting, including at least one prior platinum-based chemotherapy regimen. Results from the trial showed a 24 percent confirmed objective response rate (ORR) (95% CI; 15.9-33.3), as assessed by an independent review committee (IRC) using Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 criteria. The median duration of response (DOR) was 8.3 months (95% CI; 4.2 to not reached).
“TIVDAK’s approval as a monotherapy in the U.S. is an important milestone for women with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy, as they are in need of a new treatment option and we look forward to making it available to them,” said Jan van de Winkel, Ph.D., Chief Executive Officer, Genmab. “The journey towards the approval of TIVDAK started nearly two decades ago with innovative research by scientists at Genmab and Seagen and reflects on our purpose of making an impact in the lives of cancer patients and their families. Today’s announcement marks Genmab’s evolution into a fully integrated biotechnology company and we would like to thank patients, caregivers, investigators and our collaborators for their participation in our clinical studies.”
The Biologics License Application (BLA) for TIVDAK was submitted in February 2021 and accepted with Priority Review in April 2021. The submission was based on the results of the innovaTV 204 trial.
The FDA’s Accelerated Approval Program allows for approval of a medicine based on a surrogate endpoint that is reasonably likely to predict clinical benefit, if the medicine fills an unmet medical need for a serious condition. A global, randomized phase 3 clinical trial (innovaTV 301) is underway and is also intended to support global registrations.
About TIVDAK (tisotumab vedotin-tftv)
TIVDAK (tisotumab vedotin-tftv) is an ADC composed of Genmab’s human monoclonal antibody directed to tissue factor (TF) and Seagen’s ADC technology that utilizes a protease-cleavable linker that covalently attaches the microtubule-disrupting agent monomethyl auristatin E (MMAE) to the antibody. Nonclinical data suggests that the anticancer activity of TIVDAK is due to the binding of the ADC to TF expressing cancer cells, followed by internalization of the ADC-TF complex, and release of MMAE via proteolytic cleavage. MMAE disrupts the microtubule network of actively dividing cells, leading to cell cycle arrest and apoptotic cell death. In vitro, TIVDAK also mediates antibody-dependent cellular phagocytosis and antibody-dependent cellular cytotoxicity.1
Genmab is headquartered in Copenhagen, Denmark with locations in Utrecht, the Netherlands, Princeton, New Jersey, U.S. and Tokyo, Japan. For more information, please visit Genmab.com
View source version on businesswire.com: https://www.businesswire.com/news/home/20210920005921/en/
Source: Seagen Inc.
Sep. 20, 2021 5:26 PM ET Genmab A/S (GMAB), SGEN By: Jonathan M Block, SA News Editor
September 22, 2021
– Double-Blind Placebo-Controlled Study Evaluated the Efficacy of Early Use of Veklury IV in Non-Hospitalized Patients –
– Late-breaking Data to be Presented at IDWeek 2021 –
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced positive results from a Phase 3 randomized, double-blind, placebo-controlled trial to evaluate the efficacy and safety of a three-day course of Veklury® (remdesivir) for intravenous (IV) use for the treatment of COVID-19 in non-hospitalized patients at high risk for disease progression. This late-breaking data will be presented at the IDWeek 2021 virtual conference.
In an analysis of 562 participants randomly assigned in a 1:1 ratio to receive Veklury or placebo, Veklury demonstrated a statistically significant 87% reduction in risk for the composite primary endpoint of COVID-19 related hospitalization or all-cause death by Day 28 (0.7% [2/279]) compared with placebo (5.3% [15/283]) p=0.008. Results also showed an 81% reduction in risk for the composite secondary endpoint of medical visits due to COVID-19 or all-cause death by Day 28 for participants treated with Veklury (1.6% [4/246]) compared with placebo (8.3% [21/252]) p=0.002. In the study, no deaths were observed in either arm by Day 28.
“Antiviral medications provide maximal benefit when used early in the disease course. Last summer, data from clinical trials demonstrated the benefit of remdesivir in patients hospitalized with COVID-19, even when not yet requiring oxygen. These latest data show remdesivir’s potential to help high-risk patients recover before they get sicker and stay out of the hospital altogether,” said Robert L. Gottlieb, MD, PhD, Cardiologist and Principal Investigator at Baylor University Medical Center and Baylor Scott & White Research Institute. “We are seeing very high numbers of hospitalized patients as new COVID-19 infections surge, placing increased demands on already over-burdened healthcare systems. Remdesivir, also known as Veklury, is an effective antiviral for the treatment of hospitalized patients with COVID-19 and an essential tool to help reduce disease progression.”
The use of Veklury for the treatment of non-hospitalized patients with three days of dosing is investigational, and the safety and efficacy for this use and dosing duration have not been established or approved by any regulatory agency globally. In the United States, Veklury is indicated for adults and pediatric patients (12 years of age and older and weighing at least 40 kg) for the treatment of COVID-19 requiring hospitalization. Veklury is contraindicated in patients who are allergic to Veklury or any of its components; please see below for additional Important Safety Information for Veklury.
About the IV Outpatient Study (GS-US-540-9012)
Study GS-US-540-9012 (PINETREE) was a Phase 3, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of a three-day course of Veklury IV in reducing the rate of hospitalization or all-cause death among non-hospitalized COVID-19 patients at high risk for disease progression. The primary study endpoint was the composite of COVID-19 hospitalization or all-cause death by Day 28. The composite endpoint of medical visits due to COVID-19 or all-cause death by Day 28 was a secondary outcome measure.
The study was designed to enroll 1,264 participants, randomized in a double-blind fashion so that half of enrolled participants would receive Veklury and the other half would receive a matched placebo. Gilead decided to stop the study in April 2021, reflecting the evolution of the COVID-19 landscape and changing patient needs. At the time that enrollment was terminated, 584 participants were enrolled. The study remained blinded and participants already enrolled in the study were followed according to the protocol until the last patient visit occurred, and at that point the study was closed.
This trial was designed to evaluate the potential role of Veklury in helping patients diagnosed with COVID-19 who were considered high-risk for disease progression based on comorbidities and age but had not recently been hospitalized due to the infection. Common comorbidities in study participants included obesity, hypertension, and diabetes. A third of the participants were aged 60 or older. Participants in the study must have received a positive diagnosis within four days of initiating treatment and experienced symptoms for seven days or less.
About Veklury
Veklury (remdesivir) is a nucleotide analog invented by Gilead, building on more than a decade of the company’s antiviral research. Veklury is the antiviral standard of care for the treatment of hospitalized patients with COVID-19. At this time, more than half of patients hospitalized with COVID-19 in the United States are treated with Veklury. Veklury is approved or authorized for temporary use in approximately 50 countries worldwide and generic remdesivir, manufactured by our voluntary licensing partners, is provided to 127 middle- and low-income countries. Veklury and generic remdesivir have been made available to more than seven million patients around the world. Veklury has broad-spectrum antiviral activity both in vitro and in vivo in animal models against multiple emerging viral pathogens, including Ebola, SARS, Marburg, and MERS.
Veklury is a nucleotide analog that directly inhibits viral replication of SARS-CoV-2 by targeting the viral RNA polymerase inside of the cell. On entering the body Veklury is transformed into the active metabolite remdesivir triphosphate, which is then incorporated into the viral RNA and stops replication of the virus within the host cell. No known variations have significantly altered the viral RNA polymerase. All known novel virus variants show mutations at different locations in the SARS-CoV-2 spike protein, which is on the outer surface of the virus and can cause decreased affinity of anti-SARS-CoV-2 antibodies.
Veklury’s antiviral activity has been tested against isolates of variants of concern (VOCs), including Alpha, Beta, Gamma and Delta, and Epsilon, as well as one variant of interest (VOI) of SARS-CoV-2. These laboratory findings suggest that Veklury will continue to be active against the currently identified variations in the SARS-CoV-2 virus.
U.S. Indication for Veklury
Veklury® (remdesivir 100 mg for injection) is indicated for adults and pediatric patients (12 years of age and older and weighing at least 40 kg) for the treatment of COVID-19 requiring hospitalization. Veklury should only be administered in a hospital or in a healthcare setting capable of providing acute care comparable to inpatient hospital care.
U.S. full Prescribing Information for Veklury is available at www.gilead.com.
Veklury, Gilead and the Gilead logo are registered trademarks of Gilead Sciences, Inc., or its related companies.
For more information about Gilead, please visit the company’s website at www.gilead.com , follow Gilead on Twitter (@Gilead Sciences) or call Gilead Public Affairs at 1-800-GILEAD-5 or 1-650-574-3000.
View source version on businesswire.com: https://www.businesswire.com/news/home/20210922005622/en/
Source: Gilead Sciences, Inc.
Sep. 22, 2021 9:19 AM ET Gilead Sciences, Inc. (GILD) By: Jonathan M Block, SA News Editor1 Comment
bluebird bio Submits Biologics License Application (BLA) to FDA for betibeglogene autotemcel (beti-cel) Gene Therapy for Patients With β-thalassemia Who Require Regular Red Blood Cell Transfusions
BLA submission based on data from Phase 1/2 and Phase 3 Northstar studies, which represent more than 220 patient-years of experience with beti-cel
CAMBRIDGE, Mass.--(BUSINESS WIRE)--Sep. 21, 2021-- bluebird bio, Inc. (Nasdaq: BLUE) today announced it has completed the rolling submission of its Biologics License Application (BLA) to the U.S. Food and Drug Administration (FDA) for betibeglogene autotemcel (beti-cel) gene therapy in adult, adolescent and pediatric patients with β-thalassemia who require regular red blood cell (RBC) transfusions, across all genotypes. The FDA previously granted beti-cel Orphan Drug status and Breakthrough Therapy designation for the treatment of transfusion-dependent β-thalassemia (TDT). If approved, beti-cel will be the first hematopoietic (blood) stem cell (HSC) ex-vivo gene therapy for patients in the United States.
“With this submission, we are one step closer to bringing a potentially transformative gene therapy to people living with TDT and their families,” said Andrew Obenshain, president, severe genetic diseases, bluebird bio. “At bluebird bio, we have a deep understanding of gene therapies, built over a decade of research and development in severe genetic diseases. We look forward to working with the FDA on its review of this BLA as we realize the promise that one-time gene therapies hold for patients.”
The BLA submission for beti-cel is based on data from patients treated in bluebird bio studies, including the Phase 3 HGB-207 (Northstar-2) and HGB-212 (Northstar-3) studies, and the Phase 1/2 HGB-204 (Northstar) and HGB-205 studies. Together, these studies represent more than 220 patient-years of experience with beti-cel. As of March 9, 2021, the results include a total of 63 pediatric, adolescent and adult patients who have been treated with beti-cel across β0/β0 and non-β0/β0 genotypes. The data include two patients with up to seven years of follow-up, eight with at least six years of follow-up and 19 with at least five years of follow-up, and were most recently shared during the 26th Annual Congress of the European Hematology Association (EHA2021 Virtual).
About betibeglogene autotemcel (beti-cel)
betibeglogene autotemcel (beti-cel) is a one-time gene therapy custom-designed to treat the underlying cause of transfusion-dependent β-thalassemia (TDT). In order to correct the deficiency of adult hemoglobin that is the hallmark of TDT, beti-cel adds functional copies of a modified form of the β-globin gene (βA-T87Q-globin gene) into a patient’s own hematopoietic (blood) stem cells (HSCs). Once a patient has the βA-T87Q-globin gene, they have the potential to produce HbAT87Q, which is gene therapy-derived adult hemoglobin (Hb) at levels that may eliminate or significantly reduce the need for transfusions. In beti-cel studies, transfusion independence (TI) is defined as no longer needing red blood cell (RBC) transfusions for at least 12 months while maintaining a weighted average Hb of at least 9 g/dL. Across Phase 3 studies, 89% (32/36) of evaluable patients across ages and genotypes, including pediatric patients as young as four years of age and those with the most severe β0/β0 genotypes, achieved TI.
beti-cel is manufactured using the BB305 lentiviral vector (LVV), a third-generation, self-inactivating LVV. The BB305 LVV contains a cellular (non-viral) regulatory element, known as a promoter, that drives gene expression only in the erythroid cell lineage (RBCs and their precursors).
Adverse reactions considered related to beti-cel were uncommon and consisted primarily of infusion-related reactions (abdominal pain, hot flush, dyspnea, tachycardia and non-cardiac chest pain) and cytopenias (thrombocytopenia, leukopenia and neutropenia). Pain in extremity shortly after treatment was also documented. One of these adverse events (AE) was a serious adverse event (SAE) of thrombocytopenia considered possibly related to beti-cel.
The majority of AEs and SAEs unrelated to beti-cel were consistent with the known side effects of HSC collection and bone marrow ablation with busulfan (including several SAEs of veno-occlusive disease).
The Phase 3 Northstar-2 (HGB-207) and Northstar-3 (HGB-212) studies evaluating beti-cel are ongoing; enrollment is complete, and all patients have been treated. bluebird bio is also conducting a long-term follow-up study, LTF-303, to monitor safety and efficacy, for people who have participated in bluebird bio-sponsored beti-cel clinical studies. Patients treated with beti-cel in the commercial setting will also be able to enroll in the REG-501 registry study for long-term safety and efficacy follow-up.
Previously, the European Commission granted conditional marketing authorization for beti-cel, marketed as ZYNTEGLO™ gene therapy, for patients 12 years and older with TDT who do not have a β0/β0 genotype, for whom HSC transplantation is appropriate, but a human leukocyte antigen-matched related HSC donor is not available. In August 2021, bluebird bio announced its decision to focus its severe genetic disease business in the U.S.
For more information, visit bluebirdbio.com.
View source version on businesswire.com: https://www.businesswire.com/news/home/20210921005978/en/
Source: bluebird bio, Inc.
Sep. 21, 2021 4:27 PM ET bluebird bio, Inc. (BLUE)
By: Jonathan M Block, SA News Editor2 Comments
September 16, 2021
– Patients Whose Disease Changed to Triple-Negative Had Similar Positive Outcomes to Overall Metastatic TNBC Population in ASCENT Study –
– Trodelvy Is the First Treatment to Demonstrate Survival Benefit in Metastatic TNBC –
FOSTER CITY, Calif.--(BUSINESS WIRE)-- Gilead Sciences, Inc. (Nasdaq: GILD) today announced new data from the Phase 3 ASCENT study evaluating Trodelvy® (sacituzumab govitecan-hziy) in patients with relapsed or refractory metastatic triple-negative breast cancer (TNBC) who received two or more prior systemic therapies, at least one of them for metastatic disease. In a retrospective subgroup analysis, Trodelvy improved progression-free survival (PFS), overall survival (OS) and objective response rate (ORR) compared with chemotherapy chosen by the patients’ physicians in patients who were not initially diagnosed with TNBC. The results were presented at the European Society of Medical Oncology (ESMO) Congress 2021 from September 16-21, 2021 (Poster #258P).
“In the metastatic stage of breast cancer, it is not uncommon for people to change from one subtype to another,” said Javier Cortés, MD, Head of the International Breast Cancer Center (IBCC), Madrid and Barcelona, Spain. “Roughly one-third of patients with TNBC in the ASCENT study were not originally diagnosed with TNBC, and they still experienced a survival benefit with Trodelvy compared with chemotherapy. For treating physicians, this reinforces Trodelvy’s efficacy in more complex patients.”
This analysis included 146 chemotherapy-eligible brain metastasis-negative patients with an original breast cancer diagnosis that was not TNBC, of which 70 received Trodelvy and 76 received physician’s choice of chemotherapy. Among these patients, Trodelvy improved median PFS compared with chemotherapy (4.6 months vs. 2.3 months; HR: 0.48; P=0.0004), median OS (12.4 months vs. 6.7 months; HR: 0.44; P<0.0001) and ORR (31% vs. 4%). Outcomes were similar to those of the overall ASCENT trial population.
About the ASCENT Study
The ASCENT study is a global, open-label, randomized Phase 3 study that enrolled more than 500 patients across 230 study locations. The study evaluated the efficacy and safety of Trodelvy compared with a single-agent chemotherapy of the physician’s choice in patients with unresectable, locally advanced or metastatic TNBC who had received at least two prior systemic treatments. Patients were randomized to receive either Trodelvy or a chemotherapy chosen by the patients’ treating physicians. The primary endpoint was PFS (as determined by blinded independent central review) in patients without brain metastases. Secondary endpoints included: PFS for full study population or intention-to-treat (ITT) population, OS in both the ITT population and in the subgroup without brain metastasis, independently determined ORR, duration of response, time to onset of response according to Response Evaluation Criteria in Solid Tumors (RECIST 1.1), quality of life and safety. More information about ASCENT is available at http://clinicaltrials.gov/show/NCT02574455.
About Trodelvy
Trodelvy (sacituzumab govitecan-hziy) is a first-in-class antibody and topoisomerase inhibitor conjugate directed to the Trop-2 receptor, a protein overexpressed in multiple types of epithelial tumors, including metastatic TNBC and metastatic UC, where high expression is associated with poor survival and relapse. Beyond the approvals of Trodelvy in the United States, it is also approved in Australia, Switzerland, and the UK for adults with metastatic TNBC. Trodelvy is also under regulatory review in the EU andCanada, as well as in Singapore through our partner Everest Medicines. Trodelvy is also being investigated as a potential treatment for hormone receptor-positive/human epidermal growth factor receptor 2-negative metastatic breast cancer and metastatic non-small cell lung cancer. Additional evaluation across multiple solid tumors is also underway.
In the United States, Trodelvy is indicated for the treatment of:
Please see full Prescribing Information, including BOXED WARNING.
U.S. Prescribing Information for Trodelvy including BOXED WARNING, is available at www.gilead.com.
Trodelvy, Gilead and the Gilead logo are trademarks of Gilead Sciences, Inc., or its related companies.
For more information about Gilead, please visit the company’s website at www.gilead.com, follow Gilead on Twitter (@GileadSciences) or call Gilead Public Affairs at 1-800-GILEAD-5 or 1-650-574-3000.
View source version on businesswire.com: https://www.businesswire.com/news/home/20210916005030/en/
Source: Gilead Sciences, Inc.
https://seekingalpha.com/symbol/GILD
September 19, 2021 at 11:30 AM EDT Back
TARRYTOWN, N.Y. and PARIS, Sept. 19, 2021 /PRNewswire/ --
Phase 3 trial met its primary and key secondary endpoints
Libtayo is one of two PD-(L)1 inhibitors to demonstrate positive Phase 3 results in first-line advanced NSCLC irrespective of histology both as monotherapy and in combination with chemotherapy
Trial enrolled patients with varied baseline characteristics, including squamous and non-squamous histologies and all PD-L1 expression levels; 84% had an ECOG 1 performance status (reduced daily functioning)
Regeneron will host investor webcast on Monday, September 20 to discuss results and broader oncology portfolio
Regeneron Pharmaceuticals, Inc. (NASDAQ: REGN) and Sanofi announced positive Phase 3 results for a Libtayo® (cemiplimab) combination treatment were presented today during a late-breaking session at the European Society for Medical Oncology Virtual Congress 2021. The trial, which met its primary overall survival (OS) endpoint and all key secondary endpoints, assessed the investigational use of PD-1 inhibitor Libtayo in combination with a physician's choice of platinum-doublet chemotherapy (Libtayo combination) in patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) irrespective of histology and across all PD-L1 expression levels, compared to chemotherapy alone. These results were also achieved in a patient population with varied baseline characteristics and will form the basis of regulatory submissions, including in the U.S. and European Union (EU).
"Libtayo added to chemotherapy significantly improved patient outcomes, extending median overall survival to 22 months and median progression-free survival to 8 months," said Miranda Gogishvili, M.D., an oncologist at the High Technology Medical Center University Clinic, in Tbilisi, Georgia and a trial investigator. "Exploratory analyses showed that survival improvements were seen across squamous and non-squamous histologies and in patients with reduced daily functioning, with 43% of patients having squamous disease and 84% having an ECOG 1 performance status. Furthermore, in another exploratory analysis, the Libtayo combination helped delay deterioration in patient-reported quality of life and pain symptoms."
This Phase 3 trial was stopped early because Libtayo significantly improved overall survival compared to chemotherapy, a milestone also achieved by our Phase 3 trial for Libtayo monotherapy as a first-line treatment for advanced non-small cell lung cancer with high PD-L1 expression," said Israel Lowy, M.D., Ph.D., Senior Vice President, Translational and Clinical Sciences, Oncology at Regeneron. "Both trials were designed to reflect everyday clinical practice by allowing for the enrollment of patients with difficult-to-treat disease characteristics. And this is the second Libtayo trial to demonstrate significant improvement in its primary and key secondary endpoints for these patient populations, compared to chemotherapy."
Lung cancer is the leading cause of cancer death worldwide. In 2020, an estimated 2.2 million and 225,000 new cases were diagnosed globally and in the U.S., respectively. Approximately 84% of all lung cancers are NSCLC, with 75% of these cases diagnosed in advanced stages. While PD-1 inhibitor monotherapy has primarily advanced the treatment of NSCLC with ≥50% PD-L1 expression, approximately 70% of all NSCLC cases will have <50% PD-L1 expression, making it the most common treatment setting.
"These data add to the growing body of evidence supporting the use of Libtayo in patients with advanced non-small cell lung cancer," said Peter C. Adamson, M.D., Global Development Head, Oncology and Pediatric Innovation at Sanofi. "With additional trials underway investigating Libtayo as the backbone in combinations with conventional and novel therapeutic approaches, we are encouraged by the potential to further improve outcomes for patients with difficult-to-treat cancers."
The use of Libtayo in combination with chemotherapy for advanced NSCLC is investigational, and its safety and efficacy have not been fully evaluated by any regulatory authority.
About Libtayo
Libtayo is a fully human monoclonal antibody targeting the PD-1 immune checkpoint receptor on T-cells. By binding to PD-1, Libtayo has been shown to block cancer cells from using the PD-1 pathway to suppress T-cell activation.
The generic name for Libtayo in its approved U.S. indications is cemiplimab-rwlc, with rwlc as the suffix designated in accordance with Nonproprietary Naming of Biological Products Guidance for Industry issued by the U.S. FDA. Libtayo is being jointly developed by Regeneron and Sanofi under a global collaboration agreement.
The extensive clinical program for Libtayo is focused on difficult-to-treat cancers. Libtayo is currently being investigated in advanced cervical cancer, as well as in trials combining Libtayo with either conventional or novel therapeutic approaches for other solid tumors and blood cancers. These potential uses are investigational, and their safety and efficacy have not been evaluated by any regulatory authority.
What is the most important information I should know about Libtayo?
Libtayo is a medicine that may treat certain cancers by working with your immune system. Libtayo can cause your immune system to attack normal organs and tissues in any area of your body and can affect the way they work. These problems can sometimes become severe or life-threatening and can lead to death. You can have more than one of these problems at the same time. These problems may happen anytime during treatment or even after your treatment has ended.
Please see full Prescribing Information, including Medication Guide.
For additional information about the company, please visit www.regeneron.com
SOURCE Regeneron Pharmaceuticals, Inc.
Sep. 20, 2021 12:38 AM ET Sanofi (SNY), REGN By: Mamta Mayani, SA News Editor1 Comment
September 20, 2021
- In three pivotal Phase 3 studies, risankizumab-rzaa demonstrated significant improvements in clinical remission and endoscopic response as both induction and maintenance therapy[1],[2],[3]
- The overall safety findings in these pivotal studies were generally consistent with the known safety profile of risankizumab-rzaa[1-7]
- This submission represents AbbVie's ongoing commitment to bring risankizumab-rzaa to more people living with immune-mediated diseases
NORTH CHICAGO, Ill., Sept. 20, 2021 /PRNewswire/ -- AbbVie (NYSE: ABBV) today announced that it has submitted an application to the U.S. Food and Drug Administration (FDA) seeking approval for risankizumab-rzaa (600 mg intravenous (IV) induction and 360 mg subcutaneous (SC) maintenance therapy), an interleukin-23 (IL-23) inhibitor, for the treatment of patients 16 years and older with moderate to severe Crohn's disease. The submission is supported by safety and efficacy data from three Phase 3 studies – ADVANCE, MOTIVATE and FORTIFY.
"While there have been advancements in care, many people with Crohn's disease do not achieve lasting remission," said Tom Hudson, senior vice president of research and development, chief scientific officer, AbbVie. "This submission is an important step forward in our commitment to providing an additional treatment option for those who struggle with this debilitating and often unpredictable disease."
In the analysis plans for the U.S. submission of the ADVANCE and MOTIVATE induction studies, a significantly greater proportion of patients with Crohn's disease treated with either dose of risankizumab-rzaa IV induction therapy (600 mg or 1200 mg) met the co-primary endpoints of clinical remission and endoscopic response at week 12 compared to placebo.1,2
In the analysis plans for the U.S. submission of the FORTIFY trial, a randomized-withdrawal maintenance trial of patients with clinical response to risankizumab-rzaa induction therapy, a significantly greater proportion of participants achieved the co-primary endpoints of endoscopic response and clinical remission with risankizumab-rzaa SC maintenance therapy at one year (52 weeks) for both assessed doses (360 mg or 180 mg), compared to those who were randomized to the withdrawal arm and received placebo SC (control group).3
The safety profile of all tested doses of risankizumab-rzaa in moderate to severe Crohn's disease in the ADVANCE, MOTIVATE and FORTIFY studies was generally consistent with the known safety profile of risankizumab-rzaa.1-7
SKYRIZI is part of a collaboration between Boehringer Ingelheim and AbbVie, with AbbVie leading development and commercialization globally.
About Crohn's Disease
Crohn's disease is a chronic, systemic disease that manifests as inflammation within the gastrointestinal (or digestive) tract, causing persistent diarrhea, abdominal pain, and rectal bleeding.9-11 It is a progressive disease, meaning it can get worse over time.10,11 Because the signs and symptoms of Crohn's disease are unpredictable, it causes a significant burden on people living with the disease—not only physically, but also emotionally and economically.8
About the ADVANCE and MOTIVATE Studies12-14
The ADVANCE and MOTIVATE studies are Phase 3, multicenter, randomized, double-blind, placebo-controlled induction studies designed to evaluate the efficacy and safety of two doses of risankizumab-rzaa, 600 mg and 1200 mg, in adults with moderate to severe Crohn's disease, compared to placebo. The ADVANCE study included a mixed population of patients who had responded inadequately or are intolerant to conventional and/or biologic therapy. The MOTIVATE study evaluated patients who had responded inadequately or were intolerant to biologic therapy. Topline results of the studies were shared in January 2021, and additional analyses were presented at Digestive Disease Week® (DDW) Virtual Conference 2021. More information can be found on www.clinicaltrials.gov (ADVANCE: NCT03105128; MOTIVATE: NCT03104413).
About the FORTIFY Study3,15
The FORTIFY study is a Phase 3, multicenter, randomized, double-blind, control group, 52-week maintenance study designed to evaluate the efficacy and safety of risankizumab-rzaa 180 mg and 360 mg as maintenance therapy versus withdrawal who responded to risankizumab-rzaa induction treatment in the ADVANCE and MOTIVATE studies. Topline results were announced in June 2021. An open label extension of FORTIFY will continue to assess the long-term safety of risanzikumab-rzaa in subjects who completed participation in FORTIFY. More information can be found on www.clinicaltrials.gov (NCT03105102).
About Risankizumab-rzaa (SKYRIZI®)
SKYRIZI is an interleukin-23 (IL-23) inhibitor that selectively blocks IL-23 by binding to its p19 subunit.16 IL-23, a cytokine involved in inflammatory processes, is thought to be linked to a number of chronic immune-mediated diseases, including Crohn's disease.16,17 In April 2019, SKYRIZI received U.S. Food and Drug Administration approval for the treatment of moderate to severe plaque psoriasis in adults who are candidates for systemic therapy or phototherapy. The approved dose for SKYRIZI is 150 mg, administered by a prefilled pen or prefilled syringe at week 0 and 4, and every 12 weeks thereafter. SKYRIZI was also approved by the European Commission in April 2019. Phase 3 trials of SKYRIZI in psoriatic arthritis, Crohn's disease, and ulcerative colitis are ongoing.13-15, 18-21 The use of risankizumab-rzaa in Crohn's disease is not approved and its safety and efficacy have not been established by regulatory authorities.
About Risankizumab-rzaa (SKYRIZI®) in the United States16
SKYRIZI is a prescription medicine used to treat adults with moderate to severe plaque psoriasis who may benefit from taking injections or pills (systemic therapy) or treatment using ultraviolet or UV light (phototherapy).
For more information about AbbVie, please visit us at www.abbvie.com
Sep. 20, 2021 9:55 AM ET AbbVie Inc. (ABBV)
By: Jonathan M Block, SA News Editor2 Comments
Monday, Sep 20, 2021
South San Francisco, CA -- September 20, 2021 --Genentech, a member of the Roche Group (SIX: RO, ROG; OTCQX: RHHBY), today presented new data from the Phase III IMpower010 study at the European Society for Medical Oncology (ESMO) Congress 2021 Presidential Symposium, reinforcing the significant disease-free survival (DFS) benefit offered by Tecentriq ® (atezolizumab) for people with Stage II-IIIA non-small cell lung cancer (NSCLC) whose tumors express PD-L1≥1%. Data from the IMpower010 trial were published simultaneously in The Lancet. In IMpower010, treatment with Tecentriq, following surgery and chemotherapy, reduced the risk of disease recurrence or death (DFS) by 34% (hazard ratio [HR]=0.66, 95% CI: 0.50–0.88) in people with Stage II-IIIA NSCLC whose tumors express PD-L1≥1%, compared with best supportive care (BSC). Safety data for Tecentriq were consistent with its known safety profile and no new safety signals were identified.“Today more than half of all people with early-stage NSCLC experience recurrence following surgery,” said Levi Garraway, M.D., Ph.D., chief medical officer and head of Global Product Development. “IMpower010 shows how, for the first time, a cancer immunotherapy may help many of these patients live longer without their disease returning. The data presented at ESMO and WCLC further contribute to our understanding of Tecentriq in this treatment setting.” Genentech has an extensive development program for Tecentriq, including multiple ongoing and planned Phase III studies across different settings in lung, genitourinary, skin, breast, gastrointestinal, gynecological, and head and neck cancers. This includes studies evaluating Tecentriq both alone and in combination with other medicines, as well as studies in metastatic, adjuvant and neoadjuvant settings across various tumor types.
About the IMpower010 studyIMpower010 is a Phase III, global, multicenter, open-label, randomized study evaluating the efficacy and safety of Tecentriq compared with BSC, in participants with Stage IB-IIIA NSCLC (UICC 7th edition), following surgical resection and up to 4 cycles of adjuvant cisplatin-based chemotherapy. The study randomized 1,005 people with a ratio of 1:1 to receive either Tecentriq (up to 16 cycles) or BSC. The primary endpoint is investigator-determined DFS in the PD-L1-positive Stage II-IIIA, all randomized Stage II-IIIA and intent-to-treat (ITT) Stage IB-IIIA populations. Key secondary endpoints include overall survival (OS) in the overall study population, ITT Stage IB-IIIA NSCLC.
About Tecentriq® (atezolizumab)Tecentriq is a monoclonal antibody designed to bind with a protein called PD-L1. Tecentriq is designed to bind to PD-L1 expressed on tumor cells and tumor-infiltrating immune cells, blocking its interactions with both PD-1 and B7.1 receptors. By inhibiting PD-L1, Tecentriq may enable the re-activation of T cells. Tecentriq may also affect normal cells.
Tecentriq U.S. IndicationsTecentriq is a prescription medicine used to treat adults with:A type of lung cancer called non-small cell lung cancer (NSCLC).
A type of lung cancer called small cell lung cancer (SCLC).
It is not known if Tecentriq is safe and effective in children.
Please see http://www.Tecentriq.com for full Prescribing Information and additional Important Safety Information.
For more information visit http://www.gene.com/cancer-immunotherapy.
For additional information about the company, please visit http://www.gene.com.
Sep. 20, 2021 9:42 AM ET
By: Aakash Babu, SA News Editor
PUBLISHED18 September 2021
Updated results from the CASPIAN Phase III trial showed AstraZeneca’s Imfinzi (durvalumab) in combination with a choice of chemotherapies, etoposide plus either carboplatin or cisplatin, demonstrated a sustained, clinically meaningful overall survival (OS) benefit at three years for adults with extensive-stage small cell lung cancer (ES-SCLC) treated in the 1st-line setting.
These data, which show the longest survival update ever reported for an immunotherapy treatment in this setting, were presented during a mini-oral session on 18 September 2021 at the European Society of Medical Oncology (ESMO) Congress 2021.
The CASPIAN trial met the primary endpoint of OS in June 2019, reducing the risk of death by 27% (based on a hazard ratio [HR] of 0.73; 95% confidence interval [CI] 0.59-0.91; p=0.0047), which has formed the basis of regulatory approvals in many countries around the world. Updated results were previously presented during the ASCO20 Virtual Scientific Program in May 2020 with a median follow up of more than two years.
The latest results for Imfinzi plus chemotherapy showed sustained efficacy after a median follow up of more than three years for censored patients, with a 29% reduction in the risk of death versus chemotherapy alone (based on an HR of 0.71; 95% CI 0.60-0.86; nominal p=0.0003). Updated median OS was 12.9 months versus 10.5 for chemotherapy.
The results included a planned exploratory analysis, where an estimated 17.6% of patients treated with Imfinzi plus chemotherapy were alive at three years, versus 5.8% of patients treated with chemotherapy alone. The survival benefits were consistent across all subgroups, in line with previous analyses.
Luis Paz-Ares, MD, PhD, Chair, Medical Oncology Department, Hospital Universitario 12 de Octubre, Madrid, Spain and principal investigator in the CASPIAN Phase III trial said: “Patients with extensive-stage small cell lung cancer historically have had limited treatment options and still face a dire prognosis, which makes these data showing that three times as many patients survive three years following Imfinzi treatment especially meaningful. These results reinforce Imfinzi plus platinum chemotherapy as an important standard of care in this setting.”
Susan Galbraith, Executive Vice President, Oncology R&D, said: “This remarkable improvement in survival is an unprecedented achievement at three years for patients with extensive-stage small cell lung cancer. We are deeply committed to helping improve survival rates in this disease with research into new treatment options to transform outcomes at various stages, not only with the CASPIAN trial, but also with the ADRIATIC trial in limited-stage disease.”
Imfinzi plus chemotherapy continued to demonstrate a well-tolerated safety profile consistent with the known profiles of these medicines. Results showed 32.5% of patients experienced a serious adverse event (all causality) with Imfinzi plus chemotherapy versus 36.5% with chemotherapy alone.
Imfinzi in combination with etoposide and either carboplatin or cisplatin is approved in the 1st-line setting of ES-SCLC in more than 55 countries, including the US, Japan, China and across the EU.
CASPIAN
CASPIAN was a randomised, open-label, multi-centre, global Phase III trial in the 1st-line treatment of 805 patients with ES-SCLC. The trial compared Imfinzi in combination with chemotherapy (etoposide and either carboplatin or cisplatin), or Imfinzi and chemotherapy with the addition of a second immunotherapy, tremelimumab, versus chemotherapy alone.
In the two experimental arms, patients were treated with four cycles of chemotherapy. In comparison, the control arm allowed up to six cycles of chemotherapy and optional prophylactic cranial irradiation.
The trial was conducted in more than 200 centres across 23 countries, including the US, Europe, South America, Asia and the Middle East. The primary endpoint was OS in each of the two experimental arms.
In June 2019, AstraZeneca announced the CASPIAN Phase III trial had met one primary endpoint of demonstrating OS for Imfinzi plus chemotherapy at a planned interim analysis. In March 2020, however, it was announced that the second experimental arm with tremelimumab did not meet its primary endpoint of OS.
Imfinzi
Imfinzi (durvalumab) is a human monoclonal antibody that binds to PD-L1 and blocks the interaction of PD-L1 with PD-1 and CD80, countering the tumour’s immune-evading tactics and releasing the inhibition of immune responses.
In addition to approvals in ES-SCLC and unresectable, Stage III NSCLC, Imfinzi is approved for previously treated patients with advanced bladder cancer in several countries. Since the first approval in May 2017, more than 100,000 patients have been treated with Imfinzi.
As part of a broad development programme, Imfinzi is being tested as a single treatment and in combinations with other anti-cancer treatments for patients with NSCLC, SCLC, bladder cancer, hepatocellular carcinoma, biliary tract cancer (a form of liver cancer), oesophageal cancer, gastric and gastroesophageal cancer, cervical cancer, ovarian cancer, endometrial cancer, and other solid tumours.
Please visit astrazeneca.com
Sep. 20, 2021 6:46 AM ET AstraZeneca PLC (AZN) By: Mamta Mayani, SA News Editor
NEW YORK and MAINZ, Germany, September 20, 2021—Pfizer Inc. (NYSE: PFE, “Pfizer”) and BioNTech SE (Nasdaq: BNTX, “BioNTech”) today announced results from a Phase 2/3 trial showing a favorable safety profile and robust neutralizing antibody responses in children 5 to 11 years of age using a two-dose regimen of 10 µg administered 21 days apart, a smaller dose than the 30 µg dose used for people 12 and older. The antibody responses in the participants given 10 µg doses were comparable to those recorded in a previous Pfizer-BioNTech study in people 16 to 25 years of age immunized with 30 µg doses. The 10 µg dose was carefully selected as the preferred dose for safety, tolerability and immunogenicity in children 5 to 11 years of age. These are the first results from a pivotal trial of a COVID-19 vaccine in this age group.
“Over the past nine months, hundreds of millions of people ages 12 and older from around the world have received our COVID-19 vaccine. We are eager to extend the protection afforded by the vaccine to this younger population, subject to regulatory authorization, especially as we track the spread of the Delta variant and the substantial threat it poses to children,” said Albert Bourla, Chairman and Chief Executive Officer, Pfizer. “Since July, pediatric cases of COVID-19 have risen by about 240 percent in the U.S. – underscoring the public health need for vaccination. These trial results provide a strong foundation for seeking authorization of our vaccine for children 5 to 11 years old, and we plan to submit them to the FDA and other regulators with urgency.”
“We are pleased to be able to submit data to regulatory authorities for this group of school-aged children before the start of the winter season,” said Dr. Ugur Sahin, CEO and co-founder of BioNTech. “The safety profile and immunogenicity data in children aged 5 to 11 years vaccinated at a lower dose are consistent with those we have observed with our vaccine in other older populations at a higher dose.”
The data summarized from this Phase 2/3 study, which is enrolling children 6 months to 11 years of age, was for 2,268 participants who were 5 to 11 years of age and received a 10 µg dose level in a two-dose regimen. In the trial, the SARS-CoV-2–neutralizing antibody geometric mean titer (GMT) was 1,197.6 (95% confidence interval [CI, 1106.1, 1296.6]), demonstrating strong immune response in this cohort of children one month after the second dose. This compares well (was non-inferior) to the GMT of 1146.5 (95% CI: 1045.5, 1257.2) from participants ages 16 to 25 years old, used as the control group for this analysis and who were administered a two-dose regimen of 30 µg. Further, the COVID-19 vaccine was well tolerated, with side effects generally comparable to those observed in participants 16 to 25 years of age.
Pfizer and BioNTech plan to share these data with the U.S. Food and Drug Administration (FDA), European Medicines Agency (EMA) and other regulators as soon as possible. For the United States, the companies expect to include the data in a near-term submission for Emergency Use Authorization (EUA) as they continue to accumulate the safety and efficacy data required to file for full FDA approval in this age group. A request to the EMA to update the EU Conditional Marketing Authorization is also planned. Topline readouts for the other two age cohorts from the trial – children 2-5 years of age and children 6 months to 2 years of age – are expected as soon as the fourth quarter of this year.
Pfizer and BioNTech plan to submit data from the full Phase 3 trial for scientific peer-review publication.
About the Phase 1/2/3 Trial in Children
The Phase 1/2/3 trial initially enrolled up to 4,500 children ages 6 months to 11 years of age in the United States, Finland, Poland, and Spain from more than 90 clinical trial sites. It was designed to evaluate the safety, tolerability, and immunogenicity of the Pfizer-BioNTech vaccine on a two-dose schedule (approximately 21 days apart) in three age groups: ages 5 to 11 years; ages 2 to 5 years; and ages 6 months to 2 years. Based on the Phase 1 dose-escalation portion of the trial, children ages 5 to 11 years received two-dose schedule of 10 µg each while children under age 5 received a lower 3 µg dose for each injection in the Phase 2/3 study. The trial enrolled children with or without prior evidence of SARS-CoV-2 infection.
COMIRNATY, which is based on BioNTech’s proprietary mRNA technology, was developed by both BioNTech and Pfizer. BioNTech is the Marketing Authorization Holder in the United States, the European Union, the United Kingdom, Canada and the holder of emergency use authorizations or equivalents in the United States (jointly with Pfizer) and other countries. Submissions to pursue regulatory approvals in those countries where emergency use authorizations or equivalent were initially granted are planned.
U.S. Indication & Authorized Use
COMIRNATY® (COVID-19 Vaccine, mRNA) is an FDA-approved COVID-19 vaccine made by Pfizer for BioNTech.
The Pfizer-BioNTech COVID-19 Vaccine has received EUA from FDA to:
The FDA-approved COMIRNATY® (COVID-19 Vaccine, mRNA) and the EUA-authorized Pfizer-BioNTech COVID-19 Vaccine have the same formulation and can be used interchangeably to provide the COVID-19 vaccination series. An individual may be offered either COMIRNATY® (COVID-19 Vaccine, mRNA) or the Pfizer-BioNTech COVID-19 Vaccine to prevent coronavirus disease 2019 (COVID-19) caused by SARS-CoV-2.
Please click here for full Prescribing Information (16+ years of age). Please click here for Fact Sheet for Vaccination Providers (12+ years of age).
For more information, please visit www.BioNTech.de.
Sep. 20, 2021 7:08 AM ET Pfizer Inc. (PFE), BNTX
By: Mamta Mayani, SA News Editor8 Comments
09/20/2021CATEGORY:
The application is based on Phase 2/3 RELATIVITY-047 trial, in which the relatlimab and nivolumab fixed-dose combination demonstrated a statistically significant and clinically meaningful progression-free survival benefit over Opdivo monotherapy
U.S. Food and Drug Administration assigns a target action date of March 19, 2022
PRINCETON, N.J.--(BUSINESS WIRE)-- Bristol Myers Squibb (NYSE: BMY) today announced that the U.S. Food and Drug Administration (FDA) has accepted for priority review the Biologics License Application (BLA) for the LAG-3-blocking antibody relatlimab and nivolumab fixed-dose combination, administered as a single infusion, for the treatment of adult and pediatric patients (12 years and older and weighing at least 40 kg) with unresectable or metastatic melanoma. The FDA assigned a Prescription Drug User Fee Act (PDUFA) goal date of March 19, 2022.
“Although we’ve seen significant advances in the treatment of melanoma since the introduction of immune checkpoint inhibitors, there continue to be patients who could benefit from a novel dual immunotherapy approach,” said Jonathan Cheng, senior vice president and head of oncology development, Bristol Myers Squibb. “Based on the results of the RELATIVITY-047 trial, we believe that the relatlimab and nivolumab fixed-dose combination has the potential to improve outcomes for patients with metastatic or unresectable melanoma. We look forward to potentially introducing the first LAG-3-blocking antibody, and Bristol Myers Squibb’s third distinct checkpoint inhibitor, to help patients in need.”
The BLA submission was based on the efficacy and safety results of the Phase 2/3 RELATIVITY-047 trial, which demonstrated a statistically significant and clinically meaningful progression-free survival benefit of a combination therapy over standard of care anti-PD-1 monotherapy in metastatic melanoma. Relatlimab is the first LAG-3-blocking antibody to demonstrate a clinical benefit for patients with Phase 3 data. Primary results from the RELATIVITY-047 trial were presented in an oral session during the American Society of Clinical Oncology (ASCO) Annual Meeting in June 2021 and were selected for the official ASCO press program.
The fixed-dose combination of relatlimab and nivolumab is an investigational therapy and is not approved for use in any country.
Bristol Myers Squibb thanks the patients and investigators involved in the RELATIVITY-047 clinical trial.
About RELATIVITY-047 (CA224-047)
RELATIVITY-047 (CA224-047) is a global, randomized, double-blind Phase 2/3 study evaluating the fixed-dose combination of relatlimab and nivolumab in patients with previously untreated metastatic or unresectable melanoma versus Opdivo alone. The primary endpoint of the trial is progression-free survival (PFS) by Blinded Independent Central Review (BICR) and the secondary endpoints are overall survival (OS) and objective response rate (ORR). A total of 714 patients were randomized 1:1 to receive a fixed-dose combination of relatlimab 160 mg and nivolumab 480 mg or Opdivo 480 mg by intravenous infusion every four weeks (Q4W) until disease recurrence, unacceptable toxicity or withdrawal of consent. Follow-up for the secondary endpoints of OS and ORR is ongoing.
About Opdivo
Opdivo is a programmed death-1 (PD-1) immune checkpoint inhibitor that is designed to uniquely harness the body’s own immune system to help restore anti-tumor immune response. By harnessing the body’s own immune system to fight cancer, Opdivo has become an important treatment option across multiple cancers.
Opdivo’s leading global development program is based on Bristol Myers Squibb’s scientific expertise in the field of Immuno-Oncology and includes a broad range of clinical trials across all phases, including Phase 3, in a variety of tumor types. To date, the Opdivo clinical development program has treated more than 35,000 patients. The Opdivo trials have contributed to gaining a deeper understanding of the potential role of biomarkers in patient care, particularly regarding how patients may benefit from Opdivo across the continuum of PD-L1 expression.
In July 2014, Opdivo was the first PD-1 immune checkpoint inhibitor to receive regulatory approval anywhere in the world. Opdivo is currently approved in more than 65 countries, including the United States, the European Union, Japan and China. In October 2015, the Company’s Opdivo and Yervoy combination regimen was the first Immuno-Oncology combination to receive regulatory approval for the treatment of metastatic melanoma and is currently approved in more than 50 countries, including the United States and the European Union.
INDICATIONS
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of patients with unresectable or metastatic melanoma.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of patients with unresectable or metastatic melanoma.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) whose tumors express PD-L1 (≥1%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab) and 2 cycles of platinum-doublet chemotherapy, is indicated for the first-line treatment of adult patients with metastatic or recurrent non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
OPDIVO® (nivolumab) is indicated for the treatment of patients with metastatic non-small cell lung cancer (NSCLC) with progression on or after platinum-based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving OPDIVO.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of adult patients with unresectable malignant pleural mesothelioma (MPM).
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the first-line treatment of patients with intermediate or poor risk advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab), in combination with cabozantinib, is indicated for the first-line treatment of patients with advanced renal cell carcinoma (RCC).
OPDIVO® (nivolumab) is indicated for the treatment of patients with advanced renal cell carcinoma (RCC) who have received prior anti-angiogenic therapy.
OPDIVO® (nivolumab) is indicated for the treatment of adult patients with classical Hodgkin lymphoma (cHL) that has relapsed or progressed after autologous hematopoietic stem cell transplantation (HSCT) and brentuximab vedotin or after 3 or more lines of systemic therapy that includes autologous HSCT. This indication is approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab) is indicated for the treatment of patients with recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN) with disease progression on or after platinum-based therapy.
OPDIVO® (nivolumab) is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
OPDIVO® (nivolumab), as a single agent, is indicated for the adjuvant treatment of patients with urothelial carcinoma (UC) who are at high risk of recurrence after undergoing radical resection of UC.
OPDIVO® (nivolumab), as a single agent, is indicated for the treatment of adult and pediatric (12 years and older) patients with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of adults and pediatric patients 12 years and older with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
OPDIVO® (nivolumab), in combination with YERVOY® (ipilimumab), is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of patients with melanoma with involvement of lymph nodes or metastatic disease who have undergone complete resection.
OPDIVO® (nivolumab) is indicated for the treatment of patients with unresectable advanced, recurrent or metastatic esophageal squamous cell carcinoma (ESCC) after prior fluoropyrimidine- and platinum-based chemotherapy.
OPDIVO® (nivolumab) is indicated for the adjuvant treatment of completely resected esophageal or gastroesophageal junction cancer with residual pathologic disease in patients who have received neoadjuvant chemoradiotherapy (CRT).
OPDIVO® (nivolumab), in combination with fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the treatment of patients with advanced or metastatic gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma.
Please see US Full Prescribing Information for OPDIVO and YERVOY.
For more information about Bristol Myers Squibb, visit us at BMS.com
Sep. 20, 2021 7:30 AM ET Bristol-Myers Squibb Company (BMY)
By: Dulan Lokuwithana, SA News Editor
- With the MONALEESA-2 final analysis, only Kisqali has reported statistically significant overall survival (OS) benefit with an aromatase inhibitor for postmenopausal women with HR+/HER2- advanced breast cancer in the first-line (1L) setting2
- Kisqali plus letrozole achieved median OS of over five years (63.9 months), a survival benefit of over 12 months vs. placebo plus letrozole in postmenopausal women (HR=0.76; p=0.004)2
- Kisqali is the only CDK4/6 inhibitor with proven OS benefit across all three Phase III trials of the MONALEESA program with different endocrine therapy partners, regardless of menopausal status or line of therapy2-4
- MONALEESA-2 OS data to be presented at ESMO Congress 2021 as a late-breaker in an oral session
NEWS PROVIDED BY
B2/Novartis Pharmaceuticals Corporation
Sep 19, 2021, 07:35 ET
EAST HANOVER, N.J., Sept. 19, 2021 /PRNewswire/ -- Novartis today announced results of the final overall survival (OS) analysis of the Phase III MONALEESA-2 study, which evaluated Kisqali® (ribociclib) in combination with letrozole compared to placebo plus letrozole in postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative (HR+/HER2-) advanced or metastatic breast cancer with no prior systemic treatment for advanced disease. These data will be presented as a late-breaker oral presentation at the European Society for Medical Oncology (ESMO) Congress 2021 on September 19 (#LBA17).
Kisqali in combination with letrozole met its key secondary endpoint of OS, demonstrating a statistically significant and clinically meaningful improvement in survival (median 63.9 vs. 51.4 months; HR=0.76; 95% CI: 0.63-0.93; p=0.004)2. The analysis found that after a median follow-up of over six and a half years, the longest for any CDK4/6 inhibitor trial to date, the improvement in the median OS was over one year2. MONALEESA-2 showed that after five years, patients treated with Kisqali in combination with letrozole had more than a 50% chance of survival (52.3% vs. 43.9%; 95% CI: 46.5-57.7 vs. 38.3-49.4)2.
"These remarkable ribociclib overall survival data are highly encouraging and represent the longest reported median survival from a randomized trial in HR+/HER2- advanced breast cancer. This extension of life is great news for our patients and the building block for further progress," said Gabriel N. Hortobagyi, MD, FACP, professor of medicine with The University of Texas MD Anderson Cancer Center. "I have spent the last 45 years researching and increasing our scientific understanding of breast cancer, so it is incredibly rewarding to see just how far we've come."
In MONALEESA-2, a 12-month delay in time to chemotherapy was observed with Kisqali (median 50.6 vs. 38.9 months; HR=0.74; 95% CI: 0.61-0.91) compared to those taking letrozole alone2. With this longer follow-up, no new safety signals were observed; adverse events were consistent with previously reported Phase III trial results for Kisqali.
Visit https://www.hcp.novartis.com/virtual-congress/esmo-2021/ for the latest information from Novartis, including our commitment to the Oncology community, and access to our ESMO2021 Virtual Scientific Program data presentations (for registered participants).
About Kisqali® (ribociclib)
Kisqali is the CDK4/6 inhibitor with the largest body of clinical trial evidence demonstrating consistent and superior overall survival benefit compared to endocrine therapy alone. Overall survival results from MONALEESA-7 and MONALEESA-3 were presented at ASCO 2019 and ESMO 2019 respectively, as well as published in the New England Journal of Medicine, with updated exploratory analyses presented at SABCS 2020 and ASCO 2021, demonstrating Kisqali plus endocrine therapy significantly extends life in pre/perimenopausal or postmenopausal women with HR+/HER2- advanced breast cancer3,4,6,7.
Kisqali is approved by the US Food and Drug Administration (FDA) and by the European Commission (EC) as initial endocrine-based therapy for postmenopausal women with HR+/HER2- locally advanced or metastatic breast cancer in combination with an aromatase inhibitor. Kisqali in combination with an aromatase inhibitor is approved for the treatment of pre-, peri- or postmenopausal women as initial endocrine-based therapy, and also indicated for use in combination with fulvestrant as both first- or second-line therapy in postmenopausal women by the FDA and by the EC9. Kisqali is approved in over 95 countries1.
Novartis is continuing to reimagine cancer with additional trials of Kisqali. NATALEE is a large confirmatory clinical trial of Kisqali with endocrine therapy in the adjuvant treatment of HR+/HER2- early breast cancer being conducted in collaboration with Translational Research In Oncology (TRIO)10. Novartis is also collaborating with SOLTI, who is leading the Phase III HARMONIA clinical trial evaluating Kisqali compared to palbociclib in patients with HR+/HER2- advanced breast cancer with aggressive tumor biology, defined as HER2-enriched1.
Kisqali was developed by the Novartis Institutes for BioMedical Research (NIBR) under a research collaboration with Astex Pharmaceuticals.
Please see full Prescribing Information for KISQALI, available at www.kisqali.com.
For more information, please visit https://www.novartis.us.
http://www.pharma.novartis.com
Sep. 20, 2021 5:38 AM ET Novartis AG (NVS)
By: Mamta Mayani, SA News Editor
September 19, 2021 7:30 am ET
First Anti-PD-1 Therapy in Combination With Chemotherapy to Demonstrate Statistically Significant Overall Survival for These Patients
Data From Phase 3 KEYNOTE-355 Trial Presented at ESMO Congress 2021
KENILWORTH, N.J.--(BUSINESS WIRE)-- Merck (NYSE: MRK), known as MSD outside the United States and Canada, today announced the final overall survival (OS) results from the pivotal Phase 3 KEYNOTE-355 trial investigating KEYTRUDA, Merck’s anti-PD-1 therapy, in combination with chemotherapy (paclitaxel, nab-paclitaxel or gemcitabine/carboplatin) for the first-line treatment of patients with metastatic triple-negative breast cancer (mTNBC). KEYTRUDA is the first anti-PD-1 therapy in combination with chemotherapy to demonstrate a statistically significant and clinically meaningful improvement in OS for these patients.
In this study, KEYTRUDA plus chemotherapy reduced the risk of death by 27% (HR=0.73 [95% CI, 0.55-0.95]; p=0.0093) in patients with mTNBC whose tumors expressed PD-L1 (Combined Positive Score [CPS] ≥10), as compared to chemotherapy alone. There was an increase of 6.9 months in median OS with KEYTRUDA plus chemotherapy compared to chemotherapy alone (23.0 months [95% CI, 19.0-26.3] vs. 16.1 months [95% CI, 12.6-18.8], respectively). Although the trial was not powered to compare efficacy between treatment groups by different chemotherapy regimens, the increase in OS was observed for KEYTRUDA plus chemotherapy across the three chemotherapy choices. These data were presented today in an oral presentation at the European Society for Medical Oncology (ESMO) Congress 2021 (Abstract #LBA16).
“Metastatic TNBC has the worst survival prognosis among breast cancer subtypes, and there is an urgent need for treatment options that improve survival,” said Dr. Hope Rugo, director, Breast Oncology and Clinical Trials Education, University of California San Francisco (UCSF) Helen Diller Family Comprehensive Cancer Center. “I am very encouraged to see these new overall survival data for the KEYTRUDA combination, demonstrating a 27% relative reduction in the risk of death compared to chemotherapy alone in patients with mTNBC whose tumors expressed PD-L1 (CPS ≥10).”
About KEYNOTE-355
KEYNOTE-355 is a randomized, two-part, placebo-controlled Phase 3 trial (ClinicalTrials.gov, NCT02819518) evaluating KEYTRUDA in combination with one of the three different chemotherapies compared with placebo plus one of the three chemotherapy regimens for the first-line treatment of mTNBC that has not been previously treated with chemotherapy in the advanced setting. The study endpoints include OS and PFS in patients whose tumors expressed PD-L1 (CPS ≥1 and CPS ≥10) and in all participants (ITT population). The other endpoints include objective response rate, duration of response, disease control rate, patient-reported outcomes and safety. Part 2 enrolled 847 patients who were randomized 2:1 to receive KEYTRUDA (200 mg every three weeks) plus chemotherapy (investigator’s choice of paclitaxel, nab-paclitaxel or gemcitabine/carboplatin) or placebo plus paclitaxel, nab-paclitaxel or gemcitabine/carboplatin.
The study population characteristics were: median age of 53 years (range, 22 to 85), 21% age 65 or older; 100% female; 68% white, 21% Asian and 4% Black; 59% Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0, and 41% ECOG PS of 1; and 68% were post-menopausal. Among enrolled patients in each treatment group, approximately 75% had tumors expressing PD-L1 with CPS ≥1 (n=425/566 for KEYTRUDA plus chemotherapy; n=211/281 for chemotherapy alone), and approximately 38% had tumors expressing PD-L1 with CPS ≥10 (n=220/566 for KEYTRUDA plus chemotherapy; n=103/281 for chemotherapy alone).
Immune-mediated adverse reactions (AEs) of any grade occurred in 26.5% of patients receiving KEYTRUDA plus chemotherapy and 6.4% of patients receiving chemotherapy alone. For patients receiving KEYTRUDA plus chemotherapy, the most common immune-mediated AE (occurring in ≥10% of patients) was hypothyroidism (15.8%). There were two treatment-related deaths due to acute kidney injury and pneumonia in patients receiving KEYTRUDA plus chemotherapy; neither was considered immune-mediated.
About KEYTRUDA® (pembrolizumab) Injection, 100 mg
KEYTRUDA is an anti-programmed death receptor-1 (PD-1) therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. KEYTRUDA is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2, thereby activating T lymphocytes which may affect both tumor cells and healthy cells.
Merck has the industry’s largest immuno-oncology clinical research program. There are currently more than 1,600 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. The KEYTRUDA clinical program seeks to understand the role of KEYTRUDA across cancers and the factors that may predict a patient's likelihood of benefitting from treatment with KEYTRUDA, including exploring several different biomarkers.
Selected KEYTRUDA® (pembrolizumab) Indications in the U.S.
Melanoma
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic melanoma.
KEYTRUDA is indicated for the adjuvant treatment of patients with melanoma with involvement of lymph node(s) following complete resection.
Non-Small Cell Lung Cancer
KEYTRUDA, in combination with pemetrexed and platinum chemotherapy, is indicated for the first-line treatment of patients with metastatic nonsquamous non-small cell lung cancer (NSCLC), with no EGFR or ALK genomic tumor aberrations.
KEYTRUDA, in combination with carboplatin and either paclitaxel or paclitaxel protein-bound, is indicated for the first-line treatment of patients with metastatic squamous NSCLC.
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with NSCLC expressing PD-L1 [tumor proportion score (TPS) ≥1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is:
KEYTRUDA, as a single agent, is indicated for the treatment of patients with metastatic NSCLC whose tumors express PD-L1 (TPS ≥1%) as determined by an FDA-approved test, with disease progression on or after platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving KEYTRUDA.
Head and Neck Squamous Cell Cancer
KEYTRUDA, in combination with platinum and fluorouracil (FU), is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent head and neck squamous cell carcinoma (HNSCC).
KEYTRUDA, as a single agent, is indicated for the first-line treatment of patients with metastatic or with unresectable, recurrent HNSCC whose tumors express PD-L1 [combined positive score (CPS ≥1)] as determined by an FDA-approved test.
KEYTRUDA, as a single agent, is indicated for the treatment of patients with recurrent or metastatic HNSCC with disease progression on or after platinum-containing chemotherapy.
Classical Hodgkin Lymphoma
KEYTRUDA is indicated for the treatment of adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL).
KEYTRUDA is indicated for the treatment of pediatric patients with refractory cHL, or cHL that has relapsed after 2 or more lines of therapy.
Primary Mediastinal Large B-Cell Lymphoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with refractory primary mediastinal large B-cell lymphoma (PMBCL), or who have relapsed after 2 or more prior lines of therapy. KEYTRUDA is not recommended for treatment of patients with PMBCL who require urgent cytoreductive therapy.
Urothelial Carcinoma
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic urothelial carcinoma (mUC):
KEYTRUDA is indicated for the treatment of patients with Bacillus Calmette-Guerin-unresponsive, high-risk, non-muscle invasive bladder cancer (NMIBC) with carcinoma in situ with or without papillary tumors who are ineligible for or have elected not to undergo cystectomy.
Microsatellite Instability-High or Mismatch Repair Deficient Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with MSI-H central nervous system cancers have not been established.
Microsatellite Instability-High or Mismatch Repair Deficient Colorectal Cancer
KEYTRUDA is indicated for the treatment of patients with unresectable or metastatic MSI-H or dMMR colorectal cancer (CRC).
Gastric Cancer
KEYTRUDA, in combination with trastuzumab, fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of patients with locally advanced unresectable or metastatic HER2-positive gastric or gastroesophageal junction (GEJ) adenocarcinoma.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Esophageal Cancer
KEYTRUDA is indicated for the treatment of patients with locally advanced or metastatic esophageal or GEJ (tumors with epicenter 1 to 5 centimeters above the GEJ) carcinoma that is not amenable to surgical resection or definitive chemoradiation either:
Cervical Cancer
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy whose tumors express PD-L1 (CPS ≥1) as determined by an FDA-approved test. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Hepatocellular Carcinoma
KEYTRUDA is indicated for the treatment of patients with hepatocellular carcinoma (HCC) who have been previously treated with sorafenib. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merkel Cell Carcinoma
KEYTRUDA is indicated for the treatment of adult and pediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma (MCC). This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Renal Cell Carcinoma
KEYTRUDA, in combination with axitinib, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma.
Tumor Mutational Burden-High Cancer
KEYTRUDA is indicated for the treatment of adult and pediatric patients with unresectable or metastatic tumor mutational burden-high (TMB-H) [≥10 mutations/megabase] solid tumors, as determined by an FDA-approved test, that have progressed following prior treatment and who have no satisfactory alternative treatment options. This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The safety and effectiveness of KEYTRUDA in pediatric patients with TMB-H central nervous system cancers have not been established.
Cutaneous Squamous Cell Carcinoma
KEYTRUDA is indicated for the treatment of patients with recurrent or metastatic cutaneous squamous cell carcinoma (cSCC) or locally advanced cSCC that is not curable by surgery or radiation.
Triple-Negative Breast Cancer
KEYTRUDA is indicated for the treatment of patients with high-risk early-stage triple-negative breast cancer (TNBC) in combination with chemotherapy as neoadjuvant treatment, and then continued as a single agent as adjuvant treatment after surgery.
KEYTRUDA, in combination with chemotherapy, is indicated for the treatment of patients with locally recurrent unresectable or metastatic TNBC whose tumors express PD-L1 (CPS ≥10) as determined by an FDA-approved test.
Merck’s Focus on Cancer
Our goal is to translate breakthrough science into innovative oncology medicines to help people with cancer worldwide. At Merck, the potential to bring new hope to people with cancer drives our purpose and supporting accessibility to our cancer medicines is our commitment. As part of our focus on cancer, Merck is committed to exploring the potential of immuno-oncology with one of the largest development programs in the industry across more than 30 tumor types. We also continue to strengthen our portfolio through strategic acquisitions and are prioritizing the development of several promising oncology candidates with the potential to improve the treatment of advanced cancers. For more information about our oncology clinical trials, visit www.merck.com/clinicaltrials.
For more information, visit www.merck.com
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and Medication Guide for KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf.
Sep. 20, 2021 5:46 AM ET Merck & Co., Inc. (MRK) By: Mamta Mayani, SA News Editor1 Comment
New Data on RYBREVANT® (amivantamab-vmjw) in Combination with Lazertinib Show Early Activity in Patients with Non-Small Cell Lung Cancer Whose Disease Has Progressed After Both Osimertinib and Platinum-Based ChemotherapyCHRYSALIS-2 findings presented at ESMO Annual Congress 2021 suggest that RYBREVANT® and lazertinib combination has encouraging anti-tumor activity in this population that has exhausted standard-of-care treatments
September 19, 2021 (RARITAN, N.J.) – The Janssen Pharmaceutical Companies of Johnson & Johnson today announced preliminary results from the Phase 1b CHRYSALIS-2 (NCT04077463) study evaluating RYBREVANT® (amivantamab-vmjw) in combination with lazertinib in the treatment of patients with non-small cell lung cancer (NSCLC) characterized by epidermal growth factor receptor (EGFR) exon 19 deletion or L858R mutations whose disease had progressed after treatment with osimertinib and platinum chemotherapy.[i] While previously reported results have demonstrated durable responses with RYBREVANT® in combination with lazertinib in chemotherapy-naïve patients previously treated with osimertinib, these new data suggest that intervening chemotherapy does not impact activity with the combination.1 These data were featured for the first time in a mini-oral presentation at the European Society for Medical Oncology (ESMO) Annual Congress 2021 virtual meeting on Sunday, September 19 (Abstract #1193MO).
“Patients with non-small cell lung cancer whose disease has progressed despite receiving standard of care treatments have a tremendous need for additional treatment options,” said Catherine A. Shu, M.D., Clinical Director of the Thoracic Medical Oncology Service, Columbia University Herbert Irving Comprehensive Cancer Center, and presenting study investigator.† “We are encouraged by these data showing that the combination of amivantamab and lazertinib elicited antitumor activity, even in a heavily pretreated patient population.”
In Cohort A of the CHRYSALIS-2 study, patients with NSCLC with EGFR exon 19 deletion or L858R mutations whose disease had progressed after treatment with osimertinib and platinum chemotherapy received the recommended combination dose of RYBREVANT® at 1050 mg (for patients who weigh <80kg) or 1400 mg (for patients who weigh ≥80 kg) and oral lazertinib at 240 mg.1 The study also included a heavily pretreated population (n=56), who received platinum-based chemotherapy, osimertinib and other therapies, with no prespecified number or sequence of prior treatment.1 A protocol amendment created a target population (n=80), which specified progression on osimertinib and platinum-based chemotherapy, in that order.1
In May, the U.S. Food and Drug Administration (FDA) approved RYBREVANT®, a fully human bispecific antibody, as the first targeted treatment for patients with NSCLC with EGFR exon 20 insertion mutations.[iii] Ongoing studies, including CHRYSALIS-2, are evaluating the potential of RYBREVANT® in combination with lazertinib, a third-generation EGFR tyrosine kinase inhibitor (TKI), across multiple cohorts and for different driver and resistance genetic mutations. Lazertinib was approved earlier this year in South Korea for patients with NSCLC with EFGR mutations and T90M mutations.
“These findings build on previous results showing the potential of RYBREVANT and lazertinib combination therapy in patients with EGFR-mutated non-small cell lung cancer and provide further insights supporting our comprehensive clinical development program in lung cancer,” said Craig Tendler, M.D., Vice President, Late Development and Global Medical Affairs, Oncology, Janssen Research & Development, LLC. “As we continue to evaluate RYBREVANT as a monotherapy and in combination with lazertinib, we look forward to continuing to advance science and improve outcomes for people living with advanced NSCLC.”
*RECIST (version 1.1) refers to Response Evaluation Criteria in Solid Tumors, which is a standard way to measure how well solid tumors respond to treatment and is based on whether tumors shrink, remain the same or increase in size.2
About RYBREVANT®
RYBREVANT® (amivantamab-vmjw) received accelerated approval by the U.S. FDA in May 2021 for the treatment of adult patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.[iv] Shortly after FDA approval, the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Small Cell Lung Cancer* included amivantamab-vmjw (RYBREVANT®) as a subsequent therapy option with a Category 2A recommendation for patients that have progressed on or after platinum-based chemotherapy with or without immunotherapy and have EGFR exon 20 insertion mutation-positive advanced NSCLC.[v] Janssen has filed regulatory submissions for RYBREVANT® with health authorities in Europe and other markets.
RYBREVANT® is being studied in multiple clinical trials, including the Phase 1 CHRYSALIS (NCT02609776) study to evaluate the safety, pharmacokinetics and preliminary efficacy of RYBREVANT® as a monotherapy and in combination, including with lazertinib, in patients with advanced NSCLC with various EGFR mutations; the Phase 1/1b, CHRYSALIS-2 study (NCT04077463) assessing the combination of RYBREVANT® and lazertinib in patients who have progressed after treatment with osimertinib and chemotherapy; as first-line therapy in untreated advanced EGFR-mutated NSCLC in the Phase 3 MARIPOSA (NCT04487080NCT04487080) study assessing amivantamab in combination with lazertinib; the planned Phase 3 MARIPOSA-2 (NCT04988295) study assessing the efficacy of lazertinib, RYBREVANT® and carboplatin-pemetrexed versus carboplatin-pemetrexed in patients with locally advanced or metastatic EGFR exon 19 deletion or exon 21 L858R substitution NSCLC after osimertinib failure; the Phase 3 PAPILLON (NCT04538664) study assessing RYBREVANT® in combination with carboplatin-pemetrexed versus chemotherapy alone in patients with advanced or metastatic EGFR-mutated NSCLC and exon 20 insertion mutations; and the Phase 1 PALOMA (NCT04606381) study assessing the feasibility of subcutaneous (SC) administration of RYBREVANT® based on safety and pharmacokinetics and to determine a dose, dose regimen and formulation for RYBREVANT® SC delivery.[vi],[vii],[viii],[ix],[x],[xi]
*Referenced with permission from the NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Non-Small Cell Lung Cancer V.5.2021. © National Comprehensive Cancer Network, Inc. 2021. All rights reserved. Accessed June 15, 2021. To view the most recent and complete version of the guidelines, visit NCCN.org.
NCCN makes no warranties of any kind whatsoever regarding their content, use or application and disclaims any responsibility for their application or use in any way.
About Lazertinib
Lazertinib is an oral, third-generation, brain-penetrant, EGFR TKI that targets both the T790M mutation and activating EGFR mutations while sparing wild type-EGFR.[xii] Interim safety and efficacy results from the lazertinib Phase 1-2 study were published in The Lancet Oncology in 2019. In 2018, Janssen Biotech, Inc. entered into a license and collaboration agreement with Yuhan Corporation for the development of lazertinib.
Please read full Prescribing Information for RYBREVANT®.
Learn more at www.janssen.com.
Sep. 20, 2021 3:26 AM ET Johnson & Johnson (JNJ) By: Mamta Mayani, SA News Editor
09/19/2021
- Tisotumab Vedotin in Combination with Carboplatin Showed Encouraging, Durable Anti-Tumor Activity as First-Line Treatment -
- Tisotumab Vedotin in Combination with Pembrolizumab Showed Encouraging, Durable Anti-Tumor Activity in Previously Treated Patients -
BOTHELL, Wash. & COPENHAGEN, Denmark--(BUSINESS WIRE)-- Seagen Inc. (Nasdaq: SGEN) and Genmab A/S (Nasdaq: GMAB) today presented interim data from two cohorts of the phase 1b/2 innovaTV 205 multi-cohort, open-label trial of tisotumab vedotin in recurrent or metastatic cervical cancer at the European Society for Medical Oncology (ESMO) Virtual Congress 2021 as part of a featured mini oral presentation. Initial results from these two dose expansion cohorts of the study showed encouraging and durable anti-tumor activity with tisotumab vedotin in combination with carboplatin (Cohort D) as first-line therapy for patients with advanced cervical cancer who had not received prior systemic therapy, with a 55% objective response rate (ORR) and with tisotumab vedotin in combination with pembrolizumab (Cohort F) for patients with advanced cervical cancer who experienced disease progression after 1-2 lines of prior systemic therapy, with a 38% ORR. Both combinations demonstrated a manageable and acceptable safety profile, with no new safety signals identified.
“For patients diagnosed with recurrent or metastatic cervical cancer, there is a need for additional treatment options in the first-, second- and third-line settings,” said Ignace B. Vergote, M.D., Ph.D., co-founder of European Network of Gynaecological Oncological Trial groups (ENGOT), and lead investigator on the innovaTV 205/ENGOT-cx8/GOG-3024 clinical trial. “Interim results from the innovaTV 205 study show the potential for tisotumab vedotin to treat these patients with encouraging response rates in combination with carboplatin and also in combination with pembrolizumab.”
“As we advance our clinical development program for tisotumab vedotin into earlier lines of therapy in cervical cancer, we’re encouraged by these interim results of the combination cohorts with tisotumab vedotin,” said Roger Dansey, M.D., Chief Medical Officer, Seagen. “Based on these results from the innovaTV 205 study, we also plan to evaluate tisotumab vedotin further in various combinations in first-line metastatic or recurrent cervical cancer.”
“We are pleased to share the initial results from the innovaTV 205 study, as these data build upon our understanding of the potential for tisotumab vedotin as a combination therapy in first- and second-line treatment of recurrent or metastatic cervical cancer,” said Jan van de Winkel, Ph.D., Chief Executive Officer, Genmab. “We recognize the need for new therapies for patients with cervical cancer globally and are committed to advancing the tisotumab vedotin development program.”
Tisotumab Vedotin (TV) + Carboplatin (Carbo) in First-line (1L) or + Pembrolizumab (Pembro) in Previously Treated (2L/3L) Recurrent or Metastatic Cervical Cancer (r/mCC): Interim Results of ENGOT-cx8/GOG-3024/innovaTV 205 Study (Presentation #723MO, mini oral presentation on Sunday, September 19)
1L TV + Carbo Dose Expansion Cohort Interim Results
Within this cohort, recurrent or metastatic cervical cancer patients who had not received any prior systemic therapy were given the recommended phase 2 dose of tisotumab vedotin 2.0 mg/kg plus carboplatin AUC 5 Q3W.
Efficacy:
For more information about the innovaTV 205 clinical trial and the study collaborators, visit here, and to learn more about other clinical trials with tisotumab vedotin, visit clinicaltrials.gov.
About Tisotumab Vedotin
Tisotumab vedotin is an antibody-drug conjugate (ADC) composed of Genmab’s fully human monoclonal antibody specific for tissue factor and Seagen’s ADC technology that utilizes a protease-cleavable linker that covalently attaches the microtubule-disrupting agent monomethyl auristatin E (MMAE) to the antibody and releases it upon internalization, inducing programmed cell death. In cancer biology, tissue factor is a cell-surface protein and is associated with tumor growth, angiogenesis, metastasis and poor prognosis.11 Tissue factor was selected as a target for an ADC approach based on its increased levels of expression on multiple solid tumors and its rapid internalization.
Tisotumab vedotin is being evaluated in a global phase 3, randomized clinical trial called innovaTV 301 versus investigator’s choice of chemotherapy in recurrent or metastatic cervical cancer. The primary endpoint is overall survival, and secondary endpoints include progression-free survival, duration of response, objective response rate, safety and tolerability. Enrollment is ongoing and the study is intended to support global registrations. More information about the innovaTV 301 clinical trial, including enrolling sites, is available here. In addition, tisotumab vedotin is being evaluated in ongoing clinical trials as monotherapy in recurrent or metastatic cervical cancer, ovarian cancer and other solid tumors and in combination with commonly used therapies in recurrent or metastatic cervical cancer.
Additional clinical studies for tisotumab vedotin include phase 2 studies in second-/third-line recurrent or metastatic cervical cancer as monotherapy (innovaTV 204), phase 2 study in first-/second-line recurrent or metastatic cervical cancer as monotherapy or in combination with other agents (innovaTV 205) and additional studies in various solid tumors.
For more information on our marketed products and robust pipeline, visit www.seagen.com
For more information, please visit Genmab.com.
About the Seagen and Genmab Collaboration
Tisotumab vedotin is being co-developed by Seagen and Genmab, under an agreement in which the companies share costs and profits for the product on a 50:50 basis.
View source version on businesswire.com: https://www.businesswire.com/news/home/20210919005022/en/
Source: Seagen Inc.
Sep. 20, 2021 4:38 AM ET Seagen Inc. (SGEN), GMAB By: Mamta Mayani, SA News Editor
Sep. 17, 2021 4:00 PM ETIncyte Corporation (INCY)
MONTREAL, Sept. 17, 2021 /CNW/ - Incyte (INCY) today announced that Health Canada has granted a Notice of Compliance with conditions for Pemazyre® (pemigatinib), a selective fibroblast growth factor receptor (FGFR) inhibitor, for the treatment of adults with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with a fibroblast growth factor receptor 2 (FGFR2) fusion or other rearrangement.
"For appropriate patients with cholangiocarcinoma, the approval of Pemazyre in Canada offers patients hope with a new potential treatment option after their cancer has returned, following the failure of first-line treatment," said Durhane Wong-Rieger, Ph.D., President and CEO, Canadian Organization for Rare Disorders.
The conditional approval is based on data from the FIGHT-202 study evaluating pemigatinib as a treatment for patients with previously treated, locally advanced or metastatic cholangiocarcinoma. Study results demonstrated that in patients harbouring FGFR2 fusions or rearrangements (Cohort A), pemigatinib monotherapy resulted in an overall response rate (ORR) of 35.5 percent (primary endpoint) and median duration of response (DOR) of 9.1 months (secondary endpoint) with a median follow-up of 15.4 months. Adverse events observed included serous retinal detachment (SRD) and hyperphosphatemia. The most common adverse reactions (incidence ≥15%) are hyperphosphatemia, alopecia, diarrhea, fatigue, nail toxicity, dysgeusia, nausea, constipation, stomatitis, dry mouth, decreased appetite, vomiting, dry eye, arthralgia, abdominal pain, hypophosphatemia, dry skin, edema peripheral, weight decreased, headache, urinary tract infection, dehydration, hypercalcemia, and palmar-plantar erythrodysaesthesia syndrome.
"We are pleased that Health Canada has granted conditional marketing authorization for Pemazyre and Incyte welcomes the opportunity to serve the cholangiocarcinoma community with this much-needed treatment option," said Josée Brisebois, Ph.D.., Head of Medical Affairs, Incyte Biosciences Canada. "Incyte will strive to secure rapid access across Canada to this innovative targeted therapy to patients suffering from this difficult disease."
About FIGHT-202
The FIGHT-202 is a multi-center, open-label, single-arm, Phase 2 study (NCT02924376) that evaluated the safety and efficacy of pemigatinib – a selective fibroblast growth factor receptor (FGFR) inhibitor – in adult (age ≥18 years) patients with previously treated, locally advanced or metastatic cholangiocarcinoma with documented FGFR2 fusion or rearrangement.
Patients were enrolled into one of three cohorts – Cohort A (FGFR2 fusions or rearrangements), Cohort B (other FGF/FGFR genomic alterations) or Cohort C (no FGF/FGFR genomic alterations). All patients received 13.5 mg pemigatinib orally once daily (QD) on a 21-day cycle (two weeks on/one week off) until radiological disease progression or unacceptable toxicity.
The primary endpoint of FIGHT-202 was overall response rate (ORR) in Cohort A, assessed by independent review per RECIST v1.1. Secondary endpoints include ORR in Cohorts B, A plus B, and C; and duration of response (DOR).
For more information about FIGHT-202, visit https://clinicaltrials.gov/ct2/show/NCT02924376.
About FIGHT
The FIGHT (Fibroblast Growth factor receptor in oncology and Hematology Trials) clinical trial program includes ongoing Phase 2 and 3 studies investigating the safety and efficacy of pemigatinib therapy across several FGFR-driven malignancies. Phase 2 monotherapy studies include FIGHT-202, as well as FIGHT-201 investigating pemigatinib in patients with metastatic or surgically unresectable bladder cancer, including with activating FGFR3 mutations or fusions/rearrangements; FIGHT-203 in patients with myeloproliferative neoplasms with activating FGFR1 fusions/rearrangements; FIGHT-207 in patients with previously treated, locally-advanced/metastatic or surgically unresectable solid tumour malignancies harbouring activating FGFR mutations or translocations, irrespective of tumour type. FIGHT-205 is a Phase 2 study investigating pemigatinib plus pembrolizumab combination therapy and pemigatinib monotherapy as first-line treatment for metastatic or unresectable bladder cancer harbouring FGFR3 mutations or rearrangements who are not eligible to receive cisplatin. FIGHT-302 is a Phase 3 study investigating pemigatinib as a first-line treatment for patients with cholangiocarcinoma with FGFR2 fusions or rearrangements.
About Pemazyre® (pemigatinib)
Pemazyre is a kinase inhibitor indicated for the treatment of adults with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with a fibroblast growth factor receptor 2 (FGFR2) fusion or other rearrangement.6
In the United States, Pemazyre is approved for the treatment of adults with previously treated, unresectable locally advanced or metastatic cholangiocarcinoma with an FGFR2 fusion or other rearrangement as detected by an FDA-approved test. This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
In Japan, Pemazyre is approved for the treatment of patients with unresectable biliary tract cancer (BTC) with a fibroblast growth factor receptor 2 (FGFR2) fusion gene, worsening after cancer chemotherapy.
In Europe, Pemazyre is approved for the treatment of adults with locally advanced or metastatic cholangiocarcinoma with a fibroblast growth factor receptor 2 (FGFR2) fusion or rearrangement that have progressed after at least one prior line of systemic therapy.
Pemazyre is a potent, selective, oral inhibitor of FGFR isoforms 1, 2 and 3, which, in preclinical studies, has demonstrated selective pharmacologic activity against cancer cells with FGFR alterations.
Pemazyre is marketed by Incyte in the United States, Europe and Japan. Incyte has granted Innovent Biologics, Inc. rights to develop and commercialize pemigatinib in hematology and oncology in Mainland China, Hong Kong, Macau and Taiwan. Incyte has retained all other rights to develop and commercialize pemigatinib outside of the United States.
Pemazyre is a trademark of Incyte.
For additional information on Incyte, please visit Incyte.com
To learn more about Incyte Biosciences Canada, visit https://incytebiosciences.ca.
SOURCE Incyte Biosciences Canada
https://seekingalpha.com/symbol/INCY
Exelixis Announces U.S. FDA Approval of CABOMETYX® (cabozantinib) for Patients with Previously Treated Radioactive Iodine-Refractory Differentiated Thyroid CancerPDF Version
– FDA approval based on phase 3 COSMIC-311 pivotal trial, which demonstrated significant improvement in progression-free survival with CABOMETYX versus placebo –
– Exelixis is prepared to fully support expanded indication immediately –
– Application approved well ahead of Prescription Drug User Fee Act target action date of December 4, 2021 –
ALAMEDA, Calif.--(BUSINESS WIRE)--Sep. 17, 2021-- Exelixis, Inc. (Nasdaq: EXEL) today announced that the U.S. Food and Drug Administration (FDA) approved CABOMETYX® (cabozantinib) for the treatment of adult and pediatric patients 12 years of age and older with locally advanced or metastatic differentiated thyroid cancer (DTC) that has progressed following prior vascular endothelial growth factor receptor (VEGFR)-targeted therapy and who are radioactive iodine-refractory or ineligible. The FDA granted Breakthrough Therapy designation and Priority Review to CABOMETYX and its approval comes more than two months ahead of the Prescription Drug User Fee Act (PDUFA) target action date of December 4, 2021. DTC is the most common type of thyroid cancer in the U.S., and patients who are resistant to radioactive iodine treatment face a poor prognosis.1,2,3,4
“Before today, patients with radioactive iodine-refractory differentiated thyroid cancer who have progressed following prior VEGFR-targeted therapy were facing aggressive disease and no standard treatment option,” said Marcia S. Brose, M.D., Ph.D., Chief, Cancer Center Operation Sidney Kimmel Cancer Center at Jefferson Torresdale Hospital, Co-Director, Community Based Clinical Trials, Sidney Kimmel Cancer Center at Thomas Jefferson University, and principal investigator of COSMIC-311. “In the COSMIC-311 pivotal phase 3 trial, CABOMETYX extended the time patients live without progression of their cancer. The FDA approval of CABOMETYX is an important advancement for these patients who are badly in need of new treatment options.”
The approval is based on results from COSMIC-311, a phase 3 pivotal trial evaluating CABOMETYX versus placebo in patients with radioactive iodine-refractory DTC who progressed after up to two prior VEGFR-targeted therapies. At a planned interim analysis, CABOMETYX significantly reduced the risk of disease progression or death versus placebo (p<0.0001) in the intent-to-treat population. At a follow-up analysis with a median follow-up of 10.1 months, the median progression-free survival (PFS) as assessed by blinded independent radiology committee was 11.0 months for patients treated with CABOMETYX (n=170) compared with 1.9 months for patients treated with placebo (n=88); hazard ratio (HR): 0.22; 95% confidence interval (CI): 0.15–0.31. These results will be presented at the 2021 European Society of Medical Oncology (ESMO) Congress this month.
“This approval of CABOMETYX builds on our existing legacy of delivering transformational medicines for patients with difficult-to-treat forms of cancer,” said Michael M. Morrissey, Ph.D., President and Chief Executive Officer, Exelixis. “We would like to thank the clinical trial participants, the physicians and their staff who participated in the COSMIC-311 trial and to acknowledge the team at the FDA for their collaboration during the quick review of our application.”
About CABOMETYX® (cabozantinib)
In the U.S., CABOMETYX tablets are approved for the treatment of patients with advanced RCC; for the treatment of patients with hepatocellular carcinoma who have been previously treated with sorafenib; for patients with advanced RCC as a first-line treatment in combination with nivolumab; and for adult and pediatric patients 12 years of age and older with locally advanced or metastatic DTC that has progressed following prior VEGFR-targeted therapy and who are radioactive iodine-refractory or ineligible. CABOMETYX tablets have also received regulatory approvals in the European Union and additional countries and regions worldwide. In 2016, Exelixis granted Ipsen exclusive rights for the commercialization and further clinical development of cabozantinib outside of the U.S. and Japan. In 2017, Exelixis granted exclusive rights to Takeda Pharmaceutical Company Limited for the commercialization and further clinical development of cabozantinib for all future indications in Japan. Exelixis holds the exclusive rights to develop and commercialize cabozantinib in the U.S.
Please see accompanying full Prescribing Information https://www.cabometyx.com/downloads/CABOMETYXUSPI.pdf.
For more information about Exelixis, please visit www.exelixis.com
View source version on businesswire.com: https://www.businesswire.com/news/home/20210917005517/en/
Source: Exelixis, Inc.
Sep. 17, 2021 4:32 PM ETExelixis, Inc. (EXEL)By: Aakash Babu, SA News Editor3 Comments
Exelixis Announces Final Results from Phase 3 COSMIC-311 Pivotal Trial of CABOMETYX® in Patients with Previously Treated Radioactive Iodine-Refractory Differentiated Thyroid Cancer Presented at ESMO 2021PDF Version
– At a median follow-up of 10.1 months, median progression-free survival was 11.0 months for CABOMETYX compared with 1.9 months for placebo –
– Results served as a basis for the recent U.S. FDA approval of CABOMETYX for adult and pediatric patients 12 and older with locally advanced or metastatic DTC that has progressed following prior VEGFR-targeted therapy and who are radioactive iodine-refractory or ineligible –
ALAMEDA, Calif.--(BUSINESS WIRE)--Sep. 20, 2021-- Exelixis, Inc. (Nasdaq: EXEL) today announced final results from the phase 3 COSMIC-311 pivotal trial of CABOMETYX® (cabozantinib) in patients with previously treated radioactive iodine-refractory differentiated thyroid cancer (DTC). Following a previous announcement that the trial met one of the two primary endpoints of significant improvement versus placebo in progression-free survival (PFS) assessed by blinded independent radiology committee (BIRC; p<0.0001), the results of the final analysis are being presented during the Mini Oral Session – NETs and Endocrine Tumours at 5:30 p.m. CEST on Monday, September 20 at the 2021 European Society of Medical Oncology (ESMO) Congress (LBA67). At a median follow-up of 10.1 months, the significant improvement in PFS with CABOMETYX was maintained, with consistent benefit in subgroups based on prior treatment.